

Abstract
Background
Idiopathic pulmonary fibrosis (IPF) is progressive and rapidly fatal. Improved understanding of pathogenesis is required to prosper novel therapeutics. Epigenetic changes contribute to IPF; therefore, microRNAs may reveal novel pathogenic pathways.
Objectives
We sought to determine the regulatory role of microRNA (miR)-155 in the profibrotic function of murine lung macrophages and fibroblasts, IPF lung fibroblasts, and its contribution to experimental pulmonary fibrosis.
Methods
Bleomycin-induced lung fibrosis in wild-type and miR-155−/− mice was analyzed by histology, collagen, and profibrotic gene expression. Mechanisms were identified by in silico and molecular approaches and validated in mouse lung fibroblasts and macrophages, and in IPF lung fibroblasts, using loss-and-gain of function assays, and in vivo using specific inhibitors.
Results
miR-155−/− mice developed exacerbated lung fibrosis, increased collagen deposition, collagen 1 and 3 mRNA expression, TGF-β production, and activation of alternatively activated macrophages, contributed by deregulation of the miR-155 target gene the liver X receptor (LXR)α in lung fibroblasts and macrophages. Inhibition of LXRα in experimental lung fibrosis and in IPF lung fibroblasts reduced the exacerbated fibrotic response. Similarly, enforced expression of miR-155 reduced the profibrotic phenotype of IPF and miR-155−/− fibroblasts.
Conclusions
We describe herein a molecular pathway comprising miR-155 and its epigenetic LXRα target that when deregulated enables pathogenic pulmonary fibrosis. Manipulation of the miR-155/LXR pathway may have therapeutic potential for IPF.
Key words: MicroRNA-155, lung fibrosis, liver X receptor, fibroblasts, alternatively activated macrophages
Abbreviations used: Arg2, Arginase 2; Col, Collagen; IPF, Idiopathic pulmonary fibrosis; LXRα, Liver X receptor alpha; miR-155, MicroRNA-155; 3′UTR, 3-Prime untranslated region; 22(S)HC, 22(S)-hydroxycholesterol; WT, Wild-type
Graphical abstract
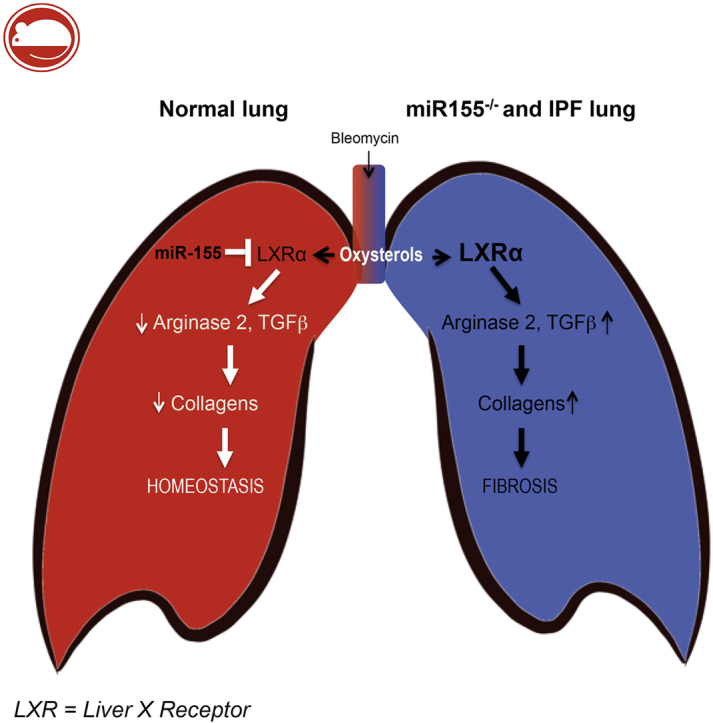
Idiopathic pulmonary fibrosis (IPF) affects more than 5 million people worldwide and its incidence is increasing.1 Histology of IPF includes interstitial fibroblastic foci and deposition of collagen-rich extracellular matrix,2 and pirfenidone-targeting tissue remodeling has improved therapeutic options.3 However, mechanisms controlling IPF progression remain poorly understood. IPF is associated with age, male sex, and cigarette smoking,4 suggesting an epigenetic contribution to pathogenesis.
MicroRNAs (miRs) are 22-nucleotide noncoding RNAs that regulate gene expression.5 Single miRs bind 6 to 8 nucleotide complementary sequences, mainly in the 3-prime untranslated region (3′UTR) of target mRNAs, causing degradation or translation inhibition6 and can finetune diverse mRNA often within the same biological pathway.7 Identifying disease-specific miRs can reveal novel target mRNA/pathways and provide insight into pathogenesis and identify therapeutic targets.
MicroRNA-155 (miR-155) is required for normal immune function8, 9; its overexpression is associated with inflammation, autoimmunity,8, 10 and cancer,11 whereas miR-155–deficient mice develop age-related airway fibrosis.12 miR-155 may therefore act as a homeostatic rheostat contributing to the onset and duration of inflammation and remodeling. Our hypothesis was that miR-155 attenuates pathways that induce lung remodeling. We revealed exacerbated experimental fibrosis in miR-155−/− mice upon lung injury. A novel miR-155–regulated pathway identified in this context was the liver X receptor alpha (LXR)α, which is an oxysterol-activated transcription factor (NR1H3).13
Methods
Bleomycin-induced lung fibrosis was induced in miR-155−/− and control mice as described.14, 15 Mouse lung fibroblasts and macrophages were derived from wild-type (WT) and miR-155−/− mice by lung digestion followed by fluorescence-activated cell sorting. Primary lung fibroblasts from patients with IPF (n = 7) and normal controls (n = 8) were obtained and cultured as described.16 Experimental interventions included transfecting cells with miR-155 mimic or LXRα siRNA, or incubating with LXR agonist/antagonist or various alarmins. Comprehensive details are provided in this article's Methods section in the Online Repository at www.jacionline.org.
