

SUMMARY
Understanding immunological memory formation depends on elucidating how multipotent memory precursor (MP) cells maintain developmental plasticity and longevity to provide long-term immunity while other effector cells develop into terminally differentiated effector (TE) cells with limited survival. Profiling active (H3K27ac) and repressed (H3K27me3) chromatin in naïve, MP and TE CD8+ T cells during viral infection revealed increased H3K27me3 deposition at numerous pro-memory and pro-survival genes in TE relative to MP cells, indicative of fate restriction, but permissive chromatin at both pro-memory and pro–effector genes in MP cells, indicative of multipotency. Polycomb repressive complex 2-deficiency impaired clonal expansion and TE cell differentiation, but minimally impacted CD8+ memory T cell maturation. Abundant H3K27me3 deposition at pro-memory genes occurred late during TE cell development, likely from diminished transcription factor FOXO1 expression. These results outline a temporal model for loss of memory cell potential through selective epigenetic-silencing of pro-memory genes in effector T cells.
Keywords: CD8+ T cell differentiation, epigenetics, H3K27me3, H3K27ac, Polycomb repressive complex 2, PRC2, EZH2, FOXO1, terminal differentiation, plasticity
eTOC
Cytotoxic CD8+ T cells either terminally differentiate and die or form a rapidly responding population of memory T cells following pathogen clearance. Gray et al. define a temporal model for how effector T cells lose memory cell potential through selective epigenetic-silencing of pro-memory genes.
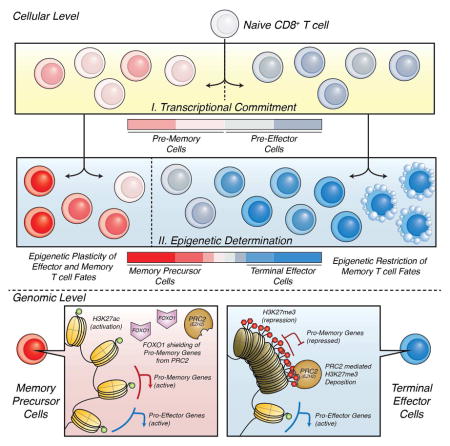
INTRODUCTION
Cytotoxic CD8+ T cells help clear intracellular bacterial and viral infections and protect against future infections by forming a long-lived, rapidly responding population of memory T cells. During an acute viral infection, the pathogen-specific CD8+ T cells develop into a heterogeneous population of cytotoxic effector and memory T cells, comprised of subsets discernable by their phenotypes, functions, anatomical locations and long-term fates(Kaech and Cui, 2012). Many anti-viral effector CD8+ T cells differentiate into terminal effector (TE) cells (distinguished by stable expression of killer cell lectin like receptor G1 (KLRG1) and repression of interleukin 7 receptor α (IL-7Rα) [KLRG1Hi IL7RLo]) that migrate into the periphery as interferon γ (IFNγ)-producing cytotoxic T lymphocytes (CTLs) (Kaech and Cui, 2012). However, the TE cells have the least memory cell potential and display the greatest rates of contraction following infection. TE cells are considered terminally differentiated because they proliferate poorly in response to homeostatic cytokines (e.g., IL-15) or antigen, and maintain their phenotypic and functional properties upon restimulation (i.e., they do not adopt properties of other effector subsets)(Joshi et al., 2007). In contrast, subsets of memory precursor (MP) effector cells (distinguished by higher expression of IL-7Rα [IL7RHi]) are intrinsically more fit to persist and self-renew (Joshi et al., 2007, Best et al., 2013, Sarkar et al., 2008). The MP cells are also multipotent, developing into diverse types of memory CD8+ T cells (e.g., central, effector and resident memory T cells) that form recalled effector cells upon secondary infection (Sarkar et al., 2008, Mackay et al., 2013, Joshi et al., 2007). Given that these CD8+ T cell fate decisions determine the quantity and quality of immunological memory that forms following vaccination and infection, it is critical to understand how and when they are specified.
Temporal regulation of effector CD8+ T cell differentiation and the factors involved have begun to be delineated. Even as early as the first cell division, asymmetric partitioning of the Mechanistic Target of Rapamycin Complex 1 (mTORC1) and 2 and the transcription factors (TFs) c-Myc and T-bet biases the daughters cells towards effector or memory cell fates (Chang et al., 2011, Verbist et al., 2016, Pollizzi et al., 2016). Other studies have shown that commitment to a TE cell fate can be visualized in early effector CD8+ T cells ~3.5 to 4.5 days post infection (p.i.) by increased expression of KLRG1 or the interleukin 2 receptor α (IL-2Rα) chain or reduced expression of the transcription factor ID3 (see references within (Kaech and Cui, 2012, Best et al., 2013, Kalia et al., 2010)). The KLRG1Lo early effector cells remain uncommitted at this stage, and continue to give rise to KLRG1Hi IL7RLo TE cells as well as KLRG1Hi IL7RHi and KLRG1Lo IL7RHi cells that differ in their long-term fates and the types of memory cells they form (Kaech and Cui, 2012). CD62LHi central memory (Tcm) and CD103Hi tissue resident memory (Trm) cells progressively form several weeks after infection, demonstrating that effector and memory CD8+ T cell specification is dynamic (Mackay et al., 2013, Kaech and Cui, 2012). Further, recent work has shown that these memory cells have distinct roles in immune surveillance and homeostasis, distinguished by expression of the chemokine receptor CX3CR1 (Gerlach et al., 2016).
TE vs. MP cell differentiation is influenced by cytokines such as type-1 interferons, IL-12, and IL-2, which are transduced through transcription factors STAT4, STAT5, and the AKT and mTOR pathways to induce effector gene expression and repress quiescence and pro-memory genes (see references within (Kaech and Cui, 2012)). Increasing levels of inflammation induce graded expression of TFs, including T-bet, ZEB2 and Blimp-1, that cooperatively promote TE-signature gene expression and terminal differentiation (Kaech and Cui, 2012, Dominguez et al., 2015, Shin et al., 2013, Omilusik et al., 2015). Simultaneously, the AKT-mediated inactivation of the TF FOXO1 represses expression of MP-signature genes, including Il7r, Sell, Tcf7, Lef1, Bach2 and other memory promoting genes (see references within (Kaech and Cui, 2012, Kim et al., 2013)). While the above studies offer insight into the molecular control of differentiation of diverse effector T cell subsets during infection, they do not explain how the changes in gene expression are stably inherited in daughter cells to generate terminally differentiated TE cells fated to die or multipotent MP cells fated to persist and generate memory cells.
Dynamic regulation of epigenetic and chromatin states will influence how T cells acquire or lose plasticity and/or how particular T cell fates are determined. To better elucidate the epigenetic mechanisms by which TE cells become committed to a terminal fate and MP cells remain multipotent in the context of changing environments during acute lymphocytic choriomeningitis virus (LCMV) infection, we profiled active chromatin associated histone 3 lysine 27 acetyl (H3K27ac) and repressed chromatin associated histone 3 lysine 27 trimethyl (H3K27me3) genome-wide in MP and TE effector CD8+ T cells. This demonstrated biased deposition of repressive H3K27me3 at MP-signature genes in TE cells indicating preferential repression of pro-memory genes as TE cells terminally differentiate. Conversely, MP cells did not contain greater amounts of H3K27me3 at TE-signature genes despite lower transcriptional activity, illuminating their epigenetic plasticity. Additionally, we found that inactivity of the Polycomb Repressive Complex 2 (PRC2), which catalyzes de novo H3K27 trimethylation, via deletion of the methyltransferase Enhancer of Zeste Homolog 2 (EZH2) or its cofactor Embryonic Ectoderm Development (EED) (Margueron and Reinberg, 2011) in virus-specific CD8+ T cells impaired the formation of terminally differentiated TE cells. While having minimal impact on memory CD8+ T cell maturation, Ezh2-deficiency impaired secondary responses of memory cells to reinfection. Finally, we found that abundant deposition of H3K27me3 occurs relatively late during effector development to stably silence pro-memory genes specifically in TE cells, likely in a FOXO1 regulated manner. These results outline a sophisticated model for how memory cell potential is lost as effector CD8+ T cells terminally differentiate through epigenetic silencing of pro-memory genes.
