

Abstract
Purpose
The success of hematopoietic stem cell transplantation (HSCT) depends on donor cell homing to the bone marrow (BM). However, there is no reliable method of noninvasively monitoring the kinetics and distribution of transferred cells. Using Zirconium-89 (89Zr)-oxine cell labeling combined with positron emission tomography (PET) imaging, we sought to visualize and quantify donor cell homing in a mouse BM transplantation model.
Experimental Design
The effect of 89Zr-oxine labeling on BM cell viability and differentiation was evaluated in vitro. 89Zr-labeled BM cells (2×107 cells, 16.6 kBq/106 cells) were transferred intravenously and serial microPET images were obtained (n=5). The effect of a CXCR4 inhibitor, plerixafor, (5 mg/kg) and granulocyte-colony stimulation factor (G-CSF, 2.5 μg) on BM homing and mobilization were examined (n=4). Engraftment of the transferred 89Zr-labeled cells was evaluated (n=3).
Results
89Zr-oxine-labeled BM cells showed delayed proliferation, but differentiated normally. Transferred BM cells rapidly migrated to the BM, spleen, and liver (n=5). Approximately 36% of donor cells homed to the BM within 4 h, irrespective of prior BM ablation. Inhibition of CXCR4 by plerixafor alone or with G-CSF significantly blocked the BM homing (p<0.0001, vs non-treated, at 2 h), confirming a crucial role of the CXCR4-CXCL12 system. Mobilization of approximately 0.64% of pre-transplanted BM cells induced a 3.8-fold increase of circulating BM cells. 89Zr-labeled donor cells engrafted as well as non-labeled cells.
Conclusions
89Zr-oxine PET imaging reveals rapid BM homing of transferred BM cells without impairment of their stem cell functions, and thus, could provide useful information for optimizing HSCT.
Keywords: Hematopoietic stem cell transplantation, Cell tracking, Bone marrow cells, Positron emission tomography, Zirconium-89
Introduction
Hematopoietic stem cell transplantation (HSCT) is an important therapy for a number of medical conditions. It has allowed patients with lymphoma, leukemia and multiple myeloma to receive more aggressive myeloablative treatments than would otherwise be tolerated, resulting in improved survival (1). However, a number of issues remain. For instance, mortality from infection during the prolonged lymphopenia after myeloablation remains problematic, reaching 25% in the case of cytomegalovirus infections (1). Acute graft-versus-host-disease (GVHD) incidence occurs in 30–60% of HSCT in the context where donor and recipient are HLA-matched siblings (1,2). To speed HSC engraftment and reduce GVHD, parameters such as the optimal number of cells to transfer, frequency of transplantation (single or repeated HSCT), and best route of cell delivery can be adjusted. For example, intra-osseous injection of hematopoietic stem cells (HSC) as opposed to intravenous (i.v.) administration has been suggested as a means to minimize the loss of transferred cells by avoiding trapping within the lungs and to reduce GVHD (3–6).
The efficacy of BM homing and engraftment of HSCs directly correlates with the success of HSCT. This becomes a particularly important issue in the case of cord blood transplants, in which the number of HSCs contained is limited (7–9). Migration of transferred cells has been studied mainly by tissue biopsies in humans or harvesting organs in animals to determine the presence of donor cells. Unfortunately, these methods do not provide kinetic information on the trafficking of the cells in the whole-body. Such knowledge would help optimize and improve the efficiency of HSCT. Furthermore, in HSCT, mobilized HSCs have become the major source for donor cells (10); however, it is unclear as to what fraction of HSCs are mobilized by the mobilization treatments. Investigators continue searching for more effective mobilizers.
We have previously reported development of a cell labeling agent, zirconium-89 (89Zr)-oxine complex, for tracking cells with positron emission tomography (PET) imaging (11). To better understand the trafficking of HSCs in transplantation and mobilization in vivo, we applied this 89Zr-oxine cell tracking method and employed microPET/computed tomography (CT) imaging in a mouse bone marrow (BM) transplant model. This method enabled non-invasive assessment of HSC migration kinetics and quantification of cell distribution.
Materials and Methods
Mice
Female and male C57BL/6 wild type (expressing CD45.2) and congenic (expressing CD45.1) mice and green fluorescence protein (GFP) transgenic mice were purchased from Jackson Laboratories (Bar Harbor, ME). All animal experiments were performed in accordance with a protocol approved by the institutional animal care and use committee. Mice were 8–12 week old at the time of experiments.
Cell culture and cytokines
BM was flushed from femurs and tibias of mice. RPMI 1640 media (Life Technologies, Grand Island, NY) supplemented with 2 mM L-glutamine, 100 IU/mL penicillin, 100 μg/mL streptomycin (Life Technologies), 10% fetal calf serum (Gemini Bio Products, Sacramento, CA) and 50 μM 2-mercaptoethanol (Sigma Chemical, St. Louis, MO) was used for cell culture. Murine recombinant stem cell factor (SCF), Fms-related tyrosine kinase 3 ligand (FLT3L) and thrombopoietin (TPO) were purchased from R&D Systems (Minneapolis, MN). Murine recombinant granulocyte-colony stimulation factor (G-CSF) and granulocyte macrophage-colony stimulation factor (GM-CSF), and human recombinant interleukin 15 (IL-15) were purchased from Peprotech (Rocky Hill, NJ).
Flow cytometry analysis
All antibodies and fluorescein isothiocyanate conjugated annexin V were purchased from eBioscience (San Diego, CA). Propidium iodide (PI) was purchased from Sigma-Aldrich (St. Louis, MO). For Ki67 staining, cells were fixed and permeabilized using −20°C 70% ethanol and then washed with PBS with 5% fetal calf serum before proceeding with the staining. Flow cytometry data was acquired using FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA) and analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
89Zr-oxine synthesis
89Zr-oxalate solution and 89ZrCl4 solution were produced at the institutional cyclotron facility. 89Zr-oxine complex was synthesized from 89ZrCl4 and oxine following the method previously reported (11). Briefly, 4 μl of 20% tween 80 solution (Sigma-Aldrich) and 102 μl of 20 mM oxine in 0.04 N HCl were mixed in a tube, and then, 60 μl of 89ZrCl4 (37 to 74 kBq) was added and vortexed. The resulting acidic solution was neutralized by adding 500 mM NaHCO3 with incremental vortexing (pH 7.0–7.5).
89Zr-oxine BM cell labeling and determination of cell viability and cellular retention of 89Zr
BM cells were incubated with 89Zr-oxine complex at 11.0 kBq–5.55 MBq/106 cells in PBS at 25:1 volume ratios for 20 minutes, and washed twice in RPMI media. The cell-associated radioactivity was 3.6 kBq-1.7 MBq, yielding the labeling efficiency of 26–30%. To determine cell viability and retention of 89Zr after the labeling, BM cells were labeled with 2 different radioactivity doses indicated, and cultured either without exogenous cytokine (n=3), with SCF, FLT3L, and TPO (100 ng/ml each, n=6), or with GM-CSF (20 ng/ml, n=6). The number of live cells was counted using 0.4% trypan blue dye (Life Technologies) at indicated time points. At each time point, radioactivity of the cells was measured by a γ-counter (WIZARD2 automatic gamma-counter, Perkin Elmer, Waltham, MA). The cells were also stained with annexin V and PI or with anti-Ki67 antibody at the time points indicated.
