

Abstract
Oncolytic virotherapy represents an attractive option for the treatment of a variety of aggressive or refractory tumors. While this therapy is effective at rapidly debulking directly injected tumor masses, achieving complete eradication of established disease has proven difficult. One method to overcome this challenge is to use oncolytic viruses to induce secondary anti-tumor immune responses. Unfortunately, while the initial induction of these immune responses is typically robust, their subsequent efficacy is often inhibited through a variety of immunoregulatory mechanisms, including the PD1/PDL1 T-cell checkpoint pathway. To overcome this inhibition, we generated a novel recombinant myxoma virus (vPD1) which inhibits the PD1/PDL1 pathway specifically within the tumor microenvironment by secreting a soluble form of PD1 from infected cells. This virus both induced and maintained anti-tumor CD8+ T-cell responses within directly treated tumors and proved safer and more effective than combination therapy using unmodified myxoma and systemic αPD1 antibodies. Localized vPD1 treatment combined with systemic elimination of regulatory T cells had potent synergistic effects against metastatic disease that was already established in secondary solid organs. These results demonstrate that tumor-localized inhibition of the PD1/PDL1 pathway can significantly improve outcomes during oncolytic virotherapy. Furthermore, they establish a feasible path to translate these findings against clinically relevant disease.
Keywords: Oncolytic virotherapy, myxoma virus, checkpoint blockade, PD1
Introduction
Oncolytic virotherapy (OV) has long been seen as an attractive option for the treatment of a number of human cancers (1, 2). Recent results have suggested that OV likely functions through the induction of anti-tumor T-cell responses (3). Unfortunately, while the initial induction of these responses can be observed in both preclinical models (4–7) and human trials (8–12), they frequently result in only modest long-term efficacy. Identifying mechanisms and/or pathways which restrict the efficacy of OV is therefore highly significant.
One of the most exciting recent breakthroughs in cancer immunotherapy has been the discovery of T-cell checkpoint inhibitors, such as the PD1-blocking antibodies nivolumab and pembrolizumab. These antibodies are believed to function by sterically inhibiting the binding of PD1 to its primary ligand PDL1. This prevents exhaustion of anti-tumor T-cells and significantly enhances the efficacy of existing anti-tumor immune responses. Unfortunately, while PD1 blockade can result in complete regression of even large established tumors, its efficacy relies on the enhancement of existing anti-tumor responses. Due to this requirement, only 10–40% of patients typically respond to PD1-based treatment (13–18). A variety of studies have demonstrated that this response rate can be increased by combining PD1-blockade with additional therapies which improve initial immune activation, such as chemotherapeutic agents (19–21) or immune modulating cytokines (22, 23). These combination therapies, however, are often associated with unacceptably high rates of grade-3 and grade-4 autoimmune-like toxicities (22, 23).
Due to the known propensity for OV to induce initially robust anti-tumor immune responses, and the ability of PD1-blockade to maintain these responses, the combination of OV and checkpoint blockade is viewed as therapeutically attractive (24, 25). Unfortunately, preliminary data from clinical trials combining IMLYGIC® with PD1-blockade suggests that this combination likely results in the same increases in autoimmune-like toxicity seen with other combination therapies (26). In order to overcome this increased toxicity, while maintaining an improved response rate, we generated a recombinant oncolytic myxoma virus (MYXV) designed to inhibit the PD1/PDL1 pathway specifically within the local tumor microenvironment by secreting a soluble form of PD1 from infected cells. Our subsequent studies indicate that, not only is this virus capable of completely eradicating large, established tumors, but monotherapy actually outperforms the combination of parental MYXV and antibody-based PD1-blockade. Additionally, localized treatment with this virus, in combination with elimination of regulatory T-cells, displays potent synergistic effects against metastatic disease already established in secondary solid organs. These results demonstrate that tumor localized inhibition of the PD1/PDL1 checkpoint can significantly improve outcomes during OV and also provide a feasible path to translate these findings against clinically relevant disease.
Methods
Cell Lines and Reagents
B16/F10 cells were a kind gift from Dr. Chrystal Paulos at the Medical University of South Carolina in 2014. Cells were authenticated through morphological examination, expression of melanin, and ability to form subcutaneous tumors in syngeneic C57/B6 mice. PDL1 deficient B16/F10 cells (B16/F10-PDL1−/−) were generated using the CRISPR/CAS9 system (Genscript, Piscataway, NJ, USA) per the manufacturer’s recommendations. In short, B16/F10-WT cells were transfected with the plasmid pSpCas9BB-24-2A-Puro containing CD274 guide RNA 1 (seq GTATGGCAGCAACTGCACG). 48 hours after transfection, cells were transferred to media containing 1μg/ml puromycin for one week to select for transfected cells. Cells were then removed from selective media and expanded. Ablation of PDL1 expression was confirmed using flowcytometry and western blot (Figure S1). Lewis Lung Carcinoma (LLC) cells (Cat# CRL-1642, purchased 5/2016) and BSC40 cells (Cat# CRL-2761, purchased 3/2013) were purchased from ATCC (Manassas, VA, USA) and passaged for less than six months prior to use. All cells were cultured in DMEM + 10% Fetal Bovine Serum + 1x Penicillin-Streptomycin-L-Glutamine (Mediatech, Manassas, VA, USA). Cell viability was determined using the CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTT) assay (Promega, Madison, WI, USA) per manufacturer’s recommendations. Secretion of soluble human PD1 was measured using the human PD-1 DuoSet ELISA (R&D Systems, Minneapolis, MN, USA) per manufacturer’s recommendations. Secretion of IFNγ from stimulated murine splenocytes was measured using the mouse interferon gamma OPTEIA duo ELISA (BD Biosciences, Franklin Lakes, NJ, USA) per manufacturer’s recommendations. The following antibodies were used in these studies. For flowcytometry: PDL1 (clone MIH5), PD1 (clone J43), CD3 (clone 145-2c11), CD4 (clone RM4–5), CD8 (clone 53–6.7), CD69 (clone H1.2F3), CD25 (clone 3C7), CD11b (clone M1/70), NK1.1 (clone PK136), H2-Kb (clone AF6-88.5) (BD Biosciences, Franklin Lakes, NJ, USA). For western blot: PD1 (clone NAT105) (Abcam, Cambridge MA, USA), PDL1 (Cat# AF1019) (R&D Systems, Minneapolis, MN, USA), actin (clone I19) (Santa Cruz Biotechnology, Dallas, TX, USA).
Virus Generation, Amplification, Purification, and Infection
To construct vPD1, the extracellular region of human PD1 (amino acids 1–168) was amplified from a preconstructed template plasmid (PlasmID database, clone HsCD00345685) by PCR using the following primers:
For: ATCGCCCGGGAAAAATTGAAATTTTATTTTTTTTTTTTGGAATATAAATAACCATGCAGATCCCACAGGCGCC
Rev: ATCGGAATTCTCAGGTTTGGAACTGGCCGGCTG
The PCR fragment was then digested with XmaI and EcoR1 and ligated into the existing pBlueScript-M135/GFP/M136 poxviral recombination plasmid (27). The resulting plasmid (pBlueScript-M135/GFP/PD1/M136) was then transfected into BSC40 cells. After 24 hours, the transfected cells were infected with wild-type MYXV (strain Lausanne) at a multiplicity of infection (MOI)=10. After allowing 48 hours for recombination, cells were harvested and recombinant virus was clonally purified through four rounds of GFP+ foci selection. vGFP was constructed by recombining parental pBlueScript-M135/GFP/M136 with MYXV (strain Lausanne) as detailed above. Virus was amplified in BSC40 cells and purified using gradient centrifugation as previously described (28). Viral titer was determined through serial infection of BSC40 cells. Unless otherwise noted, experiments were carried out by infecting cells for 60 minutes using an MOI = 10.