Results
Experimental pulmonary fibrosis is exacerbated by miR-155 deficiency
To evaluate miR-155 epigenetic control of lung fibrosis, we used the murine model of bleomycin-induced inflammation and pulmonary fibrosis.17 Bleomycin or control PBS was given to miR-155 gene-deleted (miR-155−/−) mice and WT controls. Bleomycin-induced weight loss (Fig 1, A), lung collagen deposition (Fig 1, B), and biomarkers of inflammation (see Table E1 in this article's Online Repository at www.jacionline.org) were exacerbated in miR-155−/− mice compared with WT mice on day 18. This was accompanied by increased lung tissue expression of mRNA for collagen (Col)1a (mainly Col1a1 isoform) and Col3a1 (Fig 1, C; see Fig E1 in this article's Online Repository at www.jacionline.org), TGF-β (Tgf-β) expression, and lung collagen protein (Table E1). The increased bronchoalveolar lavage cell counts in bleomycin-treated miR-155−/− mice (Table E1) were predominantly macrophages with the repair-associated, alternatively activated (M2) phenotype (Fig 1, D) confirmed by increased arginase 2 (Arg2), chitinase, and IL-13 receptor α2 expression, whereas the expression of the classically activated macrophage (M1) phenotype marker, inducible nitric oxide synthase (Nos2), remained unchanged. Together, these data demonstrate that miR-155 deficiency exacerbated the pulmonary fibrotic response to bleomycin.
Fig 1.

Deficiency of miR-155 exacerbates experimental model of pulmonary fibrosis. A, Bleomycin (1 dose intranasal) exacerbated a decrease in body weight. B, Lung collagen deposition (turquoise staining) in miR-155−/− mice (n = 8). C, miR-155−/− bleo mice show an increase in lung Col1a1 and Col3a1 mRNA. D, M2 macrophage polarization. E, miR-155 is dynamically regulated by bleomycin in WT mice. F, Precursor (pre-)miR-155 is decreased in lung fibroblasts of WT mice (pooled n = 5) cultured with bleomycin. bleo, Bleomycin. Data shown as mean ± SEM or median and interquartile range. *P < .05.
We next investigated the kinetics of lung tissue miR-155 expression in WT mice given bleomycin (Fig 1, E). Expression of miR-155 in mice given PBS remained constant, whereas in response to bleomycin, miR-155 expression decreased at day 1, increased at day 7, and normalized by day 18. To investigate the factors that might regulate these changes, we established that bleomycin incubated in vitro with WT murine primary lung fibroblasts was sufficient to dose dependently downregulate the expression of precursor miR-155 at 8 hours (Fig 1, F) and mature miR-155 at 24 hours (see Fig E2, A, in this article's Online Repository at www.jacionline.org). To mimic the effect of exposure to cytokines generated in the damaged lung,17, 18 miR-155 expression was measured in WT murine primary lung fibroblasts incubated with exogenous alarmins IL-33,17 IL-25,19 IL-1α,18 or High mobility group box 1 (HMGB-1)20 released in response to injury. There was no change in response to IL-33, IL-25, or HMGB-1 (Fig E2, B), but IL-1α increased miR-155 expression (Fig E2, C). Thus, the dynamic expression of miR-155 in vivo may reflect a homeostatic role in inflammation and repair in response to tissue injury.
Prediction analysis identified LXRα as an miR-155 target in the lung
Identifying mRNA targets under the epigenetic control of miR-155 was our strategy to identify cryptic pathways involved in lung fibrosis. We performed in silico analysis of predicted and validated conserved mouse and human miR-155 targets (TargetScan and miRTarBase) along with targets expressed in lungs or described in respiratory or fibrotic diseases (Ingenuity Pathway Analysis database). This integrated approach identified target mRNAs (Table E2), including hypoxia and TGF-β pathways,2, 21 among which we validated increased expression of Hif1a, Tgfβr2, and Smad1 mRNA in lung tissue of miR-155−/− mice given bleomycin (see Fig E3 in this article's Online Repository at www.jacionline.org). In addition to these recognized profibrotic pathways, we identified LXRα, which has not hitherto been described in lung fibrosis. LXRα has a conserved 3′UTR seed-region sequence (GCAUUAA) complementary to miR-155; therefore, we highlighted this as a potential novel pathway to pathogenic fibrosis and this provides the basis of our study.
Endogenous miR-155 targets human LXRα
We recently demonstrated using a reporter assay that synthetic miR-155 could bind mouse Lxrα mRNA.22 To confirm that endogenous miR-155 targets human LXRα mRNA, we used an MS2-TRAP RNA affinity purification assay.23 Expression constructs encoding luciferase genes tagged with the MS2-binding domain motif with either intact LXRα, or LXRα mutated in the 3′UTR microRNA recognition element as a negative control, were transfected into HEK293 cells together with the MS2GFP-expressing plasmid. The empty vector and a construct containing a tandem of 9 miR-155 binding sites (ie, an miR-155 “sponge”) were used as negative and positive controls, respectively. MS2-binding domain–containing transcripts were isolated from transfected cells by immunoprecipitation of green fluorescent protein and the enrichment of miR-155 in precipitates was measured by quantitative PCR (Fig 2, A). Transcripts containing WT LXRα 3′UTRs showed significant enrichment in miR-155 compared with the mutated sequence, which showed miR-155 levels similar to the empty vector control; thus, endogenous miR-155 could bind to human LXRα mRNA. To confirm and extend this observation, we reintroduced miR-155 into miR-155−/− murine lung fibroblasts by transfection with a synthetic miR-155 mimic. After 24-hour culture, cytosolic LXRα protein concentrations (Fig 2, B) were reduced 60% by miR-155. Together, these findings support a functional interaction between miR-155 and LXRa mRNA.