Determination of the effects of labeling on the phenotype of BM cells and differentiation capability in vitro
The surface expressions of CD117, sca-1 and lineage markers (CD3, NK1.1, Ly6G, CD2, CD5 and B220) on BM cells were examined before and after 89Zr-oxine labeling (13.7–22.2 kBq/106 cells) and analyzed by flow cytometry (n=6). BM cell differentiation capability was examined by culturing the 89Zr-labeled and non-labeled cells with GM-CSF (20 ng/ml) and IL-15 (25 nM) for differentiation to DCs and NK/NK-T cells, respectively (n=6). On day 10, the expression of CD11c, CD86, and NK1.1 was analyzed by flow cytometry. We also assessed differentiation function of the labeled BM cells by colony forming cell (CFC) assay using methylcellulose-based media, following the manufacturer’s instructions (R&D Systems). 89Zr-labeled and non-labeled cells (5×104 cells) were plated in 35 mm dishes and incubated at 37°C and 5% carbon dioxide for 10 days. The number of formed colonies were counted under the microscope (n=3).
BM cell tracking by microPET/CT
89Zr-oxine-labeled BM cells (2×107 cells at 16.6 kBq/106 cells) were transferred to mice i.v. For BM ablation, host mice received a 9.5 Gy lethal whole-body irradiation 24 h prior to cell transfer (n=5). Additionally, a CXCR4 inhibitor, plerixafor (12) (Adooq Bioscience, Irvine, CA), was used to interrogate the role of CXCR4 in BM cell migration (n=4). Mice received an i.v. injection of plerixafor (5 mg/kg) 15 min before the BM cell transfer. Another group of mice received concurrent i.v. injection of G-CSF (2.5 μg, n=4). In addition, some mice received deferoxamine (DFO), a clinically used chelator, (Hospira, Inc., Lake Forest, IL) at 660 μg intramuscularly 15 min before and 1, 2, 3 and 4 h after the cell injection. Mice were serially imaged using a microPET/CT imager (BioPET, Bioscan, Washington, DC) up to 7 days after the cell transfer. A 400–700 keV energy window was used and 5 min-emission scan per bed position for a total of two bed positions were acquired at 0, 2 and 4 h. On day 1, 2, 5 and 7, scan time per bed position was increased to 6.5, 7.5, 12.5 and 15.5 min, respectively, to account for radioactive decay. The acquired images were reconstructed using a 3-dimensional ordered-subsets expectation maximization algorithm. VivoQuant software (inviCRO LLC, Boston, MA) was used to fuse the PET images with CT images. Whole body activity was quantitated on 0 h images (injected dose) and the 89Zr activity in the bone/BM at various time points was quantitated by carefully erasing the 89Zr signal observed out of bone/BM area using coronal, sagittal and axial slices on the VivoQuant, leaving only the 89Zr signals in the bone/BM to quantitate. MIMvista (MIM software, Cleveland, OH) was used to quantify cells migrated to the lungs, liver, spleen and spine by setting volumes of interest along the edge of the organs at various time points and also to cover the whole body on the axial images at 0 h (injected dose).
Quantification of BM cell mobilization
BM cells collected from GFP transgenic mice were labeled with 89Zr-oxine and transferred to wild type mice (2×107 cells at 3.7–11.1 kBq/106 cells, n=4). Mice received i.v. injections of plerixafor and G-CSF 3 h and 1 d following the cell transfer. Two hours after the second mobilization treatment, mice were sacrificed and the blood was collected by cardiac puncture into EDTA-coated tubes (BIOTANG Inc., Lexington, MA). The volume was measured, then the blood was centrifuged and the radioactivity of the cell fraction was measured with a γ-counter. Radioactivity of the total blood was calculated using the following formula that uses 1.58 ml average blood volume for a 20-g mice (13); (Total blood radioactivity) = (radioactivity of the blood sample) × 1.58 (ml)/(sample blood volume [ml]) × body weight (g)/20. GFP+ cells in the blood was counted by flow cytometer.
Flow cytometry analysis for engraftment of 89Zr-oxine-labeled BM cells and differentiation in vivo
The engraftment and differentiation of the 89Zr-oxine-labeled BM cells was examined by transferring CD45.2-expressing donor cells to CD45.1-expressing hosts (n=3). Ten-weeks later, the BM and splenocytes collected from the recipients were analyzed by flow cytometry using antibodies against CD117, Sca-1, CD3, NK1.1, Ly6G, CD2, CD5, B220, CD8, and CD11c combined with CD45.1 and CD45.2 congenic markers.
Statistical analysis
Effect of 89Zr-oxine labeling on BM cells survival was analyzed by repeated measure two-way analysis of variance followed by Dunnett’s correction. Specific activity was analyzed by multiple t-tests with Holm-Sidak correction. Unpaired two-tailed t-test with Welch’s correction was used to examine the effect of 89Zr-labeling on cellular phenotype and in vitro and in vivo differentiation and in the GFP+ BM cell mobilization study. Repeated measure two-way analysis of variance followed by Tukey’s multiple comparisons test correction was used for examining the effect of plerixafor and G-CSF on BM homing. GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA) was used and P values, corrected for multiple comparison where applicable, less than 0.05 were considered significant.
Results
89Zr-oxine labeling does not alter cellular phenotype and survival
When tracking ex vivo labeled cells, it is crucial that the labeling does not alter the cellular phenotype and functionality. Thus, we first examined the effects of labeling on surface phenotype. Expression of lineage markers (CD3, NK1.1, Ly6G, CD2, CD5 and B220) and presence of sca-1 and CD117 expressing cells within lineage negative cells were similar between non-labeled and 89Zr-oxine-labeled BM cells (Fig. 1A). Approximately 4.2% of lineage negative cells were HSCs expressing both sca-1 and CD117, which corresponded to approximately 0.35% of the total BM. The survival of non-labeled and 89Zr-labeled BM cells (28.8 and 13.7 kBq/106 cells) was examined by culturing them without exogenous cytokines. The cells followed a similar trend of decline in number over the 3-day period (Fig. 1B). Cell-associated 89Zr activity, with decay correction, showed a slight decrease at the early time points and then almost plateaued thereafter (Fig. 1C). Annexin V and PI staining revealed that comparable fractions of non-labeled and labeled cells were apoptotic or necrotic (Fig. 1D). The live cells showed negligible Ki67 staining on day 2 (Fig. 1E).
Figure 1. 89Zr-oxine labeling does not alter cellular phenotype and survival.