RESULTS
Epigenetic repression of pro-memory genes in terminally differentiated effector CD8+ T cells
TE and MP cells differ in their developmental potential (i.e., multipotency) and long-term fates after viral infection, and while they express several genes in common, they also have distinct gene expression signatures (referred to as MP- and TE-signature genes throughout)(Dominguez et al., 2015, Joshi et al., 2007). To understand how these developmental differences arise, we characterized the epigenetic states of these virus-specific effector CD8+ T cell subsets via genome-wide profiling of the histone modifications H3K27ac or H3K27me3 to infer regions of active- or repressed- chromatin, respectively. To this end, 1 × 105 naïve Thy1.1+ P14 (LCMV GP33-41 specific) T cell receptor (TCR) transgenic CD8+ T cells were transferred into naïve wild-type (WT) C57BL/6 recipient mice that were subsequently infected with lymphocytic choriomeningitis virus (LCMV)-Armstrong strain, which causes an acute systemic infection. Ten days post-infection (d10 p.i.), pure populations of KLRG1Hi IL7RLo (TE) and KLRG1Lo IL7RHi (MP) CD8+ T cells were sorted and the chromatin was immunoprecipitated using antibodies against H3K27ac and H3K27me3 followed by high-throughput sequencing (ChIP-Seq). A total of 8,296 H3K27me3 and 9,076 H3K27ac high-quality peaks (p-value < 10−5) were identified across MP and TE ChIP-Seq samples (Fig S1A–C). These peaks were annotated to the nearest gene transcriptional start site (TSS) (Fig S1D). From these annotations, H3K27ac deposition exhibited a positive correlation with gene expression across MP and TE cells (Fig S1G, left), while H3K27me3 deposition correlated negatively with gene expression (Fig S1G, right).
Next, we identified consensus peaks that contain significant differences in the amount of H3K27me3 or H3K27ac deposition (referred to as differentially modified regions (DMRs)) between MP and TE cells. DMRs were defined as having a fold-change greater than 1.2 with an FDR less than 0.1. Regions failing to meet these criteria were labeled as “Common” regions between MP and TE cells. As above, these DMRs and Common regions were annotated to the nearest TSS (Fig S1E–F) to identify related patterns of mRNA expression in MP and TE cells. Volcano plots of the log2 (fold-change) of H3K27ac deposition showed that, as expected, DMRs with increased H3K27ac in MP cells (Cluster 1) or TE cells (Cluster 2) were associated with increased mRNA expression in each respective cell population (MP- and TE-signature genes are labeled red and blue, respectively) (Fig 1A). In contrast, differential deposition of H3K27me3 showed that MP cells possessed few differentially methylated loci (Cluster 3), whereas TE cells had an abundance of highly methylated loci relative to MP cells (Cluster 4), many of which were associated with MP-signature genes (Fig 1B).
Figure 1. Higher levels of H3K27me3 deposition are a defining feature of TE cells compared to MP cells.

LCMV-specific KLRG1Hi IL7RLo (TE) and KLRG1Lo IL7RHi (MP) P14 CD8+ T cells were purified at d10 post LCMV-Armstrong infection and ChIP-Seq was performed for H3K27me3 and H3K27ac. See Supplemental Methods for details, but briefly, consensus peaks from replicate TE and MP samples were compared to identify “Common” regions or those that contain significantly differentially modified regions (DMRs) of H3K27me3 or H3K27ac (FDR (Benjamini-Hochberg) < 0.1 and fold-change > 1.2).
A–B) Volcano plots comparing differential deposition of (A) H3K27ac and (B) H3K27me3 between MP and TE cells were used to identify DMRs. Differential abundance of H3K27ac or H3K27me3 deposition [log2(Fold-change)] is plotted by [−log10(FDR)], where positive fold-change values represent higher deposition of the histone modification in MP cells and negative values represent higher deposition in TE cells. Horizontal dashed line denotes −log10(FDR) of 0.1, while vertical dashed lines denote a +/− log2 transformed fold-change of 1.2. DMRs associated to MP and TE gene expression signatures are labeled as blue and red dots, respectively. All remaining consensus peaks are referred to as “Common” regions between MP and TE cells (labeled as light gray dots).
C) Deposition of H3K27me3 and H3K27ac in MP and TE cells centered on DMRs +/−10kb as identified in volcano plots (A and B). Cluster 1 (dark blue) = H3K27ac deposition higher in MP than TE, Cluster 2 (light blue) = H3K27ac deposition higher in TE than MP, Cluster 3 (green) = H3K27me3 deposition higher in MP than TE, Cluster 4 (orange) = H3K27me3 deposition higher in TE than MP. Line plots at top show the summary distributions across each cluster for each H3K27ac and H3K27me3 in MP and TE cells, respectively. Scatter plots on far right show Log2(fold-change) of mRNA expression between MP and TE cells for the DMR-associated genes and summaries of entire gene expression distributions across Clusters 1–4 or Common consensus peaks are shown in boxplots.
D–E) Line plots show the ratios of H3K27ac or H3K27me3 deposition (normalized to Common consensus peaks, see Fig S1I) between MP and TE cells or within MP or TE cells for each cluster. Data shown contain the union of significant consensus peaks identified across two independent biological replicates of ChIP-Seq experiments for H3K27ac and H3K27me3 (A–E; n=10–20 mice/group/replicate). See also Figure S1.
We next plotted a 20 kb window centered on each significant H3K27ac or H3K27me3 DMR and clustered the loci based on the four quadrants defined by the volcano plots (Fig 1C). Clusters 1 and 2 show the significant H3K27ac DMRs in MP and TE cells, respectively, and the mRNA expression at many of these loci correlates with the relative greater abundance of H3K27ac within each cell population (Fig 1C, right). However, note that the relative ratio of H3K27ac between MP and TE cells at the DMRs in Cluster 2 was more similar than those in Cluster 1 (Fig 1D), signifying that MP cells have comparable amounts of histone acetylation at many TE-signature gene loci despite the lowered mRNA expression (possibly indicative of transcriptional poising). Clusters 3 and 4 show the significant H3K27me3 DMRs (Fig 1C), wherein one can see that many H3K27me3 DMRs in TE cells correlated with transcriptional repression of MP-signature genes (Cluster 4). Similarly, comparing the ratio of H3K27ac to H3K27me3 in TE and MP cells showed that TE cells generally have a bias towards a more repressed state (lower ac/me3 ratio) than MP cells (Fig 1E). In summary, these data demonstrated that the most significant, numerous, and substantial changes in H3K27me3 and H3K27ac deposition preferentially occurred at pro-memory, MP-signature genes that were epigenetically and transcriptionally repressed in the TE cells (Fig 1).
