

Abstract
Multiple Sclerosis (MS) is an autoimmune disorder where T cells attack neurons in the central nervous system (CNS) leading to demyelination and neurological deficits. A driver of increased MS risk is the soluble form of the interleukin-7 receptor alpha chain gene (sIL7R), produced by alternative splicing of IL7R exon 6. Here, we identified the RNA helicase DDX39B as a potent activator of this exon and consequently a repressor of sIL7R, and found strong genetic association of DDX39B with MS risk. Indeed, we showed that a genetic variant in the 5′ UTR of DDX39B reduces translation of DDX39B mRNAs and increases MS risk. Importantly, this DDX39B variant showed strong genetic and functional epistasis with allelic variants in IL7R exon 6. This study establishes the occurrence of biological epistasis in humans and provides mechanistic insight into the regulation of IL7R exon 6 splicing and its impact on MS risk.
eTOC Blurb
Increased risk for Multiple Sclerosis (MS), an autoimmune disease that damages the central nervous system, is associated with altered alternative splicing of Interleukin 7 receptor (IL7R) mRNAs that leads to elevated soluble IL7R. Here, we identify the RNA helicase DDX39B as a potent regulator of IL7R splicing, a repressor of soluble IL7R formation and a modifier of MS risk. We show that an MS-associated DDX39B 5′ UTR variant reduces DDX39B protein levels and shows strong genetic and functional epistasis with variants in IL7R.
Introduction
MS is characterized by self-reactive T cell mediated damage to neuronal myelin sheaths in the CNS that leads to axonal demyelination, neuronal death and progressive neurological dysfunction. This breach of immunological tolerance is thought to originate from complex interactions between environmental and genetic factors. Addressing the latter, we and others uncovered a role for IL7R in MS susceptibility (Gregory et al., 2007; International Multiple Sclerosis Genetics et al., 2007; Lundmark et al., 2007). Together with the common gamma chain (γc), IL7R forms a functional cell surface receptor for IL7, which is essential for survival, proliferation, maintenance and homeostasis of T cells (Fry and Mackall, 2005; Mazzucchelli and Durum, 2007), and may also be required for optimal TCR-mediated activation of CD4+ T cells (Lawson et al., 2015) thought to drive the initial inflammatory phase of MS (Sospedra and Martin, 2005). Importantly, IL7R expression is precisely and dynamically controlled throughout lymphopoiesis and upon T cell activation (Alves et al., 2008; Mazzucchelli and Durum, 2007; Munitic et al., 2004; Park et al., 2004), and its modulation has profound effects on immunological function as knockout of IL7R in mice and loss-of-function mutations in humans cause lymphopaenia and severe combined immunodeficiency (Maraskovsky et al., 1996; Peschon et al., 1994; Puel et al., 1998; Roifman et al., 2000). Relevant to the establishment of self-tolerance, dynamic regulation of IL7R throughout lymphopoiesis seems critical for selection of self-tolerant T cells (Dooms, 2013).
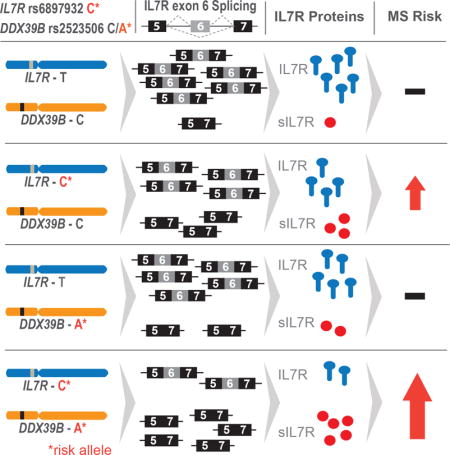
The single nucleotide polymorphism (SNP) rs6897932 within exon 6 of IL7R is strongly and reproducibly associated with MS, where the C allele of the variant is associated with elevated risk (Gregory et al., 2007; International Multiple Sclerosis Genetics et al., 2007; Lundmark et al., 2007). This variant introduces a non-synonymous threonine to isoleucine change at amino acid position 244, but this change does not appear to alter IL7 signaling (unpublished results). Importantly, we showed that the risk allele enhances skipping of exon 6 (Evsyukova et al., 2013; Gregory et al., 2007), increasing the fraction of mRNAs that code for a secreted form of the receptor (sIL7R), and this correlates with increased plasma levels of sIL7R (Hoe et al., 2010; Lundstrom et al., 2013). Elevated levels of sIL7R have been shown to exacerbate the severity of experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, presumably by enhancing the activity or bioavailability of IL7 (Lundstrom et al., 2013). These results link alternative splicing of IL7R to the pathogenesis of MS.
To better understand how IL7R exon 6 is regulated, we pursued the discovery of both cis-acting elements and trans-acting factors controlling splicing of the exon. Previously we identified several important cis-acting elements, among them a critically important exonic splicing enhancer (ESE2), immediately downstream of rs6897932, whose mutation (ΔESE2) inhibited inclusion of exon 6 (Evsyukova et al., 2013). By combining RNA affinity chromatography with mass spectrometry, we discovered factors interacting with exon 6 and flanking sequences (Evsyukova et al., 2013) and here we identified those that required ESE2 to bind. We showed that knockdown of one of these factors, the DEAD Box Polypeptide 39B (DDX39B, also known as UAP56/BAT1), increased IL7R exon 6 skipping in cell lines and in primary CD4+ T cells, and up-regulated sIL7R secretion. This is relevant in vivo since here we established that genetic variants within the DDX39B locus were associated with increased genetic risk of MS. We further demonstrated that the risk allele of one of these variants, rs2523506, reduces DDX39B protein level by diminishing the efficiency of DDX39B mRNA translation. Importantly, the increased risk associated with this variant showed significant epistasis with rs6897932 in IL7R. This study demonstrates the occurrence of genetic and functional epistasis of two MS risk loci in humans and provides a mechanistic explanation for the regulation of IL7R alternative splicing as a driver of MS risk. Furthermore, considering that variants in both IL7R and DDX39B have been associated with other autoimmune diseases (Anderson et al., 2011; Cheong et al., 2001; Degli-Esposti et al., 1992; Nakamura et al., 2012; Paternoster et al., 2012; Quinones-Lombrana et al., 2008; Todd et al., 2007), the results of this study could have a broader impact on shared mechanisms in autoimmunity.
Results
DDX39B is a potent activator of IL7R exon 6
To identify trans-acting factors controlling splicing of IL7R exon 6, we conducted an unbiased proteomic screen consisting of RNA affinity chromatography and mass spectrometry. Using different sets of IL7R exon 6 RNAs and HeLa nuclear extracts we identified a total of 89 candidate trans-factors (Evsyukova et al., 2013). Importantly, we showed that both HeLa and Jurkat T cells recapitulate the SNP rs6897932- and ESE2-dependent changes in IL7R exon 6 splicing (Fig. S1), implying exon 6 is similarly regulated in HeLa and T cells. In the experiments presented here, we used RNAs encompassing the first 40 nucleotides (nt) of exon 6, either wild type or ΔESE2 (both containing the C allele of rs6897932), to identify factors binding in the vicinity of rs6897932 and the critical ESE2. We identified 66 candidate factors with these short RNAs, 12 of which showed dependency on ESE2 (Table S1). The two top candidates whose binding was ESE2-dependent were the microtubule-associated protein 4 (MAP4) and the RNA helicase DDX39B. Functional studies ruled out a role for MAP4 in IL7R exon 6 splicing (data not shown) and thus we focused on DDX39B.
To determine whether DDX39B regulates splicing of IL7R exon 6, we silenced its expression in HeLa cells using two independent siRNAs (Fig. 1A), and determined the impact on exon 6 splicing in transcripts from both an IL7R reporter minigene and the endogenous IL7R gene. Knockdown of DDX39B caused a significant increase in exon 6 skipping in transcripts from both the minigene (Fig. 1B) and endogenous gene (Fig. 1C). This effect cannot be explained by differential effects of DDX39B on stability of IL7R transcript isoforms as both isoforms decayed with similar rates in control and DDX39B-depleted cells (data not shown). Importantly, we were able to rescue exon 6 splicing in endogenous IL7R transcripts by complementing DDX39B-depleted cells with a siRNA-resistant cDNA trans-gene encoding wild type DDX39B (Fig. 1E) but not a helicase-defective mutant (Fig. 1F). In these experiments, DDX39B depletion also led to elevated abundance of overall IL7R transcripts (confirmed by RT-qPCR; data not shown), which may be a secondary effect of DDX39B depletion, as it appeared only at later time points during knockdown. Similar rescue experiments conducted with an ESE2 mutant reporter strongly suggested there is no DDX39B effect in the absence of this ESE (Fig. S2). These results indicated the splicing phenotype observed upon transfection of DDX39B siRNAs was specifically driven by DDX39B protein depletion rather than an off-target effect of the siRNAs, and that DDX39B requires its helicase activity and an intact ESE2 to activate exon 6 inclusion. Most importantly, we showed that DDX39B knockdown elevated the secretion of sIL7R (Fig. 1D).
Figure 1. DDX39B regulates alternative splicing of IL7R exon 6.

A–D, Knockdown of DDX39B in HeLa cells using two independent DDX39B siRNAs (DDX_3 and DDX_4) and a non-silencing control siRNA (NSC). A, Western blot analysis illustrating depletion of DDX39B. B–C, RT-PCR analysis of IL7R exon 6 splicing (+E6 = exon included; −E6 = exon skipped) in transcripts from a reporter minigene (B) or the endogenous gene (C). D, Quantification of sIL7R secretion by ELISA. E–F, Rescue experiments with HeLa cell lines stably expressing siRNA-resistant DDX39B trans-gene, either wild type (E, WT) or helicase mutant (F, D199A), under the control of the tetracycline operator. Top panels illustrate DDX39B western blot analysis, whereas lower panels show RT-PCR analysis of endogenous IL7R transcripts. In all panels the data is shown as mean ± s.d., and statistical significance was assessed using Student’s t-test (*** p ≤ 0.0005; ** p ≤ 0.005; * p ≤ 0.05). See also Figures S2 and S3.
Previous experiments suggested that DDX39B interacts with U2AF65 in the vicinity of the branchpoint sequence (BP) to promote constitutive pre-mRNA splicing (Fleckner et al., 1997; Shen et al., 2008; Shen et al., 2007). Since our data revealed a role for DDX39B in alternative splicing, we wondered whether DDX39B silencing affects other alternative splicing events. A pilot investigation in HeLa cells identified 75 alternative splicing events that were changed upon DDX39B depletion (Table S2), and a significant fraction were exon skipping events, consistent with DDX39B acting as a splicing activator. We also investigated how sensitive inclusion of IL7R exon 6 was to silencing of other RNA helicases. Knockdown of DDX5, a helicase implicated in both transcriptional and post-transcriptional control (Huang et al., 2015), and DDX23, the U5 snRNP-associated human homologue of PRP28 (Teigelkamp et al., 1997), caused small effects in exon 6 skipping, whereas knockdown of DDX17, another RNA helicase with alternative splicing activity (Honig et al., 2002) had no effect (Fig. S3). These results suggest that IL7R exon 6 is particularly sensitive to DDX39B levels and indicate that this RNA helicase impacts multiple alternative splicing events. Collectively, these experiments demonstrated that DDX39B is an important activator of IL7R exon 6 splicing, and a potent repressor of sIL7R.
DDX39B is genetically associated with MS risk
To test whether genes encoding regulators of IL7R exon 6 are associated with MS susceptibility, we performed parallel genetic association analyses of autosomal genes encoding candidate trans-acting factors identified in our proteomics screen. When candidate factors form part of macromolecular complexes, we added the other components of such complexes for a total of 116 candidate genes. We combined data from six genetic cohorts of non-overlapping subjects of European descent from previously published meta-analyses (Patsopoulos et al., 2011), which included 4,088 MS cases and 7,444 controls. Genotype data were available for 4,882 SNPs in 96 of the 116 candidate genes (minor allele frequency (MAF) ≥ 1%; imputation information score ≥ 80%). Variants within +/− 10 kilobases (kb) of candidate genes were analyzed using meta-analytic logistic regression models adjusted for population stratification and cohort origin. A total of 58 SNPs reached genome-wide statistical significance (p ≤ 5.0 × 10−8), all of which resided within the DDX39B gene locus (Table S3).