In vivo tumor models
For subcutaneous (SQ) tumor models, 6–8 week old, female C57/B6 mice (Charles River Laboratories, Raleigh, NC, USA) were injected SQ with 4x105 B16/F10 cells or 4x105 LLC cells in 100ul sterile PBS. For metastatic tumor models, 6–8 week old, female C57/B6 mice (Charles River Laboratories, Raleigh, NC, USA) were injected the same day with both 4x105 B16/F10 cells SQ in 100μl sterile PBS and 1x105 B16/F10 cells in 200μl sterile PBS intravenously (IV) through the lateral tail vein. In all models, treatment began 7 days post-injection (a time at which SQ tumors were typically ~40mm2 in size). Unless otherwise noted, treatment consisted of three intratumoral injections of either saline or the indicated virus (1x107 total foci forming units (FFU)) in 100ul PBS on days 7, 9, and 11 post tumor injection. In vivo depletion of immune cell subsets was accomplished by injecting depleting antibodies intraperitoneally (IP) on days 5 and 12 post-tumor injection. The following antibodies were used for in vivo depletion studies: CD4 (clone GK1.5, 100ug/injection), CD8 (clone 53–6.72, 100ug/injection), NK1.1 (clone PK136, 200ug/injection), control IgG (clone 2A3) (BioXcell, West Lebanon, NH, USA). Efficiency and specificity of depletion was confirmed in control animals using flowcytometry (Figure S2). Antibody-based blockade of PD1 was accomplished by injecting PD1-blocking antibody (clone RMP1-14, 100ug/injection) (BioXcell, West Lebanon, NH, USA) IP on days 7, 11, 14, and 18 post tumor injection. For SQ survival studies, tumor burden was monitored using calipers and mice were euthanized when tumors reached 15mm in any direction. For metastatic survival studies, mice were monitored daily for body condition and weight and euthanized when they displayed either a body condition score of 2.0 (29) or a loss of >20% of their starting body weight. Alopecia was scored according to the following scoring system: 0 = no hair loss, 1 = hair loss <100mm2 in 1 location, 2 = hair loss either 100–300mm2 in 1 location or <100mm2 in 2 locations, 3 = hair loss >300mm2. Size of each metastasis in the lung was scored post-mortem according to the following scoring system: 1 = <1mm, 2 = ~1mm, 3 = 1–1.5mm, 4 = 1.5–2mm, 5 = >2mm. Antitumor memory of mice was assessed by rechallenging mice with 2x105 B16/F10 cells injected SQ on the contralateral flank from initial tumor site on day 100 post initial tumor injection. All experiments were conducted in accordance with the MUSC Institutional Animal Care and Use Committee (IACUC).
Tumor Infiltrating Lymphocyte Analysis
Excised tumors were transferred onto a 40μM nylon mesh filter and mechanically separated into a single cell suspension in PBS. Cells were then pelleted at 1200rpm for 5min at 4°C and resuspended in 3mls of PBS. Cell suspension was then layered onto 6mls of warmed Histopaque 1083 and centrifuged for 30min at 400g at 25°C. Cells banding at the Histopaque/PBS interface were extracted, pelleted, and stained for flowcytometry using standard methodologies.
Histology
Freshly excised tissue was fixed in 2% formalin overnight and then transferred to 70% ethanol. Tissues were then sectioned, mounted on slides and stained using the Vector- Vectastain rabbit kit (Vector Laboratories, Burlingame, CA, USA) according to manufacturer’s recommendations. In short, sections were deparaffinized and hydrated using xylene followed by a graded alcohol series. Peroxidase activity was quenched using 3% hydrogen peroxide and then sections were incubated for one hour with normal blocking serum. After blocking, sections were incubated with the appropriate antibody for one hour followed by incubation with appropriate secondary antibody for 30 minutes. Sections were incubated with VECTASTAIN® ABC Reagent for 30 minutes and then visualized using peroxidase substrate solution (DAB) until desired intensity is observed. Sections were then hydrated, cleared, and mounted. Hematoxylin and Eosin staining was done according to standard methodologies.
Results
MYXV synergizes with PD1-blockade
Previous studies have demonstrated that MYXV treatment can stabilize but not regress a variety of established tumors (30, 31). We hypothesized that this failure might be due to an inhibition of virally-induced anti-tumor immunity caused by activation of the PD1/PDL1 T-cell checkpoint. To address this question, we used the B16/F10 murine melanoma model which has been shown to be relatively resistant to both MYXV and PD1-based monotherapy. Consistent with previous reports (32), we found that B16/F10 cells constitutively expressed low levels of PDL1 both intracellularly (Figure 1A) and at the cell surface (Figure 1B). This low level expression was not significantly altered following infection with MYXV in vitro. Similarly, no obvious differences in PDL1 expression were observed on tumor cells following MYXV treatment of B16/F10 tumors in vivo (Figure S3) suggesting that viral infection does not significantly alter expression of PDL1 in B16/F10 cells either in vitro or in vivo. In contrast, viral treatment of established melanomas in vivo led to a marked increase in the numbers of PD1+/CD8+ cells in the spleens of treated animals (Figure 1C). The numbers of PD1+/CD4+ splenocytes were not significantly altered following viral treatment. Since these data indicates that both PD1 and PDL1 were likely present during MYXV therapy, we next wish to determine whether this pathway functionally impacted the outcomes of viral therapy. To test this, we genetically eliminated expression of PDL1 in B16/F10 murine melanoma cells using CRISPR (Figure S1) and then compared the efficacy of MYXV treatment in these PDL1−/− cells to that seen in parental PDL1+ cells. Consistent with previous reports (30, 31), MYXV treatment of parental B16/F10 tumors resulted in a significant delay of tumor growth; however, treatment failed to completely eradicate disease and all animals eventually succumbed to tumor burden by day 38. In stark contrast, MYXV treatment of PDL1−/− tumors resulted in the complete eradication of disease in 13 out of 14 treated animals (Figure 1D) suggesting that tumor expressed PDL1 played a critical role in determining the outcome of MYXV therapy. To identify whether this finding might be clinically translatable, we next asked whether antibody-based blockade of the PD1/PDL1 pathway would also improve the efficacy of MYXV treatment. PDL1+ B16/F10 tumors were treated with either saline or MYXV in either the presence or absence of systemically injected αPD1 antibody. The results indicated that, while neither MYXV nor αPD1 monotherapy could eradicate established tumors, the combination of virus plus PD1 blockade was capable of inducing long-term tumor regression in some animals. The efficacy of this combination therapy, however, was significantly less than that observed for MYXV monotherapy in genetically PDL1−/− tumors (compare Figure 1E to Figure 1D). Additionally, mice treated with the combination of MYXV and αPD1 antibody also displayed symptoms of an autoimmune-like toxicity characterized by progressive alopecia and infiltration of lymphocytes into the skin (Figures 1F and 1G). While MYXV and αPD1 monotherapies failed to prolong the survival of PDL1+ tumor bearing animals sufficiently to analyze their individual impact on alopecia, hair loss was not observed in PDL1−/− tumor bearing animals treated solely with MYXV (Figure 1G) suggesting it was not caused by either viral monotherapy or simply the presence of a melanoma tumor.