To understand how the epigenetic profiles of individual genes differed between MP and TE cells, we examined several important and well-studied MP- and TE-signature genes. At many MP-signature genes, such as Id3, Tcf7, and Bach2, and pro-survival genes, such as Bcl2, the TE cells contained substantially more H3K27me3 and less H3K27ac deposition than MP cells (Fig 2A, Fig S2). A similar pattern was also observed at Trm-signature genes (Mackay et al., 2013) including Sik1, Skil, and Cdh1 (Fig S2), suggesting that epigenetic silencing of these loci in TE cells accounted for their decline in plasticity and longevity. In contrast, there was relatively little H3K27me3 deposition at most pro-effector, TE-signature genes, such as Klrg1, Tbx21 (T-bet) and Prdm1 (Blimp-1), in either TE or MP cells (Fig 2B).
Figure 2. TE cells restrict memory cell potential by epigenetically repressing MP-signature genes.

Alignment tracks of H3K27ac and H3K27me3 deposition across MP (red) and TE (blue) cells at (A) MP-signature and (B) TE-signature genes. MP- and TE-signature genes were defined based on differential mRNA expression (>1.5 fold-change, FDR < 0.1). Statistically significant differentially modified regions (DMRs) are marked by rectangles below tracks, with red bars representing DMRs where the modification is higher in MP cells and blue bars representing DMRs where the modification is higher in TE cells. Black bars demarcate common consensus peaks that are not differentially modified in one cell population over the other.
Data shown contain the union of significant consensus peaks identified across two independent biological replicates of ChIP-Seq experiments for H3K27ac and H3K27me3 (A–B; n=10–20 mice/group/replicate). See also Figure S2.
Altogether, these data outlined an epigenetic dichotomy between TE and MP cells and illustrated that as effector CD8+ T cells terminally differentiated into TE cells, many pro-memory genes were selectively remodeled into a repressive state by the accumulation of H3K27me3. This provided a genomic understanding for how memory cell potential was lost as effector CD8+ T cells terminally differentiated through epigenetic silencing of pro-memory genes. The reciprocal process did not appear to occur in MP cells as they maintained permissive or active chromatin states at both MP- and TE-signature genes. Given that memory cells derived from the MP subset will need to express pro-effector, TE-signature genes quickly upon antigen re-exposure, our data supports a view where the TEfate is not epigenetically repressed in MP cells, but rather remains open or poised. These data argue that the MP vs. TE cell fate decision process differs from a conventional binary cell fate choice where each cell type represses the fate-determining genes of the “alternative” fate. Rather, MP cells maintain multipotency for both memory and effector fates, while TE cells restrict memory fates.
EZH2 is required for the anti-viral CD8+ T cell response
To better understand how H3K27me3 deposition was regulated in CD8+ T cells, we generated mice that conditionally deleted Ezh2, the de novo methyltransferase of PRC2, in thymocytes by breeding mice containing floxed Ezh2 alleles to mice expressing Cre-recombinase under the control of the CD4 promoter, enhancer, and silencer, generating Ezh2f/f CD4Cre+ mice. Cre deficient littermate Ezh2f/f mice were used as controls. Western blotting of EZH2 and H3K27me3 in naïve and activated CD8+ T cells from Ezh2f/f CD4Cre+ mice or littermate controls showed that (1) EZH2 protein is substantially induced in activated T cells and (2) H3K27me3 was virtually undetectable in activated Ezh2f/f CD4Cre+ CD8+ T cells (Fig 3A). The latter point demonstrated that EZH2-containing PRC2 was the exclusive writer of de novo H3K27me3 in activated CD8+ T cells.
Figure 3. EZH2 is required for H3K27me3 deposition and antiviral CD8+ T cell clonal expansion.

A) Naïve CD8+ T cells from Ezh2f/f mice and CD8+ T cells from Ezh2f/f and Ezh2f/f CD4Cre+ mice activated in vitro with αCD3 and αCD28 for 3 day were purified using FACS and the amounts of EZH2, β-Actin, H3K27me3 and total H3 were measured by western blot (data from 2 different experiments). Note, H3K27me3 is virtually undetectable in activated Ezh2f/f CD4Cre+ CD8+ T cells.
B) Ezh2f/f and Ezh2f/f GzmBCre+ mice were infected with LCMV Armstrong and the number of splenic DbGP33-41 and DbNP396-404 MHC class I tetramer+ CD8+ T cells combined were enumerated at d8 p.i.
C) Bar graph shows viral titer in the serum of Ezh2f/f and Ezh2f/f GzmBCre+ mice at d8 p.i.
D) Naïve P14 Ezh2f/f (red) and Ezh2f/f CD4Cre+ (black line) CD8+ T cells were labeled with CellTrace Violet and stimulated for 72 hours in vitro with GP33-41 peptide. Unstimulated naïve P14 CD8 T cells are shown in gray.
E) Congenically mismatched naïve P14+ Thy1.1+Ly5.2+ Ezh2f/f (red) and Thy1.2+Ly5.2+ Ezh2f/f CD4Cre+ (black line) CD8+ T Cells were pulsed with CellTrace Violet and adoptively co-transferred into Thy1.2+ Ly5.1+ WT recipient mice that were subsequently infected with LCMV-Armstrong and analyzed for cell division 60 hours later. P14+ CD8+ T cells from an uninfected recipient are shown in gray.
F) Bar graphs show IFNγ, TNFα, and IL2 production in GP33-41 peptide-stimulated Ezh2f/f and Ezh2f/f GzmBCre+ CD8+ T cells at d8 p.i.
Data shown are representative of two (C–E), three (A), or five (F) independent experiments (n=4–10 mice/group/experiment for C and F), or cumulative of five independent experiments (n=21 mice/group) (B). Data are expressed as mean ± SD. *p=0.02, ****p<0.0001
To examine the role of EZH2 in the anti-viral CD8+ T cell response, we generated mice that conditionally delete Ezh2 in activated CD8+ T cells by crossing Ezh2f/f mice to those expressing Cre under the control of the Granzyme B promoter, generating Ezh2f/f GzmBCre+ mice. In this system, Ezh2 was not deleted in naïve CD8+ T cells, but was deleted within 48–72 hours after activation or infection (Fig S3A–B)(Dominguez et al., 2015). The Ezh2f/f and Ezh2f/f GzmBCre+ LCMV-specific CD8+ T cells were then analyzed during infection and this showed that Ezh2 was necessary for normal effector CD8+ T cell expansion and viral control. Ezh2f/f GzmBCre+ mice formed ~10 fold fewer LCMV-specific anti-viral CD8+ T cells at d8 p.i. and had delayed viral clearance compared to Cre− (Ezh2f/f) littermate controls (Fig 3B–C). The defect in effector CD8+ T cell expansion in the absence of Ezh2 was likely due to increased apoptosis as opposed to defects in cell division since Ezh2f/f CD4Cre+ CD8+ T cells divided at similar rates to Ezh2f/f controls early after activation both in vitro and in vivo during LCMV infection (Fig 3D–E). Examination of cytokine production in a 5-hour ex vivo peptide stimulation assay revealed that Ezh2f/f GzmBCre+ CD8+ T cells were still polyfunctional, with equivalent percentages of IFNγ+ and IL-2+ cells, but modestly fewer tumor necrosis factor-α+ (TNFα+) cells, as compared to the Ezh2f/f controls (Fig 3F). Therefore, EZH2 was required for efficient clonal expansion of activated CD8+ T cell during an acute viral infection, similar to what has been observed in CD4+ T cells following Toxoplasma gondii infection(Yang et al., 2015).