The DDX39B gene is located within the major histocompatibility complex (MHC). This region harbors the human leukocyte antigen (HLA) genes, the primary genetic drivers of MS susceptibility (International Multiple Sclerosis Genetics et al., 2015; Patsopoulos et al., 2013), and exhibits extended linkage disequilibrium (LD) (de Bakker et al., 2006). Accordingly, it was imperative to establish whether DDX39B variants were associated with increased MS risk independent of the known HLA risk factors, rather than reporting on HLA risk variants in LD. To this end, we further refined our genetic model to adjust for all known HLA MS risk variants: HLA-DRB1*15:01, HLA-DRB1*03:01, HLA-DRB1*13:01, HLA-DRB1*04:04, HLA-DRB1*04:01, HLA-DRB1*14:01, HLA-A*02:01, rs9277489, HLA-B*37:01, and HLA-B*38:01 (hereafter referred to as HLA-adjusted model). There were 15 variants in DDX39B with strong association with MS risk after correction for HLA risk alleles (p ≤ 5.0 × 10−8) (Fig. 2; also see Table S3 and Fig. S4). We used this HLA-adjusted model to inform subsequent functional analyses of DDX39B variants displaying strong association with MS risk. While there were four genes within the associated region (Fig. 2), the functional experiments demonstrating robust repression of sIL7R by DDX39B (Fig. 1) strongly suggested that DDX39B drives this association and reduces MS risk by decreasing sIL7R expression.
Figure 2. Variants within or adjacent to the DDX39B locus strongly associated with MS risk.

Each diamond represents a variant analyzed, and the color of the diamonds indicates no association (gray), marginal association (yellow) or strong association (red) with MS risk. The variant rs2523506 is indicated with a larger diamond (see Figure 3). Black and blue dotted lines indicate thresholds for marginal (p ≤ 1.0 × 10−2) and strong (p ≤ 5.0 × 10−8) association, respectively. The location of the four genes present in this region of chromosome 6 is illustrated at the bottom. See also Figure S5 and Table S3.
Allele-specific DDX39B protein expression
We next investigated whether any of the DDX39B MS-associated variants altered its activity. Because none of the associated variants are located in the coding region, we hypothesized that the functional SNP(s) act by regulating DDX39B expression. Three of the associated variants are located within the transcriptional unit of the gene: rs2523506 (exon 1), rs2523512 (intron 3) and rs2516478 (intron 9) (Fig. 3A). We tested whether rs2523512 in intron 3 could impact splicing of exons 3 and 4, and found no effect (data not shown). We next focused on rs2523506 located in the 5′ UTR of DDX39B transcripts where it could alter mRNA levels and/or their translation efficiency. We first asked whether rs2523506 genotype correlated with changes in DDX39B transcript levels in peripheral blood mononuclear cells (PBMCs) isolated from relapsing-remitting MS patients and healthy controls. Quantification of the DDX39B mRNAs showed that they did not (Fig. 3B, left), indicating that MS risk association could not be explained by decreased DDX39B mRNA levels.
Figure 3. The DDX39B 5′ UTR variant rs2523506 displays allele-specific DDX39B protein expression.

A, Schematic representation of the DDX39B gene (black), spliced mRNA isoforms (red) and location of MS-associated variants rs2523506, rs2523512 and rs2516478 (asterisks). B, RT-qPCR quantification of DDX39B mRNA levels in human PBMCs (left) and African (YRI/ESN, middle) and European (IBS, right) LCLs stratified by rs2523506 genotype. Each symbol represents cells from one individual, and red lines indicate median and interquartile range for each group. Samples sizes were: PBMC, CC=32, AC=31, AA=23; YRI/ESN, CC=12, AC=11, AA=2; and IBS, CC=12, AC=12, AA=3. C, Western blot analysis of DDX39B protein abundance in African (YRI/ESN, left) and European (IBS, right) LCLs stratified by rs2523506 genotype. Panels in C were assembled with different portions of the same gel. Statistical significance of all measurements was assessed using Student’s t-test (two-sided; *** p ≤ 0.0005; ** p ≤ 0.005; * p ≤ 0.05). See also Figures S7, S8 and S9.
We next asked whether rs2523506 could influence translational efficiency of DDX39B mRNAs, which would lead to differential abundances of DDX39B protein. To address this, we initially mined a proteomics database where relative protein abundances were quantified for 5,953 genes in lymphoblastoid cell lines (LCLs) (Wu et al., 2013). We found reduced DDX39B protein levels in cell lines heterozygous at rs2523506 (AC) compared to cells homozygous for the protective allele (CC) in LCLs from an African population (Yoruba in Ibadan, Nigeria [YRI]) (Fig. S5A). Unfortunately, no data were available for the homozygous risk allele (AA). This correlation with DDX39B levels was not detected in LCLs from an European population (Utah residents with ancestry from northern and western Europe [CEU]), which also lacked data for the AA genotype (Fig. S5B), suggesting that regulation of DDX39B levels is complex and may be differentially modified by other loci or non-genetic factors between these LCL populations. Nonetheless, this analysis supported the hypothesis that the risk allele of rs2523506 can reduce DDX39B protein expression.
To rigorously test the correlation between DDX39B protein levels and rs2523506 alleles we experimentally analyzed 25 LCLs of African origin (12 CC, 11 AC and 2 AA) and 27 LCLs of European origin (12 CC, 12 AC and 3 AA). We used LCLs from two African populations, YRI and Esan in Nigeria (ESN), which were entirely distinct from the previously analyzed cell lines (Wu et al., 2013), thereby allowing us to test for replication of the correlation between rs2523506 and DDX39B protein levels. Given the lack of correlation in CEU LCLs and concerns associated with these cell lines being more extensively passaged than the other LCLs (Yuan et al., 2015), we analyzed LCLs established more recently from Iberian populations in Spain (IBS). We first checked levels of DDX39B mRNAs and, similar to findings in PBMCs, found no significant differences in DDX39B mRNA levels by rs2523506 genotype in either the African (YRI/ESN) or European (IBS) LCLs (Fig. 3B). Additionally, we assessed whether rs2523506 altered expression of genes in the vicinity of DDX39B (MICB, ATP6V1G2, NFKBIL1, LTA, TNF and LTB) in the African LCLs and found no difference in RNA levels by rs2523506 genotype (Fig. S6A). Furthermore, silencing of vicinal genes that were expressed in HeLa cells had no effect on IL7R exon 6 splicing (Fig. S6B). These analyses indicated rs2523506 does not affect RNA levels of other genes within the associated region, consistent with this variant driving MS risk via its impact on DDX39B protein expression.
We next analyzed DDX39B protein levels by western blot in both populations of LCL (Fig. 3C). Analysis in the African LCLs revealed a dose-dependent correlation of the risk allele of rs2523506 with reduced DDX39B protein levels, with approximately 25% reduction in AC lines and 50% reduction in AA lines compared to CC lines (Fig. 3C, left). We observed a similar correlation in DDX39B protein levels in the European LCLs, albeit to a lesser extent, and statistically significant when comparing CC and AA lines (Fig. 3C, right). Thus, we concluded that the A risk allele of rs2523506 is associated with decreased DDX39B protein levels. Moreover, given that DDX39B mRNA levels were unaltered by rs2523506 genotype, we hypothesized that the rs2523506 risk allele functions by reducing the DDX39B mRNA translation.
rs2523506 controls translation efficiency of DDX39B mRNAs
To investigate whether rs2523506 influences translational efficiency of DDX39B transcripts, we generated luciferase reporters containing the DDX39B 5′ UTR variants. In addition to the alternative alleles of rs2523506 (C/A), the 5′ UTR of DDX39B transcripts is further modified by alternative 3′ splice sites (3′ss) within exon 2 (3′ss-1 and 3′ss-2 in Figure 3A). Therefore, to precisely mimic the naturally occurring DDX39B 5′ UTR variants, we generated reporters reflecting the use of the different 3′ss in exon 2 and the alternative alleles of rs2523506 (C/A) (Fig. 4A). These reporters were transiently transfected into HeLa cells together with a Firefly luciferase (F-Luc) transfection control, and translational efficiency was determined by quantifying mRNA and luciferase levels, each normalized to the F-Luc control. This analysis revealed approximately 20–30% reduction in translational efficiency of the reporters containing the A risk allele compared to the C allele (Fig. 4B). Importantly, this effect of rs2523506 on translational efficiency largely explains the reduction in DDX39B protein levels observed between CC and AA LCLs. Taken together with the results in Figure 3, we concluded that the A risk allele of rs2523506 decreases DDX39B protein levels by reducing the translational efficiency of DDX39B transcripts.
Figure 4. The risk allele of rs2523506 reduces translational efficiency mediated by DDX39B 5′ UTRs.
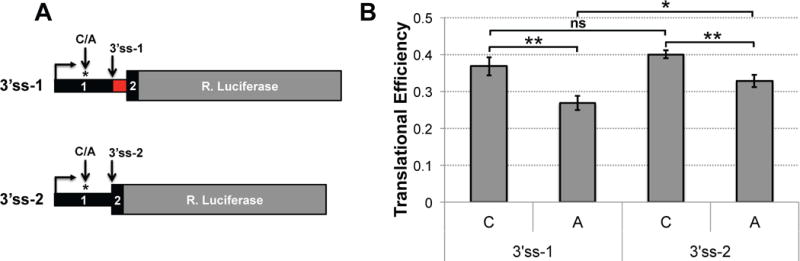
A, Schematic representation of the different DDX39B 5′ UTR luciferase reporters, which differ by alternative 3′ss in exon 2 and the single nucleotide change at rs2523506 (C/A). B, Measurements of translational efficiency in transfected HeLa cells. RNA levels and luciferase activity were measured by RT-qPCR and dual Luciferase assays, respectively. Translational efficiency was determined by dividing luciferase activity by RNA levels. Statistical significance was assessed using Student’s t-test (two-sided; ** p ≤ 0.005; * p ≤ 0.05).
Gene-gene interaction between DDX39B and IL7R
Centered on the findings that the risk allele of rs2523506 reduces DDX39B protein level, and both the risk allele of IL7R rs6897932 and knockdown of DDX39B increased skipping of IL7R exon 6, we hypothesized a functional relationship between rs6897932 in IL7R and rs2523506 in DDX39B. This hypothesis predicted that carriers of risk alleles in both genes (C in IL7R, and A in DDX39B) would exhibit higher risk of MS. We therefore tested for evidence of multiplicative interaction (additive increase on the log odds scale) between rs2523506 and rs6897932 using logistic regression modeling. The interaction model included the main effects for rs2523506 and rs6897932, and was adjusted for population ancestry, cohort origin and all MHC risk variants outside the DDX39B locus (HLA-adjusted model) (see Table S4). There was a significant interaction between rs2523506 and rs6897932 (p = 0.029), which was further investigated in rs6897932-stratified analyses with equivalent model adjustments. This approach revealed strong association between rs2523506 and rs6897932 with MS risk (Table 1, top). We observed no elevated MS risk associated with DDX39B rs2523506 among individuals homozygous for the IL7R rs6897932 protective allele (TT, N=742, odds ratio (OR) = 0.98, p = 0.904), however an effect was observed among IL7R rs6897932 heterozygotes (CT, N=4251, OR = 1.26, p = 7.3 × 10−4), and the magnitude and significance of the effect was strongest among individuals homozygous for the IL7R rs6897932 risk allele (CC, N=6239, OR = 1.39, p = 4.4 × 10−9). When testing for the reciprocal relationship, that is the risk associated with IL7R rs6897932 in DDX39B rs2523506 stratified analyses (Table 1, middle), we observed a subtle association of IL7R in non-carriers of the rs2523506 risk allele (CC, N=7834, OR = 1.10, p = 0.021), and this effect was drastically modified by the presence of the rs2523506 risk allele (AC, N=3099, OR = 1.20, p = 5.1 × 10−3; AA, N=299, OR = 2.19, p = 7.5 × 10−4). We further tested our hypothesis by determining the joint genotypic effect for all combinations of rs2523506 and rs6897932 genotypes (Table 1, bottom). This analysis uncovered a strong epistasis between these variants as we observed increased MS risk only in carriers of at least one copy of the risk alleles at both loci (IL7R CT, DDX39B AC, N=1167, OR = 1.32, p = 0.023), and this effect was strongest in individuals homozygous for the risk allele at both loci (IL7R CC, DDX39B AA, N=156, OR = 2.75, p = 4.5 × 10−7).
Table 1.