Fig 2.

LXRα is regulated by miR-155. A, miR-155 binds to human LXRα. HEK293 were transfected with either empty vector (pmiRGLO-MS2BD) or miR-155 sponge (pmiRGLO-MS2BD-miR155Sp) or 3′UTR-LXRα (pmiRGLO-MS2BD-LXRα WT), or MS2 mutated in MRE 3′UTR-LXRα (pmiRGLO-MS2BD-Lxrα-MT), and miR-155 captured in the immunoprecipitate quantified by quantitative PCR. Data presented as mean ± SEM of 2 technical replicates; representative of 3 experiments. B, miR-155−/− fibroblasts show downregulation of LXRα protein after transfection with miR-155 mimic. C, Time course of Lxrα mRNA expression in lungs of WT mice after bleomycin (n = 4-7 per group). D, Lung fibroblast gating strategy. Representative histograms (E) and quantitative evaluation (F) of an increase in LXRα expression in lung fibroblasts during fibrosis. Expression of Lxrα(G) and Abca1(H) in lungs of WT and miR-155−/− mice on day 18. I, Constitutive expression of Abca1 in lung fibroblasts (n = 4) and in alveolar macrophages (n = 4). Constitutive expression of Arg2 in alveolar macrophages (n = 5) (J) and after transfection with Lxrα siRNA (K) or treatment with 22(S)HC (30 μM) (L). Data presented as mean ± SEM or median and interquartile range. bleo, Bleomycin; MRE, microRNA recognition element; DMSO, dimethyl sulfoxide; MS2BD, MS2-binding domain. *P < .05.
LXRα expression and activity are increased in miR-155−/− mice with lung fibrosis
Compared with WT mice given PBS, the expression of Lxrα mRNA in lung tissue of WT mice given bleomycin was upregulated on day 1 and had normalized by day 7 (Fig 2, C). This increase was confirmed at the protein level in lung fibroblasts of WT mice given bleomycin, peaking at days 2 and 3 and normalizing to control PBS levels at day 7 (Fig 2, D-F; see Fig E4 in this article's Online Repository at www.jacionline.org). This in vivo expression pattern of Lxrα was reciprocal to that of miR-155 (Fig 1, E) in WT mice. Consistent with the homeostatic molecular interaction between miR-155 and Lxrα mRNA, miR-155−/− mice given bleomycin maintained higher levels of lung Lxrα expression compared with WT mice (Fig 2, G). This increased expression was associated with an increase in Lxrα activity as measured by the expression of its specific functional reporter Abca1 in lung tissue mRNA (Fig 2, H). Together, these data demonstrate that the lack of epigenetic homeostatic regulation in miR-155−/− mice was associated with a sustained increase in Lxrα expression and activity in response to bleomycin.
Serum concentrations of LXR oxysterol ligands are unchanged in experimental fibrosis
Oxidized derivatives of cholesterol, oxysterols, for example, 24(S) hydroxycholesterol and 27-hydroxycholesterol, are natural ligands that stimulate the expression and activation of LXRα.24 We showed previously that miR-155−/− mice have higher serum cholesterol concentrations while on a high fat diet22; therefore, to test whether different oxysterol concentrations in miR-155−/− mice treated with bleomycin were responsible for the Lxra activation and exacerbated lung fibrosis, we profiled serum oxysterols using mass spectrometry (Table E3). We found no differences between any of the known LXRα ligands,25, 26 suggesting that the increased activation of the Lxrα pathway in miR-155−/− mice was due to normal activation of more available LXRα.
miR-155−/− lung fibroblasts and macrophages have an LXR-dependent profibrotic phenotype
We next investigated the role of LXR pathway activation in primary lung fibroblasts and alveolar macrophages. Compared with WT cells, miR-155−/− fibroblasts and macrophages had greater and constitutive expression of the LXRa reporter gene, Abca1 (Fig 2, I), suggesting that the LXRα pathway itself was constitutively activated. In miR-155−/− macrophages, this was associated with an increased profibrotic (M2) phenotype characterized by increased expression of Arg2, a key enzyme controlling the bioavailability of hydroxyproline for collagen synthesis27 (Fig 2, J). We demonstrated that this increased Arg2 expression in miR-155−/− macrophages was restored to normal by Lxrα-siRNA (Fig 2, K; see Fig E5, A, in this article's Online Repository at www.jacionline.org) and by LXR antagonist 22(S)-hydroxycholesterol (22(S)HC)28 (Fig 2, L). To extend this to human cells, we investigated the regulatory interrelationship between LXRα and miR-155 in the expression of ARG2 in human macrophages. Healthy human monocyte–derived macrophages were transfected with control siRNA or LXRα siRNA, each with miR-155 inhibitor or control inhibitor (Fig E5, C). To induce LXRα and ARG2 expression, the cells were cultured with LXR agonist GW3965 or control dimethyl sulfoxide. The LXR agonist–induced increase in ARG2 expression was further increased by inhibition of miR-155, and this increase was restored to normal by LXRα-specific siRNA (see Fig E6 in this article's Online Repository at www.jacionline.org). Together, these data suggest that LXRα-dependent regulation of ARG2 was governed by miR-155 in human and mouse macrophages.
We next explored whether miR-155 influenced the profibrotic function of fibroblasts in an LXRα-dependent manner. In vitro proliferation, migration, and collagen production were compared in primary lines derived from mouse lung tissue. miR-155−/− fibroblasts displayed greater proliferation to serum supplementation than did WT fibroblasts (Fig 3, A), which was restored to the normal proliferation observed in WT cells by the LXR antagonist 22(S)HC in a dose-dependent manner (Fig 3, B). miR-155−/− fibroblasts also displayed increased migration compared with WT fibroblasts into the scratch space of an in vitro wound-healing assay, which was normalized by 22(S)HC (Fig 3, C and D). The increased fibroblast infiltration was not due to proliferation because the culture medium was supplemented with 0.3% FCS, a concentration that did not support fibroblast proliferation (Fig 3, A). miR-155−/− fibroblasts produced approximately 40-fold increased concentration of soluble collagen in culture than did WT fibroblasts in response to 3% FSC (Fig 3, E), which was normalized in a dose-dependent manner by 22(S)HC to concentrations produced by WT fibroblasts (Fig 3, F).