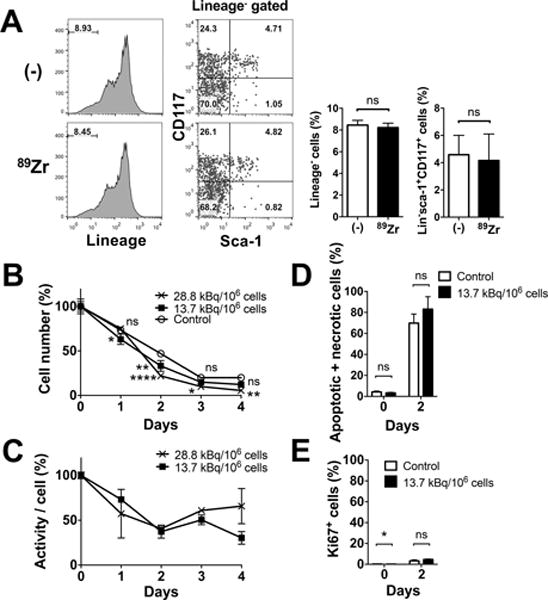
A. 89Zr-labeling (13.7–22.2 kBq/106 cells) did not alter the expression of lineage marker in BM cells or Sca-1 and CD117 expression of lineage marker− cells. Representative flow cytometry data of 3 independent experiments and the average of all data (white: control, black: 89Zr-labeled) are shown. Approximately 4.2% of lineage negative cells were HSCs expressing both sca-1 and CD117 (n=6). B. BM cells not labeled (open circle) and labeled at 13.7 (filled circle) and 28.8 (cross) kBq/106 cells were cultured without exogenous cytokines. Live cell number decreased in a similar manner in all the groups (n=3, 13.7 kBq/106 cellls: p=0.0156 and 0.0016 at day 1 and 2, 28.8 kBq.106 cells : p<0.0001, p=0.0186 and 0.0011 on day 2, 3 and 4, vs control). C. Cell associated 89Zr activity, with decay correction, showed slight decrease at the early time points and then almost plateaued (n=3). No significant difference was observed between the two labeling doses. D. The 89Zr-labeled cells (13.7 kBq/106 cells, black) showed slightly higher fraction of apoptotic and/or necrotic cells (i.e. annexin V+ and/or PI+ cells) by flow cytometry analysis compared to non-labeled control (white) on day 2, but the difference was not significant (70–83% of total cells, n=3). E. Expression of Ki67 in live cells on day 2 was negligible (n=3).
89Zr-oxine-labeled BM cells retain differentiation function in vitro
We next cultured 89Zr-labeled BM cells with various cytokines. In contrast to cytokine free culture, when a combination of SCF, FLT3L and TPO, which is known to support survival of BM cells with minimum induction of proliferation or differentiation, was used, the survival of 89Zr-labeled cells was suppressed in a dose-dependent manner (Fig. 2A). Cells labeled at 10.9 (8.14–13.7) kBq/106 cells showed a subtle increase in the cell number at later time points, whereas cells labeled at 28.5 (28.1–28.8) kBq/106 cells failed to proliferate (Fig. 2A, p=0.0001 on day 7, both doses vs control). As a result, the decay-corrected specific activity declined in cells labeled at the lower dose, reflecting the proliferation, while the specific activity of cells labeled at the higher dose remained plateaued (Fig. 2B). A higher cell death was observed with the 89Zr-labeled cells compared to the control (Fig. 2C), but the live labeled cells expressed Ki67, indicating their proliferation (Fig. 2D). GM-CSF culture of BM cells labeled at 28.8 and 13.7 kBq/106 cells demonstrated a dose-dependent delay of cell number increase compared to the non-labeled cells, with the cells labeled at the lower dose reaching the same level as non-labeled cells by day 10 (Fig. 2E). As the cell number increased, the specific activity of the cells started to decline (Fig. 2F). These changes can be explained by the sustained cell death observed with the labeled cells (Fig. 2G, day 6) and delayed proliferation of the labeled cells shown by the Ki67 staining that peaked on day 8 (Fig. 2H). After 10 days of culture, the labeled cells (13.7–22.2 kBq/106 cells) differentiated to CD11c+ DCs as well as control cells, with about 40% of the cells indicating matured phenotype (CD86+) (Fig. 2I, S1A). When BM cells were cultured with IL-15, the BM cells became NK1.1+ cells, suggesting their differentiation into NK/NK-T cells (Fig. 2J, S1B).
Figure 2. 89Zr-oxine-labeled BM cells retain differentiation function.
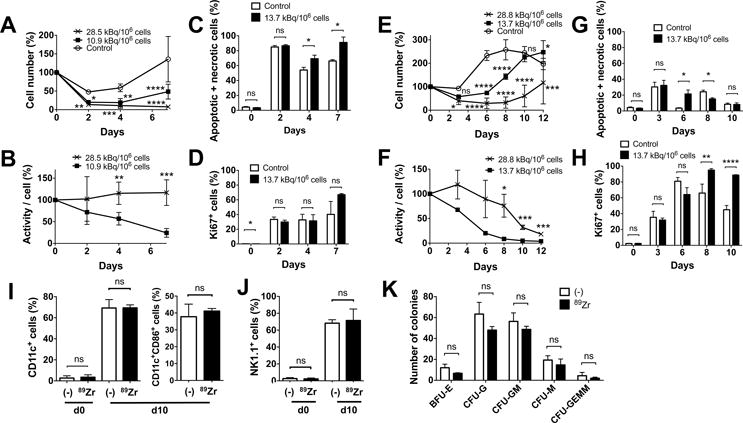
A. BM cells were labeled with 89Zr at 10.9 (8.14–13.7, filled circle) and 28.5 (28.1–28.8, cross) kBq/106 cells and cultured with SCF, FLT3-L and TPO (100 ng/ml each, n=6). The cells labeled at the lower dose slightly proliferated after day 4, while the cells labeled at the higher dose decreased in number (10.9 kBq/106 cells vs control (open circle): P = 0.0305, 0.0015 and 0.0001, 28.1 kBq/106 cells vs control: P = 0.0068, 0.0002, and 0.00001, at 2, 4, and 7 days, respectively). B. Decay corrected specific activity of the 89Zr-oxine labeled BM cells decreased in the cells labeled with the lower dose, reflecting the cellular proliferation, but remained constant in cells labeled with the higher dose (n=6) (P = 0.00197, 0.00035 at day 4 and 7, respectively). C. Higher fractions of annexin V+ and/or PI+ cells were detected by flow cytometry in the 89Zr-labeled cells (13.7 kBq/106 cells, black, n=3) on day 4 and 7 than the non-labeled cells (white). D. The labeled cells proliferated as indicated by Ki67 staining (n=3). E. BM cells were labeled with 89Zr at 13.7 (filled circle) or 28.8 (cross) kBq/106 cells and cultured with 20 ng/ml GM-CSF (n=6). Compared to the non-labeled control (open circle), the labeled cells showed delayed increase in cell number (13.7 kBq/106 cellls: p<0.0001 on day 6 and 8, p=0.0168 on day 12, 28.8 kBq.106 cells: p=0.0121 and 0.0001 on day 3 and 12, p<0.0001 on day 6, 8 and 10, vs control). F. Decay corrected cell associated 89Zr activity gradually decreased in cells labeled at the lower dose, but was plateaued with the higher dose until around day 8 (p=0.0177, 0.0008 and 0.0006 on day 8, 10, 12, 13.7 vs 28.8 kBq/106 cells). G. Higher fraction of annexin V+ and/or PI+ cells were detected in the 89Zr-labeled cells (13.7 kBq/106 cells, black) on day 6 and in the non-labeled cells (white) on day 8 (p=0.0176 on day 6 and 8). H. Proliferation of the cells indicated by the expression of Ki67 was delayed in the labeled cells reaching the peak on day 8, whereas the peak in non-labled cells was observed on day 6 (p=0.0041 and p<0.0001 on day 8 and 10). I. 89Zr-oxine-labeled BM cells (13.7–22.2 kBq/106 cells, black) differentiated to DCs (CD11c+) comparable to the non-labeled cells (white) after a 10 day-culture in GM-CSF (20 ng/ml). Around 40% of CD11c+ cells expressed a maturation marker CD86 (n=6). J. When cultured with IL-15 (25 nM), 89Zr-labeled BM cells (black) differentiated to NK1.1+ NK/NK-T cells in 10 days similar to the non-labeled cells (white) (n=6). K. Colony forming cell assay of non-labeled (white) and labeled (14.8 kBq/106 cells, black) BM cells showed no significant difference in the number of various hematopoietic cell colonies on day10 (n=3, BFU-E: burst forming unit-erythroid, CFU-G: colony forming unit-granulocyte, CFU-GM: colony forming unit-granulocyte macrophage, CFU-M: colony forming unit-macrophage, CFU-GEMM; Colony forming unit-granulocyte, erythrocyte, macrophage, megakaryocyte).