Robust Epistasis between Risk Alleles of IL7R rs6897932 and DDX39B rs2523506
| Association of rs2523506 and MS risk in individuals stratified by rs6897932 genotype | |||||||
|---|---|---|---|---|---|---|---|
| IL7R rs6897932 | |||||||
| no strat. | TT | CT | CC | ||||
| p-value | OR (95% CI) |
p-value | OR (95% CI) |
p-value | OR (95% CI) |
p-value | |
|
DDX39B rs2523506 |
0.029 | 0.98 (0.70, 1.37) N=742 |
0.904 | 1.26 (1.10, 1.45) N=4251 |
7.3 × 10−4 | 1.39 (1.25, 1.55) N=6239 |
4.4 × 10−9 |
| Association of rs6897932 and MS risk in individuals stratified by rs2523506 genotype | |||||||
| DDX39B rs2523506 | |||||||
| no strat. | CC | AC | AA | ||||
| p-value |
OR (95% CI) |
p-value |
OR (95% CI) |
p-value |
OR (95% CI) |
p-value | |
|
IL7R rs6897932 |
0.029 | 1.10 (1.02, 1.20) N=7834 |
0.021 | 1.20 (1.06, 1.37) N=3099 |
5.1 × 10−3 | 2.19 (1.39, 3.46) N=299 |
7.5 × 10−4 |
| Joint genotypic effect of rs697932 and rs2523506 on MS risk | |||||||
| IL7R rs6897932 | |||||||
| TT | CT | CC | |||||
|
OR (95% CI) |
p-value |
OR (95% CI) |
p-value |
OR (95% CI) |
p-value | ||
|
DDX39B rs2523506 |
CC | ref N=498 |
ref | 1.02 (0.82, 1.27) N=2963 |
0.888 | 1.15 (0.93, 1.43) N=4373 |
0.193 |
| AC | 1.00 (0.69, 1.45) N=322 |
0.986 | 1.32 (1.04, 1.69) N=1167 |
0.023 | 1.53 (1.21, 1.92) N=1710 |
3.2 × 10−4 | |
| AA | 0.83 (0.29, 2.41) N=22 |
0.738 | 1.36 (0.86, 2.15) N=121 |
0.189 |
2.75 (1.86, 4.08) N=156 |
4.5 × 10−7 | |
Interaction between rs2523506 and rs6897932 and association with MSrisk was investigated using a logistic regression model adjusted for population stratification, cohort origin, and HLA risk variants. The interaction model was further investigated using analyses stratified by rs6897932 (top) and rs2523506 (middle) genotypes. The bottom panel shows joint genotypic effect of rs6897932 and rs2523506 on MS risk. Bolded is the joint effect in homozygous carriers of the risk allele at both loci. See also Tables S4 and S5.
It should be noted that interaction between DDX39B and IL7R was not due to an interaction between IL7R and HLA-DRB1*15:01, the main genetic driver of MS (International Multiple Sclerosis Genetics et al., 2015; Patsopoulos et al., 2013), as we observed no evidence for a multiplicative interaction between rs6897932 and HLA-DRB1*15:01 with MS risk (Table S5). Taken together, we concluded that the risk alleles in DDX39B and IL7R show strong evidence of epistasis, with a robust dose-dependent effect, suggesting that the MS risk associated with low levels of DDX39B can be explained by its action on IL7R.
The epistasis between DDX39B rs2523506 and IL7R rs6897932 suggested the possibility of a functional interplay that could enhance skipping of IL7R exon 6 in carriers of the risk alleles at both loci. Indeed, knockdown of DDX39B revealed higher skipping of exon 6 in transcripts from a reporter carrying the risk allele of rs6897932 (IL7R-C) compared to the protective allele (IL7R-T) (Fig. 5). This result is consistent with exon 6 skipping being augmented by two insults: strengthening of a weak silencer by the risk allele of rs6897932 (Evsyukova et al., 2013; Gregory et al., 2007) and reduced expression of the activator DDX39B. Collectively, our genetic and functional studies indicate that carriers of both IL7R and DDX39B risk alleles are at highest risk of developing MS, very likely due to increased expression of sIL7R.
Figure 5. Functional interaction between DDX39B and IL7R exon 6 variants.

The interaction between DDX39B and IL7R was functionally tested in DDX39B-depleted HeLa cells using IL7R splicing reporters carrying either the risk C allele (IL7R-C; left) or the protective T allele (IL7R-T; right) of rs6897932. RT-PCR analysis of exon 6 splicing (mean ± s.d.) reveals higher exon 6 skipping when the levels of DDX39B are reduced in the context of the risk C allele than of the protective T allele of rs6897932 (Student’s t-test, two-sided; ** p ≤ 0.005).
DDX39B regulates IL7R exon 6 inclusion in primary CD4+ T cells
We then explored the effect of DDX39B knockdown on IL7R exon 6 splicing in CD4+ T cells isolated from healthy human donors who were either CC (Fig. 6A) or CT (Fig. 6B) at rs6897932 in IL7R. We transduced cells from six donors with lentiviruses expressing two independent shRNAs against DDX39B or a non-targeting control shRNA. Even though DDX39B knockdown was less efficient in primary CD4+ T cells than in HeLa cells, we observed similar increase in exon 6 skipping upon DDX39B depletion (Fig. 6). We also measured sIL7R levels by ELISA and found increased levels of sIL7R upon DDX39B knockdown in cells from all donors with one DDX39B shRNA (sh3) but only in cells from two donors with the second shRNA (sh5) (Fig. S10). While the lack of correlation of sIL7R levels with exon 6 skipping in cells transduced with sh5 remains unexplained, we note it could be due to the fact that the antibody used can detect all IL7R protein isoforms. Importantly, we observed higher levels of exon 6 skipping upon DDX39B depletion in CD4+ T cells from donors homozygous for the risk allele in IL7R (CC) than those from heterozygous (CT) (Fig. 6A and 6B), corroborating the results obtained with the IL7R splicing reporters (Fig. 5). These data are consistent with the observed DDX39B—IL7R epistasis above (Table 1). We concluded DDX39B plays an important role in promoting IL7R exon 6 inclusion in primary CD4+ T cells, thereby underscoring the importance of DDX39B in MS pathogenesis.
Figure 6. DDX39B regulates IL7R exon 6 splicing in primary CD4+ T cells.
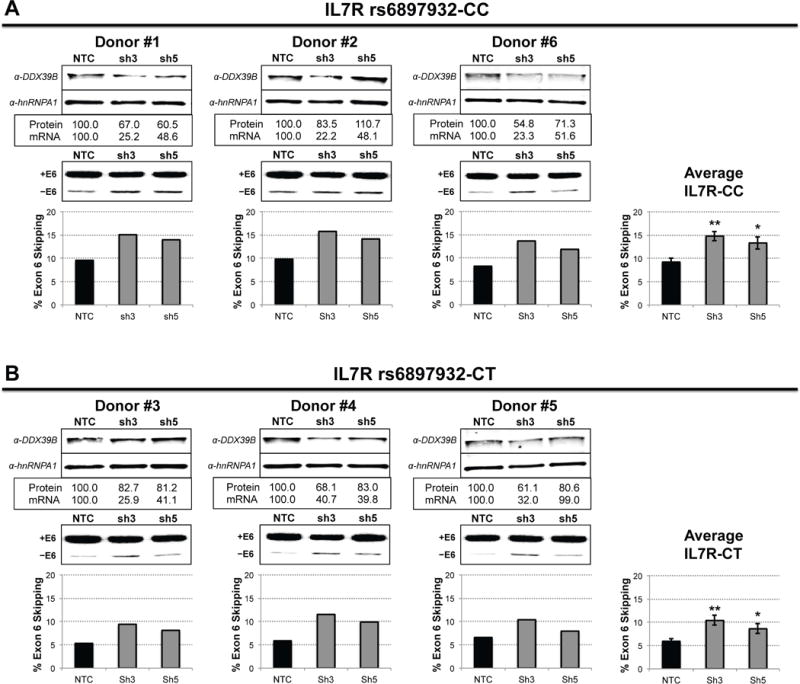
Knockdown of DDX39B in primary CD4+ T cells from six donors via lentiviral transduction with two independent shRNA against DDX39B (sh3 and sh5) and a non-targeting control shRNA (NTC). Donors are grouped by IL7R genotype: IL7R-CC (A) and IL7R-CT (B), and each panel illustrates DDX39B western blot analysis (top) and RT-PCR analysis of IL7R exon 6 splicing (bottom) for each donor individually. In each panel, the plots on the right show the average of % exon 6 skipping for each IL7R genotype (mean ± s.d., Student’s t-test: ** p ≤ 0.005; * p ≤ 0.05).
Discussion
We previously showed that the rs6897932 C allele in exon 6 of IL7R, which is strongly associated with MS susceptibility, increases skipping of exon 6 (Evsyukova et al., 2013; Gregory et al., 2007), and this correlates with elevated levels of circulating sIL7R (Hoe et al., 2010; Lundstrom et al., 2013). This is predicted to have important repercussions on the pathogenesis of MS as sIL7R has been shown to exacerbate the severity of EAE, a mouse model of MS (Lundstrom et al., 2013). Based on these findings, we proposed that trans-acting factors controlling exon 6 splicing would be candidate genes for MS susceptibility. Using a multidisciplinary approach consisting of biochemical, molecular genetic, and human genetic investigations, we validated this proposal and showed that the RNA helicase DDX39B is 1) a potent trans-activator of IL7R exon 6 in cell lines and primary human CD4+ T cells, 2) a repressor of sIL7R, and 3) a novel risk factor for MS. Of the numerous DDX39B variants found to be strongly associated with MS risk, we identified rs2523506 as a putative functional variant that exhibits strong correlation with reduced DDX39B protein expression in LCLs. Indeed, functional studies with luciferase reporters showed that the risk allele of this 5′ UTR variant diminishes the translational efficiency mediated by DDX39B 5′ UTRs, thereby directly linking the SNP to the observed changes in protein levels. Additionally, rs2523506 does not affect expression of nearby genes, including pro-inflammatory cytokines of the tumor necrosis factor superfamily (TNF, LTA and LTB). These findings suggest that increased MS risk associated with DDX39B could largely result from its function on IL7R splicing.
DDX39B is a DEAD-box protein with known functions in constitutive pre-mRNA splicing (Shen et al., 2008; Shen et al., 2007) and nuclear export of mRNAs (Luo et al., 2001; Masuda et al., 2005; Strasser et al., 2002). We have not ruled out the possibility that the increased skipping of exon 6 observed upon DDX39B knockdown results from other functions of DDX39B, in particular mRNA nuclear export. This is unlikely, however, as we have previously shown that the effects of rs6897932 and ESE2 are recapitulated in in vitro splicing assays with HeLa nuclear extracts (Evsyukova et al., 2013), which implies a direct effect on exon 6 splicing rather than nuclear export. Moreover, the role of DDX39B in promoting exon 6 inclusion appears to be independent of its function in constitutive pre-mRNA splicing, wherein DDX39B facilitates recruitment of U2 snRNP to the branchpoint through its interaction with U2AF65. Here, we showed that DDX39B requires an intact ESE2 to promote inclusion of exon 6, as functionally supported by failure to pull-down DDX39B with ΔESE2 exon 6 RNAs, and the inability of DDX39B to affect exon 6 inclusion in the absence of ESE2. Based on these results, we propose that DDX39B binds to ESE2, directly or indirectly, to promote inclusion of IL7R exon 6, and by doing so it decreases sIL7R expression and reduces MS risk.
Using powerful genetic association analyses that interrogated candidate trans-acting factors identified in our proteomic screen, we uncovered DDX39B as the lone candidate trans-factor exhibiting strong association with MS risk independently of HLA risk variants. Interestingly, none of the associated variants in the HLA-adjusted model had an effect before correction for HLA risk factors, suggesting masking by HLA associations. A similar phenomenon was previously reported for rs2516489, a variant located in an intergenic region within 10 kb of DDX39B, which showed association only after adjusting for other HLA risk variants (Patsopoulos et al., 2013). The authors proposed Simpson’s paradox as a likely explanation, wherein HLA and DDX39B variants are associated independently but the effect of DDX39B variants is likely masked by the effects of HLA variants. This masking by HLA variants may explain why associations of DDX39B with autoimmune diseases have not been as prominent as we would expect. Importantly, we observed dramatic allele-specific variation of DDX39B protein levels (up to two-fold) due to rs2523506 genotype among LCLs of African origin, and to a lesser extent among LCLs of European origin, which mainly results from an effect of rs2523506 on translational efficiency. A perplexing result is the stronger correlation of rs2523506 genotype with DDX39B protein levels in African than in European LCLs. While a complete explanation of this finding is not yet at hand, the data indicate that regulation of DDX39B levels is likely complex, and other genetic or epigenetic modifiers could titer DDX39B expression or activity. Nonetheless, it is remarkable that the correlation between DDX39B protein levels and rs2523506 genotype was observed by two distinct experimental methods, namely western blot (this work) and mass spectrometry (Wu et al., 2013), and in two populations of LCLs (YRI/ESN and IBS).