Figure 1. PD1 blockade enhances MYXV treatment of established melanoma.

(A) B16/F10 cells were either mock infected or infected with MYXV at MOI=10. 24 hours post infection cells were harvested and the total level of PDL1 was analyzed using westernblot (band representing PDL1 is indicated by arrow). Expression of β Actin is shown as a loading control. Data shown is representative is three independent experiments. (B) B16/F10 cells were either mock infected or infected with MYXV at MOI=10. 24 hours post infection cells were harvested and the cell surface expression of PDL1 was analyzed using flowcytometry. Data shown is representative of two independent experiments. (C) C57/B6 mice were injected SQ with 4x105 B16/F10 cells. On days 7, 9, and 11 post injection mice were given IT injection of either saline (n=9) or 1x107 FFU of MYXV (n=9). On day 12, mice were euthanized and the percentage of either CD4+ or CD8+ splenocytes expressing PD1 was determined using flowcytometry. Data represents the summation of four independent experiments. Significance was determined using unpaired students T-Test. (D) C57/B6 mice were injected SQ with either 4x105 B16/F10-wt or B16/F10-PDL1−/− cells. On days 7, 9, and 11 post injection mice were given IT injection of either saline (B16/F10 n=5, B16/F10-PDL1−/− n=5) or 1x107 FFU of MYXV (B16/F10 n=5, B16/F10-PDL1−/− n=14). Animals were then monitored for tumor burden and euthanized when tumors reached 15mm in any direction. Data is representative of two independent experiments. (E) C57/B6 mice were injected SQ with 4x105 B16/F10 cells. On day 7 post injection, mice were randomly separated into one of four cohorts: saline + isotype (n=9), saline + αPD1 antibody (n=8), MYXV + isotype (n=5), MYXV + αPD1 antibody (n=7). Viral injections consisted of 1x107 FFU of MYXV and were given on days 7, 9, and 11. Antibody injections consisted of 100μg/injection and were given on days 7, 11, 14, and 18. Animals were then monitored for tumor burden and euthanized when tumors reached 15mm in any direction. Data is representative >four independent experiments. (F) Example of alopecia in mice on day 70 post-treatment and H&E stain from the belly skin of displayed animal. (G) Average alopecia score from MYXV + αPD1 antibody treated animals (n=7, shown in 1E) and B16/F10-PDL1−/− bearing animals treated with MYXV (n=14, shown in 1D). * denotes significance of p<0.05 for the indicated day and was determined using unpaired students T-Test.
Generation of recombinant MYXV expressing soluble PD1
Since our data indicated that tumor-expressed PDL1 was the primary mechanism restricting the efficacy of MYXV treatment, we hypothesized that a tumor localized blockade of the PD1/PDL1 pathway might duplicate the improved outcomes seen in our previous experiments without the associated systemic toxicities. It has been previously reported that, similar to αPD1 antibodies, soluble splice variants of PD1 can also inhibit the PD1/PDL1 checkpoint and improve overall T-cell function (33, 34). In contrast to αPD1 antibodies, however, these PD1 splice variants are small single chain polypeptides which can be easily encoded into the genome of an OV. We therefore sought to generate a MYXV construct (vPD1) which would demonstrate improved efficacy against established PDL1+ tumors by secreting a soluble form of PD1 from infected cells. To accomplish this, we incorporated the extracellular region of human PD1 (aa 1–168), as well as the marker gene GFP, into the intergenic region of the MYXV genome between the viral M135R and M136R open reading frames (Figure 2A). A matched control virus (vGFP) which incorporated GFP, but not PD1, into the same intergenic locus was also generated. In vitro analysis demonstrated that inclusion of the PD1 transgene did not alter the production of new infectious progeny virus within infected B16/F10 cells (Figure 2B) or the acute oncolytic capacity of MYXV to kill directly infected cells (Figure 2C). Western blot analysis against PD1, however, revealed that infection of B16/F10 cells with vPD1 resulted in the novel expression of a protein of approximately 30kDa, which corresponds to the predicted size of a properly glycosylated fragment of PD1 (35) (Figure 3A). This fragment could also be detected in the supernatant of vPD1 infected B16/F10 cells by ELISA beginning six hours post-infection, with levels continuing to increase the longer infection progressed (Figure 3B). In vivo, expression of the PD1 transgene could be observed in vPD1 infected tumor cells using immunohistochemistry (Figure 3C) and soluble PD1 transgene could be detected in the extracellular medium following vPD1 treatment using ELISA (Figure 3D). Analysis of the bio distribution of the soluble PD1 transgene indicated that a single viral injection resulted in long-term PD1 expression with transgene being readily observable up to a week after injection. This expression was largely localized within the treated tumor although low concentrations of PD1 transgene could also be detected in the serum as well as in peripheral tissues such as the lung. No PD1 was detected in immunological organs such as the spleen (Figure 3D).
Figure 2. Construction of MYXV expressing soluble PD1.
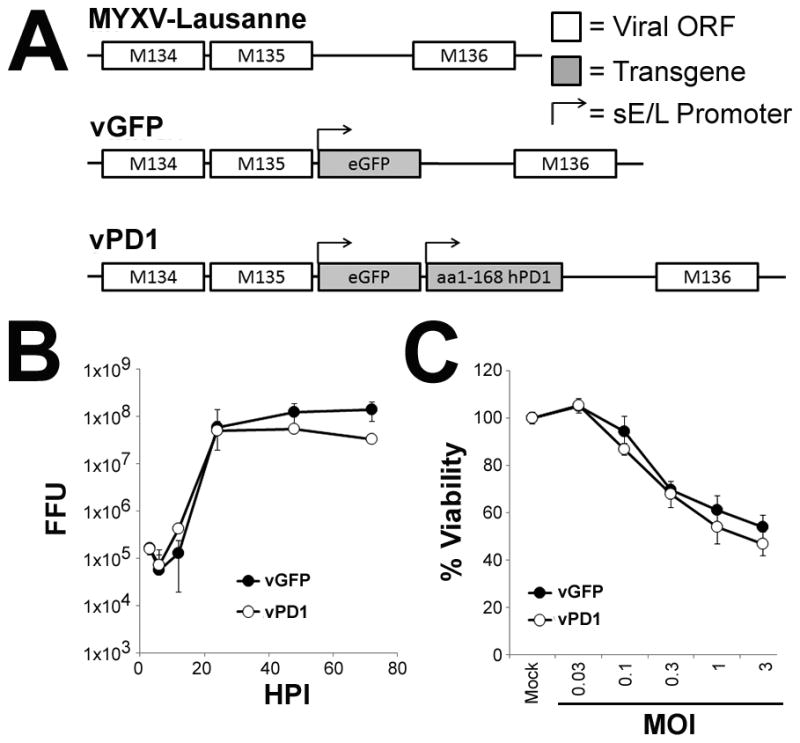
(A) Schematic of the genomic structure of unmodified MYXV and the derivative recombinant MYXV’s vGFP and vPD1. (B) B16/F10 cells were infected with either vGFP or vPD1 at MOI=10. At the indicated times post-infection, cells were harvested and frozen. Amount of infectious virus present at each time point was then determined by mechanically lysing cells and counting the number of GFP+ foci formed following a serial titration onto BSC40 cells. Data represents summation of two independent experiments. (C) B16/F10 cells were mock infected or infected with the indicated MOI’s of either vGFP or vPD1. 24 hours post-infection, cellular viability was determined using MTT assay. Data is shown as percent viability compared to mock-infected control and represents the summation of two independent experiments. Abbreviations: FFU (foci forming units), HPI (hours post infection), MOI (multiplicity of infection), sE/L (poxviral synthetic early/late promoter).