We performed a CFC assay to further assess the differentiation capacity of the labeled BM cells. After a 10 day-culture, BM cells labeled at 14.8 kBq/106 cells formed various hematopoietic cell colonies similar to the non-labeled cells (Fig. 2K).
These results indicate that 89Zr-oxine-labeled cells retained the capacity to differentiate into various mature hematopoietic cells in vitro.
BM cells labeled with 89Zr-oxine show rapid homing to bone marrow
To examine the dynamics of BM cell trafficking, we transferred 2×107 cells (16.6 kBq/106 cells) i.v. to mice and serially imaged them with a microPET/CT scanner. The donor cells quickly passed through the lungs and started trafficking to the BM, spleen and liver almost immediately following transfer (Fig. 3A, B). Within 4 h, the majority of cells homed to the BM, spleen and liver and remained there until day 7 at the conclusion of the experiment. We analyzed the cell migration kinetics at early time points by quantitating the activity in the bone/BM area (Fig. 3C, upper left), and the lungs, liver and spleen (Fig. 3C, upper right). The BM cell trafficking pattern was nearly identical between the hosts that received or had not received BM ablation prior to the cell transfer (Fig. 3C). Of the 2×107 cells transferred, approximately 36% (7.2×106 cells) homed to the BM and approximately 8% (1.6×106 cells) and 36% (7.2×106 cells) migrated to the spleen and liver, respectively, by 4 h.
Figure 3. Serial microPET/CT imaging reveals rapid trafficking of 89Zr-oxine-labeled BM cells through the lungs to the BM, spleen and liver.
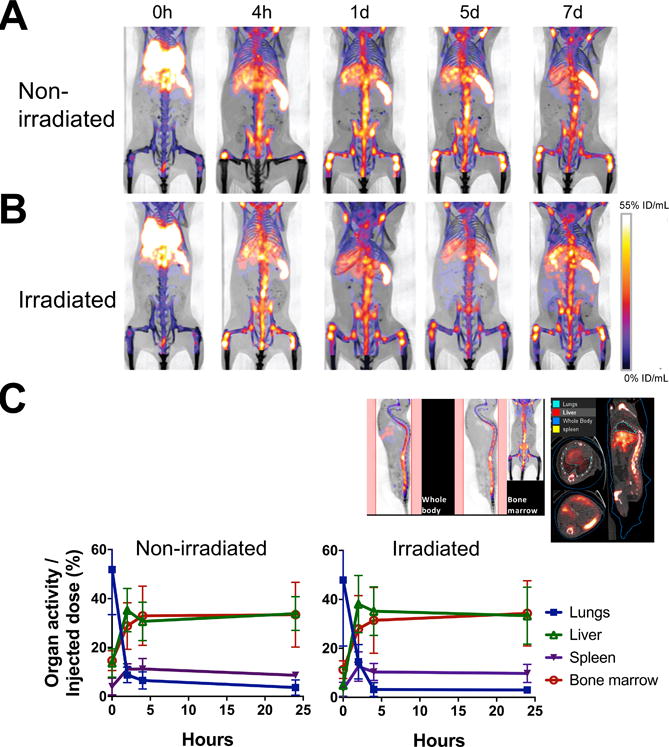
A. 89Zr-oxine-labeled BM cells (2×107 cells at 16.6 kBq/106 cells) were transferred via the tail vein to non-BM ablated (A) and BM ablated (9.5 Gy whole-body irradiation) (B) mice (n=5). PET images indicate that immediately after the injection (0 h), cells started to migrate to the lungs, liver, spleen and the BM (%ID/mL: percent of injected dose per mL of tissue). C. Quantitative analysis of the microPET/CT images revealed similar migration kinetics to the BM, lungs, liver and spleen between non-irradiated and irradiated recipient mice (n=3, blue squire: lungs, green triangle: liver, purple triangle: spleen, red circle: bone marrow). The example of quantitated area/regions of interest set on the images are indicated in the upper left (bone marrow) and right (lungs, liver, and spleen).
Homing to the BM and retention in the BM are CXCR4 dependent
It has been suggested that the CXCR4-CXCL12 system plays an important role in the BM homing of cells. We observed that blockade of CXCR4 by plerixafor (15 min before the transfer) inhibited the migration of the labeled cells to the BM at 2 h (Fig. 4A, B), indicating that CXCR4 signaling is critical for BM homing. Co-administration of G-CSF with plerixafor sustained the inhibition of BM homing (Fig. 4A, C). Quantitation of the spinal 89Zr activity indicated the significant suppression of BM homing by plerixafor treatment (p<0.0001 at 2 h), especially when combined with G-CSF (p<0.0001 at 4 h, p=0.0005 at 1 d, Fig. 4D).
Figure 4. 89Zr-oxine microPET/CT imaging demonstrates CXCR4 dependent BM homing of transferred BM cells.