A critical finding in this study is the epistatic interaction between rs2523506 in DDX39B and rs6897932 in IL7R. This interaction was missed in a recent study that investigated potential polygenic interactions in MS between HLA risk alleles and established non-HLA MS risk variants, including IL7R, because non-HLA risk variants within the MHC, such as rs2516489 and rs2523506, were not interrogated (International Multiple Sclerosis Genetics et al., 2015). It has been long thought that functional polymorphisms affecting genes within the same gene network could enhance disease risk in complex genetic disorders but this has been difficult to prove. For instance, several studies have reported suggestive evidence for genetic interactions in several autoimmune diseases including Systemic Lupus Erythematosus (Zhou et al., 2012), Rheumatoid Arthritis (Briggs et al., 2010; Perdigones et al., 2010) and MS (Shahbazi et al., 2011). Nonetheless, all these cases lack functional validation and understanding of the underlying molecular mechanisms. Here, we report strong evidence for epistasis between DDX39B rs2523506 and IL7R rs6897932, which confers higher susceptibility to MS in a dose-dependent manner. More importantly, we functionally demonstrated that the risk alleles at both loci work in concert to increase skipping of IL7R exon 6, thereby enhancing the biogenesis of sIL7R. This study represents a rare example where the molecular underpinnings of a pathogenic epistatic interaction are understood and have been functionally validated.
Lastly, our studies uncovered a protective role for DDX39B in the pathogenesis of MS, wherein decreased DDX39B expression results in up-regulation of sIL7R via skipping of IL7R exon 6. Although our multidisciplinary study centers on MS, other genetic studies have suggested associations of variants within DDX39B with other autoimmune diseases including Rheumatoid Arthritis (Kilding et al., 2004; Okamoto et al., 2003; Quinones-Lombrana et al., 2008), Type 1 Diabetes (Barrett et al., 2009; Cheong et al., 2001; Price et al., 2004; Wong et al., 2003) and Atopic Dermatitis (Paternoster et al., 2012); therefore, it is likely that DDX39B play a similar protective role in these diseases. This is supported by findings of elevated plasma levels of sIL7R in patients of Rheumatoid Arthritis (Badot et al., 2011), Type 1 Diabetes (Monti et al., 2013), and Systemic Lupus Erythematosus (Lauwerys et al., 2014). In addition, DDX39B has also been proposed to negatively regulate production of pro-inflammatory cytokines TNFa, IL1, and IL6 (Allcock et al., 2001). Collectively, these data imply that DDX39B may play a larger role than previously appreciated in the regulation of immune function and in the development of autoimmunity.
STAR Methods Text
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for reagents, resources and/or additional information should be directed to the Lead Contact, Mariano A. Garcia-Blanco (maragarc@utmb.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture conditions
HeLa and Jurkat T cells were obtained from the Duke University Cell Culture Facility. This facility provides authenticated cell lines free of mycoplasma contamination. HeLa Flp-In T-Rex cell line (Kaiser et al., 2008) was kindly provided by Dr. E Dobrikova (Duke University). LCLs were obtained from the Coriell Institute Cell Repositories. HeLa cells were grown in DMEM medium (Life Technologies), whereas Jurkat T cells and LCLs were grown in Advanced 1640 RPMI medium (Life Technologies); both medium were supplemented with 10% heat-inactivated FBS (Gemini Bio-Products) and 1% antibiotics (Pen Strep; Life Technologies).
Primary human CD4+ T cells collection and culture
Informed consent was obtained from healthy volunteers for blood donation. Collection of blood samples was in accordance with the research protocol entitled “Characterizing human immune response signatures that predict clinical outcomes by isolating PBMCs, serum and plasma from normal donors which will be used for optimization, validation and serve as normal controls for immune profiling assays” (Protocol ID: Pro00070584, Kent Weinhold, PI) approved by the Institution Review Board at Duke University. Peripheral blood mononucleated cells (PBMCs) were extracted from whole blood using the Ficoll method and CD4+ T cells were further isolated using CD4+ T cell isolation kit (Miltenyi Biotec). Isolated primary CD4+ T cells were cultured in Advanced 1640 RPMI medium supplemented with 20% heat-inactivated FBS, 1% antibiotics and 100 ng/mL human recombinant IL-2 (Peprotech). 48 hours prior transduction, CD4+ T cells were activated with anti-CD3 (50 ng/mL; eBioscience) and anti-CD28 (100 ng/mL; BD Biosciences) antibodies in the media.
DNA and RNA collection from human PBMC and LCL
Informed consent was obtained from RRMS patients and healthy volunteers enrolled in the MURDOCK-MS study. Blood samples were drawn from these individuals for DNA and RNA isolation. Sample collection was in accordance with the research protocol associated with this study as approved by the Institution Review Board at Duke University. Genomic DNA and total RNA were isolated from whole blood using the QIAamp DNA Blood mini kit (QIAGEN) and PAXgene Blood RNA kit (QIAGEN), respectively. We point out that although these RNA samples were isolated from whole blood, throughout the text we referred to them as isolated from PBMCs, given that the majority of nucleated cells in whole blood are PBMCs. Additional samples from individuals enrolled at Duke University were isolated from PBMCs using Trizol reagent. For LCLs, total RNA was harvested using the ReliaPrep RNA Cell Miniprep System (Promega). All RNA samples were treated with TURBO DNase (Ambion) according to the manufacturer’s protocol.
METHOD DETAILS
Generation of stable cell line expressing DDX39B
HeLa cells stably expressing an inducible DDX39B cDNA trans-gene were generated using the Flp-In T-Rex system (Life Technologies) as recommended by the manufacturer. HeLa Flp-In T-Rex cell line (Kaiser et al., 2008) was kindly provided to our group by Dr. E Dobrikova (Duke University). The coding sequence of DDX39B was amplified with Phusion High-Fidelity DNA polymerase (New England BioLabs) using as template cDNA prepared from total RNA isolated from human PBMCs. Forward and reverse primers contained HindIII and XhoI restriction sites, respectively. The resulting PCR amplicon was cloned into pcDNA5/FRT/TO plasmid using HindIII and XhoI restriction sites and verified by Sanger sequencing. This plasmid was co-transfected with pOG44 plasmid, which encodes the Flp recombinase, into HeLa Flp-In T-Rex cells using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions. Transfected cells were grown in DMEM medium supplemented with 10% FBS free of tetracycline (GE Healthcare Life Sciences) under blasticidin/hygromycinB selection for 15 days, and resistant cells were expanded and used for subsequent experiments. Expression of the trans-gene was induced by addition of tetracycline at 50 μg/ml.
Genotyping of human primary cells and LCLs
DNA samples from human PBMCs and primary CD4+ T cells were genotyped for DDX39B rs2523506 and IL7R rs6897932 using TaqMan genotyping assays (Applied Biosystems) or by sequencing. LCLs were previously genotyped in the HapMap project (International HapMap, 2003, 2005; International HapMap et al., 2007).
Transfection of IL7R splicing reporters
IL7R reporter minigenes, previously described in (Gregory et al., 2007), consisted of the genomic region of IL7R encompassing 614 bp of intron 5, exon 6 and 573 bp of intron 6, cloned in between constitutive upstream (U) and downstream (D) exons in the pI-11 plasmid. Three versions of the IL7R reporter minigene were used containing the alternative alleles of rs6897932 (C or T) or mutation of ESE2 in the context of the risk C allele (ΔESE2). These IL7R splicing reporters (25 ng per well in 24-well format) were transfected into HeLa cells or Jurkat T cells using Lipofectamine 2000 or FuGENE 6 (Roche), respectively, according to the manufacturer’s recommendations. Total RNA was harvested 48 hours after transfection using Trizol Reagent (Life Technologies). Exon 6 skipping was determined for each reporter by RT-PCR as delineated below.
DDX39B RNAi-mediated knockdown and rescue
siRNA duplexes against DDX39B were purchased from Qiagen (DDX_3 = Hs_BAT1_11; DDX_4 = Hs_BAT1_13) and diluted to a final concentration of 10 pM upon arrival. Two independent siRNAs were used to account for potential off-target effects. AllStars Negative Control siRNA (Qiagen) was used as negative control in all knockdown experiments. Transfections were performed in biological triplicates for each siRNA. 24 hours prior to siRNA transfection, 5 × 104 HeLa cells were seeded in 500 pL of DMEM medium per well in 24-well plate format. siRNAs were diluted in Opti-MEM I medium (Life Technologies) and transfected at final concentration of 50 nM on days 1 and 3 post-seeding using Lipofectamine RNAiMax (Life Technologies), following the manufacturer’s recommendations. On day 5 post-seeding, the cells were transfected with 25 ng of the corresponding IL7R reporter minigene using Lipofectamine 2000 (Life Technologies) under the manufacturer’s recommendations. Two versions of the IL7R reporter minigene were used containing the alternative alleles of rs6897932 (IL7R-C and IL7R-T): for the experiments in Figure 1 we used the IL7R-C reporter, whereas for the experiments in Figure 5 we used both IL7R-C and IL7R-T reporters. On day 7, cells were harvested with Trizol Reagent (Life Technologies) or ReliaPrep RNA Cell Miniprep System (Promega) for RNA isolation, or with 1X RIPA buffer for protein extraction. DDX39B depletion was determined by western blotting as delineated below. Percentage IL7R exon 6 skipping was determined by RT-PCR (see section RT-PCR analysis of IL7R splicing). Knockdown experiments were performed three times with similar results.
For the rescue experiment in Figures 1 and S3, HeLa cells stably expressing a DDX39B cDNA trans-gene, either wild type (wt) or a helicase mutant (D199A, previously characterized in (Shen et al., 2008; Shen et al., 2007)), were grown in the absence or the presence of tetracycline to control expression of the trans-gene. Cells were transfected with the corresponding siRNAs on days 1, 3 and 5 post-seeding. For this experiment we used DDX39B siRNA DDX_4 because it targets the 3′ UTR of the endogenous DDX39B transcripts, which is not present in transcripts from the cDNA trans-genes. Cells were harvested on day 9 with the ReliaPrep RNA Cell Miniprep System (Promega) for RNA isolation or with 1X RIPA buffer for western blot analysis. These experiments were performed at least two times with similar results. For the experiment with IL7R-ΔESE2 splicing reporter, this reporter was transfected into cells on day 7 and cells were harvested on day 9 as above.
In both knockdown and rescue experiments the relative level of DDX39B protein was verified by western blotting. Total protein was harvested using 1X RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris-HCl at pH 7.5) freshly supplemented with 1X protease inhibitors (Roche). 10 μg of total protein were loaded per lane on NuPAGE 4%–12% Bis-Tris pre-cast gels (Life Technologies), transferred to nitrocellulose membranes (Whatman), and blotted using standard protocols with anti-DDX39B rabbit polyclonal antibody (Abcam, ab47955) and anti-hnRNPA1 mouse monoclonal antibody (Abcam, ab5832) as loading control.
RNAi-mediated knockdown of other genes products
Knockdown of RNA helicases DDX5, DDX17 and DDX23, and of genes in the vicinity of the DDX39B locus (MICB, NFKBIL1, LTA and TNF) were performed in HeLa cells with at least two independent siRNAs and the non-silencing control siRNA (NSC). Three additional genes in this region (MCCD1, ATP6V1G2 and LTB) are not expressed in HeLa cells and thus were not tested. siRNA transfections were carried out as before but at a final siRNA concentration of 30 nM. Cells were harvested on day 5 post-seeding for RNA isolation using the ReliaPrep RNA Cell Miniprep System. Knockdown levels were quantified by RT-qPCR and normalized to GAPDH as detailed below. IL7R exon 6 skipping was determined by RT-PCR as delineated below. These experiments were performed once in biological triplicates.
Lentiviral packaging
DDX39B shRNA pLKO.1 vector (sh3 = TRCN0000286976; sh5 = TRCN0000294383) and the Mission non-targeting control shRNA pLKO.1 vector (SHC002) were purchased from Sigma-Aldrich. Lentiviral packaging of these shRNA constructs was carried out in 293T cells using Polyethylenimine (PEI) method. In brief, 7.5 × 106 cells were seeded in 15-cm dishes in DMEM media supplemented with 10% FBS and 1% antibiotics 24 hours prior transfection (3 15-cm dishes per construct). Cells were co-transfected with 17 μg of the corresponding shRNA pLKO.1 vectors or GFP control vector (pLCE), 17 μg of packaging plasmid (pCMVR8.74) and 7 μg of VSV-G envelope plasmid (pMD2.G) in serum-free media and 120 μg PEI. DNA-PEI mixes were added drop-wise to the corresponding dishes and 18 hours after the medium was replaced with 20 mL fresh DMEM media. After 72 hours, supernatants were collected, filtered through 0.45 μm filters and concentrated to 6 mL in Amicon Ultra 100K centrifugal filter units (EMD Millipore). Concentrated lentiviral particles were stored at 4°C overnight.