Figure 3. vPD1 secretes soluble PD1 specifically within the tumor microenvironment.

(A) B16/F10 cells were either mock infected or infected with vGFP or vPD1 at MOI=10. 24 hours post infection cells were harvested and the expression of intracellular PD1 was analyzed using westernblot. Expression of β Actin is shown as a loading control. Data shown is representative of two independent experiments. (B) B16/F10 cells were infected with either vGFP or vPD1 at MOI=10. At the indicated times post-infection, supernatant was collected from each infection and frozen. The concentration of soluble human PD1 in each sample was then determined using ELISA. Dotted line indicates approximate detection limit of assay. Data shown is representative of two independent experiments. (C) C57/B6 mice were injected SQ with 4x105 B16/F10 cells. On day 7 post-injection, mice were treated with a single IT injection of either saline or 1x107 FFU of vGFP or vPD1. 48 hours post-treatment, tumors were harvested and stained for either GFP or human PD1 using immunohistochemistry. Data shown is representative of two independent experiments each with n>5. (D) C57/B6 mice were injected SQ with 4x105 B16/F10 cells. On day 7 post-injection, mice were treated with a single IT injection of either saline (n=3) or 1x107 FFU of vGFP (n=3) or vPD1 (n=3). At the indicated times post infection, animals were euthanized and serum as well as the indicated tissues were harvested. Tissues were disassociated over a 40μM mesh and separated into cell and non-cell fractions. The concentration of human PD1 in the non-cell-associated fraction was then determined using ELISA. Dotted line indicates approximate detection limit of assay. Data is representative of two independent experiments.
vPD1 monotherapy eradicates established melanoma
To determine the efficacy of vPD1 at treating established tumors, we next performed a series of experiments in which we compared the efficacy of vPD1 monotherapy to that of vGFP combined with antibody-based PD1-blockade in both the B16/F10 and LLC models. Consistent with our previous results, MYXV monotherapy (vGFP + isotype antibody) resulted in only a minor delay in B16/F10 tumor growth (Figure 4A) with an overall response (OR) rate of 70% (Figure 4B). No animals given MYXV monotherapy displayed a complete response (CR) (Figure 4C). In contrast, while mice treated with vGFP and αPD1 antibody displayed only a small trend towards improved OR rates (70% vs 90%, p=0.40), this combination therapy resulted in the complete eradication of established melanomas in ~30% (5/17) of mice. Unfortunately, these mice also developed the progressive alopecia observed in our previous experiments (Figures 4D and 4E). In contrast, monotherapy with recombinant vPD1 induced tumor eradication in ~59% (12/22) of mice, which was a significant improvement over vGFP/αPD1 combination therapy (p=0.04) (Figures 4A–4C). Additionally, while some mice treated with vPD1 did display minor alopecia, the severity of this hair loss was significantly reduced compared to that observed following combination therapy (Figures 4D and 4E). Similar results were observed following treatment of established murine LLC tumors, although the overall efficacy of treatment in this model was much less pronounced and no mice displayed any evidence of alopecia (Figure S4).
Figure 4. Monotherapy with vPD1 can eradicate established melanomas.

(A–C) C57/B6 mice were injected SQ with 4x105 B16/F10 cells. On day 7 post injection, mice were randomly separated into one of four cohorts: saline + isotype (n=12), vGFP + isotype (n=10), vGFP + αPD1 antibody (n=17), or vPD1 (n=22). Viral injections consisted of 1x107 FFU of MYXV and were given on days 7, 9, and 11. Antibody injections consisted of 100μg/injection and were given on days 7, 11, 14, and 18. (A) Animals were monitored for tumor burden and euthanized when tumors reached 15mm in any direction. Data represents summation of four independent experiments. Significance was determined using log-rank test. (B) Average overall response rates for each treatment group (defined as the number of animals surviving when the last saline treated animal was euthanized). Significance was determined by averaging response rates from four experiments and analyzing using unpaired students T-Test. (C) Average complete response rates for each treatment group (defined as animals displaying no remaining tumor burden 80 days post treatment). Significance was determined by averaging response rates from four experiments and analyzing using unpaired students T-Test. (D) Average alopecia score from vGFP + αPD1 antibody (n=12) and vPD1 (n=22) treated animals. * denotes significance of p<0.05 for the indicated day and was determined using unpaired students T-Test. (E) Example of alopecia in mice on day 70 post treatment. Abbreviations: OR (overall response), CR (compete response).
vPD1 functions through enhancing CD8+ T cell activation
To begin to elucidate the mechanism(s) through which secretion of soluble PD1 enhanced MYXV therapy, we next asked whether vPD1’s increased efficacy was due to virally induced immunotherapy or to changes in the viruses direct oncolytic capacity. To analyze the direct oncolytic capacity of our viral treatments, tumor bearing animals were treated with either: saline, vGFP + isotype, vGFP + αPD1, or vPD1. 24 hours after the final viral treatment, tumors were excised and the percent of viable cells as well as the rate of viral infection in these cells was analyzed using flowcytometry. Viral monotherapy (vGFP+isotype antibody) resulted in a ~50% reduction in tumor viability (29% vs 15%, p=0.04) (Figure 5A) with a relatively small number of the remaining viable tumor cells (3.2%) still displaying evidence of viral infection (Figure 5B). Statistically identical rates of both acute reductions in tumor viability as well as viral infection were observed following treatment with either vGFP+αPD1 antibody or vPD1 suggesting that inhibition of the PD1/PDL1 pathway neither increased nor decreased MYXV’s direct oncolytic capacity. Therefore, to analyze the impact of soluble PD1 on virally induced immunotherapy, we next asked how efficacious vPD1 would be in the absence of an adaptive T cell response. Tumor bearing animals were depleted of both CD4+ and CD8+ T-cells and subsequently treated with either saline, vGFP, or vPD1 (Figure 5C). Consistent with our previous findings, the results indicated that vPD1 was capable of inducing complete tumor regression in 60% (3/5) of immune replete control animals. In contrast, vPD1 treatment in CD4/CD8 double depleted animals was incapable of inducing complete tumor regression in any animal (0/5) and instead caused only a slight (~15 day) delay in tumor growth identical to that observed following treatment with vGFP. Also, consistent with vPD1 inducing anti-tumor immunotherapy, splenocytes from immune replete mice whose tumors had been eradicated following vPD1 treatment secreted increased levels of IFNγ following stimulation with the melanoma specific gp100209–217 peptide and were capable of rejecting virus-naïve B16/F10 tumors upon rechallenge (Figure S5). To further determine which immunological cell populations were critical for vPD1 therapy, we next asked how single elimination of either CD4+ or CD8+ T cells, as well as NK1.1+ natural killer cells (NK cells), would impact the efficacy of vPD1 treatment. Tumor bearing animals were injected with depleting antibodies against either CD4, CD8, or NK1.1 to eliminate specific immunological subsets (Figure S2). Animals were then treated with vPD1 and monitored for survival (Figure 5D). Consistent with our previous results, 50% (4/8) of immune replete animals treated with vPD1 displayed complete regression of their established melanomas. In contrast, vPD1 treated mice depleted of CD8+ cells displayed a highly reduced CR rate (10%, 1/10, p=0.02) indicating that CD8+ cells were necessary for vPD1-based eradication of established melanomas. Interestingly, mice depleted of CD4+ cells actually showed a significantly enhanced (p=0.04) response to therapy with 90% of animals (9/10) displaying a CR. Neither of these effects was due to changes in the direct oncolytic capacity of MYXV since depletion of either CD4+ or CD8+ T cells had only minimal effect on the outcome of vGFP therapy (Figure S6). Depletion of NK cells had no obvious effect on the efficacy of vPD1 therapy (Figure 5D).