EZH2-containing PRC2 is required for differentiation of Terminal Effector CD8+ T cells
To investigate how EZH2 controls effector CD8+ T cell differentiation, we assessed the phenotype of LCMV-specific Ezh2f/f GzmBCre+ CD8+ T cells at days 4–8 p.i. During the early effector phase (d4.5 p.i.), despite reduced expansion, a similar percentage of KLRG1Hi effector CD8+ T cells formed between Ezh2f/f and Ezh2f/f GzmBCre+ cells (Fig 4A). However, by d8 p.i., very few KLRG1HiIL7RLo TE-like cells were observed in the virus-specific Ezh2f/f GzmBCre+ CD8+ T cells relative to Ezh2f/f littermate control cells (Fig 4B–C). Rather, the CD8+ T cells lacking Ezh2 generated effector cells that expressed lower amounts of KLRG1, IL-7R and increased amounts of CD27 and CD62L, compared to the littermate controls (Fig. 4B–C). In addition, the Ezh2-deficient anti-viral CD8+ T cells expressed higher amounts of pro-memory TFs including TCF1 (TCF7), FOXO1, and Eomes, and lower amounts of the pro-effector TF T-bet (Fig 4D). Similarly, in different genetic models, Ezh2f/f CD4Cre+ mice formed few virus-specific KLRG1Hi IL7RLo TE-like cells and many more KLRG1Lo IL-7RHi CD27Hi CD62LHi cells compared to Ezh2f/f controls following infection (Fig S3C), and replication of this phenotype in mixed Ezh2f/f: Ezh2f/f GzmBCre+ bone marrow chimeras at d8 p.i. confirmed that EZH2 functions in a CD8+ T cell intrinsic manner to generate TE-like cells (Fig S3D). Lastly, because EZH2 is known to have non-histone targets in cells (Gunawan et al., 2015), we examined the phenotypes of virus-specific CD8+ T cells lacking the PRC2 subunit EED by generating Eedf/f CD4Cre+ mice that were infected with LCMV-Armstrong. This showed that the phenotypes of the CD8+ T cells lacking Eed were very similar to those lacking Ezh2 at day 8 p.i. (Fig S4). Together, these data suggested that PRC2 (containing both EZH2 and EED) was intrinsically required for the expansion and terminal differentiation of effector CD8+ T cells, and that in the absence of PRC2 effector CD8+ T cells acquired more MP-like qualities.
Figure 4. EZH2 is required for KLRG1Hi CD27Lo TE CD8+ T cell differentiation.

A) Ezh2f/f (solid) and Ezh2f/f GzmBCre+ (open) mice were infected with LCMV-Armstrong, and at d4.5 p.i. the percentage of KLRG1Hi virus-specific CD8+ T cells was determined in the peripheral blood using DbGP33-41 and DbNP396-404 MHC class I tetramer+ staining and flow cytometry.
B–D) The same mice as in (A) were examined at d8 p.i. for expression of pro-effector and pro-memory receptors and TFs on splenic DbGP33-41 tetramer+ CD8+ T cells. B) Histograms show the relative surface expression of the indicated receptors in Ezh2f/f (black) and Ezh2f/f GzmBCre+ (gray) CD8+ T cells. C) Bar graph shows KLRG1/IL7R subsets as a percentage of DbGP33-41 + Ezh2f/f (solid) and Ezh2f/f GzmBCre+ (open) CD8+ T cells. D) Bar graphs show intracellular mean fluorescence intensity (MFI) of the indicated TFs in Ezh2f/f (solid) and Ezh2f/f GzmBCre+ (open) CD8+ T cells.
Data shown are representative of three (A) or five (B,D) or cumulative of five (C) independent experiments (n=4–10 mice/group/experiment). Data are expressed as mean ± SD. **p=0.006, ***p=0.0001,****p<0.0001. See also Figure S3 and S4.
EZH2 is not required for memory CD8+ T cell formation, but is required for protective immunity
We next examined whether EZH2 was required for memory CD8+ T cell formation by two approaches. First, we created mice in which we could inducibly delete Ezh2 conditionally in CD8+ T cells using the GranzymeB-ERT2Cre system (which regulates expression of the estrogen-receptor (ER)-Cre fusion protein by the GzmB promoter and ER-Cre nuclear localization via tamoxifen treatment)(Bannard et al., 2009). These Ezh2f/f GzmB-ERT2Cre mice also contained a ROSA26floxSTOPfloxYFP cassette to report ER-Cre activity in the virus-specific CD8+ T cells. Briefly, Ezh2f/f GzmB-ERT2Cre mice and Ezh2f/+ GzmB-ERT2Cre littermate controls were infected with LCMV-Armstrong and 8 days later, after effector CD8+ T cell formation, the mice were gavaged with 2 mg of tamoxifen every 3 days from d8 to 23 p.i., then sacrificed at d30 p.i. to measure the numbers and phenotypes of LCMV-specific (YFP+) memory CD8+ T cells. Ezh2 deletion in this system was confirmed after completion of tamoxifen treatment by genomic DNA PCR analysis in virus-specific CD8+ T cells, which showed near complete deletion of Ezh2 (Fig S5). This experiment showed that loss of Ezh2 in anti-viral CD8+ T cells after d8 p.i. did not have a major impact on the quantity or quality of memory CD8+ T cells that formed (Fig 5A–C). In agreement, similar numbers and types of memory CD8+ T cells formed in Ezh2f/f GzmBCre+ mice (the non-inducible Cre system described in Fig 3) as in the littermate controls at d30 p.i. (Fig 5D–F). Thus, EZH2 was critical for naïve→effector CD8+ T cell differentiation, but not effector→memory differentiation, in line with the expression pattern of EZH2 in antiviral CD8+ T cells (data not shown).
Figure 5. EZH2 is not required for memory formation, but is required for secondary responses.

A–C) Ezh2f/+GzmBERT2Cre+ and Ezh2f/fGzmBERT2Cre+ mice were infected with LCMV-Armstrong, then starting at d8 p.i., EZH2 was inducibly deleted in virus-specific CD8+ T cells by tamoxifen (2mg) administration every three days for three weeks. A) The splenic YFP+ DbGP33-41 + CD8+ T cells were examined at d30 p.i. by flow cytometry and contour plots (left) show expression of KLRG1 and IL7R. Bar graphs (right) show the numbers of Ezh2f/+GzmBERT2Cre+ (solid) and Ezh2f/fGzmBERT2Cre+ (open) virus-specific YFP+ CD8+ T cells in each of the four KLRG1/IL-7R subsets. B) Bar graph shows percentage of YFP+ DbGP33-41 + CD8+ T cells expressing CD62L. C) Bar graph shows FOXO1 protein levels in YFP+ DbGP33-41 + CD8+ T cells.
D–H) Ezh2f/f and Ezh2f/f GzmBCre+ mice (the non-inducible Cre system described in Fig 3) were infected with LCMV-Armstrong and at d30 p.i. the phenotype (D–F) and protective capacity (G–H) of the GP33-41-specific memory CD8+ T cells were examined. D) Contour plots (left) show expression of KLRG1 and IL7R and bar graphs (right) show the numbers of Ezh2f/f (solid) and Ezh2f/f GzmBCre+ (open) virus-specific CD8+ T cells in each of the four KLRG1/IL-7R subsets. E) Bar graph shows percentage of DbGP33-41 + CD8+ T cells expressing CD62L. F) Bar graph shows FOXO1 protein levels in DbGP33-41 + CD8+ T cells. G–H) 50,000 GP33-41-specific Ezh2f/f or Ezh2f/f GzmBCre+ memory CD8+ T cells (from D) were transferred into naïve B6 mice that were then infected with recombinant Listeria monocytogenes expressing the GP33-41 epitope (LM-33). G) At d4.5 post-challenge, the numbers of recalled GP33-41-specific Ezh2f/f and Ezh2f/f GzmBCre+ CD8+ T cells were enumerated in the spleen. H) LM-33 bacterial titers in the liver were determined at d3 post-challenge.