Transduction of primary human CD4+ T cells
Primary CD4+ T cells from 6 different donors were transduced following modifications to the method in (Bilal et al., 2015). In brief, 4.0 × 106 activated primary CD4+ T cells from each donor were transduced with 1 mL of the corresponding concentrated lentivirus in the presence of 8 μg/mL hexadimethrine bromide (Sigma-Aldrich) at a final density of 1.0 × 106 cells/mL in RPMI media supplemented with 20% FBS, 1% antibiotics and 100 ng/mL recombinant human IL-2. Transductions were carried out for 72 hours, followed by antibiotic selection with puromycin (1.5 μg/mL) for 96 hours. Prior to the start of puromycin selection, GFP-transduced cells were assayed for GFP expression by flow cytometry. Cells were transduced at 50–60% efficiency as determined by % GFP positive cells, and were enriched to >90% by puromycin selection. 24 hours prior to collection, cells were resuspended in RPMI media without puromycin and IL-2 for analysis. Supernatants were collected for quantification of sIL7R secretion, and cells were harvested for RNA isolation with the ReliaPrep RNA Cell Miniprep System (Promega) or for protein extraction with 1X RIPA buffer as before. Knockdown of DDX39B was determined by western blotting as before, and by RT-qPCR with normalization to GAPDH. IL7R exon 6 skipping and sIL7R secretion were quantified by RT-PCR and ELISA, respectively, as described below.
RT-PCR analyses of IL7R splicing
Total RNA was harvested using either Trizol Reagent (Life Technologies) or the ReliaPrep RNA Cell Miniprep System (Promega) according to the manufacturer’s instructions. Isolated RNAs were treated with TURBO DNase (Ambion) or DNase I (Promega) to degrade contaminating DNA following the company’s recommendations. 0.2–1.0 μg of DNase-treated total RNA was used as template for reverse transcription using either the ImProm-II Reverse Transcription System (Promega) or the High Capacity cDNA Reverse Transcription System (Applied Biosystems) and random hexamer primers. PCR reactions were prepared as follows: 5 μL of the corresponding RT reaction (diluted 1:5) was mixed with 200 nM of forward and reverse primers (see below), 100 nM dNTPs, 50 mM KCl, 2 mM MgCl2, 0.3 μL of Taq polymerase, and [α-32P]dCTP (3000 Ci/mmol, 10 mCi/mL, PerkinElmer) at a final concentration of 0.1 μCi/μL. PCR primers for minigene constructs (IL7R-C, IL7R-T and IL7R-ΔESE2) were complementary to T7 (forward) and SP6 (reverse) promoter sequences present in the construct, respectively (Gregory et al., 2007). PCR primers for endogenous IL7R were complementary to exon 5 (forward) and exon 7 (reverse). PCR products were electrophoresed on 6% non-denaturing polyacrylamide/TBE gels, dried in a gel drier and exposed to a Molecular Dynamics Phosphorimager screen. Quantifications were performed using ImageQuant software (Molecular Dynamics). Percentage exon 6 skipping was determined as: [(skipped product)/(included + skipped products) × 100]. The data are presented as the mean of triplicate samples and error bars represent standard deviations. Statistical significance in knockdown and rescue experiments was assessed using Student’s t-test (two-sided).
Quantification of sIL7R by ELISA
Supernatants from the DDX39B knockdown experiments in HeLa cells and primary CD4+ T cells described above were collected and concentrated to 100 μl by centrifugation in Amicon Ultra 3K centrifugal filters (Millipore, UFC500324). Secreted sIL7R was quantified from the concentrated supernatants by ELISA as in (Crawley et al., 2010). In brief, 96-wells plate (R&D Systems, DY990) were coated at 4°C overnight with a mouse anti-human IL7R monoclonal antibody (R&D Systems, MAB306). The next day, the plates were blocked with 3% BSA in PBS for one hour (room temp), followed by two-hour incubation (room temp) with the concentrated supernatants, and four washes with PBS-Tween (0.05%). Detection of bound sIL7R was carried out by one-hour incubation (room temp) with biotinylated goat anti-human IL7R polyclonal antibody (R&D Systems, BAF306), followed by 30-minute incubation (room temp) with streptavidin-horseradish peroxidase (Millipore, #18-152), and 20-minute incubation (room temp) with TMB peroxidase substrate (SurModics BioFX, TMBW-1000-01), with four washes with PBS-Tween (0.05%) between manipulations. The reaction was stopped with 1N H2SO4, and the product was visualized in a plate reader at 450 nm. The concentration of samples was extrapolated from standard curves of recombinant human IL7R-Fc chimera (R&D Systems, 306-IR-050). Given that knockdown of DDX39B increases skipping of IL7R exon 6, we expected secretion of sIL7R to increase in the knockdown. For this reason, we used a one-sided Student’s t-test to assess statistical significance, comparing each independent siRNA against the negative control (Fig. 1D). This experiment was performed twice with similar results.
RT-qPCR expression analyses
Transcript expression analyses in PBMCs and LCLs were carried out by RT-qPCR. We used 0.2–0.7 pg of DNase-treated RNA as template for reverse transcription using the ImProm-II Reverse Transcription System (Promega) and oligo(dT) primers. cDNAs were diluted 1:5 and RT-qPCR reactions were carried out using Power SYBR Green PCR Master Mix (Applied Biosystems), with 5 pL of the corresponding cDNA, and 200 nM of the corresponding forward and reverse RT-qPCR primers. The relative abundance of the target genes (DDX39B, MICB, MCCD1, ATP6V1G2, NFKBIL1, LTA, TNF and LTB) was determined with gene-specific assays spanning an exon junction and normalized to GAPDH. The data were stratified by rs2523506 genotypes and normalized to rs2523506 CC genotype. Each point in the box plots represents measurements in PBMCs or LCLs from one individual and is shown as the average of technical triplicates. Statistical significance was established using the Student’s t-test (two-sided).
Overall abundance of IL7R transcripts in rescue experiment and knockdown in primary CD4+ T cells was determined by RT-qPCR as before with primers complementary to the constitutively spliced exons 3 (forward) and 4 (reverse). IL7R levels were normalized to GAPDH, and the data is shown as fold-change over the negative control. Statistical significance was established using the Student’s t-test (two-sided).
Global splicing analysis by RNA-seq
Global assessment of alternative splicing events regulated by DDX39B was carried out by DDX39B knockdown followed by RNA-seq. Knockdown was performed as before with DDX39B siRNAs DDX_3 and DDX_4, and control siRNA (NSC) in two independent experiments. Poly-A+ RNA was enriched from 1 pg of total RNA and used as template to generate libraries using the Illumina TruSeq platform as recommended by the manufacturer. Libraries were sequenced on a 2 × 100 format on an Illumina Hi-Seq 1500. Splicing analysis was carried out using Vast-tools program version 0.2.1 (Irimia et al., 2014) by aligning the paired-end reads to the vast-tools human database (vastdb.hsa.7.3.14) using the default parameters. Two replicates of each condition were compared with the diff function of vast-tools to determine differential splicing. Two parameters were used to establish differential splicing events: E[ΔPsi], which refers to the difference in splicing between the experimental siRNAs (DDX_3 or DDX_4) and control siRNA (NSC) (positive and negative values indicate up-regulation and down-regulation, respectively), and max(x)@P(|ΔPsi|>x)>0.95, which indicates the change in splicing at 95% confidence level. To control for off-target effects, we only considered changes in splicing with max(x)@P(|ΔPsi|>x)>0.95 ≥ 10.0 with at least one DDX39B siRNA but E[ΔPsi] ≥ +/− 20.0 with both DDX39B siRNAs.
DDX39B western blot analyses in LCLs
Cell lysates were harvested from each cell line at two independent times using 1X RIPA buffer as before. Each cell lysate (10 μg of total protein per lane) was blotted twice, and the values presented correspond to the average of the independent measurements. To obtain more accurate measurements of relative protein abundance, DDX39B protein levels were normalized to either hnRNPA1 (Fig. 3C) or PTBP1 (not shown), yielding similar results. DDX39B and hnRNPA1 were blotted as before, whereas PTBP1 was blotted using custom-made anti-PTB rabbit serum (Wagner and Garcia-Blanco, 2002). We used the Student’s t-test (two-sided) to assess statistical significance of the findings.
Luciferase translation efficiency assays
Luciferase reporters were generated by cloning the different DDX39B 5′ UTR variants into pcDNA5 plasmid containing the coding sequence of Renilla luciferase (R-Luc). DDX39B 5′ UTR variants were synthesized as gBlocks (Integrated DNA Technologies) and cloned immediately upstream of the R-Luc ORF using HindIII (5′) and KpnI (3′) restriction sites. All constructs were verified by sequencing. DDX39B 5′ UTR-R-Luc reporters (25 ng/well) were independently co-transfected with a Firefly luciferase control (F-Luc, 10 ng/well) into HeLa cells in 24-well format using Lipofectamine 2000 as indicated by the manufacturer. 24 hours post-transfection cell lysates were collected for luciferase assays with Passive Lysis Buffer and Luciferase assays were immediately performed using the Dual Luciferase Reporter Assay System (Promega). Total RNA was isolated from independent wells and converted into cDNA as above. RNA level for experimental and control reporters was quantified by RT-qPCR as before using primers complementary to the R-Luc and F-Luc ORFs, respectively. Luciferase activity and RNA levels were determined by normalizing R-Luc signals to F-Luc signals (R-Luc/F-Luc). Translational efficiency was then determined by dividing normalized Luciferase activity by normalized RNA levels. Statistical significance was assessed by Student’s t-test (two-sided).
Genetic association and gene-gene interaction analyses
We used genotypic data from six genetic cohorts of non-overlapping case and control subjects of European descent, imputed to >37.4 million SNPs across the genome using BEAGLE and 1000 Genomes Interim Phase I haplotypes using standard procedures (Patsopoulos et al., 2011). We analyzed SNPs within 10 kilobases of autosomal candidate genes with ≥ 80% information and minor allele frequency > 1% (4882 SNPs in 96 of the 116 candidate genes). EIGENSOFT, a principle components algorithm, identified genetic outliers and calculated the top 10 eigenvectors of the genotype data within each cohort; the first 5 eigenvectors sufficiently accounted for population stratification (Patsopoulos et al., 2011). Each variant was analyzed using a meta-analytic logistic regression model as implemented in PLINK (Purcell et al., 2007), assuming alleles have an additive effect on the log-odds scale within each cohort. Each model included the top 5 principal components. We assumed the genetic effects were fixed across all cohorts, as previously performed for these data (Patsopoulos et al., 2013; Patsopoulos et al., 2011). One candidate gene (DDX39B) resides within the MHC, therefore meta-analyses for this locus were also adjusted for known HLA risk variants (HLA-adjusted model, included HLA-DRB1*15:01, HLA-DRB1*03:01, HLA-DRB1*13:01, HLA-DRB1*04:04, HLA-DRB1*04:01, HLA-DRB1*14:01, HLA-A*02:01, rs9277489, HLA-B*37:01, and HLA-B*38:01), and subsequently for an additional non-HLA MHC risk variant, rs2516489 (MHC-adjusted model). These risk variants within MHC region were imputed using BEAGLE (Browning and Browning, 2009; Patsopoulos et al., 2013) by leveraging a collection of 2,767 individuals of the Type 1 Diabetes Genetics Consortium with typed classical HLA alleles as previously described and validated (International et al., 2010; Patsopoulos et al., 2013; Raychaudhuri et al., 2012). We used a logistic regression model in STATA (StataCorp) to investigate a multiplicative interaction between DDX39B rs2523506 and IL7R rs6897932, which was characterized further in IL7R rs6897932 and DDX39B rs2523506 stratified analyses. Similar methodology was employed to study interaction between HLA-DRB1*15:01 and IL7R rs6897932. The joint genotypic effect of rs2523506 and rs6897932 on MS risk was determined by comparing each genotypic combination to the reference (non-carriers of risk alleles at both loci). All models were adjusted for population stratification, cohort origin and HLA risk variants.
QUANTIFICATION AND STATISTICAL ANALYSIS
Molecular analyses
Statistical significance of all molecular analyses in this study was assessed by Student’s t-test (two-sided; *** p ≤ 0.0005; ** p ≤ 0.005; * p ≤ 0.05). Sample sizes for expression analyses in PBMCs and LCLs are indicated in the text and in the legend of the corresponding figures. Data on expression analyses in PBMCs and LCLs is shown as median with interquartile range, whereas data on knockdown and rescue experiments is presented as mean ± s.d. These measurements are indicated in the legend of the corresponding figures.
Genetic Association Analyses and gene-gene interaction
Logistic regression models were used to examine the relationship between genetic variants and multiple sclerosis disease status (binary trait); we report odds ratios and 95% confidence intervals in the text. Genome-wide statistical significance was imposed for these meta-analytic models (two-sided; p ≤ 5 × 10−8). Models for variants residing within the MHC: having met the significance threshold, were subsequently adjusted for all other known MS MHC risk variants; statistical significance of p ≤ 0.05 was imposed for these fully parameterized models. Similar significance threshold of p ≤ 0.05 was imposed for the interactions between IL7R rs6897932 and DDX39B rs2523506, and HLA-DRB1*15:01 and IL7R rs6897932. Sample sizes for these analyses are reported in the text.