Figure 5. vPD1 functions through an immunotherapeutic mechanism.

(A and B) C57/B6 mice were injected SQ with 4x105 B16/F10 cells. On day 7 post injection, mice were randomly separated into one of four cohorts: saline (n=9), vGFP + isotype (n=7), vGFP + αPD1 antibody (n=6), or vPD1 (n=11). Viral injections consisted of 1x107 FFU of MYXV and were given on days 7, 9, and 11. Antibody injections consisted of 100μg/injection and were given on days 7 and 11. On day 12, mice were euthanized and tumors were extracted and disassociated into single cells for analysis by flowcytometry. (A) Percent of tumor cells staining as viable. (B) Percent of viable tumor cells expressing detectable GFP fluorescence. Data represents summation of four independent experiments. Significance was determined using unpaired students T-Test. (C) C57/B6 mice were injected SQ with 4x105 B16/F10 cells. On day 5, animals were split into two groups and given either mock injection or IP injection of depleting antibodies against both CD4 and CD8. Antibody depletion treatment was repeated on day 12. Depleted animals were separated into three cohorts and given IT injections of either: saline (n=5), or 1x107 FFU of vGFP (n=5) or vPD1 (n=5) on days 7, 9, and 11. Non-depleted animals injected IT with either saline (n=5) or 1x107 FFU vPD1 (n=5) were used as controls. Animals were then monitored for tumor burden and euthanized when tumors reached 15mm in any direction. (D) C57/B6 mice were injected SQ with 4x105 B16/F10 cells. On day five, animals were given either mock injection (n=12) or IP injection of depleting antibodies against CD4 (n=10), CD8 (n=10), or NK1.1 (n=15). Antibody depletion treatment was repeated on day 12. Animals were then treated with IT injection of 1x107 FFU of vPD1 on days 7, 9, and 11. Non-depleted animals treated with IT injection of saline (n=8) were included as a control. Animals were then monitored for tumor burden and euthanized when tumors reached 15mm in any direction. Data represents summation of two independent experiments. Significance was determined using log-rank test.
It has been previously demonstrated that the efficacy of MYXV treatment can be enhanced by increasing the numbers of CD8+ tumor infiltrating lymphocytes (TILs) (30) while PD1 blockade often relies on the improved function of existing CD8+ TILs. To determine which of these mechanisms mediated the increased efficacy of vPD1 we next analyzed the numbers and phenotype of TILs present after treatment with either vGFP or vPD1. Gross histological examination indicated that mock treated tumors displayed a clear boundary with virtually no lymphocytic cells either in or around the tumor mass (Figure S7). In contrast, tumors treated with either vGFP, vGFP + αPD1 antibodies, or vPD1 displayed significant regions of necrosis within the tumor as well as large numbers of lymphocytic cells both within the tumor boundaries and in surrounding tissues. Flow cytometry indicated that these newly infiltrating TILs were predominantly CD8+ T-cells although minor trends towards increased numbers of CD4+ T-cells were also observed (Figure 6A). Interestingly, while no significant differences in the numbers of either CD8+ or CD4+ cells were observed following treatment with any of the different viral-based regimes, surface expression of the early activation marker CD69 was more highly upregulated on CD8+ TILs from vPD1 treated mice than on CD8+ TILs from other treatments (Figure 6B). Additionally, a population of fully activated (CD25+/CD69hi) CD8+ TILs could be identified in tumors treated with vPD1 but not in tumors treated with other regimes (Figure 6C).
Figure 6. vPD1 enhances activation of CD8+ TILs.

(A–C) C57/B6 mice were injected SQ with 4x105 B16/F10 cells. On day 7, mice were randomly separated into one of four cohorts: saline (n=9), vGFP + isotype (n=7), vGFP + αPD1 antibody (n=6), or vPD1 (n=11). Viral injections consisted of 1x107 FFU and were given on days 7, 9, and 11. Antibody injections consisted of 100μg/injection and were given on days 7 and 11. On day 12, mice were euthanized, tumors disassociated into single cell suspensions, and TIL’s extracted using Histopaque. TILs were then analyzed using flowcytometry. (A) Numbers of CD4+ and CD8+ cells as percentage of total viable cells. (B) Surface expression of the early activation marker CD69 on CD8+ TILs. (C) Surface expression of the early and late activation markers CD69 and CD25 on the surface of CD8+ TILS. Data represents summation of four independent experiments. Significance was determined using unpaired students T-Test.
Localized vPD1 therapy generates an abscopal effect against established metastasis
Our previous findings indicated that vPD1 treatment is effective at eradicating a single established SQ tumor which can be directly injected with virus. The outcome for many cancers; however, is dependent on treatment of disease which has already metastasized to secondary solid organs. We therefore wished to determine whether localized vPD1 treatment would also be effective against pre-established metastatic disease. To test this, mice were injected on the same day with B16/F10 cells both SQ, to establish a large primary lesion, as well as IV, to establish disease metastatic to the lung. Tumors at both sites were then allowed to establish for 7 days to create a model of severe, stage IV melanoma. Mice were then treated by injecting vPD1 IT into the single SQ tumor with or without systemic depletion of CD4+ cells. Animals were then either euthanized on day 16, to visualize metastatic tumor burden in the lungs (Figures 7A–7C), or monitored for overall health and euthanized when they displayed acute weight loss resulting from severe metastatic disease (Figures 7D and 7E). The results indicated that mice treated with IT saline displayed high numbers of large metastasis in the lungs 16 days post tumor injection. Consistent with these metastasis mimicking severe clinical disease, these mice also displayed rapid weight loss beginning ~day 15 which required euthanasia of 100% of animals by day 21. Identical results were observed in mice treated with IT saline and then depleted of CD4+ cells. Localized monotherapy with vPD1 reduced the average size of individual metastatic lesions; however, it failed to reduce the total number of these lesions in the lungs and had only a minimal effect on clinical progression. In contrast, localized treatment with vPD1 combined with systemic CD4 depletion resulted in a highly significant reduction in both the total number of lung metastasis as well as the average size of the remaining lesions. This combination therapy also significantly improved overall health of animals and significantly delayed disease progression. Consistent with the mechanisms observed during localized treatment, systemic therapy involving vPD1 appeared to be mediated by CD8+ T cells since depletion of these cells eliminated the reductions in both numbers of metastasis and the size of remaining tumors. Depletion of NK cells had no obvious effect on the treatment of systemic disease (Figures 7B and 7C).