Data shown are representative of two (B–C, E–F, H) or cumulative of two (G) or three (A, D) independent experiments (n=9/group (A), n=9–13/group (D, G), n=3–5 mice/group/experiment (B–C, E–F, H)). Data are expressed as mean ± SD. *p=0.02, **p=0.01, ***p=0.0005. See also Figure S5.
Second, to examine the protection provided by Ezh2-deficient memory CD8+ T cells upon secondary infection, small numbers of Ezh2-deficient or Ezh2-sufficient memory CD8+ T cells specific for the LCMV-epitope GP33-41 (isolated from Ezh2f/f GzmBCre+ or Ezh2f/f mice, respectively), were transferred into naïve B6 recipients that were subsequently infected with a recombinant strain of Listeria monocytogenes that expresses the GP33-41 epitope (LM-33). Similar to what was observed during the primary infection, the Ezh2-deficient memory CD8+ T cells did not expand or clear bacteria as effectively as the control memory CD8+ T cells (Fig 5G–H). These data demonstrated that EZH2 was not required for the formation of memory CD8+ T cells in general, but was required for their protective recall responses and generation of secondary effector cells.
TE cell fates are determined during late effector cell development
Prior work has shown that effector CD8+ T cells transcriptionally repress pro-memory genes early after activation and commitment to effector or memory cell fates can be observed prior to d4.5 p.i. (Kalia et al., 2010, Best et al., 2013, Sarkar et al., 2008), but when are these cell fates precisely determined? We reasoned that delineating when the pro-memory loci are selectively remodeled by PRC2 would help to better temporally define TE cell determination. Therefore, we first examined if EZH2 was required for the early transcriptional repression of MP-signature genes, such as Tcf7, Foxo1, Bach2 and Id3, in CD8+ T cells during acute infection (Fig S6). This showed that at day 4.5 p.i., the mRNA expression was reduced equivalently in both the Ezh2-deficient and Ezh2-sufficient CD8+ T cells relative to naïve CD8+ T cells (Fig 6A), indicating that H3K27me3 deposition was not required for the initial transcriptional repression of pro-memory genes during the early effector phase.
Figure 6. Increased H3K27me3 deposition occurs specifically in late-stage TE cells.

A) Tbx21, Tcf7, Foxo1, Bach2, and Id3 mRNA were measured in purified naïve (day 0) Ezh2f/f (solid) P14+ CD8+ T cells, and in Ezh2f/f (solid) and Ezh2f/f GzmBCre+ (open) P14+ CD8+ T cells from day 4.5 p.i. using qRT-PCR. Data are representative of 3 independent experiments (n=2–3 mice/group/experiment).
B) WT P14+ CD8+ T cells were purified from naïve (day 0) and LCMV-Armstrong infected mice at days 1.5, 4.5 (KLRG1Hi and KLRG1Lo populations), 10 (KLRG1HiIL7RLo TE and KLRG1LoIL7RHi MP populations), and 30 p.i., and then ChIP-qPCR was performed for H3K27me3 using primers to the Tcf7 TSS and Bach2 intron 1 (black). Region within Ttn served as a negative control (white) for H3K27me3 based on genome-wide H3K27me3 ChIP-seq analysis. Data are cumulative of four independent experiments (n=2–4 mice/group/experiment) with % of input for each sample normalized internally to the % of input for the day 30 sample, thereby calculating fold enrichment relative to the d30 sample as plotted. Data are expressed as mean ± SD. *p<0.05, **p<0.01. See also Figure S6.
Next, we monitored when H3K27me3 deposition increased at MP-signature loci (Tcf7 and Bach2) in effector CD8+ T cells and found that it was lower in naïve CD8+ T cells and early effector cells at days 1.5 and 4.5 p.i. compared to day 10 TE cells (Fig 6B). There was no difference observed between KLRG1Hi and KLRG1Lo effector cells at day 4.5 p.i.; this is when KLRG1Hi cells begin to appear during infection and demonstrate commitment to KLRG1Hi IL-7RLo TE cell fates (Joshi et al., 2007) (Fig 6B). However, by day 10 p.i., H3K27me3 dramatically increased preferentially in KLRG1Hi IL-7RLo TE cells, but not KLRG1Lo IL-7RHi MP cells (Fig 6B). This result suggested that increased H3K27me3 occurred during a late stage of effector T cell differentiation to stabilize repression of pro-memory genes whose transcription was already shut-down in developing TE cells. Moreover, d30 p.i. memory CD8+ T cells displayed low levels of H3K27me3 deposition, comparable to the MP cell population from which most memory cells descended (Fig 6B). Altogether, these results delineated a molecular timeline between commitment (early effector phase) and determination (late effector phase) of TE cell fates by revealing that EZH2-epigenetic remodeling was not required for early transcriptional repression of pro-memory genes, but was required later to sustain repression. This significant increase in H3K27me3 observed in the late effector phase may represent the quintessential step in TE cell fate determination.
FOXO1 regulates deposition of H3K27me3 in CD8+ T cells
What controls PRC2-mediated H3K27me3 deposition at pro-memory genes in TE cells? We reasoned that key transcription factors involved in MP and TE cell differentiation, such as T-bet, STAT4 and FOXO1, may be involved(Joshi et al., 2007, Kim et al., 2013). To examine this idea, we performed H3K27me3 ChIP-qPCR at the pro-memory genes Tcf7 and Bach2 from P14+ KLRG1Hi IL7RLo CD8+ T cells from d10 p.i. that were lacking (1) Foxo1 or (2) Stat4 or (3) retrovirally overexpressing (OE) T-bet. Control (WT) P14+ KLRG1Hi IL7RLo CD8+ T cells were infected with empty-vector-GFP retroviruses. Despite the profound effects of T-bet OE and Stat4-deficiency on promoting and repressing TE cell development, respectively, neither of these genetic alterations affected abundance of H3K27me3 at these loci relative to control (WT) cells. In contrast, KLRG1Hi IL7RLo cells lacking Foxo1 had significantly higher amounts (>2 fold) of H3K27me3 at the pro-memory genes Tcf7 and Bach2 (Fig 7A). This result suggested that FOXO1 restrained PRC2 activity at such pro-memory loci.
Figure 7. FOXO1 regulates H3K27me3 deposition.

A) WT (control cells infected with empty-vector-GFP retrovirus), Foxo1f/f CD4Cre+, Tbet-overexpressing (TbetOE), and Stat4−/− KLRG1Hi IL7RLo (TE) Thy1.1 P14+ CD8+ T cells were purified by FACS from infected mice from day 10 p.i. and ChIP-qPCR was performed for H3K27me3 using primers to the Tcf7 TSS and Bach2 intron 1 (black). A region within Ttn served as a negative control (white) for H3K27me3 based on genome-wide H3K27me3 ChIP-seq analysis. Data are cumulative from two (Stat4−/−), three (TbetOE), or four (Foxo1f/f CD4Cre+) independent experiments (n=2 [Stat4−/−], 7 [TbetOE], or 11 [Foxo1f/f CD4Cre+] mice/per group) with percentage of input for each sample normalized internally to the percentage of input of WT, thereby calculating fold enrichment relative to WT as plotted.