DATA AND SOFTWARE AVAILABILTY
The RNA-seq dataset has been deposited in GEO under accession number GSE94730.
ADDITIONAL RESOURCES
None.
Supplementary Material
Figure S1. Regulation of IL7R exon 6 splicing in HeLa cells and Jurkat T cells (Related to Figure 1 and Table S1). IL7R reporters carrying the alternative alleles of rs6897932 (C or T) or mutation of the critical exonic splicing enhancer 2 (ΔESE2) were transiently transfected into HeLa (left) or Jurkat (right) cells and exon 6 splicing was determined by RT-PCR as in Figure 1. The data is presented as mean ± s.d. Statistical significance was assessed using Student’s t-test (two-sided; *** p ≤ 0.0005; ** p ≤ 0.005; * p ≤ 0.05).
Figure S2. DDX39B requires intact ESE2 to regulate IL7R exon 6 splicing (Related to Figure 1). Ectopic expression of siRNA-resistant DDX39B trans-gene in DDX39B-depleted cells cannot rescue splicing of IL7R in transcripts from a reporter mutant of ESE2 (IL7R-ΔESE2). The experiment was performed as in Figure 1 followed by transfection of IL7R-ΔESE2 reporter. A, Western blot analysis of DDX39B. B, RT-PCR analysis of exon 6 skipping in transcripts from the IL7R-ΔESE2 reporter (+E6 = exon 6 included; −E6 = exon 6 skipped). The data is presented as mean ± s.d. and statistical significance was determined using Student’s t-test (two-sided; *** p ≤ 0.0005; ** p ≤ 0.005; * p ≤ 0.05).
Figure S3. Knockdown of additional RNA helicases has a small or no effect on IL7R exon 6 splicing (Related to Figure 1). Knockdown of RNA helicases DDX5, DDX23 and DDX17 was carried out in HeLa cells using three independent siRNAs against each target and a control siRNA (NSC). A, Quantification of knockdown by RT-qPCR (normalized to GAPDH). B, RT-PCR analysis of IL7R exon 6 splicing in endogenous transcripts as in Figure 1C (+E6 = exon 6 included; −E6 = exon 6 skipped). The data is shown as mean ± s.d., and statistical significance was assessed using Student’s t-test (two-sided; *** p ≤ 0.0005; ** p ≤ 0.005; * p ≤ 0.05).
Figure S4. Regional plot showing MS-associated variants within the DDX39B locus adjusted for all known MHC risk variants (Related to Figure 2 and Table S3). Variants were adjusted for population stratification, cohort of origin, the 10 known HLA risk variants (HLA-adjusted model) and the non-HLA risk variant rs2516489 (MHC-adjusted model). Each diamond represents a variant analyzed: yellow diamonds passed the threshold for statistical significance (p ≤ 0.05; black dotted line), whereas gray diamonds did not. The location of the four genes present in this region of chromosome 6 is shown at the bottom. Notably, rs2523506 and several other DDX39B variants showed a modest but significant association independent of rs2516489 and all other HLA risk variants. In healthy controls from each cohort, rs2516489 was in strong LD (r2 > 0.9) with several of the DDX39B associated variants, which explains the attenuated association after correction for rs2516489.
Figure S5. Relative abundance of DDX39B protein in lymphoblastoid cell lines (LCLs) (Related to Figure 3). We mined DDX39B protein levels in a large proteomic database where the relative protein abundance for 5,953 genes was determined by mass spectrometry in 95 LCLs derived from mainly European (CEU) and African (YRI) individuals (Wu et al., 2013). We extracted the relative DDX39B protein levels for YRI (A) and CEU (B) LCLs and stratified those by the genotypes of rs2523506 [data was only available for LCLs homozygous for the protective allele (CC) and heterozygous (AC)]. Sample sizes were as follow: YRI (CC n=24; AC n=7) and CEU (CC n=34; AC n=16). Each symbol represents an individual LCL, whereas the red lines indicate median and interquartile range. Statistical significance was determined using Student’s t-test (two-sided; * p ≤0.05).
Figure S6. SNP rs2523506 does not affect expression of genes in the vicinity of DDX39B and knockdown of these genes does not affect IL7R exon 6 splicing (Related to Fig. 1 and Fig. 3). A, RT-qPCR quantification of genes in the vicinity of DDX39B (MICB, DDX39B, ATP6V1G2, NFKBIL1, LTA, TNF and LTB) in YRI LCLs stratified by rs2523506 genotype (sample sizes: CC=12, AC=11, AA=2). Expression of an additional gene (MCCD1) was not detected. Each symbol represents the average of triplicate measurements from one individual, and red lines indicate median and interquartile range for each group. B–C, Knockdown of DDX39B vicinal genes that were expressed in Hela cells (MICB, NFKBIL1, LTA and TNF). The three other genes (MCCD1, ATP6V1G2 and LTB) are not expressed in HeLa cells and thus were not analyzed. B, RT-qPCR quantification of mRNA knockdown (normalized to GAPDH). C, RT-PCR analysis of IL7R exon 6 splicing as in Figure 1C (+E6 = exon 6 included; −E6 = exon 6 skipped). Plots in B and C are shown as mean ± s.d. Statistical significance was assessed using Student’s t-test (two-sided: *** p ≤ 0.0005; ** p ≤ 0.005; * p ≤ 0.05).
Figure S7. Quantification of sIL7R in primary CD4+ T cells (Related to Figure 6). A, Quantification of sIL7R secretion by ELISA in control (NTC) and DDX39B-depleted (sh3 and sh5) primary CD4+ T cells from Figure 6. The results are shown grouped by IL7R genotypes: IL7R-CC (top) and IL7R-CT (bottom). B, Correlation of sIL7R levels with relative abundance of IL7R transcripts that skip exon 6 (IL7R −E6) for each shRNA. The relative abundance of IL7R −E6 transcripts was extrapolated from % exon 6 skipping and RT-qPCR quantification of overall abundance of IL7R transcripts for each condition.
Table S1. Trans-acting factors exhibiting dependence on ESE2 for binding to IL7R exon 6 (Related to Figure 1).
Trans-acting factors were pulled-down from HeLa nuclear extracts by Tobramycin RNA affinity chromatography using an RNA spanning the first 40 nt of IL7R exon 6, either wild type or mutant ESE2 (ΔESE2), and a no RNA control (Evsyukova et al., 2013). RNA-Protein complexes were eluted with excess tobramycin and the bound proteins were identified by mass spectrometry. The table indicates the total number of unique peptides corresponding to each factor that were pulled-down with no RNA control, wild type or ESE2 RNAs in two independent experiments, and the fold-change decrease in binding observed with mutation of ESE2.
Table S2. Alternative splicing events regulated by DDX39B in HeLa cells (Related to Figure 1).
Alternative splicing analysis was carried out using the Vast-tools program. The table illustrates alternative splicing events that were up-regulated or down-regulated upon DDX39B depletion. “Gene” indicates genes whose splicing was altered; “Event” indicates the type of alternative splicing event (INT = Intron, EX = Exon, ALTD = alternative donor site or 5′ splice site, ALTA = alternative acceptor site or 3′ splice site); “NSC”, “DDX_3” and “DDX_4” indicate % inclusion in cells transfected with non-silencing control, DDX_3 or DDX_4 siRNAs, respectively; “E[ΔPsi]” indicates the difference in splicing between DDX_3 or DDX_4 and NSC; “max(x)@P(|ΔPsi|>x)>0.95” indicates splicing change upon knockdown at 95% confidence level. The table only shows splicing changes with “max(x)@P(|ΔPsi|>x)>0.95” ≥ 10.0 with at least one DDX39B siRNA and “E[ΔPsi]” ≥ +/− 20.0 with both DDX39B siRNAs.
Table S3. All variants in the DDX39B locus analyzed in this study (Related to Figure 2 and Figure S5).
Variants within +/− 10 kb of the DDX39B gene (chromosome 6) were analyzed using a meta-analytic logistic regression model adjusted for population stratification and cohort origin (PCA-adjusted model). Given that DDX39B is located in the MHC, the adjustment model was sequentially refined to establish independence from the known HLA risk variants (HLA-adjusted model), and an additional non-HLA risk variant (rs2516489) within the MHC (MHC-adjusted model). Bolded variants passed the threshold for statistical significance in the corresponding model (p ≤ 5.0 × 10−8 in PCA- and HLA-adjusted models; p ≤ 0.05 in MHC-adjusted model). The asterisk indicates the only variant in the vicinity of the DDX39B gene (rs2516489) previously associated with MS (Patsopoulos et al., 2013). Abbreviations: bp, base pair in the genome; SNP, variants imputed; Alleles, major and minor alleles, respectively (the alleles shown correspond to the sense strand but both DDX39B and ATP6V1G2 genes are in the antisense strand); location, indicates the gene or intergenic region where the variant resides; and OR, odds ratio. The dash lines in the last two columns indicate variants that were in complete LD with rs2516489 in the cohorts and thus no data was available after correcting for this variant.
Table S4: Multivariable logistic meta-analysis identifies a significant multiplicative interaction between IL7R rs6897932 and DDX39B rs2523506 (Related to Table 1).
Multivariable logistic regression meta-analyses demonstrate evidence for a significant multiplicative interaction between IL7R rs6897932 and DDX39B rs2523506, adjusted for population substructure, known HLA risk variants, and cohort of origin. The table illustrates the output for each adjusted variable with comparison between the two models: the HLA adjusted model without (left) and with (right) the interaction term between IL7R rs6897932 and DDX39B rs2523506 (bolded parameters) [Beta = beta coefficient; Se = standard error; OR = odds ratio]. Both models are stable and model parameters are consistent, with the exception of the significant statistical interaction.
Table S5: Lack of evidence for a multiplicative interaction between risk alleles of IL7R rs6897932 and HLA-DRB1*15:01 (Related to Table 1).
We tested for evidence of interaction between HLA-DRB1*15:01 and rs6897932 using a logistic regression model adjusted for population stratification, cohort origin and HLA risk variants, including the DDX39B rs2523506 variant (Table 1); results were not significant (no strat.). Stratification of individuals by rs6897932 genotypes (IL7R — TT (N=742), CT (N=4251) and CC (N=6239)) for analysis of DRB1*15:01 revealed strong association with MS across all three strata (note the overlap in 95% confidence intervals). The results indicate the interaction between DDX39B and IL7R is not due to an interaction between IL7R and HLA-DRB1*15:01, and thus, it is likely that this interaction between DDX39B and IL7R could explain part of the heretofore unaccounted heritable risk of MS.
Table S6: Oligonucleotide primers used in this study (Related to STAR Methods Section)
Table S7: siRNAs used in this study (Related to STAR Methods Section).
Acknowledgments
We thank Drs. M. Skeen (Duke), L. K. Newby (Duke MURDOCK study) for providing DNA and RNA samples from patients and healthy individuals, and S. Arvai (Duke) for technical assistance with these samples, the IMSGC for sharing imputed whole genome genotype data, Dr. T. G. Wood and the UTMB NGS Facility for RNA-seq data, and Drs. B. R. Cullen and S. M. Horner (Duke) for reagents, advice and generosity related to experiments with primary CD4+ T cells. Finally, we express our appreciation for individuals participating in the MURDOCK-MS study. This work was supported by NIH grants R01-NS060925 (S.S.G. and M.A.G.B.) and R01-ES017080 (L.F.B.), National MS Society Pilot Award and Duke University Whitehead Scholarship (D.C.K. and L.W.), Ruth and A. Morris Williams Faculty Research Prize funds from Duke University School of Medicine (G.D.T.), start-up funds from UTMB (M.A.G.B.), funds from Mr. H. Stone and family for MS research (S.G.G.), and NIH F32-NS087899 postdoctoral fellowship (G.G.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions:
Author contributions were as follow: Conceptualization, G.G.M., S.S.B., F.B.S.B., L.F.B., S.G.G. and M.A.G.B.; Methodology, G.G.M., F.B.S.B., L.F.B., G.D.T., D.C.K., S.S.B., S.G.G. and M.A.G.B.; Investigation, G.G.M., F.B.S.B., I.E., G.S.L., E.M.K., S.G.W., T.N., L.W. and L.B.; Writing (Original Draft), G.G.M. and M.A.G.B.; Writing (Review/Editing), F.B.S.B., D.C.K., S.S.B. and S.G.G.; Funding Acquisition, L.F.B., D.C.K., G.D.T., S.G.G. and M.A.G.B; Resources, E.M.K., S.G.W., G.D.T., D.C.K. and S.G.G.; Supervision, S.S.B., L.F.B., S.G.G. and M.A.G.B.