Figure 7. Localized vPD1 therapy can treat metastatic melanoma.

(A–C) C57/B6 mice were injected with B16/F10 cells both SQ (4x105 cells) and IV (1x105 cells). On day five, animals were given either mock injection or IP injection of depleting antibodies against CD4 and either CD8 or NK1.1. Animals were then separated further and given IT injection of either saline or 1x107 FFU of vPD1 into the SQ lesion on days 7, 9, and 11. This resulted in six cohorts: saline (n=7), saline + αCD4 (n=5), vPD1 (n=8), vPD1 + αCD4 (n=10), vPD1 + αCD4 + αCD8 (n=8), vPD1 + αCD4 + αNK1.1 (n=8). Antibody depletion was repeated on day 12. On day 16, mice were euthanized and the lungs examined for the presence of melanin+ lesions. (A) Pictures of representative lungs from the indicated cohorts. (B) Average number of melanin+ lesions (regardless of size) present in the lungs of each mouse. (C) Average size of each melanin+ lesion (regardless of number) present in the lungs of each mouse. Data represents summation of two independent experiments. Significance was determined using unpaired students T-Test (* = p<0.05 and *** =p<0.001). (D and E). C57/B6 mice were injected with B16/F10 cells both SQ (4x105 cells) and IV (1x105 cells). On day five, animals were given either mock injection or IP injection of a depleting antibody against CD4. Each group was then separated into two cohorts and given IT injection of either saline or 1x107 FFU of vPD1 into the SQ lesion on days 7, 9, and 11. Animals were then monitored daily for body weight and euthanized when they displayed either a body weight <80% of starting weight or a BCS=2.0. (D) Average animal body weight for each group as a percentage of starting weight. (E) Overall survival of animals in each cohort. Data represents summation of two independent experiments. Significance was determined using log-rank test.
Discussion
It is becoming increasingly clear that OV functions more through induction of anti-tumor immune responses then through acute viral killing of infected tumor cells. The initial induction of these responses can be seen in both animal models and human patients (4–12), unfortunately, their subsequent efficacy often fails to provide significant clinical benefit (2). Here we demonstrate that oncolytic MYXV treatment induces CD8+ T-cell infiltration into directly injected melanoma tumors (Figures 6 and S4) but that the subsequent efficacy of this response is inhibited by the presence of PDL1 expressed on tumor cells (Figure 1D). Interestingly, these results suggest a relatively straight forward function for the PD1/PDL1 pathway during MYXV treatment in which PD1 on T cells is engaged primarily by PDL1 expression on tumor cells. Previous work had indicated that PD1 can also inhibit T cell function by engaging with either PDL2 or PDL1 expressed on a variety of non-tumor cells, such as Tregs (36), myeloid derived suppressor cells (37), or dermal mast cells (38). Our results, however, suggest that these alternative sources of PD1 ligands play only a minimal role during MXYV therapy in the B16/F10 model. Whether these alternative sources of PD1 ligands play a larger role during treatment of more complex patient tumors remains to be determined. Regardless, our findings support the hypothesis that blockade of the PD1/PDL1 pathway represents a feasible approach to improve the efficacy of MYXV-based OV in some settings.
Interestingly, while traditional PD1-blockade, using systemically delivered antibodies, clearly increases the efficacy of MYXV therapy, this combination displays a much lower response rate then was observed in genetically PDL1−/− tumors and also causes an apparent autoimmune like toxicity (Figure 1). These results mimic early reports from clinical trials combined IMLYGIC® with PD1-blockade in humans (26) and suggest that methods to improve combination therapy using OV and αPD1 antibodies are still needed. To overcome the disadvantages associated with systemically administered αPD1 antibody (Figure 1 and (22, 23)), we therefore generated a recombinant MYXV (vPD1) designed to inhibit the PD1 checkpoint specifically within the tumor microenvironment through secretion of soluble PD1 (Figures 2 and 3). Biodistribution analysis of the soluble PD1 transgene indicated that it accumulated primarily in the tumor suggesting that the vPD1 virus achieved its primary goal of a tumor localized PD1 blockade (Figure 3). This biodistribution is in striking contrast to that of systemically injected αPD1 antibodies, where less than 5% of injected antibody is typically seen accumulating in the tumor (39). Interestingly, previous studies utilizing tumor-localized PD1 inhibition have typically observed decreased response rates compared to systemically delivered controls (40–42). This includes two studies similar to ours in which αPD1 blocking antibodies were encoded into the genomes of either oncolytic measles (43) or vaccinia virus (44). In contrast, our incorporation of soluble PD1 into MYXV actually enhanced efficacy compared to MYXV combined with systemically administered αPD1 antibody (Figure 4). While this could be due to some inherently beneficial property of MYXV, it seems more likely that achieving PD1-blockade using soluble PD1 provides observable benefits over more traditional methods. Such benefits might be due to a variety of mechanisms including the simultaneous inhibition of both PDL1 and PDL2 or the increased dispersion of soluble PD1, which is significantly smaller then PD1 blocking antibodies, through the complex tumor microenvironment.
In addition to the poor efficacy of systemically administered αPD1 antibodies, combination of these antibodies with MYXV also induced an apparent autoimmune like toxicity which presented as progressive alopecia and lymphocytic infiltration into the skin (Figure 1). The exact nature of this alopecia was not examined in our work; however, it appears similar to dermal autoimmune-like toxicities which are observed following administration of αPD1 antibodies in humans (45). The relative contributions of MYXV and αPD1 antibodies to the development of this disorder were impossible to define in our studies since animals treated with either monotherapy had to be euthanized due to tumor burden prior to developing symptoms. It is interesting to note, however, that autoimmune-like toxicities have generally not been reported in previous animal studies utilizing αPD1 antibodies (46) and that the most common skin-based autoimmune like toxicity reported in both mice and humans is vitiligo, which was not observed in our experiments (45, 46). This suggests that the combination of αPD1 antibodies with either, any oncolytic virus or MYXV specifically, might induce unique autoimmune like pathologies. In this regard, our results demonstrate that the severity of alopecia can be significantly reduced through use of the vPD1 virus (Figure 4). While this could be due to differences in the forms of checkpoint blockade used previous reports have indicated that intratumoral delivery of αCTLA4 antibodies also alleviates some of the toxicities associated with checkpoint blockade (41) suggesting that the primary reason behind vPD1’s reduced toxicity is likely the tumor localization of the soluble PD1 transgene (Figure 3).
In our experiments, maximal PD1 expression was observed ~48 hours post viral treatment. Transgene, however, could still be readily detected for at least one week after a single viral injection. These kinetics are consistent with previously published reports indicating that oncolytic MYXV infection, as well as associated transgene expression, is maintained in B16/F10 melanomas for ~7–10 days (47). It should be noted that our typical treatment regimen, three viral injections over five days, was chosen to maximize the amount of virus delivered to the tumor prior to the potential development of an anti-MYXV humoral response. While this regime was obviously successful, our biodistribution results indicate that it is likely suboptimal in terms of maximizing the longevity of PD1 expression within the tumor. Optimized dosing regimens might therefore further enhance the efficacy of viral treatment.