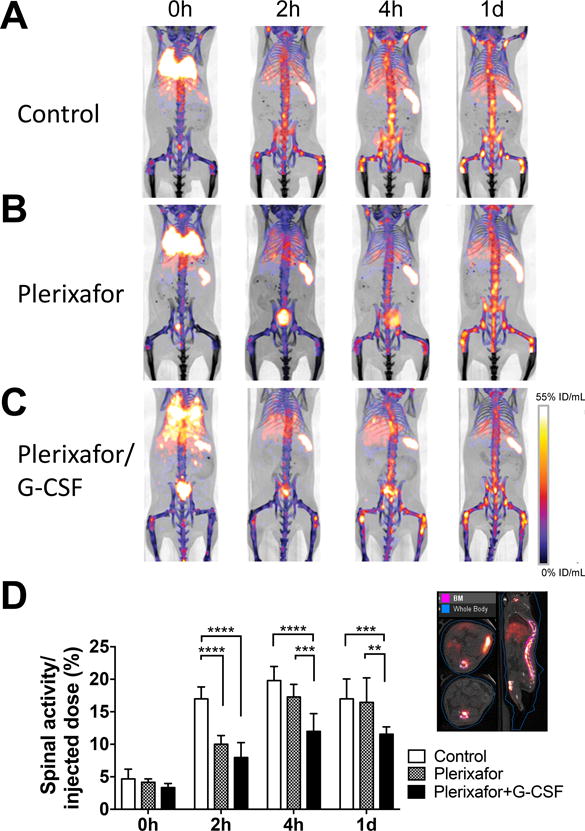
Groups of mice received 89Zr-oxine-labeled BM cells (2×107 cells at 16.6 kBq/106 cells)); A. Control mice (n=4), B. mice receiving plerixafor (5 mg/kg) i.v. 15 min before the BM cell transfer, and C. mice receiving plerixafor (5 mg/kg) and G-CSF (2.5ug) i.v. 15 min before the BM transfer. All recipient mice received a lethal whole-body irradiation at 9.5 Gy 24h prior to cell transfer. PET images indicates that in comparison to the control, plerixafor alone or plerixafor/G-CSF inhibited the initial donor cell migration to the BM. Addition of G-CSF to plerixafor sustained the suppression of BM homing (%ID/mL: percent of injected dose per mL of tissue, Representative images of 4 experiments). D. Quantification of spinal 89Zr activity demonstrated inhibition of BM homing with plerixafor at the 0–2 h time points and prolonged inhibition with plerixafor/G-CSF. An example of regions of interest set on the images are shown at the right. Asterisks indicate statistical significance within each time point. *:P < 0.05, **:P < 0.01, ***:P < 0.001, ****:P< 0.0001. White bar: control, patterned bar: plerixafor, and black bar: plerixafor/G-CSF.
The combination of plerixafor/G-CSF is known to have a stronger HSC mobilization effect than plerixafor alone and has been used in the clinic as a method to collect HSCs for the HSCT. However, what fraction of HSCs the agents mobilize is still an open question. The strong inhibitory effect of plerixafor/G-CSF on BM homing observed above prompted us to address this question using plerixafor/G-CSF in mice that had already received BM transplantation. The injection of the agents on two consecutive days (at 3 h and 1 d after transplantation) induced a 3.8-fold increase of 89Zr activity in the circulation (Fig. 5A; P = 0.0009), corresponding to an increase from approximately 0.46×105 cells to 1.74×105 cells in the circulation, assuming that the transferred BM cells did not divide within the 1 d-experimental period. This increase in the number of circulating cells indicates that approximately 1.28 × 105 pre-transplanted donor cells were mobilized, which corresponds to 0.64% of injected cells. Flow cytometry confirmed an increase of percentage of GFP+ cells in the blood in mice treated with plerixafor/G-CSF compared to control (Fig. 5B, p=0.0009), despite the fact that the treatment might have also mobilized some endogenous radioresistant cells from the BM, lowering the percentage of GFP+ cells in the circulation. Of note, 89Zr activity in the circulation corresponded well with GFP+ cell number (Fig. S2).
Figure 5. 89Zr-labeled GFP+ pre-transplanted BM cells are mobilized into the blood by plerixafor/G-CSF treatment.
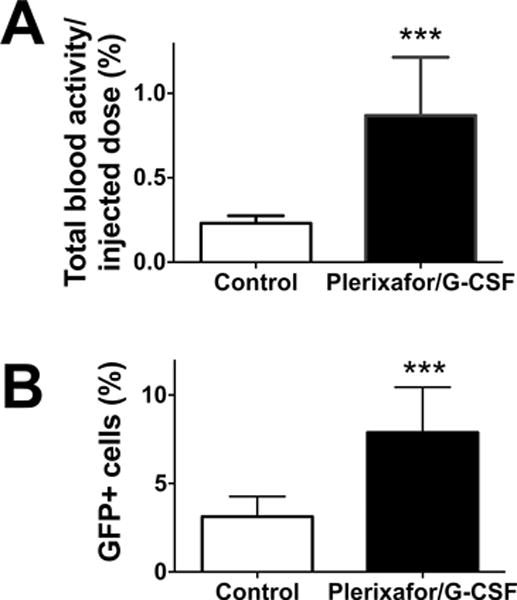
Two groups of mice were pre-transplanted with 89Zr-labeled GFP+ BM cells (2×107 cells at 37–111 kBq/106 cells). One group of mice received i.v. injections of plerixafor/G-CSF at 3 h and 1 d and blood was collected 2h after the second mobilization treatment from both experimental and control groups. A. 89Zr activity associated with cells in the blood of experimental group (black) increased by 3.8-fold compared to control (white) (n=4; P = 0.0009). B. Flow cytometry analysis confirmed that BM cell mobilization increased GFP+ cell fraction in the circulation (n=4, white: control, black: mobilized).
Transplanted BM cells labeled with 89Zr-oxine engraft and reconstitute the immune cells in BM ablated hosts
Finally, we analyzed the engraftment of 89Zr-oxine-labeled CD45.2+ BM cells in CD45.1+ recipient mice 10 weeks following cell transfer. Flow cytometry of recipient BM revealed that 89Zr-labeled donor cells had engrafted only when the host mice had received whole-body irradiation before the BM transfer (Fig. 6A, B, S3A). In the BM-ablated mice, 85% of BM cells were 89Zr-labeled donor cell origin and phenotypically similar to the control mice that received unlabeled BM cells. In the periphery, about 90% of splenocytes consisted of 89Zr-labeled donor-derived cells, which included B cells (B220+), DCs (CD11c+), NK cells (CD3−NK1.1+) and CD4 and CD8 T cells (CD4+ or CD8+CD3+NK1.1−) (Fig. 6C, D, S3B). These results suggest that critical functions of BM cells, such as homing capacity to the BM, engraftment in the BM niche, and differentiation into mature cells, were retained in 89Zr-oxine-labeled cells.
Figure 6. Donor BM cells labeled with 89Zr-oxine engraft and differentiate into mature immune cells in the recipient mice.