B) Bar graph shows FOXO1 protein levels in KLRG1Hi and KLRG1Lo WT P14+ CD8+ T cells and Foxo1f/f CD4Cre+ P14+ CD8+ T cells at d10 p.i. with LCMV-Armstrong in vivo.
Data are expressed as mean ± SD. *p<0.05, **p<0.01, ****p<0.0001
C) Significant FOXO1 binding sites (p-value < 10−5) identified by ChIP-seq from naive CD8+ T cells (GSE46943) were annotated to the nearest consensus peak. Density plots were calculated from the distribution of distances from a FOXO1 binding site to the nearest consensus peak and visualized separately for DMRs versus Common regions for H3K27ac (top) and H3K27me3 (bottom). Select genes associated with FOXO1 binding are noted below the density plots, and colored according to Cluster 1–4 or Common. See also Figure S7.
One may predict that if FOXO1 affects H3K27me3 at MP-signature genes, such as Tcf7 and Bach2, its expression may be higher in MP cells and it may preferentially bind to such loci. Using flow cytometry, we observed that KLRG1Lo IL-7RHi MP cells contained more FOXO1 protein than KLRG1Hi IL-7RLo TE cells (Fig 7B). We then examined putative FOXO1 binding sites in the four different clusters of DMRs identified in Figure 1 using previously generated FOXO1 ChIP-seq data ((Kim et al., 2013), GSE46943) from naïve CD8+ T cells. The density of FOXO1 peaks surrounding Common consensus peaks (i.e., non-DMRs), H3K27ac DMRs, or H3K27me3 DMRs in MP and TE cells (i.e., Clusters 1–4 defined in Fig 1) was measured and plotted (Fig 7C). FOXO1 binding was densely enriched near the H3K27ac Common peaks or DMRs in Cluster 1, suggesting that FOXO1 generally bound to active genes and open chromatin regions (note, FOXO1 binding was not concentrated proximal to H3K27ac DMRs in TE cells (Cluster 2)) (Fig 7C, top). There were no FOXO1 binding sites in H3K27me3 DMRs in MP cells (Cluster 3), but interestingly, FOXO1 binding sites were preferentially and densely enriched near H3K27me3 DMRs found in TE cells (Cluster 4) (Fig 7C, bottom). This analysis predicted that FOXO1 preferentially bound to gene loci that are transcriptionally active in MP cells and repressed in TE cells. Pathway analysis of the FOXO1-bound sites within 2kb of consensus peaks in CD8+ T cells showed that chromatin organization and histone modifications were the most highly enriched biological processes, suggesting a potentially important role for FOXO1 in regulating the chromatin state of CD8 T cells (Fig S7). Collectively, these results provide a deeper mechanistic understanding of how and when memory cell potential and longevity is gained or lost in effector CD8+ T cells during viral infection— decreased FOXO1 expression and binding to pro-memory genes in TE cells may allow for increased PRC2 activity and stable epigenetic repression, leading to the determination of a TE cell fate. Concurrently, increased FOXO1 expression in developing MP cells may shield pro-memory genes from H3K27me3 deposition, allowing for sustained or even increased expression as memory CD8+ T cells mature (Best et al., 2013, Kaech et al., 2002, Sarkar et al., 2008).
DISCUSSION
The mechanisms underlying differentiation and commitment of effector and memory CD8+ T cells is an active area of investigation. While the asymmetric cell division, signal-strength, and decreasing-potential models seek to explain how the TE and MP cell populations become distinct (Kaech and Cui, 2012), they do not explain how or when TE cells become committed to their identity and lose memory cell potential and longevity, nor how MP cells remain plastic to form diverse memory cell populations that can regenerate effector cells. Our study illuminated mechanisms for how this occurred by identifying that several pro-memory and pro-survival genes were selectively remodeled in developing KLRG1Hi IL-7RLo TE cells to contain greater H3K27me3 and lesser H3K27ac deposition, which more stably restricted transcription and consequently memory cell potential and lifespan. KLRG1Lo IL-7RHi MP cells, on the other hand, maintained pro-memory, pro-survival and TE-signature gene loci in active or permissive epigenetic states, allowing for the present expression of “memory” genes and the future expression of “effector” genes. Further, we showed that PRC2 complex members EZH2 and EED were required for effector cell expansion and the differentiation of KLRG1Hi IL-7RLo TE cells and that PRC2 activity may be restrained at MP-signature genes by FOXO1. These data provided mechanistic insight for how developmental plasticity was epigenetically wired in subsets of effector CD8+ T cells or lost in others as they terminally differentiated. In many respects, the epigenetic changes in TE cells resembled those in stem cells differentiating into specialized cell types— that is, the cells turned on lineage-determining genes (i.e., TE-signature genes) and silenced stemness genes (i.e., MP-signature genes). However, an important distinction in MP cells compared to embryonic stem cells is that EZH2 helps to preserve embryonic stem-cell identity by depositing H3K27me3 and repressing lineage-determining genes(Chen and Dent, 2014); contrastingly, our data showed that MP cells maintained TE-signature genes in a permissive state rather than a repressed state.
Our data more clearly delineated a step-wise process from specification to determination of TE fates during viral infection. While transcriptional repression of pro-memory genes occurs rapidly following CD8+ T cell activation (Best et al., 2013), our results showed this early transcriptional repression occurred independent of EZH2. Moreover, the pro-memory loci Tcf7 and Bach2 were not as heavily H3K27me3 methylated in KLRG1Hi cells at day 4.5 p.i. as compared to day 10 p.i. Thus, despite being committed to a TE cell fate at day 4.5, evidence of more stable epigenetic repression of pro-memory genes in TE cells did not occur until several days later; this suggested that determination of TE cell fates occurred at later stages of effector cell differentiation and H3K27me3 stabilized already repressed gene loci, similar to what has been observed in thymocyte lineage commitment (Zhang et al., 2012). Epigenetic silencing by DNA methylation also regulates CD8+ T cell effector formation (Scharer et al., 2013, Ladle et al., 2016) and thus, it will be important to study the temporal and spatial relationship between H3K27me3 and DNA methylation in effector CD8 T cells.
Prior work mapping genome-wide changes in histone acetylation and methylation in virus-specific effector and memory cells following murine influenza infection (Russ et al., 2014) demonstrated that many genes were epigenetically remodeled during the effector to memory transition. Although epigenetic differences between effector T cell subsets were not compared, as done here, it is possible that such changes occurred overtime because of selective maintenance of IL-7RHi MP cells. It is also possible that these changes occurred due to active removal of H3K27me3 in the memory cells and their progenitors by the H3K27me3-specific demethylases UTX and JMJD3 (Manna et al., 2015). Of interest, the mRNA expression of Utx and Jmjd3 is increased between the effector to memory cell transition (Kaech et al., 2002).