Not surprisingly, we observed that the efficacy of vPD1 was predominantly mediated by CD8+ T cells (Figures 5–7). Unexpectedly, however, we also observed that depletion of CD4+ cells had a profound positive impact on the outcomes of vPD1 therapy. This effect is most-likely mediated by elimination of inhibitory CD4+ Tregs; however, the exact mechanism remains unknown. One possibility is that depletion of intratumoral Tregs further increases the efficacy of existing CD8+ TILs. Alternatively, depletion of CD4+ cells might increase the numbers of CD8+ TILs present at the initiation of therapy (48). Interestingly, while depletion of CD4+ cells had profound effects during vPD1 treatment, the same depletion failed to enhance the efficacy of vGFP (Figure S6). This suggests a possible hierarchy of immune regulatory responses during OV in which the PD1 checkpoint is dominant over inhibition caused by Tregs. Whether this hierarchy is a general phenomenon during MYXV-based OV or specific to either melanoma or the B16/F10 model remains to be seen. Second, it is interesting to note that pan-depletion of CD4+ cells did not adversely affect the generation of curative CD8 responses. It has been recently demonstrated that, while CD4 help is required to generate effective T cell responses against dominant epitopes, the absence of this help actually amplifies the response against subdominant epitopes (49) and also enhances the activation of bystander CD8’s (50). Thus, it is possible that depletion of CD4 help might actually enhance the generation of anti-tumor immune responses induced by OV.
While exciting, it must be noted that there are a number of limitations associated with our studies. Most notably while vPD1 was highly effective against localized disease, monotherapy with the virus was only modestly effective against pre-established metastatic lesions. This observation represents a significant translational hurdle since prognosis for melanoma, and many other cancers, depends primarily on eradication of secondary metastasis. Several potential approaches to overcome this limitation, however, already exist. One such approach would be to deliver vPD1 directly to sites metastatic disease. Unfortunately, direct IV injection of vPD1 was largely ineffective in our metastatic B16/F10 model (our unpublished observations), likely due to poor delivery of the virus to the sites of metastasis. This is a well-established phenomenon for oncolytic poxviruses and is caused by the rapid up-take of viral particles by the liver following IV injection (51). Due to this well-known hurdle, a number of methods have been attempted to improve systemic delivery including biochemical modification to the viral particle or the use of tumor tropic carrier cells (52, 53). Combining these delivery methods with vPD1 therapy therefore appears attractive. Alternatively the preexisting tendency of systemically injected MYXV to be cleared in the liver could be used as a method to deliver virus specifically to hepatic localized disease. For example, uveal melanoma tends to metastasize primarily to the liver and patient prognosis once this occurs is extremely poor. Studies into the efficacy of systemically injected vPD1 against metastatic uveal melanoma therefore seem attractive. In our studies, we used systemic combination therapy to enhance the efficacy of localized vPD1 treatment. The results clearly demonstrate that vPD1’s efficacy against systemic disease can be significantly improved through depletion of CD4+ cells. Further studies into the precise mechanisms involved in this enhancement might therefore yield clinically relevant methods to duplicate these results.
A second potential limitation of our findings is our use of injectable syngeneic tumor models, primarily the B16/F10 model. While these models mimic some of the characteristics of clinical disease, they are highly aggressive short term models. In contrast, most cancer patients are likely to present with well-established disease which has developed over months or years. This difference might be particularly critical during checkpoint therapy as patients often present with immune systems where T-cells have expressed PD1 for long periods of time and are already functionally impaired. In contrast, splenic T cell in our studies were largely PD1− until the initiation of treatment. Thus, while vPD1 is clearly effective at preventing T cell exhaustion, whether it can effectively enhance T-cell function in the context of an already exhausted immune system remains to be determined.
Supplementary Material
Acknowledgments
Funding: This work was supported by the following grants: E.B. NIH-NIAID (1K22AI095372-01A1), NIH-NCI (1R01CA194090-01A1), and startup funding from the Medical University of South Carolina. This work was also supported in part by the Hollings Cancer Center’s Support Grant P30 CA138313.
We would like to thank Dr. Mark Rubinstein and Dr. Chrystal Paulos for valuable discussion of this project.
References
- 1.Dock G. The influence of complicating diseases upon leukemia. Am J Med Sci. 1904;127:563. [Google Scholar]
- 2.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nature clinical practice Oncology. 2007 Feb;4:101. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 3.Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Molecular therapy : the journal of the American Society of Gene Therapy. 2011 Jun;19:1008. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell MP, Pavelko KD. Enhancing the Tumor Selectivity of a Picornavirus Virotherapy Promotes Tumor Regression and the Accumulation of Infiltrating CD8+ T Cells. Molecular cancer therapeutics. 2016 Mar;15:523. doi: 10.1158/1535-7163.MCT-15-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishihara M, et al. Systemic CD8+ T cell-mediated tumoricidal effects by intratumoral treatment of oncolytic herpes simplex virus with the agonistic monoclonal antibody for murine glucocorticoid-induced tumor necrosis factor receptor. PloS one. 2014;9:e104669. doi: 10.1371/journal.pone.0104669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleijn A, et al. The in vivo therapeutic efficacy of the oncolytic adenovirus Delta24-RGD is mediated by tumor-specific immunity. PloS one. 2014;9:e97495. doi: 10.1371/journal.pone.0097495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zamarin D, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Science translational medicine. 2014 Mar 5;6:226ra32. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vassilev L, et al. Repeated intratumoral administration of ONCOS-102 leads to systemic antitumor CD8 T-cell response and robust cellular and transcriptional immune activation at tumor site in a patient with ovarian cancer. Oncoimmunology. 2015 Jul;4:e1017702. doi: 10.1080/2162402X.2015.1017702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ranki T, et al. Local treatment of a pleural mesothelioma tumor with ONCOS-102 induces a systemic antitumor CD8 T-cell response, prominent infiltration of CD8 lymphocytes and Th1 type polarization. Oncoimmunology. 2014 Nov;3:e958937. doi: 10.4161/21624011.2014.958937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kristian T, et al. T-cell subsets in peripheral blood and tumors of patients treated with oncolytic adenoviruses. Molecular therapy : the journal of the American Society of Gene Therapy. 2015 May;23:964. doi: 10.1038/mt.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hemminki O, et al. Immunological data from cancer patients treated with Ad5/3-E2F-Delta24-GMCSF suggests utility for tumor immunotherapy. Oncotarget. 2015 Feb 28;6:4467. doi: 10.18632/oncotarget.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufman HL, et al. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Annals of surgical oncology. 2010 Mar;17:718. doi: 10.1245/s10434-009-0809-6. [DOI] [PubMed] [Google Scholar]