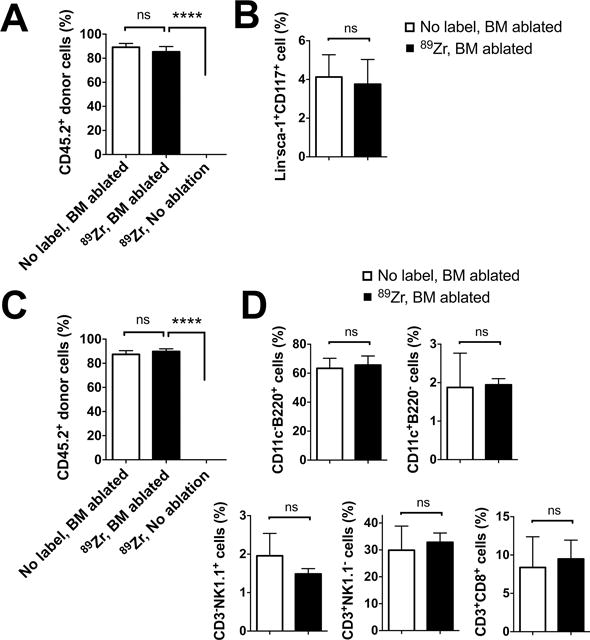
Flow cytometry analysis of BM cells and splenocytes was performed 10 weeks after the BM transfer (n=3). A. CD45.2+ donor BM cells, with or without 89Zr-labeling, reconstituted the BM of the hosts (CD45.1+) that received BM ablation prior to the transplantation, but not that of the non-BM ablated hosts. B. 89Zr-labeling did not impair the engraftment of CD45.2+lineage marker−sca-1+CD117− hematopoietic stem/progenitor cells in the BM ablated hosts. C. Similarly, CD45.2+ donor derived cells reconstituted the spleen only in the BM ablated hosts. D. Analysis of splenocytes revealed that donor BM cells had differentiated into DCs (CD11c+), B cells (B220+), NK cells (CD3−NK1.1+), T cells (CD3+NK1.1−) in BM ablated recipient mice. (white: non-labeled BM transferred to BM ablated host, black: 89Zr-labeled BM transferred to BM ablated hosts),
Discussion
HSC trafficking to the BM is crucial for the engraftment of the transferred cells and, ultimately, the success of BM transplantation therapy. In this study, we examined BM cell trafficking using 89Zr-oxine cell labeling and PET imaging to visualize and quantify donor cell migration after i.v. cell transfer.
Limited imaging modalities currently exist for cell tracking in humans. Among those being tested, magnetic resonance imaging (MRI) with iron-nanoparticle labeling has the advantages of high resolution and absence of ionizing radiation, but is neither sensitive for detecting systemically distributed cells nor is it quantitative (14). Radionuclide imaging of cells provides a higher sensitivity and is more quantitative in comparison to MRI (15). Within radionuclide imaging, PET offers great advantages over single photon emission tomography (SPECT) because of its higher resolution and sensitivity, reducing the required radioactivity doses by at least a factor of ten (16, 17), and therefore, providing the potential to reduce cytotoxicity (18–20). However, a standard method of cell tracking for PET has not been established. 18F-fluorodeoxyglucose (FDG) can be used to label cells, but it relies on high glucose metabolic activity and thus is not suitable for BM cells (21). Moreover, the short half-life of 18F (109.7 min) poses a severe limitation on the duration of tracking. Ideally, a PET cell labeling agent should readily permeate the plasma membrane and be stably retained inside the cell for a period of days, while also causing limited cytotoxicity. 89Zr-oxine complex is a novel agent that has been developed to meet these requirements based on the 3-day half-life of 89Zr and the minimal amounts of radioactivity needed for imaging (11).
We demonstrated that whole BM cells could be efficiently labeled ex vivo by 89Zr-oxine complex. 89Zr-oxine complex sufficient for PET imaging did not alter cellular phenotype (Fig. 1A). Labeled BM cells cultured with GM-CSF demonstrated a delayed proliferation, but were capable of full differentiation (Fig. 2E, H, I). As the cells labeled at low dose proliferated, the 89Zr activity per cell (specific activity) decreased rapidly (Fig. 2F). With the high dose, specific activity was maintained until the cells started to slightly increase in number on day 8. Consistently, cells cultured in conditions that do not induce strong proliferation (i.e., no cytokine or the SCF, FLT3L and TPO cocktail) showed sustained 89Zr retention (Fig. 1C, 2B). Differentiation capacity of 89Zr-labeled BM cells to a broader spectrum of hematopoietic cells, such as erythrocytes, granulocytes, and macrophages, was also proven by the CFC assay (Fig. 2K). In in vitro systems, it is difficult to create a culture condition that completely mimics the in vivo environment and thus, BM cells not responding to the added cytokines would die. However, in vivo, various stem cells/progenitor cells could survive once the cells successfully home to organs that provide microenvironments favorable to them.
The i.v. transferred BM cells labeled with 89Zr-oxine passed through the lungs, and then migrated to the BM, spleen, and liver within 4 h of injection. Some cells appeared to immediately transit the lungs and proceed to the BM in the vertebra, pelvis and long bones. It has been reported that a significant fraction of BM cells transferred i.v. are trapped in the lungs, but our data contradict these reports. When a well-prepared single-cell suspension was used, we consistently observed rapid trans-pulmonary passage of HSCs. Aggregation of cells during cell preparation may produce lung trapping, leading to loss of HSCs and inefficient engraftments. This rapid transit through the lungs seemed to be a unique property of HSCs as more mature cells often become at least temporarily trapped in the lungs (11). It is interesting that BM cells also homed to the liver and spleen, organs that are reported as initial sites for extra-medullary hematopoiesis in mice (22, 23).
CXCR4 signaling is known to regulate trafficking of BM cells by directing them to the sites of CXCL12 production, such as the BM, spleen and liver (24). Endothelial cells, osteoblasts and reticular cells produce CXCL12 in these organs (25). We observed that the CXCR4 blockade with plerixafor (12) prevented the homing of cells to the BM and the effect lasted longer when G-CSF was co-administered (Fig. 4). The effect of the CXCR4 blockade on the cell migration to the spleen and liver, however, was not evident. G-CSF has been shown to induce the release of proteolytic enzymes such as metalloproteinase-9, neutrophil proteases, and cathepsin G, which cleave various HSC supportive molecules within the BM, including CXCL12, thus reducing the CXCL12 gradient toward the BM (26, 27).
Although plerixafor and G-CSF have been widely used to mobilize and then to collect HSCs from the circulation as part of allogeneic and autologous HCST, it is unclear what fraction of BM cells become mobilized. 89Zr-oxine BM cell tracking enabled us to estimate the number of the cells that homed to the BM and then mobilized under the influence of plerixafor/G-CSF. Our data suggested that mobilization of only 0.64% of transferred BM cells was sufficient to induce a 3.8-fold increase of the circulating BM cells (Fig. 5). It is possible that plerixafor/G-CSF acted only on the cells homed to the BM niche, not on those trafficked to the liver and spleen, similar to the observation that plerixafor/G-CSF inhibited the initial homing of the donor cells to the BM, not to the liver and spleen. In this case, we can estimate that approximately 1.9% of the BM cells pre-transplanted to the BM were mobilized by the 2 doses of plerixafor/G-CSF (approximately 1.28 × 105 cells), because 89Zr-labeled cells situated in the BM at 1 d in irradiated recipients were 34.3% of injected cells (approximately 6.9×106 cells, Fig. 3C). It seemed that plerixafor/G-CSF inhibited CXCR4 in a stronger manner on circulating BM cells than the BM cells that have already situated in the organs. This could be because mobilization required plerixafor/G-CSF to compete with BM cell-anchoring molecules such as CXCL12, whereas CXCR4 on the circulating transferred cells were readily available to plerixafor to bind. As 89Zr does not transfer from labeled cells to neighboring non-labeled cells in co-culture (28), the 89Zr-labeled cells found in the blood are considered to be the 89Zr-labeled transferred BM cells. Although we used only a limited mobilization method in this study, investigations of various molecules and mechanisms suggested to be involved in the BM homing and mobilization of HSCs, such as sphingosin-1 phosphate (29, 30), CD26 (31) and circadian rhythm (32, 33), using the 89Zr-oxine cell-tracking technique would be interesting.