Fig 3.

The phenotype of miR-155−/− lung fibroblasts is driven by LXR. A, miR-155−/− fibroblasts showed higher proliferation in response to FCS (%S) than did WT fibroblasts (pooled lungs of 4 mice). B, LXR antagonist 22(S)HC reduced the proliferation of miR-155−/−. C and D, miR-155−/− fibroblasts had greater migration capacity; partially inhibited by 22(S)HC, and (E) produced more collagen than did WT fibroblasts. Collagen (F) and TGF-β production (G and H) by miR-155−/− fibroblasts was inhibited by 22(S)HC (Fig 3, G) and by miR-155 mimic (Fig 3, H). I,Arg2 was higher in miR-155−/− than in WT fibroblasts and this was reduced by LXRα siRNA. Data are presented as mean ± SEM of 4 biological replicates. DMSO, Dimethyl sulfoxide. *P < .05.
TGF-β is the principal cytokine driving collagen gene expression, and oxysterol agonists of LXR can induce TGF-β production.29, 30 Therefore, to investigate the role of miR-155 in LXRα-dependent collagen production, we quantified TGF-β in WT and miR-155−/− fibroblast supernatants cultured for 48 hours in 3% FCS, with/without 22(S)HC. miR-155−/− fibroblasts produced higher concentrations of TGF-β than did WT fibroblasts and this increase was inhibited either by LXR antagonism (Fig 3, G) or by restoring miR-155 by transfection (Fig 3, H; see Fig E5, B). To investigate whether arginase was involved in this process, we measured the expression of Arg2 in fibroblasts that were transfected with Lxrα siRNA or control siRNA (Fig E5, B). miR-155−/− fibroblasts had higher expression levels of Arg2 than did WT fibroblasts (Fig 3, I), and specific inhibition of Lxrα by siRNA restored Arg2 expression in miR-155−/− fibroblasts to the normal levels of WT fibroblasts. These observations indicate that excessive production of soluble collagen by miR-155−/− fibroblasts may be due to an LXR-dependent increase in TGF-β and increased arginase-driven production of hydroxyproline.
The exacerbated bleomycin-induced lung fibrosis in miR-155−/− mice is LXR-dependent
To test the involvement of LXR in experimental lung fibrosis, miR-155−/− and WT mice were given bleomycin or control PBS, and treated with the LXR antagonist 22(S)HC or control cyclodextrin excipient. The subsequent loss of body weights for miR-155−/− and WT mice is shown on different panels for clarity in Fig 4, A. The exacerbated bleomycin-induced weight loss in miR-155−/− mice was mitigated by treatment with 22(S)HC to the weight loss seen in WT mice given bleomycin, as was the exacerbated lung tissue collagen deposition (Fig 4, B), and the inflammatory bronchoalveolar lavage cytology (see Fig E7 in this article's Online Repository at www.jacionline.org). The miR-155−/−–associated increased lung tissue Col1a1, Col3a1, and Arg2, and the bronchoalveolar lavage cell Arg2 mRNA expression were also attenuated by 22(S)HC (Fig 4, C and D). 22(S)HC had no significant effect on weight loss in bleomycin-treated WT mice (Fig 4, B). These data demonstrate that the exacerbated inflammatory and fibrotic response to bleomycin in miR-155−/− mice is at least partly dependent on LXRα and tractable in vivo by LXR antagonism.
Fig 4.

Inhibition of LXR ameliorates lung fibrosis in miR-155−/− mice. From 2 days before the administration of bleomycin (n = 10), mice were treated with daily injections of 22(S)HC. Weight loss (A) and collagen deposition (B) (turquoise) in miR-155−/− mice was mitigated by 22(S)HC. The expression of Col1a1, Col3a1, and Arg2 in lung tissues (C) and Arg2(D) in BAL cells in miR-155−/− mice was reduced by 22(S)HC. Data presented as mean ± SEM or median (interquartile range). BAL, Bronchoalveolar lavage. *P < .05.
The exacerbated profibrotic behavior of IPF fibroblasts is normalized by neutralization of the LXR pathway
To investigate the contribution of LXR pathway activation to the exacerbated lung tissue–remodeling characteristic of IPF, we obtained primary lung fibroblast lines from patients with IPF and control subjects (details in Tables E4 and E5). The constitutive cytosolic LXRα protein concentration was greater in IPF than in normal lung fibroblasts (Fig 5, A). IPF lung fibroblasts showed increased collagen synthesis in vitro compared with control lung fibroblasts, which could be either reduced in a dose-dependent manner by LXR antagonist (Fig 5, B) or further increased by the LXR agonist GW3965 (Fig 5, C). The contribution of LXR activation to the excess collagen production by IPF fibroblasts was further confirmed by transfecting IPF lung fibroblasts with LXRα siRNA (Fig E5, D), which attenuated the collagen production (Fig 5, D).
Fig 5.

The profibrotic phenotype of IPF fibroblasts can be normalized by neutralization of LXR. Synchronized normal (n = 4) and IPF (n = 6) primary lung fibroblasts were cultured with 1% FCS (%S). A, IPF fibroblasts contained higher concentrations of LXRα protein. B, Collagen production by IPF fibroblasts was (Fig 5, B) reduced by 22(S)HC or potentiated by GW3965 (C). D,LXRα siRNA reduced collagen production by IPF fibroblasts. E, 22(S)HC inhibited TGF-β production. F, GW3965 potentiated serum-induced ARG2 expression by IPF fibroblasts. G,Lxrα siRNA reduced ARG2 expression by IPF fibroblasts. Data are presented as mean ± SEM of 4 biological replicates. *P < .05.