What drives this pro-memory gene specific, late deposition of H3K27me3 in TE cells? Given that sustained mTOR activity was found to drive TE cell differentiation and impair memory cell development(Kaech and Cui, 2012, Pollizzi et al., 2015), in part due to impaired FOXO1 activity(Kim et al., 2013), we reasoned that FOXO1 may be an interesting candidate to consider in regulating PRC2 activity. Moreover, FOXP3, another fork-head box TF family member, was found to bind to EZH2 and regulate PRC2 activity in Treg cells(DuPage et al., 2015, Arvey et al., 2014). Indeed, our data suggested that FOXO1 prevented deposition of H3K27me3 on certain pro-memory genes in CD8+ T cells. Although we were not able to demonstrate biochemical interaction between FOXO1 and PRC2 subunits in co-immunoprecipitation experiments in effector CD8+ T cells (data not shown), we found that Foxo1-deficiency, but not T-bet OE or Stat4-deficiency, led to increased H3K27me3 levels at certain pro-memory genes in KLRG1Hi IL-7RLo LCMV-specific effector CD8+ T cells. Given that FOXO1 was more highly expressed in MP than TE cells, we postulated that pro-memory genes may be shielded from H3K27me3 deposition in MP cells by FOXO1. Additionally, FOXO1 binding sites were enriched for genes involved in chromatin remodeling and organization in CD8+ T cells, suggesting that FOXO1 may actively remodel chromatin to retain a more pluripotent state in MP cells. Furthermore, increased AKT-mediated phosphorylation of FOXO1 in developing KLRG1Hi, IRF4Hi cells may lead to nuclear exclusion followed by ubiquitination, acetylation and proteosomal degradation of FOXO1, which commits the effector cells to terminal differentiation (Chang et al., 2011, Staron et al., 2014, Lin et al., 2015). Altogether, our data suggested that FOXO1 may regulate PRC2-mediated deposition of H3K27me3 in CD8+ T cells as they terminally differentiated; however, future studies are required to clarify the precise mechanism of FOXO1 regulation.
Our data also highlighted the role of epigenetic bivalency, where a locus contains both active-and repressive-associated histone modifications, in cellular differentiation. Similar to previous studies in CD8+ T cells (Russ et al., 2014, Araki et al., 2009), we found that many genes were bivalently modified during CD8+ T cell differentiation. Further, we found that the relative ratio of H3K27ac/H3K27me3 differed between MP and TE cells at numerous differentially expressed genes. TE cells had a lower relative ratio of H3K27ac/H3K27me3 than MP cells at many pro-memory and pro-survival genes, suggesting that the relative level, not simple presence or absence, of repressive to activating epigenetic modifications determined whether repressed pro-memory genes became stably epigenetically silenced. An alternative explanation of the observed bivalency is undistinguished heterogeneity in the TE and MP populations, wherein cells may differ epigenetically despite sharing the same population identifying surface markers. While 90% of TE cells appear stably terminally differentiated, we have previously shown that ~10% of these cells transform into IL-7RHi cells (Joshi et al., 2007). Thus, there may be some convertibility within this subset and it could involve epigenetic remodeling and erasing of H3K27me3 from the pro-memory gene loci in response to different stresses or tissue environments. In summary, this work identified a model for how memory cell potential was lost as effector CD8+ T cells terminally differentiated through epigenetic silencing of genes controlling T cell survival and memory cell fates, and adds to our growing understanding of the profound importance of epigenetic regulation in T cell plasticity and developmental potential (Vahedi et al., 2012).
EXPERIMENTAL PROCEDURES
Mice, infections and treatments
C57BL/6 (B6) mice were purchased from Charles River Laboratories (Fredericksburg, VA). P14 (LCMV H-2Db GP33-41 specific) T cell receptor (TCR) transgenic mice were obtained from R. Ahmed(Pircher et al., 1989). Ezh2flox/flox(Shen et al., 2008), Eedflox/flox(Xie et al., 2014), and CD4Cre mice(Lee et al., 2001) were purchased from Jackson Laboratories (Bar Harbor, ME). GranzymeB-Cre (GzmBCre) mice(Jacob and Baltimore, 1999) were provided by J. Jacob (Emory University, Atlanta, GA). GranzymeB-ERT2Cre ROSA26floxSTOPfloxYFP mice(Bannard et al., 2009) were provided by Doug Fearon (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). Foxo1flox/flox mice(Ouyang et al., 2009) were provided by Ming Li (Memorial Sloan Kettering Cancer Center, New York, NY). Stat4−/− mice(Kaplan et al., 1996) were provided by M. Grusby (Harvard Medical School, Boston, MA). “P14 chimeric mice” were generated by transferring 5–10×104 Thy1.1+ P14 CD8+ T cells to naive Thy1.2+ B6 recipients. Ezh2Δ/flox PCR was previously described(Shen et al., 2008). All animal experiments were completed according to approved Institutional Animal Care and Use Committee protocols. Details of infections and treatments are found in the Supplemental Experimental Procedures.
Gene expression by qRT-PCR and immunoblotting
Immunoblotting was performed as previously described using rabbit monoclonal antibodies from Cell Signaling: EZH2 (D2C9), β-Actin (13E5), H3K27me3 (C62B11), H3 (D1H2)(Hand et al., 2007). Details of these procedures and list of qPCR primers and antibodies are found in the Supplemental Experimental Procedures.
Chromatin immunoprecipitation (ChIP) and ChIP-Sequencing (ChIP-Seq)
ChIP was performed with anti-H3K27me3 (Abcam, ab6002), anti-H3K27ac (Abcam, ab4729), and anti-mouse IgG antibodies. Sequencing was performed on an Illumina HiSeq 2500 with 4 samples per lane (170M reads are distributed at 42.5M reads per sample with 75bp reads in single-end mode). Details of these procedures and list of qPCR primers are found in the Supplemental Experimental Procedures.
Analysis of ChIP-seq Data and Differential Modification Analysis
Details of these analyses are found in the Supplemental Experimental Procedures.
Flow cytometry, surface and intracellular staining, peptide stimulations, and antibodies
Flow cytometry and sorting were performed on an LSRII and BD FACS Aria II (Becton Dickinson), respectively, and analyzed with FlowJo software (FlowJo, LLC). Details of these procedures and list of antibodies are found in the Supplemental Experimental Procedures.
In vitro culture and CellTrace Violet proliferation assay
Details of these procedures and list of qPCR primers are found in the Supplemental Experimental Procedures.
Mixed bone marrow chimeras and Retroviral overexpression
MSCV-empty-vector-GFP and MSCV-T-bet-GFP (Tbet-overexpression)(Szabo et al., 2000) were obtained from L. Glimcher (Dana Farber, Boston, MA). Details of these procedures are found in the Supplemental Experimental Procedures.
Statistical Analysis
Results from flow cytometry and qPCR data were presented as mean ± standard deviation. Statistical significance was computed with Prism 7 (GraphPad Software) by paired or unpaired Student’s t test with a p-value of < 0.05 considered as significant. Details of statistical analysis for ChIP-Seq data are found in the Supplemental Experimental Procedures.
Supplementary Material
HIGHLIGHTS.
H3K27me3 is more abundant at certain pro-memory genes in TE CD8+ T cells
PRC2 is required for CD8+ T cell clonal expansion and TE differentiation
H3K27me3 is deposited during late effector CD8+ T cell differentiation
FOXO1 regulates H3K27me3 deposition at certain pro-memory loci in TE cells
Acknowledgments
We thank members of the Kaech and Kleinstein laboratories for helpful comments and suggestions. This study was supported in part by the Howard Hughes Medical Institute (HHMI) (S.M.K.), HHMI Gilliam Fellowship (R.A.A.), the HHMI International Student Research Fellowship (T.G.), the National Institutes of Health (NIH) grants R37 AI066232 (S.M.K.), F30 AI114090 (S.M.G), 1S10OD018521 (S. M. Mane), NIH/NIGMS T32 GM007205 (S.M.G.). This work was supported by the HPC facilities operated by the Yale Center for Research Computing and the Yale Center for Genome Analysis.
Footnotes
AUTHOR CONTRIBUTIONS
S.M.G, R.A.A, T.G. and S.M.K designed the research. S.M.G. and T.G performed the experiments. R.A.A. performed the bioinformatics analyses under the advisement of S.H.K. and S.M.K. S.M.G., R.A.A., T.G., S.H.K. and S.M.K. analyzed the results. S.M.G., R.A.A. and S.M.K. wrote the manuscript. All authors read and approved the final version of this manuscript.