M.A.G.B. and S.G.G. have filed invention disclosures with Duke University for matters related to the work described and consultant agreements with Pfizer, Inc. related to the development of animal models indirectly related to the work described. They attest that in no way did these interests influence the planning, execution or interpretation of the results, or the writing of this manuscript. The other authors have no competing interests, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
References
- Allcock RJ, Williams JH, Price P. The central MHC gene, BAT1, may encode a protein that down-regulates cytokine production. Genes Cells. 2001;6:487–494. doi: 10.1046/j.1365-2443.2001.00435.x. [DOI] [PubMed] [Google Scholar]
- Alves NL, van Leeuwen EM, Derks IA, van Lier RA. Differential regulation of human IL-7 receptor alpha expression by IL-7 and TCR signaling. Journal of immunology. 2008;180:5201–5210. doi: 10.4049/jimmunol.180.8.5201. [DOI] [PubMed] [Google Scholar]
- Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nature genetics. 2011;43:246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badot V, Durez P, Van den Eynde BJ, Nzeusseu-Toukap A, Houssiau FA, Lauwerys BR. Rheumatoid arthritis synovial fibroblasts produce a soluble form of the interleukin-7 receptor in response to pro-inflammatory cytokines. J Cell Mol Med. 2011;15:2335–2342. doi: 10.1111/j.1582-4934.2010.01228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nature genetics. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilal MY, Vacaflores A, Houtman JC. Optimization of methods for the genetic modification of human T cells. Immunol Cell Biol. 2015;93:896–908. doi: 10.1038/icb.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs FB, Ramsay PP, Madden E, Norris JM, Holers VM, Mikuls TR, Sokka T, Seldin MF, Gregersen PK, Criswell LA, et al. Supervised machine learning and logistic regression identifies novel epistatic risk factors with PTPN22 for rheumatoid arthritis. Genes Immun. 2010;11:199–208. doi: 10.1038/gene.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. American journal of human genetics. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong KY, Allcock RJ, Eerligh P, Witt CS, Christiansen FT, McCann V, Price P. Localization of central MHC genes influencing type I diabetes. Human immunology. 2001;62:1363–1370. doi: 10.1016/s0198-8859(01)00351-2. [DOI] [PubMed] [Google Scholar]
- Crawley AM, Faucher S, Angel JB. Soluble IL-7R alpha (sCD127) inhibits IL-7 activity and is increased in HIV infection. Journal of immunology. 2010;184:4679–4687. doi: 10.4049/jimmunol.0903758. [DOI] [PubMed] [Google Scholar]
- de Bakker PI, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, Ke X, Monsuur AJ, Whittaker P, Delgado M, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nature genetics. 2006;38:1166–1172. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degli-Esposti MA, Leelayuwat C, Dawkins RL. Ancestral haplotypes carry haplotypic and haplospecific polymorphisms of BAT1: possible relevance to autoimmune disease. Eur J Immunogenet. 1992;19:121–127. doi: 10.1111/j.1744-313x.1992.tb00051.x. [DOI] [PubMed] [Google Scholar]
- Dooms H. Interleukin-7: Fuel for the autoimmune attack. Journal of autoimmunity. 2013;45:40–48. doi: 10.1016/j.jaut.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Evsyukova I, Bradrick SS, Gregory SG, Garcia-Blanco MA. Cleavage and polyadenylation specificity factor 1 (CPSF1) regulates alternative splicing of interleukin 7 receptor (IL7R) exon 6. Rna. 2013;19:103–115. doi: 10.1261/rna.035410.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleckner J, Zhang M, Valcarcel J, Green MR. U2AF65 recruits a novel human DEAD box protein required for the U2 snRNP-branchpoint interaction. Genes Dev. 1997;11:1864–1872. doi: 10.1101/gad.11.14.1864. [DOI] [PubMed] [Google Scholar]
- Fry TJ, Mackall CL. The many faces of IL-7: from lymphopoiesis to peripheral T cell maintenance. Journal of immunology. 2005;174:6571–6576. doi: 10.4049/jimmunol.174.11.6571. [DOI] [PubMed] [Google Scholar]
- Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop A, Caillier SJ, Ban M, Goris A, Barcellos LF, et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nature genetics. 2007;39:1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- Hoe E, McKay FC, Schibeci SD, Gandhi K, Heard RN, Stewart GJ, Booth DR. Functionally significant differences in expression of disease-associated IL-7 receptor alpha haplotypes in CD4 T cells and dendritic cells. Journal of immunology. 2010;184:2512–2517. doi: 10.4049/jimmunol.0902900. [DOI] [PubMed] [Google Scholar]
- Honig A, Auboeuf D, Parker MM, O’Malley BW, Berget SM. Regulation of alternative splicing by the ATP-dependent DEAD-box RNA helicase p72. Mol Cell Biol. 2002;22:5698–5707. doi: 10.1128/MCB.22.16.5698-5707.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Thomas B, Flynn RA, Gavzy SJ, Wu L, Kim SV, Hall JA, Miraldi ER, Ng CP, Rigo F, et al. DDX5 and its associated lncRNA Rmrp modulate TH17 cell effector functions. Nature. 2015;528:517–522. doi: 10.1038/nature16193. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- International HapMap, C. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- International HapMap, C. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap, C. Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HIVCS, Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, Ripke S, Brumme CJ, Pulit SL, et al. The major genetic determinants of HIV–1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics, C. Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics, C., International, I.B.D.G.C., and International, I.B.D.G.C.I. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nature genetics. 2015 doi: 10.1038/ng.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M, Weatheritt RJ, Ellis JD, Parikshak NN, Gonatopoulos-Pournatzis T, Babor M, Quesnel-Vallieres M, Tapial J, Raj B, O’Hanlon D, et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159:1511–1523. doi: 10.1016/j.cell.2014.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser C, Dobrikova EY, Bradrick SS, Shveygert M, Herbert JT, Gromeier M. Activation of cap-independent translation by variant eukaryotic initiation factor 4G in vivo. Rna. 2008;14:2170–2182. doi: 10.1261/rna.1171808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilding R, Iles MM, Timms JM, Worthington J, Wilson AG. Additional genetic susceptibility for rheumatoid arthritis telomeric of the DRB1 locus. Arthritis and rheumatism. 2004;50:763–769. doi: 10.1002/art.20043. [DOI] [PubMed] [Google Scholar]
- Lauwerys BR, Husson SN, Maudoux AL, Badot V, Houssiau FA. sIL7R concentrations in the serum reflect disease activity in the lupus kidney. Lupus Sci Med. 2014;1:e000036. doi: 10.1136/lupus-2014-000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson BR, Gonzalez-Quintial R, Eleftheriadis T, Farrar MA, Miller SD, Sauer K, McGavern DB, Kono DH, Baccala R, Theofilopoulos AN. Interleukin-7 is required for CD4(+) T cell activation and autoimmune neuroinflammation. Clin Immunol. 2015;161:260–269. doi: 10.1016/j.clim.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundmark F, Duvefelt K, Iacobaeus E, Kockum I, Wallstrom E, Khademi M, Oturai A, Ryder LP, Saarela J, Harbo HF, et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nature genetics. 2007;39:1108–1113. doi: 10.1038/ng2106. [DOI] [PubMed] [Google Scholar]
- Lundstrom W, Highfill S, Walsh ST, Beq S, Morse E, Kockum I, Alfredsson L, Olsson T, Hillert J, Mackall CL. Soluble IL7Ralpha potentiates IL-7 bioactivity and promotes autoimmunity. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E1761–1770. doi: 10.1073/pnas.1222303110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ML, Zhou Z, Magni K, Christoforides C, Rappsilber J, Mann M, Reed R. Pre-mRNA splicing and mRNA export linked by direct interactions between UAP56 and Aly. Nature. 2001;413:644–647. doi: 10.1038/35098106. [DOI] [PubMed] [Google Scholar]
- Maraskovsky E, Teepe M, Morrissey PJ, Braddy S, Miller RE, Lynch DH, Peschon JJ. Impaired survival and proliferation in IL-7 receptor-deficient peripheral T cells. Journal of immunology. 1996;157:5315–5323. [PubMed] [Google Scholar]
- Masuda S, Das R, Cheng H, Hurt E, Dorman N, Reed R. Recruitment of the human TREX complex to mRNA during splicing. Genes Dev. 2005;19:1512–1517. doi: 10.1101/gad.1302205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucchelli R, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol. 2007;7:144–154. doi: 10.1038/nri2023. [DOI] [PubMed] [Google Scholar]
- Monti P, Brigatti C, Krasmann M, Ziegler AG, Bonifacio E. Concentration and activity of the soluble form of the interleukin-7 receptor alpha in type 1 diabetes identifies an interplay between hyperglycemia and immune function. Diabetes. 2013;62:2500–2508. doi: 10.2337/db12-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munitic I, Williams JA, Yang Y, Dong B, Lucas PJ, El Kassar N, Gress RE, Ashwell JD. Dynamic regulation of IL-7 receptor expression is required for normal thymopoiesis. Blood. 2004;104:4165–4172. doi: 10.1182/blood-2004-06-2484. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Nishida N, Kawashima M, Aiba Y, Tanaka A, Yasunami M, Nakamura H, Komori A, Nakamuta M, Zeniya M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. American journal of human genetics. 2012;91:721–728. doi: 10.1016/j.ajhg.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Makino S, Yoshikawa Y, Takaki A, Nagatsuka Y, Ota M, Tamiya G, Kimura A, Bahram S, Inoko H. Identification of I kappa BL as the second major histocompatibility complex-linked susceptibility locus for rheumatoid arthritis. American journal of human genetics. 2003;72:303–312. doi: 10.1086/346067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Yu Q, Erman B, Appelbaum JS, Montoya-Durango D, Grimes HL, Singer A. Suppression of IL7Ralpha transcription by IL-7 and other prosurvival cytokines: a novel mechanism for maximizing IL-7-dependent T cell survival. Immunity. 2004;21:289–302. doi: 10.1016/j.immuni.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Paternoster L, Standl M, Chen CM, Ramasamy A, Bonnelykke K, Duijts L, Ferreira MA, Alves AC, Thyssen JP, Albrecht E, et al. Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nature genetics. 2012;44:187–192. doi: 10.1038/ng.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsopoulos NA, Barcellos LF, Hintzen RQ, Schaefer C, van Duijn CM, Noble JA, Raj T, Imsgc Anzgene, Gourraud PA, et al. Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS genetics. 2013;9:e1003926. doi: 10.1371/journal.pgen.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsopoulos NA, Bayer Pharma MSGWG, Steering Committees of Studies Evaluating, I.-b., a, C.C.R.A., Consortium, A.N., GeneMsa, International Multiple Sclerosis Genetics, C. Esposito F, Reischl J, Lehr S, et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Annals of neurology. 2011;70:897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdigones N, Vigo AG, Lamas JR, Martinez A, Balsa A, Pascual-Salcedo D, de la Concha EG, Fernandez-Gutierrez B, Urcelay E. Evidence of epistasis between TNFRSF14 and TNFRSF6B polymorphisms in patients with rheumatoid arthritis. Arthritis and rheumatism. 2010;62:705–710. doi: 10.1002/art.27292. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, Park LS, Ziegler SF, Williams DE, Ware CB, et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med. 1994;180:1955–1960. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price P, Wong AM, Williamson D, Voon D, Baltic S, Allcock RJ, Boodhoo A, Christiansen FT. Polymorphisms at positions -22 and -348 in the promoter of the BAT1 gene affect transcription and the binding of nuclear factors. Human molecular genetics. 2004;13:967–974. doi: 10.1093/hmg/ddh113. [DOI] [PubMed] [Google Scholar]
- Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(−)B(+)NK(+) severe combined immunodeficiency. Nature genetics. 1998;20:394–397. doi: 10.1038/3877. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones-Lombrana A, Lopez-Soto A, Ballina-Garcia FJ, Alperi-Lopez M, Queiro-Silva R, Lopez-Vazquez A, Lopez-Larrea C, Gonzalez S. BAT1 promoter polymorphism is associated with rheumatoid arthritis susceptibility. J Rheumatol. 2008;35:741–744. [PubMed] [Google Scholar]
- Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, Alfredsson L, Padyukov L, Klareskog L, Worthington J, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nature genetics. 2012;44:291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roifman CM, Zhang J, Chitayat D, Sharfe N. A partial deficiency of interleukin-7R alpha is sufficient to abrogate T-cell development and cause severe combined immunodeficiency. Blood. 2000;96:2803–2807. [PubMed] [Google Scholar]
- Shahbazi M, Roshandel D, Omidnyia E, Rshaidbaghan A. Interaction of HLA-DRB1*1501 allele and TNF-alpha -308 G/A single nucleotide polymorphism in the susceptibility to multiple sclerosis. Clin Immunol. 2011;139:277–281. doi: 10.1016/j.clim.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Shen H, Zheng X, Shen J, Zhang L, Zhao R, Green MR. Distinct activities of the DExD/H-box splicing factor hUAP56 facilitate stepwise assembly of the spliceosome. Genes Dev. 2008;22:1796–1803. doi: 10.1101/gad.1657308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Zhang L, Zhao R. Biochemical characterization of the ATPase and helicase activity of UAP56, an essential pre-mRNA splicing and mRNA export factor. J Biol Chem. 2007;282:22544–22550. doi: 10.1074/jbc.M702304200. [DOI] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annual review of immunology. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Strasser K, Masuda S, Mason P, Pfannstiel J, Oppizzi M, Rodriguez-Navarro S, Rondon AG, Aguilera A, Struhl K, Reed R, et al. TREX is a conserved complex coupling transcription with messenger RNA export. Nature. 2002;417:304–308. doi: 10.1038/nature746. [DOI] [PubMed] [Google Scholar]
- Teigelkamp S, Mundt C, Achsel T, Will CL, Luhrmann R. The human U5 snRNP-specific 100-kD protein is an RS domain-containing, putative RNA helicase with significant homology to the yeast splicing factor Prp28p. Rna. 1997;3:1313–1326. [PMC free article] [PubMed] [Google Scholar]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nature genetics. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, Garcia-Blanco MA. RNAi-mediated PTB depletion leads to enhanced exon definition. Mol Cell. 2002;10:943–949. doi: 10.1016/s1097-2765(02)00645-7. [DOI] [PubMed] [Google Scholar]
- Wong AM, Allcock RJ, Cheong KY, Christiansen FT, Price P. Alleles of the proximal promoter of BAT1, a putative anti-inflammatory gene adjacent to the TNF cluster, reduce transcription on a disease-associated MHC haplotype. Genes Cells. 2003;8:403–412. doi: 10.1046/j.1365-2443.2002.00641.x. [DOI] [PubMed] [Google Scholar]
- Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, Tang H, Snyder M. Variation and genetic control of protein abundance in humans. Nature. 2013;499:79–82. doi: 10.1038/nature12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Tian L, Lu D, Xu S. Analysis of genome-wide RNA-sequencing data suggests age of the CEPH/Utah (CEU) lymphoblastoid cell lines systematically biases gene expression profiles. Sci Rep. 2015;5:7960. doi: 10.1038/srep07960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XJ, Lu XL, Nath SK, Lv JC, Zhu SN, Yang HZ, Qin LX, Zhao MH, Su Y, Shen N, et al. Gene-gene interaction of BLK, TNFSF4, TRAF1, TNFAIP3, and REL in systemic lupus erythematosus. Arthritis and rheumatism. 2012;64:222–231. doi: 10.1002/art.33318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Regulation of IL7R exon 6 splicing in HeLa cells and Jurkat T cells (Related to Figure 1 and Table S1). IL7R reporters carrying the alternative alleles of rs6897932 (C or T) or mutation of the critical exonic splicing enhancer 2 (ΔESE2) were transiently transfected into HeLa (left) or Jurkat (right) cells and exon 6 splicing was determined by RT-PCR as in Figure 1. The data is presented as mean ± s.d. Statistical significance was assessed using Student’s t-test (two-sided; *** p ≤ 0.0005; ** p ≤ 0.005; * p ≤ 0.05).