Control normal and IPF fibroblasts produced TGF-β in culture supernatants; however, only IPF fibroblasts increased TGF-β production in response to 1% FCS supplementation and this increased production was inhibited by LXR antagonist (Fig 5, E). Normal and IPF fibroblasts constitutively expressed similar levels of ARG2 mRNA; however, only IPF fibroblasts showed higher expression of ARG2 after stimulation with LXR agonist GW3965 (Fig 5, F) and this increased ARG2 expression was attenuated by transfection with LXRα siRNA (Fig 5, G). These data indicate that TGF-β and ARG2 are regulated in an LXRα-dependent manner in IPF fibroblasts. In addition, compared with control lung fibroblasts, IPF lung fibroblasts showed greater in vitro proliferation in response to 1% FCS supplementation, which was reduced by LXR antagonist (see Fig E8, A, in this article's Online Repository at www.jacionline.org). IPF lung fibroblasts had increased migratory capacity into the scratch space of an in vitro wound-healing assay, compared with normal lung fibroblasts, and this increased migration was reduced to levels of normal fibroblasts by LXR antagonism (Fig E8, B and C). Thus, activation of the LXR pathway may drive the excessive profibrotic phenotypic characteristics of IPF fibroblasts.
LXRα is deregulated from miR-155 in IPF lung fibroblasts
To test whether the LXRα-dependent collagen production by IPF fibroblasts was regulated by miR-155, control and IPF fibroblasts were transfected with miR-155 and stimulated by synthetic LXR agonist GW3965 in vitro. The increased collagen production by IPF fibroblasts was decreased (see Fig E9, A, in this article's Online Repository at www.jacionline.org), suggesting that collagen synthesis, as the prime exemplar of the LXRα-dependent profibrotic function of IPF fibroblasts, can be regulated by miR-155.
Because constitutively increased LXRα expression (Fig 5, A) and activity contributed to IPF fibroblast phenotype, we investigated whether this was caused by altered serum concentrations of LXRα oxysterol ligands in patients with IPF or by altered miR-155 expression. Comparing serum oxysterol concentrations in IPF and control subjects showed no differences in any of the LXRα ligands tested (Table E6). The constitutive miR-155 expression in IPF fibroblasts was similar to that of control lung fibroblasts (Fig E9, B); therefore, we investigated whether the increased LXRα expression and activation in IPF fibroblasts was due to a deregulated interaction between miR-155 and LXRα. Because the consequence of LXRa deregulation resulting in exacerbated lung fibrosis became apparent in miR-155−/− mice only when stressed with bleomycin, we compared the dynamic interaction between miR-155 and LXRα in control and IPF fibroblasts cultured under the hypoxic stress (1% O2) that mimics the lung environment in IPF.31 Compared with normoxia, miR-155 expression was increased by hypoxia in both healthy and IPF fibroblasts (Fig E9, C); however, LXRα and ABCA1 expression was increased by hypoxia only in IPF fibroblasts (Fig E9, D), suggesting selective deregulation of LXRα function. To explore the dynamics of the interaction between miR-155 and LXRα, we correlated the ratio of their relative expressions in normal and IPF lung fibroblasts. The relative expression levels of LXRα and miR-155 in normal and IPF lung fibroblasts cultured under normoxic conditions showed no significant correlation (Spearman ρ and 95% CI): normal fibroblasts r = 0.263 (−0.310 to 0.69) and IPF fibroblasts r = 0.439 (−0.072 to 0.767). However, under hypoxic conditions, there was a negative correlation in normal fibroblasts r = −0.655 (−0.868 to −0.236) that was not apparent in IPF fibroblasts r = −0.152 (−0.602 to 0.375) (Fig E9, E). This suggested that there was tight posttranscriptional control of LXRα expression by homeostatic miR-155 in response to a stressor such as hypoxia in normal fibroblasts that was lost in IPF fibroblasts, potentially contributing to the deregulated LXRα activity.
The mechanism of this deregulation may be due to increased competitive miR-155 binding by other mRNA targets that contain multiple miR-155 seed-region binding sites.7 To test this hypothesis, we evaluated the expression of a validated miR-155 target ZNF65232 that contains 7 miR-155 binding sites (HumanTargetScan v7.0) in normal and IPF fibroblasts cultured in normoxia and hypoxia. ZNF652 was upregulated by hypoxia in IPF but not in normal lung fibroblasts (Fig E9, F) and in contrast to LXRα, the expression of ZNF652 correlated negatively with miR-155 expression (Fig E9, G), suggesting that under hypoxic stress, miR-155 may be preferentially bound by the increased ZNF652 leading to derepression of LXRα in IPF fibroblasts.
Discussion
Characteristic IPF fibrosis is refractory to anti-inflammatory therapy4 and antifibrotic drugs underline the primacy of aberrant wound healing to pathogenesis.3 We provide new understanding of this process. Mouse models and IPF lung fibroblasts had constitutively increased LXRα transcription when deregulated from homeostatic miR-155, associated with LXR-dependent excessive fibrotic phenotype mediated by increased TGF-β, arginase, and collagen production that could be mitigated by LXR antagonist (Fig 6).
Fig 6.

Deregulation of the miR-155/LXRα axis contributes to exacerbated pulmonary fibrosis. TGF-β, Transforming growth factor β.
Expression of miR-155 is rapidly and transiently reduced in WT mice after bleomycin, associated with transient reciprocally increased Lxrα expression and protein, and the remodeling is self-limiting.15 In contrast, miR-155−/− mice have constitutively increased Lxrα and an exacerbated lung fibrosis, and this difference may provide novel insight into mechanisms of relentless lung remodeling. IPF lung fibroblasts also have constitutively more LXRα protein (and upregulated LXRα and ABCA1: IPF data repositories GSE205233 and GEOD-2420634), and greater LXR-dependent profibrotic activation that was normalized by miR-155 overexpression, LXRa gene silencing, or metabolic antagonism of LXRα activity using 22(S)HC.