It has been reported that prior irradiation leads to increased recruitment of cells to the BM (34, 35), however, we observed that initial BM homing was independent of prior BM ablation (Fig. 3). Similar percentages of cells homed to the BM regardless of prior ablation. By contrast, cell engraftment did require prior ablation, as is widely known (Fig. 6, S3). In recipient mice that received a lethal whole-body irradiation prior to the BM transfer, reconstitution of BM and spleen occurred irrespective of 89Zr-labeling of the donor cells. This suggested that 89Zr-labeling did not impair engraftment, proliferation and differentiation to the mature immune cells in their new hosts.
89Zr-oxine cell labeling combined with PET imaging technology enables the quantitative analysis of the dynamics of cell trafficking after transfer and provides important information that is otherwise unobtainable, while maintaining the functional properties of the cells. In the research settings, the effects of modification of stem cells to enhance BM homing capability the conditioning of the recipient with cytokines (e.g. CXCL12) or cytokine antagonists (27, 34, 36), or the altering of the cell delivery route (e.g. intrabone (3–6)), for example, could be more accurately assessed with quantitative 89Zr-oxine PET imaging. Fortunately, the extremely low radioactive doses required for this technique would make this method directly applicable to humans, thus, serving as a tool to measure new transplant strategies rather than relying on animal models. In humans, because of the large number of cells to be transplanted, labeling only a small fraction of the cells would be sufficient for tracking.
With 89Zr-oxine cell labeling technique, 89Zr is released after cell death. This minimizes the risk of 89Zr being transferred from dead cells to phagocytes such as macrophages via engulfment. We observed that the majority of released 89Zr was excreted from the kidneys; however, a small fraction of free 89Zr could be taken up in the bone matrix (37). We assessed the effect of free 89Zr uptake in the bone by injecting DFO multiple times after the transfer of BM cells labeled with 89Zr-oxine and found that bone uptake of free 89Zr was not evident until day 2 (Fig. S4). Moreover, in the clinical setting, continuous infusion of DFO is possible and would induce rapid excretion of 89Zr from the kidneys, preventing bone uptake, and thus clear identification of cellular homing would be possible. To evaluate for potential toxicity induced by 89Zr retention in the BM/bone, we carefully measured mouse body weight and did not find any significant changes even at doses higher than those used for imaging (Fig. S5). Therefore, it is unlikely that acute/semi-acute severe side effects will be observed in BM cell tracking using tracer doses of 89Zr-oxine.
In conclusion, labeling of BM cells with 89Zr-oxine complex allowed the detection of rapid BM homing and mobilization of BM cells after transplantation. 89Zr-oxine labeling of BM cells did not impair their survival, proliferation, chemotaxis and differentiation functions. Furthermore, this technique allowed quantification of the cells and revealed dynamic kinetics of the cells. Thus, 89Zr-oxine PET imaging would be a useful tool to study BM transplantation in the research and clinical trial setting.
Supplementary Material
TRANSLATIONAL RELEVANCE.
In hematopoietic stem cell transplantation (HSCT), the fate of transferred cells is largely unknown, due to a lack of methods to non-invasively monitor donor cells in vivo. 89Zr-oxine complex ex vivo cell labeling combined with positron emission tomography (PET) imaging has shown promise as a sensitive method of tracking transferred bone marrow cells. The high sensitivity of PET and the lack of intrinsic background signal in the recipient allows use of very low radiolabeling doses thus, minimizes damage to cell viability, differentiation or function. Visualization of HSCT using 89Zr-oxine PET could be a useful tool for determining the effects of optimization and improvements in HSCT.
Acknowledgments
Financial support: This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research under ZIA BC 010657. K.O. A. was an awardee of the National Institutes of Health Undergraduate Scholarship Program.
Footnotes
Conflict of interest: The authors declare no competing financial interests. The authors N.S. and P.L.C. have filed U.S. Patent Applications for generation and application of the 89Zr-oxine complex.
References
- 1.Jenq RR, van den Brink MR. Allogeneic haematopoietic stem cell transplantation: individualized stem cell and immune therapy of cancer. Nat Rev Cancer. 2010;10:213–21. doi: 10.1038/nrc2804. [DOI] [PubMed] [Google Scholar]
- 2.Li M, Sun K, Welniak LA, Murphy WJ. Immunomodulation and pharmacological strategies in the treatment of graft-versus-host disease. Expert Opin Pharmacother. 2008;9:2305–16. doi: 10.1517/14656566.9.13.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramirez PA, Wagner JE, Brunstein CG. Going straight to the point: intra-BM injection of hematopoietic progenitors. Bone Marrow Transplant. 2010;45:1127–33. doi: 10.1038/bmt.2010.39. [DOI] [PubMed] [Google Scholar]
- 4.Castello S, Podesta M, Menditto VG, Ibatici A, Pitto A, Figari O, et al. Intra-bone marrow injection of bone marrow and cord blood cells: an alternative way of transplantation associated with a higher seeding efficiency. Exp Hematol. 2004;32:782–7. doi: 10.1016/j.exphem.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 5.Frassoni F, Gualandi F, Podesta M, Raiola AM, Ibatici A, Piaggio G, et al. Direct intrabone transplant of unrelated cord-blood cells in acute leukaemia: a phase I/II study. Lancet Oncol. 2008;9:831–9. doi: 10.1016/S1470-2045(08)70180-3. [DOI] [PubMed] [Google Scholar]
- 6.Hagglund H, Remberger M, Ringden O. Twenty-year follow-up of a randomized trial comparing intraosseous and i.v. BM transplantation. Bone Marrow Transplant. 2014;49:1541–2. doi: 10.1038/bmt.2014.184. [DOI] [PubMed] [Google Scholar]
- 7.Ballen KK, Gluckman E, Broxmeyer HE. Umbilical cord blood transplantation: the first 25 years and beyond. Blood. 2013;122:491–8. doi: 10.1182/blood-2013-02-453175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaney M, Ballen KK. Umbilical cord blood transplantation: review of factors affecting the hospitalized patient. J Intensive Care Med. 2015;30:13–22. doi: 10.1177/0885066613488730. [DOI] [PubMed] [Google Scholar]