Accession Numbers
Data is available online at the Gene Expression Omnibus (GSE72408) and the Sequence Read Archive (SRA273724 and SRP101899).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Araki Y, Wang Z, Zang C, Wood WH, 3rd, Schones D, Cui K, Roh TY, Lhotsky B, Wersto RP, Peng W, Becker KG, Zhao K, Weng NP. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 2009;30:912–25. doi: 10.1016/j.immuni.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvey A, Van Der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, Rudensky AY. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol. 2014 doi: 10.1038/ni.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannard O, Kraman M, Fearon DT. Secondary replicative function of CD8+ T cells that had developed an effector phenotype. Science. 2009;323:505–9. doi: 10.1126/science.1166831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A, Dustin ML, Goldrath AW. Transcriptional insights into the CD8+ T cell response to infection and memory T cell formation. Nat Immunol. 2013;14:404–412. doi: 10.1038/ni.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JT, Ciocca ML, Kinjyo I, Palanivel VR, Mcclurkin CE, Dejong CS, Mooney EC, Kim JS, Steinel NC, Oliaro J, Yin CC, Florea BI, Overkleeft HS, Berg LJ, Russell SM, Koretzky GA, Jordan MS, Reiner SL. Asymmetric proteasome segregation as a mechanism for unequal partitioning of the transcription factor T-bet during T lymphocyte division. Immunity. 2011;34:492–504. doi: 10.1016/j.immuni.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. 2014;15:93–106. doi: 10.1038/nrg3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez CX, Amezquita RA, Guan T, Marshall HD, Joshi NS, Kleinstein SH, Kaech SM. The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J Exp Med. 2015;212:2041–56. doi: 10.1084/jem.20150186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, Zhang R, Turka L, Marson A, Bluestone JA. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity. 2015;42:227–38. doi: 10.1016/j.immuni.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach C, Moseman A, Loughhead S, Alvarez D, Von Andrian UH. CX3CR1 distinguishes three antigen-experienced CD8 T cell subsets with distinct migratory, functional and homeostatic properties. The Journal of Immunology. 2016;196:64.3–64.3. [Google Scholar]
- Gunawan M, Venkatesan N, Loh JT, Wong JF, Berger H, Neo WH, Li LYJ, Win MKL, Yau YH, Guo T, See PCE, Yamazaki S, Chin KC, Gingras AR, Shochat SG, Ng LG, Sze SK, Ginhoux F, Su IH. The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nat Immunol. 2015 doi: 10.1038/ni.3125. advance online publication. [DOI] [PubMed] [Google Scholar]
- Hand TW, Morre M, Kaech SM. Expression of IL-7 receptor α is necessary but not sufficient for the formation of memory CD8 T cells during viral infection. Proceedings of the National Academy of Sciences. 2007;104:11730–11735. doi: 10.1073/pnas.0705007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob J, Baltimore D. Modelling T-cell memory by genetic marking of memory T cells in vivo. Nature. 1999;399:593–7. doi: 10.1038/21208. [DOI] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation Directs Memory Precursor and Short-Lived Effector CD8+ T Cell Fates via the Graded Expression of T-bet Transcription Factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Cui WG. Transcriptional control of effector and memory CD8(+) T cell differentiation. Nature Reviews Immunology. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–51. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged Interleukin-2Rα Expression on Virus-Specific CD8+ T Cells Favors Terminal-Effector Differentiation In Vivo. Immunity. 2010;32:91–103. doi: 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–7. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. The Transcription Factor Foxo1 Controls Central-Memory CD8+ T Cell Responses to Infection. Immunity. 2013 doi: 10.1016/j.immuni.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladle BH, Li KP, Phillips MJ, Pucsek AB, Haile A, Powell JD, Jaffee EM, Hildeman DA, Gamper CJ. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T-cell fate decisions following activation. Proc Natl Acad Sci U S A. 2016;113:10631–6. doi: 10.1073/pnas.1524490113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–74. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- Lin WH, Adams WC, Nish SA, Chen YH, Yen B, Rothman NJ, Kratchmarov R, Okada T, Klein U, Reiner SL. Asymmetric PI3K Signaling Driving Developmental and Regenerative Cell Fate Bifurcation. Cell Rep. 2015;13:2203–18. doi: 10.1016/j.celrep.2015.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14:1294–301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- Manna S, Kim JK, Bauge C, Cam M, Zhao Y, Shetty J, Vacchio MS, Castro E, Tran B, Tessarollo L, Bosselut R. Histone H3 Lysine 27 demethylases Jmjd3 and Utx are required for T-cell differentiation. Nat Commun. 2015;6:8152. doi: 10.1038/ncomms9152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omilusik KD, Best JA, Yu B, Goossens S, Weidemann A, Nguyen JV, Seuntjens E, Stryjewska A, Zweier C, Roychoudhuri R, Gattinoni L, Bird LM, Higashi Y, Kondoh H, Huylebroeck D, Haigh J, Goldrath AW. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J Exp Med. 2015;212:2027–39. doi: 10.1084/jem.20150194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Beckett O, Flavell RA, Li MO. An Essential Role of the Forkhead-Box Transcription Factor Foxo1 in Control of T Cell Homeostasis and Tolerance. Immunity. 2009;30:358–371. doi: 10.1016/j.immuni.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–61. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, Delgoffe GM, Powell JD. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest. 2015;125:2090–108. doi: 10.1172/JCI77746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollizzi KN, Sun IH, Patel CH, Lo YC, Oh MH, Waickman AT, Tam AJ, Blosser RL, Wen J, Delgoffe GM, Powell JD. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat Immunol. 2016;17:704–11. doi: 10.1038/ni.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ BE, Olshanksy M, Smallwood HS, Li J, Denton AE, Prier JE, Stock AT, Croom HA, Cullen JG, Nguyen ML, Rowe S, Olson MR, Finkelstein DB, Kelso A, Thomas PG, Speed TP, Rao S, Turner SJ. Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8(+) T cell differentiation. Immunity. 2014;41:853–65. doi: 10.1016/j.immuni.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205:625–40. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol. 2013;191:3419–29. doi: 10.4049/jimmunol.1301395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell. 2008;32:491–502. doi: 10.1016/j.molcel.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HM, Kapoor VN, Guan T, Kaech SM, Welsh RM, Berg LJ. Epigenetic modifications induced by Blimp-1 Regulate CD8(+) T cell memory progression during acute virus infection. Immunity. 2013;39:661–75. doi: 10.1016/j.immuni.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, Cui G, Li MO, Kaech SM. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014;41:802–14. doi: 10.1016/j.immuni.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–69. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, O’shea John J. STATs Shape the Active Enhancer Landscape of T Cell Populations. Cell. 2012;151:981–993. doi: 10.1016/j.cell.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbist KC, Guy CS, Milasta S, Liedmann S, Kaminski MM, Wang R, Green DR. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature. 2016;532:389–93. doi: 10.1038/nature17442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, Orkin SH. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell. 2014;14:68–80. doi: 10.1016/j.stem.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XP, Jiang K, Hirahara K, Vahedi G, Afzali B, Sciume G, Bonelli M, Sun HW, Jankovic D, Kanno Y, Sartorelli V, O’shea JJ, Laurence A. EZH2 is crucial for both differentiation of regulatory T cells and T effector cell expansion. Sci Rep. 2015;5:10643. doi: 10.1038/srep10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell. 2012;149:467–82. doi: 10.1016/j.cell.2012.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.