LXR may exert profibrotic effects by inducing Arg2 and Tgfβ expression. The Arg2 promoter contains an LXR response element and is activated by LXR agonism in macrophages,35 and we extend this finding to mouse and human fibroblasts. Arg2 is the mitochondrial form involved in hydroxyproline production and is essential for collagen biosynthesis. Upregulated Arg2 in miR-155−/− macrophages and fibroblasts is normalized by inhibition of Lxrα by siRNA, or its activity by metabolic antagonism. LXR may also exert profibrotic effects by similarly regulating TGF-β expression, and the excessively high concentrations of TGF-β produced in vitro by miR-155−/− and IPF fibroblasts were normalized by LXR antagonism.
Our profibrotic LXR function in lung conflicts with the antifibrotic function of T0901317-LXR activation in skin during experimental systemic sclerosis model.36 This can be reconciled; synthetic ligand T0901317 locks LXR into the conformation that recruits coactivators, whereas natural oxysterol ligands and GW3965 induce the flexible conformation that binds coactivators and corepressors,37 and there are tissue-specific epigenetic changes in chromatin that determine LXR-driven gene expression.38 Furthermore, the multiple-dose bleomycin-induced skin fibrosis is driven by IL-6 from inflammatory macrophages inhibited by LXR activation,36 whereas, in contrast, our single-bolus bleomycin-induced lung fibrosis is associated with repair M2 macrophage activation (Fig 1, D17), which is enhanced by LXR activation.35, 39 Alveolar macrophages are uniquely enriched in genes of lipid metabolism that are cross-regulated by LXR, supporting their role in lung homeostasis.40
The cryptic involvement of LXRα in fibrosis became apparent when deregulated in miR-155−/− mice plus the stressor of bleomycin. The mechanism of LXRα deregulation in IPF fibroblasts may be due to ineffective regulation by miR-155, which becomes apparent under hypoxic stress equivalent to the IPF lung environment.31 IPF and control lung fibroblasts had similar miR-155 expression when cultured under normal oxygen tensions. Under hypoxic conditions, the expression levels of miR-155 correlated negatively with LXRα in control lung fibroblasts, implying tight epigenetic control, whereas there was no equivalent engagement between miR-155 and LXRα in IPF fibroblasts, thus enabling continued LXRα autoactivation41 and profibrotic behavior. This deregulation might be mediated by several mechanisms,38 including competition for available miR-155 by other targets with the AGCAUUAA seed-region7 as validated in cancer cells.42 One strong miR-155 candidate target mRNA is ZNF652, which has 7 seed-region binding sites. ZNF652 is induced by hypoxia in IPF but not normal fibroblasts. We identified that in contrast to LXRα, its increased expression negatively correlated with miR-155 in IPF fibroblasts, suggesting that ZNF652 mRNA competitively bound miR-155 leading to derepression of LXRa.
Expression of miR-155 has been identified as increased43 or reduced,44 and serum miR-155 levels were normal45 in IPF. This may reflect the dynamism of miR-155 expression in experimental IPF. In lung tissue, it is transiently downregulated by bleomycin (Fig 1, F) and TGF-β,46 and induced by inflammatory mediators, for example, IL-1α (Fig E2, C) or hypoxia,47 as a counterbalance mechanism regulating homeostatic lung tissue repair.
Fibrosis of the lung is a common comorbidity of systemic sclerosis. The pathogenesis and clinical features of the autoimmune and inflammation-driven lung pathology of systemic sclerosis differs from IPF48 and 2 recent studies describe a pathogenic role for miR-155 in the experimental skin and lung fibrosis associated with systemic sclerosis.49, 50 This reflects the dual role of miR-155 driving chronic inflammation–associated pathologies and resolving fibrosis that we found aberrant in IPF.
Key messages.
-
•
Deficiency of miR-155 exacerbates bleomycin-induced experimental pulmonary fibrosis.
-
•
In the absence of miR-155 epigenetic control, LXRα activity is deregulated in mouse primary lung fibroblasts facilitating increased collagen and TGF-β production, and in macrophages enhancing alternative activation, each inhibited by LXR antagonism, LXRα gene silencing, or exogenous miR-155 mimic.
-
•
The exacerbated bleomycin-induced pulmonary fibrosis in miR-155−/− mice was mitigated in vivo by LXR antagonism.
-
•
Primary IPF lung fibroblasts had constitutively raised LXRα, deregulated from miR-155, and their profibrotic phenotype was inhibited by LXR antagonism, LXRα gene silencing, or exogenous miR-155 mimic.
Footnotes
M.K.-S. was supported by Arthritis Research UK (grant no. 19213); A.M.M. was supported by the British Heart Foundation (grant no. FS/08/035/25309). M.K.H. was supported by the Ministry of Higher Education and Scientific Research, Republic of Iraq. B.E.M. was supported by a joint grant (grant no. 657-2012) from Medical Research Scotland and Lamellar Biomedical Ltd, Bellshill, United Kingdom. C.A.F.-B. was supported by the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant nos. P30 AR061271 and K24 AR060297). S.J. was supported by British Pigeon Fanciers' Medical Research Trust. Oxysterol measurement was supported by Biotechnology & Biological Sciences Research Council (grant nos. BB/I001735/1 and BB/L001942/1).
Disclosure of potential conflict of interest: M. K. Hasso receives research support from the Ministry of Higher Education and Scientific Research. P. J. Crick, Y. Wang, and W. J. Griffiths receive research support from the Biotechnology and Biological Sciences Research Council. The rest of the authors declare that they have no relevant conflicts of interest.