- 9.Ratajczak MZ. A novel view of the adult bone marrow stem cell hierarchy and stem cell trafficking. Leukemia. 2015;29:776–82. doi: 10.1038/leu.2014.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henon P. New developments in peripheral blood stem cell transplants. Leukemia. 1992;6(Suppl 4):106–9. [PubMed] [Google Scholar]
- 11.Sato N, Wu H, Asiedu KO, Szajek LP, Griffiths GL, Choyke PL. (89)Zr-Oxine Complex PET Cell Imaging in Monitoring Cell-based Therapies. Radiology. 2015;275:490–500. doi: 10.1148/radiol.15142849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fricker SP, Anastassov V, Cox J, Darkes MC, Grujic O, Idzan SR, et al. Characterization of the molecular pharmacology of AMD3100: a specific antagonist of the G-protein coupled chemokine receptor, CXCR4. Biochem Pharmacol. 2006;72:588–96. doi: 10.1016/j.bcp.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Harkness JE, Wagner JE. Biology and husbandry. In: Harkness JE, Wagner JE, editors. The biology and medicine of rabbits and rodents. 3rd. Philadelphia: Lea & Febiger; 1989. p. 372. [Google Scholar]
- 14.de Vries IJ, Lesterhuis WJ, Barentsz JO, Verdijk P, van Krieken JH, Boerman OC, et al. Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy. Nature biotechnology. 2005;23:1407–13. doi: 10.1038/nbt1154. [DOI] [PubMed] [Google Scholar]
- 15.Srinivas M, Aarntzen EH, Bulte JW, Oyen WJ, Heerschap A, de Vries IJ, et al. Imaging of cellular therapies. Adv Drug Deliv Rev. 2010;62:1080–93. doi: 10.1016/j.addr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 16.Rahmim A, Zaidi H. PET versus SPECT: strengths, limitations and challenges. Nucl Med Commun. 2008;29:193–207. doi: 10.1097/MNM.0b013e3282f3a515. [DOI] [PubMed] [Google Scholar]
- 17.Bhargava KK, Gupta RK, Nichols KJ, Palestro CJ. In vitro human leukocyte labeling with (64)Cu: an intraindividual comparison with (111)In-oxine and (18)F-FDG. Nucl Med Biol. 2009;36:545–9. doi: 10.1016/j.nucmedbio.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brenner W, Aicher A, Eckey T, Massoudi S, Zuhayra M, Koehl U, et al. 111In-labeled CD34+ hematopoietic progenitor cells in a rat myocardial infarction model. J Nucl Med. 2004;45:512–8. [PubMed] [Google Scholar]
- 19.Kuyama J, McCormack A, George AJ, Heelan BT, Osman S, Batchelor JR, et al. Indium-111 labelled lymphocytes: isotope distribution and cell division. Eur J Nucl Med. 1997;24:488–96. doi: 10.1007/BF01267679. [DOI] [PubMed] [Google Scholar]
- 20.Gholamrezanezhad A, Mirpour S, Ardekani JM, Bagheri M, Alimoghadam K, Yarmand S, et al. Cytotoxicity of 111In-oxine on mesenchymal stem cells: a time-dependent adverse effect. Nucl Med Commun. 2009;30:210–6. doi: 10.1097/MNM.0b013e328318b328. [DOI] [PubMed] [Google Scholar]
- 21.Farwell MD, Pryma DA, Mankoff DA. PET/CT imaging in cancer: Current applications and future directions. Cancer. 2014;120:3433–45. doi: 10.1002/cncr.28860. [DOI] [PubMed] [Google Scholar]
- 22.Morrison SJ, Uchida N, Weissman IL. The biology of hematopoietic stem cells. Annu Rev Cell Dev Biol. 1995;11:35–71. doi: 10.1146/annurev.cb.11.110195.000343. [DOI] [PubMed] [Google Scholar]
- 23.Tagaya H, Kunisada T, Yamazaki H, Yamane T, Tokuhisa T, Wagner EF, et al. Intramedullary and extramedullary B lymphopoiesis in osteopetrotic mice. Blood. 2000;95:3363–70. [PubMed] [Google Scholar]
- 24.Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23:43–52. doi: 10.1038/leu.2008.299. [DOI] [PubMed] [Google Scholar]
- 25.Yellowley C. CXCL12/CXCR4 signaling and other recruitment and homing pathways in fracture repair. Bonekey Rep. 2013;2:300. doi: 10.1038/bonekey.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohle R, Murea S, Kirsch M, Haas R. Differential expression of L-selectin, VLA-4, and LFA-1 on CD34+ progenitor cells from bone marrow and peripheral blood during G-CSF-enhanced recovery. Exp Hematol. 1995;23:1535–42. [PubMed] [Google Scholar]
- 27.Hopman RK, DiPersio JF. Advances in stem cell mobilization. Blood Rev. 2014;28:31–40. doi: 10.1016/j.blre.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davidson-Moncada J, Sato N, Hoyt RF, Reger RN,MT, Clevenger R, et al. A Novel Method to Study the in Vivo Trafficking and Homing of Adoptively Transferred NK Cells in Rhesus Macaques and Humans. Blood. 2014;124:659. [Google Scholar]
- 29.Ratajczak MZ, Lee H, Wysoczynski M, Wan W, Marlicz W, Laughlin MJ, et al. Novel insight into stem cell mobilization-plasma sphingosine-1-phosphate is a major chemoattractant that directs the egress of hematopoietic stem progenitor cells from the bone marrow and its level in peripheral blood increases during mobilization due to activation of complement cascade/membrane attack complex. Leukemia. 2010;24:976–85. doi: 10.1038/leu.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Juarez JG, Harun N, Thien M, Welschinger R, Baraz R, Pena AD, et al. Sphingosine-1-phosphate facilitates trafficking of hematopoietic stem cells and their mobilization by CXCR4 antagonists in mice. Blood. 2012;119:707–16. doi: 10.1182/blood-2011-04-348904. [DOI] [PubMed] [Google Scholar]
- 31.Christopherson KW, 2nd, Hangoc G, Mantel CR, Broxmeyer HE. Modulation of hematopoietic stem cell homing and engraftment by CD26. Science. 2004;305:1000–3. doi: 10.1126/science.1097071. [DOI] [PubMed] [Google Scholar]
- 32.Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–21. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 33.Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–7. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- 34.Ponomaryov T, Peled A, Petit I, Taichman RS, Habler L, Sandbank J, et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J Clin Invest. 2000;106:1331–9. doi: 10.1172/JCI10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kane J, Krueger SA, Dilworth JT, Torma JT, Wilson GD, Marples B, et al. Hematopoietic stem and progenitor cell migration after hypofractionated radiation therapy in a murine model. Int J Radiat Oncol Biol Phys. 2013;87:1162–70. doi: 10.1016/j.ijrobp.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 36.Lapidot T, Dar A, Kollet O. How do stem cells find their way home? Blood. 2005;106:1901–10. doi: 10.1182/blood-2005-04-1417. [DOI] [PubMed] [Google Scholar]
- 37.Abou DS, Ku T, Smith-Jones PM. In vivo biodistribution and accumulation of 89Zr in mice. Nucl Med Biol. 2011;38:675–81. doi: 10.1016/j.nucmedbio.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.