- 13.Postow MA, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015 May 21;372:2006. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber JS, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. The lancet oncology. 2015 Apr;16:375. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 15.Robert C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015 Jan 22;372:320. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 16.Topalian SL, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014 Apr 1;32:1020. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robert C, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015 Jun 25;372:2521. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 18.Robert C, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014 Sep 20;384:1109. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 19.Robert C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011 Jun 30;364:2517. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 20.Di Giacomo AM, et al. Ipilimumab and fotemustine in patients with advanced melanoma (NIBIT-M1): an open-label, single-arm phase 2 trial. The lancet oncology. 2012 Sep;13:879. doi: 10.1016/S1470-2045(12)70324-8. [DOI] [PubMed] [Google Scholar]
- 21.Di Giacomo AM, et al. Three-year follow-up of advanced melanoma patients who received ipilimumab plus fotemustine in the Italian Network for Tumor Biotherapy (NIBIT)-M1 phase II study. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2015 Apr;26:798. doi: 10.1093/annonc/mdu577. [DOI] [PubMed] [Google Scholar]
- 22.Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015 Sep 24;373:1270. doi: 10.1056/NEJMc1509660. [DOI] [PubMed] [Google Scholar]
- 23.Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013 Jul 11;369:122. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajani KR, Vile RG. Harnessing the Power of Onco-Immunotherapy with Checkpoint Inhibitors. Viruses. 2015;7:5889. doi: 10.3390/v7112914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zamarin D, Postow MA. Immune checkpoint modulation: rational design of combination strategies. Pharmacology & therapeutics. 2015 Jun;150:23. doi: 10.1016/j.pharmthera.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 26.Puzanov I, et al. ASCO Annual Meeting; 2014.2014. [Google Scholar]
- 27.Johnston JB, et al. Role of the serine-threonine kinase PAK-1 in myxoma virus replication. Journal of virology. 2003 May;77:5877. doi: 10.1128/JVI.77.10.5877-5888.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smallwood SE, Rahman MM, Smith DW, McFadden G. Myxoma virus: propagation, purification, quantification, and storage. Current protocols in microbiology. 2010 May 1;Chapter 14(Unit 14A) doi: 10.1002/9780471729259.mc14a01s17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foltz C, Ullman-Cullere M. Guidelines for assessing the health and body condition of mice. Lab Animal. 1999;28:28. [PubMed] [Google Scholar]
- 30.Tosic V, et al. Myxoma virus expressing a fusion protein of interleukin-15 (IL15) and IL15 receptor alpha has enhanced antitumor activity. PloS one. 2014;9:e109801. doi: 10.1371/journal.pone.0109801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stanford MM, et al. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Molecular therapy : the journal of the American Society of Gene Therapy. 2008 Jan;16:52. doi: 10.1038/sj.mt.6300348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature medicine. 2002 Aug;8:793. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 33.Li B, et al. Cloning and expression of human sPD-1-Fc in Cos-7 cells. Xi bao yu fen zi mian yi xue za zhi = Chinese journal of cellular and molecular immunology. 2011 Jun;27:631. [PubMed] [Google Scholar]
- 34.Song MY, Park SH, Nam HJ, Choi DH, Sung YC. Enhancement of vaccine-induced primary and memory CD8(+) T-cell responses by soluble PD-1. Journal of immunotherapy. 2011 Apr;34:297. doi: 10.1097/CJI.0b013e318210ed0e. [DOI] [PubMed] [Google Scholar]
- 35.Finger LR, et al. The human PD-1 gene: complete cDNA, genomic organization, and developmentally regulated expression in B cell progenitors. Gene. 1997 Sep 15;197:177. doi: 10.1016/s0378-1119(97)00260-6. [DOI] [PubMed] [Google Scholar]
- 36.Li Z, et al. PD-L1 Expression Is Associated with Tumor FOXP3(+) Regulatory T-Cell Infiltration of Breast Cancer and Poor Prognosis of Patient. Journal of Cancer. 2016;7:784. doi: 10.7150/jca.14549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noman MZ, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. The Journal of experimental medicine. 2014 May 5;211:781. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakae S, et al. Mast cells enhance T cell activation: importance of mast cell costimulatory molecules and secreted TNF. Journal of immunology. 2006 Feb 15;176:2238. doi: 10.4049/jimmunol.176.4.2238. [DOI] [PubMed] [Google Scholar]
- 39.Natarajan A, et al. Novel Radiotracer for ImmunoPET Imaging of PD-1 Checkpoint Expression on Tumor Infiltrating Lymphocytes. Bioconjugate chemistry. 2015 Oct 21;26:2062. doi: 10.1021/acs.bioconjchem.5b00318. [DOI] [PubMed] [Google Scholar]
- 40.Wang C, Ye Y, Hochu GM, Sadeghifar H, Gu Z. Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano letters. 2016 Mar 24; doi: 10.1021/acs.nanolett.5b05030. [DOI] [PubMed] [Google Scholar]
- 41.Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJ. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013 Oct 1;19:5381. doi: 10.1158/1078-0432.CCR-12-0781. [DOI] [PubMed] [Google Scholar]
- 42.Sandin LC, et al. Local CTLA4 blockade effectively restrains experimental pancreatic adenocarcinoma growth in vivo. Oncoimmunology. 2014 Jan 1;3:e27614. doi: 10.4161/onci.27614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Engeland CE, et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Molecular therapy : the journal of the American Society of Gene Therapy. 2014 Nov;22:1949. doi: 10.1038/mt.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kleinpeter P, et al. Vectorization in an oncolytic vaccinia virus of an antibody, a Fab and a scFv against programmed cell death -1 (PD-1) allows their intratumoral delivery and an improved tumor-growth inhibition. Oncoimmunology. 2016;5:e1220467. doi: 10.1080/2162402X.2016.1220467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costa R, et al. Toxicity profile of approved anti-PD-1 monoclonal antibodies in solid tumors: a systematic review and meta-analysis of randomized clinical trials. Oncotarget. 2016 Nov 11; doi: 10.18632/oncotarget.13315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J, Blake SJ, Smyth MJ, Teng MW. Improved mouse models to assess tumour immunity and irAEs after combination cancer immunotherapies. Clinical & translational immunology. 2014 Aug;3:e22. doi: 10.1038/cti.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doty RA, Liu J, McFadden G, Roy EJ, MacNeill AL. Histological evaluation of intratumoral myxoma virus treatment in an immunocompetent mouse model of melanoma. Oncolytic Virother. 2013 Jan;2:1. doi: 10.2147/OV.S37971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ueha S, et al. Robust Antitumor Effects of Combined Anti-CD4-Depleting Antibody and Anti-PD-1/PD-L1 Immune Checkpoint Antibody Treatment in Mice. Cancer immunology research. 2015 Jun;3:631. doi: 10.1158/2326-6066.CIR-14-0190. [DOI] [PubMed] [Google Scholar]
- 49.Freeman ML, Roberts AD, Burkum CE, Woodland DL, Blackman MA. Promotion of a subdominant CD8 T cell response during murine gammaherpesvirus 68 infection in the absence of CD4 T cell help. Journal of virology. 2014 Jul;88:7862. doi: 10.1128/JVI.00690-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monjazeb AM, et al. Bystander activation and anti-tumor effects of CD8+ T cells following Interleukin-2 based immunotherapy is independent of CD4+ T cell help. PloS one. 2014;9:e102709. doi: 10.1371/journal.pone.0102709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baril P, et al. Differential biodistribution of oncolytic poxvirus administered systemically in an autochthonous model of hepatocellular carcinoma. The journal of gene medicine. 2011 Dec;13:692. doi: 10.1002/jgm.1624. [DOI] [PubMed] [Google Scholar]
- 52.Guo ZS, et al. The combination of immunosuppression and carrier cells significantly enhances the efficacy of oncolytic poxvirus in the pre-immunized host. Gene therapy. 2010 Dec;17:1465. doi: 10.1038/gt.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Munguia A, Ota T, Miest T, Russell SJ. Cell carriers to deliver oncolytic viruses to sites of myeloma tumor growth. Gene therapy. 2008 May;15:797. doi: 10.1038/gt.2008.45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.