Contributor Information
Mariola Kurowska-Stolarska, Email: Mariola.Kurowska-Stolarska@glasgow.ac.uk.
Charles McSharry, Email: Charles.McSharry@glasgow.ac.uk.
Supplementary data
References
- 1.Navaratnam V., Fogarty A.W., Glendening R., McKeever T., Hubbard R.B. The increasing secondary care burden of idiopathic pulmonary fibrosis: hospital admission trends in England from 1998 to 2010. Chest. 2013;143:1078–1084. doi: 10.1378/chest.12-0803. [DOI] [PubMed] [Google Scholar]
- 2.Wynn T.A. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King T.E., Jr., Bradford W.Z., Castro-Bernardini S., Fagan E.A., Glaspole I., Glassberg M.K. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 4.King T.E., Jr., Pardo A., Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 5.Ruvkun G. Molecular biology: glimpses of a tiny RNA world. Science. 2001;294:797–799. doi: 10.1126/science.1066315. [DOI] [PubMed] [Google Scholar]
- 6.Lim L.P., Lau N.C., Garrett-Engele P., Grimson A., Schelter J.M., Castle J. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 7.Pasquinelli A.E. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13:271–282. doi: 10.1038/nrg3162. [DOI] [PubMed] [Google Scholar]
- 8.Kurowska-Stolarska M., Alivernini S., Ballantine L.E., Asquith D.L., Millar N.L., Gilchrist D.S. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci U S A. 2011;108:11193–11198. doi: 10.1073/pnas.1019536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thai T.H., Calado D.P., Casola S., Ansel K.M., Xiao C., Xue Y. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 10.O'Connell R.M., Kahn D., Gibson W.S., Round J.L., Scholz R.L., Chaudhuri A.A. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33:607–619. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Connell R.M., Rao D.S., Chaudhuri A.A., Boldin M.P., Taganov K.D., Nicoll J. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. 2008;205:585–594. doi: 10.1084/jem.20072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez A., Vigorito E., Clare S., Warren M.V., Couttet P., Soond D.R. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jakobsson T., Treuter E., Gustafsson J.A., Steffensen K.R. Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol Sci. 2012;33:394–404. doi: 10.1016/j.tips.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 14.Moeller A., Ask K., Warburton D., Gauldie J., Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. 2008;40:362–382. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.B Moore B., Lawson W.E., Oury T.D., Sisson T.H., Raghavendran K., Hogaboam C.M. Animal models of fibrotic lung disease. Am J Respir Cell Mol Biol. 2013;49:167–179. doi: 10.1165/rcmb.2013-0094TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilewski J.M., Liu L., Henry A.C., Knauer A.V., Feghali-Bostwick C.A. Insulin-like growth factor binding proteins 3 and 5 are overexpressed in idiopathic pulmonary fibrosis and contribute to extracellular matrix deposition. Am J Pathol. 2005;166:399–407. doi: 10.1016/S0002-9440(10)62263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li D., Guabiraba R., Besnard A.G., Komai-Koma M., Jabir M.S., Zhang L. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. 2014;134:1422–1432.e11. doi: 10.1016/j.jaci.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suwara M.I., Green N.J., Borthwick L.A., Mann J., Mayer-Barber K.D., Barron L. IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol. 2014;7:684–693. doi: 10.1038/mi.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hams E., Armstrong M.E., Barlow J.L., Saunders S.P., Schwartz C., Cooke G. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci U S A. 2014;111:367–372. doi: 10.1073/pnas.1315854111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rowe S.M., Jackson P.L., Liu G., Hardison M., Livraghi A., Solomon G.M. Potential role of high-mobility group box 1 in cystic fibrosis airway disease. Am J Respir Crit Care Med. 2008;178:822–831. doi: 10.1164/rccm.200712-1894OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wynn T.A., Ramalingam T.R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller A.M., Gilchrist D.S., Nijjar J., Araldi E., Ramirez C.M., Lavery C.A. MiR-155 has a protective role in the development of non-alcoholic hepatosteatosis in mice. PLoS One. 2013;8:e72324. doi: 10.1371/journal.pone.0072324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoon J.H., Srikantan S., Gorospe M. MS2-TRAP (MS2-tagged RNA affinity purification): tagging RNA to identify associated miRNAs. Methods. 2012;58:81–87. doi: 10.1016/j.ymeth.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janowski B.A., Willy P.J., Devi T.R., Falck J.R., Mangelsdorf D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 25.Song C., Liao S. Cholestenoic acid is a naturally occurring ligand for liver X receptor alpha. Endocrinology. 2000;141:4180–4184. doi: 10.1210/endo.141.11.7772. [DOI] [PubMed] [Google Scholar]
- 26.Griffiths W.J., Crick P.J., Wang Y., Ogundare M., Tuschl K., Morris A.A. Analytical strategies for characterization of oxysterol lipidomes: liver X receptor ligands in plasma. Free Radic Biol Med. 2013;59:69–84. doi: 10.1016/j.freeradbiomed.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 27.Endo M., Oyadomari S., Terasaki Y., Takeya M., Suga M., Mori M. Induction of arginase I and II in bleomycin-induced fibrosis of mouse lung. Am J Physiol Lung Cell Mol Physiol. 2003;285:L313–L321. doi: 10.1152/ajplung.00434.2002. [DOI] [PubMed] [Google Scholar]