Figure S2. DDX39B requires intact ESE2 to regulate IL7R exon 6 splicing (Related to Figure 1). Ectopic expression of siRNA-resistant DDX39B trans-gene in DDX39B-depleted cells cannot rescue splicing of IL7R in transcripts from a reporter mutant of ESE2 (IL7R-ΔESE2). The experiment was performed as in Figure 1 followed by transfection of IL7R-ΔESE2 reporter. A, Western blot analysis of DDX39B. B, RT-PCR analysis of exon 6 skipping in transcripts from the IL7R-ΔESE2 reporter (+E6 = exon 6 included; −E6 = exon 6 skipped). The data is presented as mean ± s.d. and statistical significance was determined using Student’s t-test (two-sided; *** p ≤ 0.0005; ** p ≤ 0.005; * p ≤ 0.05).
Figure S3. Knockdown of additional RNA helicases has a small or no effect on IL7R exon 6 splicing (Related to Figure 1). Knockdown of RNA helicases DDX5, DDX23 and DDX17 was carried out in HeLa cells using three independent siRNAs against each target and a control siRNA (NSC). A, Quantification of knockdown by RT-qPCR (normalized to GAPDH). B, RT-PCR analysis of IL7R exon 6 splicing in endogenous transcripts as in Figure 1C (+E6 = exon 6 included; −E6 = exon 6 skipped). The data is shown as mean ± s.d., and statistical significance was assessed using Student’s t-test (two-sided; *** p ≤ 0.0005; ** p ≤ 0.005; * p ≤ 0.05).
Figure S4. Regional plot showing MS-associated variants within the DDX39B locus adjusted for all known MHC risk variants (Related to Figure 2 and Table S3). Variants were adjusted for population stratification, cohort of origin, the 10 known HLA risk variants (HLA-adjusted model) and the non-HLA risk variant rs2516489 (MHC-adjusted model). Each diamond represents a variant analyzed: yellow diamonds passed the threshold for statistical significance (p ≤ 0.05; black dotted line), whereas gray diamonds did not. The location of the four genes present in this region of chromosome 6 is shown at the bottom. Notably, rs2523506 and several other DDX39B variants showed a modest but significant association independent of rs2516489 and all other HLA risk variants. In healthy controls from each cohort, rs2516489 was in strong LD (r2 > 0.9) with several of the DDX39B associated variants, which explains the attenuated association after correction for rs2516489.
Figure S5. Relative abundance of DDX39B protein in lymphoblastoid cell lines (LCLs) (Related to Figure 3). We mined DDX39B protein levels in a large proteomic database where the relative protein abundance for 5,953 genes was determined by mass spectrometry in 95 LCLs derived from mainly European (CEU) and African (YRI) individuals (Wu et al., 2013). We extracted the relative DDX39B protein levels for YRI (A) and CEU (B) LCLs and stratified those by the genotypes of rs2523506 [data was only available for LCLs homozygous for the protective allele (CC) and heterozygous (AC)]. Sample sizes were as follow: YRI (CC n=24; AC n=7) and CEU (CC n=34; AC n=16). Each symbol represents an individual LCL, whereas the red lines indicate median and interquartile range. Statistical significance was determined using Student’s t-test (two-sided; * p ≤0.05).
Figure S6. SNP rs2523506 does not affect expression of genes in the vicinity of DDX39B and knockdown of these genes does not affect IL7R exon 6 splicing (Related to Fig. 1 and Fig. 3). A, RT-qPCR quantification of genes in the vicinity of DDX39B (MICB, DDX39B, ATP6V1G2, NFKBIL1, LTA, TNF and LTB) in YRI LCLs stratified by rs2523506 genotype (sample sizes: CC=12, AC=11, AA=2). Expression of an additional gene (MCCD1) was not detected. Each symbol represents the average of triplicate measurements from one individual, and red lines indicate median and interquartile range for each group. B–C, Knockdown of DDX39B vicinal genes that were expressed in Hela cells (MICB, NFKBIL1, LTA and TNF). The three other genes (MCCD1, ATP6V1G2 and LTB) are not expressed in HeLa cells and thus were not analyzed. B, RT-qPCR quantification of mRNA knockdown (normalized to GAPDH). C, RT-PCR analysis of IL7R exon 6 splicing as in Figure 1C (+E6 = exon 6 included; −E6 = exon 6 skipped). Plots in B and C are shown as mean ± s.d. Statistical significance was assessed using Student’s t-test (two-sided: *** p ≤ 0.0005; ** p ≤ 0.005; * p ≤ 0.05).
Figure S7. Quantification of sIL7R in primary CD4+ T cells (Related to Figure 6). A, Quantification of sIL7R secretion by ELISA in control (NTC) and DDX39B-depleted (sh3 and sh5) primary CD4+ T cells from Figure 6. The results are shown grouped by IL7R genotypes: IL7R-CC (top) and IL7R-CT (bottom). B, Correlation of sIL7R levels with relative abundance of IL7R transcripts that skip exon 6 (IL7R −E6) for each shRNA. The relative abundance of IL7R −E6 transcripts was extrapolated from % exon 6 skipping and RT-qPCR quantification of overall abundance of IL7R transcripts for each condition.
Table S1. Trans-acting factors exhibiting dependence on ESE2 for binding to IL7R exon 6 (Related to Figure 1).
Trans-acting factors were pulled-down from HeLa nuclear extracts by Tobramycin RNA affinity chromatography using an RNA spanning the first 40 nt of IL7R exon 6, either wild type or mutant ESE2 (ΔESE2), and a no RNA control (Evsyukova et al., 2013). RNA-Protein complexes were eluted with excess tobramycin and the bound proteins were identified by mass spectrometry. The table indicates the total number of unique peptides corresponding to each factor that were pulled-down with no RNA control, wild type or ESE2 RNAs in two independent experiments, and the fold-change decrease in binding observed with mutation of ESE2.
Table S2. Alternative splicing events regulated by DDX39B in HeLa cells (Related to Figure 1).
Alternative splicing analysis was carried out using the Vast-tools program. The table illustrates alternative splicing events that were up-regulated or down-regulated upon DDX39B depletion. “Gene” indicates genes whose splicing was altered; “Event” indicates the type of alternative splicing event (INT = Intron, EX = Exon, ALTD = alternative donor site or 5′ splice site, ALTA = alternative acceptor site or 3′ splice site); “NSC”, “DDX_3” and “DDX_4” indicate % inclusion in cells transfected with non-silencing control, DDX_3 or DDX_4 siRNAs, respectively; “E[ΔPsi]” indicates the difference in splicing between DDX_3 or DDX_4 and NSC; “max(x)@P(|ΔPsi|>x)>0.95” indicates splicing change upon knockdown at 95% confidence level. The table only shows splicing changes with “max(x)@P(|ΔPsi|>x)>0.95” ≥ 10.0 with at least one DDX39B siRNA and “E[ΔPsi]” ≥ +/− 20.0 with both DDX39B siRNAs.
Table S3. All variants in the DDX39B locus analyzed in this study (Related to Figure 2 and Figure S5).
Variants within +/− 10 kb of the DDX39B gene (chromosome 6) were analyzed using a meta-analytic logistic regression model adjusted for population stratification and cohort origin (PCA-adjusted model). Given that DDX39B is located in the MHC, the adjustment model was sequentially refined to establish independence from the known HLA risk variants (HLA-adjusted model), and an additional non-HLA risk variant (rs2516489) within the MHC (MHC-adjusted model). Bolded variants passed the threshold for statistical significance in the corresponding model (p ≤ 5.0 × 10−8 in PCA- and HLA-adjusted models; p ≤ 0.05 in MHC-adjusted model). The asterisk indicates the only variant in the vicinity of the DDX39B gene (rs2516489) previously associated with MS (Patsopoulos et al., 2013). Abbreviations: bp, base pair in the genome; SNP, variants imputed; Alleles, major and minor alleles, respectively (the alleles shown correspond to the sense strand but both DDX39B and ATP6V1G2 genes are in the antisense strand); location, indicates the gene or intergenic region where the variant resides; and OR, odds ratio. The dash lines in the last two columns indicate variants that were in complete LD with rs2516489 in the cohorts and thus no data was available after correcting for this variant.
Table S4: Multivariable logistic meta-analysis identifies a significant multiplicative interaction between IL7R rs6897932 and DDX39B rs2523506 (Related to Table 1).
Multivariable logistic regression meta-analyses demonstrate evidence for a significant multiplicative interaction between IL7R rs6897932 and DDX39B rs2523506, adjusted for population substructure, known HLA risk variants, and cohort of origin. The table illustrates the output for each adjusted variable with comparison between the two models: the HLA adjusted model without (left) and with (right) the interaction term between IL7R rs6897932 and DDX39B rs2523506 (bolded parameters) [Beta = beta coefficient; Se = standard error; OR = odds ratio]. Both models are stable and model parameters are consistent, with the exception of the significant statistical interaction.
Table S5: Lack of evidence for a multiplicative interaction between risk alleles of IL7R rs6897932 and HLA-DRB1*15:01 (Related to Table 1).
We tested for evidence of interaction between HLA-DRB1*15:01 and rs6897932 using a logistic regression model adjusted for population stratification, cohort origin and HLA risk variants, including the DDX39B rs2523506 variant (Table 1); results were not significant (no strat.). Stratification of individuals by rs6897932 genotypes (IL7R — TT (N=742), CT (N=4251) and CC (N=6239)) for analysis of DRB1*15:01 revealed strong association with MS across all three strata (note the overlap in 95% confidence intervals). The results indicate the interaction between DDX39B and IL7R is not due to an interaction between IL7R and HLA-DRB1*15:01, and thus, it is likely that this interaction between DDX39B and IL7R could explain part of the heretofore unaccounted heritable risk of MS.
Table S6: Oligonucleotide primers used in this study (Related to STAR Methods Section)
Table S7: siRNAs used in this study (Related to STAR Methods Section).