- 28.Tranheim Kase E., Nikolic N., Pettersen Hessvik N., Fjeldheim A.K., Jensen J., Thoresen G.H. Dietary supplementation with 22-S-hydroxycholesterol to rats reduces body weight gain and the accumulation of liver triacylglycerol. Lipids. 2012;47:483–493. doi: 10.1007/s11745-012-3663-4. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz Y., Khoshchenko O.M., Dushkin M.I., Feofanova N.A. Effects of cholesterol and nuclear hormone receptor agonists on the production of transforming growth factor-beta in macrophages. Bull Exp Biol Med. 2009;148:406–409. doi: 10.1007/s10517-010-0724-7. [DOI] [PubMed] [Google Scholar]
- 30.Kikuchi T., Sugiura H., Koarai A., Ichikawa T., Minakata Y., Matsunaga K. Increase of 27-hydroxycholesterol in the airways of patients with COPD: possible role of 27-hydroxycholesterol in tissue fibrosis. Chest. 2012;142:329–337. doi: 10.1378/chest.11-2091. [DOI] [PubMed] [Google Scholar]
- 31.Tzouvelekis A., Harokopos V., Paparountas T., Oikonomou N., Chatziioannou A., Vilaras G. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am J Respir Crit Care Med. 2007;176:1108–1119. doi: 10.1164/rccm.200705-683OC. [DOI] [PubMed] [Google Scholar]
- 32.Neilsen P.M., Noll J.E., Mattiske S., Bracken C.P., Gregory P.A., Schulz R.B. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene. 2013;32:2992–3000. doi: 10.1038/onc.2012.305. [DOI] [PubMed] [Google Scholar]
- 33.Wang X.M., Zhang Y., Kim H.P., Zhou Z., Feghali-Bostwick C.A., Liu F. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med. 2006;203:2895–2906. doi: 10.1084/jem.20061536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meltzer E.B., Barry W.T., D'Amico T.A., Davis R.D., Lin S.S., Onaitis M.W. Bayesian probit regression model for the diagnosis of pulmonary fibrosis: proof-of-principle. BMC Med Genomics. 2011;4:70. doi: 10.1186/1755-8794-4-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marathe C., Bradley M.N., Hong C., Lopez F., Ruiz de Galarreta C.M., Tontonoz P. The arginase II gene is an anti-inflammatory target of liver X receptor in macrophages. J Biol Chem. 2006;281:32197–32206. doi: 10.1074/jbc.M605237200. [DOI] [PubMed] [Google Scholar]
- 36.Beyer C., Huang J., Beer J., Zhang Y., Palumbo-Zerr K., Zerr P. Activation of liver X receptors inhibits experimental fibrosis by interfering with interleukin-6 release from macrophages. Ann Rheum Dis. 2015;74:1317–1324. doi: 10.1136/annrheumdis-2013-204401. [DOI] [PubMed] [Google Scholar]
- 37.Albers M., Blume B., Schlueter T., Wright M.B., Kober I., Kremoser C. A novel principle for partial agonism of liver X receptor ligands: competitive recruitment of activators and repressors. J Biol Chem. 2006;281:4920–4930. doi: 10.1074/jbc.M510101200. [DOI] [PubMed] [Google Scholar]
- 38.Steffensen K.R., Jakobsson T., Gustafsson J.A. Targeting liver X receptors in inflammation. Expert Opin Ther Targets. 2013;17:977–990. doi: 10.1517/14728222.2013.806490. [DOI] [PubMed] [Google Scholar]
- 39.Hasty A.H., Yvan-Charvet L. Liver X receptor alpha-dependent iron handling in M2 macrophages: the missing link between cholesterol and intraplaque hemorrhage? Circ Res. 2013;113:1182–1185. doi: 10.1161/CIRCRESAHA.113.302613. [DOI] [PubMed] [Google Scholar]
- 40.Gautier E.L., Shay T., Miller J., Greter M., Jakubzick C., Ivanov S. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laffitte B.A., Joseph S.B., Walczak R., Pei L., Wilpitz D.C., Collins J.L. Autoregulation of the human liver X receptor alpha promoter. Mol Cell Biol. 2001;21:7558–7568. doi: 10.1128/MCB.21.22.7558-7568.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar M.S., Armenteros-Monterroso E., East P., Chakravorty P., Matthews N., Winslow M.M. HMGA2 functions as a competing endogenous RNA to promote lung cancer progression. Nature. 2014;505:212–217. doi: 10.1038/nature12785. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Pandit K.V., Corcoran D., Yousef H., Yarlagadda M., Tzouvelekis A., Gibson K.F. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:220–229. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho J.H., Gelinas R., Wang K., Etheridge A., Piper M.G., Batte K. Systems biology of interstitial lung diseases: integration of mRNA and microRNA expression changes. BMC Med Genomics. 2011;4:8. doi: 10.1186/1755-8794-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 45.Li P., Zhao G.Q., Chen T.F., Chang J.X., Wang H.Q., Chen S.S. Serum miR-21 and miR-155 expression in idiopathic pulmonary fibrosis. J Asthma. 2013;50:960–964. doi: 10.3109/02770903.2013.822080. [DOI] [PubMed] [Google Scholar]
- 46.Pottier N., Maurin T., Chevalier B., Puissegur M.P., Lebrigand K., Robbe-Sermesant K. Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: implication in epithelial-mesenchymal interactions. PLoS One. 2009;4:e6718. doi: 10.1371/journal.pone.0006718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O'Connell R.M., Rao D.S., Chaudhuri A.A., Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 48.Herzog E.L., Mathur A., Tager A.M., Feghali-Bostwick C., Schneider F., Varga J. Review: interstitial lung disease associated with systemic sclerosis and idiopathic pulmonary fibrosis: how similar and distinct? Arthritis Rheum. 2014;66:1967–1978. doi: 10.1002/art.38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Christmann R.B., Wooten A., Sampaio-Barros P., Borges C.L., Carvalho C.R., Kairalla R.A. miR-155 in the progression of lung fibrosis in systemic sclerosis. Arthritis Res Ther. 2016;18:155. doi: 10.1186/s13075-016-1054-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan Q., Chen J., Li W., Bao C., Fu Q. Targeting miR-155 to treat experimental scleroderma. Sci Rep. 2016;6:20314. doi: 10.1038/srep20314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.