

Abstract
GLYCOGEN STORAGE DISEASE TYPE III (GSD III) was diagnosed in 4 Inuit children (3 confirmed, 1 suspected case) at our institution over the last decade. This rare autosomal recessive disease, which results from a deficiency of the debranching enzyme required for complete degradation of the glycogen molecule, has not been previously described in this population. The possible clinical presentations are heterogeneous, as is the spectrum of severity of this disease. The long-term sequelae can be severe, including recurrent hypoglycemia, hepatic cirrhosis and progressive muscle weakness. These 4 cases would suggest an increased prevalence of GSD III in the Inuit population. Therefore, it is important for health care providers caring for this population to consider and recognize this rare but serious disease.
Glycogen storage disease type III (GSD III) is part of a rare group of inherited enzyme defects that affect the glycogen synthesis and degradation cycle. It is caused by an autosomal recessive deficiency of the glycogen debranching enzyme, amylo-1,6-glucosidase, which results in incomplete degradation of the glycogen molecule. This enzyme is critical in both liver and muscle tissue. Deficiency of the enzyme in both these tissues produces a variant known as GSD IIIa, which can involve skeletal and cardiac muscle. Another common variant, known as GSD IIIb, is caused by a deficiency of the enzyme in the liver only and is not associated with any muscle involvement.1 In their severest forms, both variants can present in infancy and early childhood with hepatomegaly, hypoglycemia and growth retardation. Milder cases may present only in adulthood, with asymptomatic hepatomegaly, occult liver disease or myopathy.2 Although fasting tolerance for hypoglycemia generally improves with age and hepatomegaly frequently regresses,3 chronic fibrosis leading to overt cirrhosis and end-stage liver disease may occur in a small portion of patients with GSD III.4,5,6
To our knowledge, there are no published cases of GSD III in the Aboriginal population of North America. We describe 3 confirmed cases and 1 suspected case of GSD III in Inuit children from northern Quebec and eastern Nunavut, with different clinical presentations.
The cases
The clinical information for each of the 4 cases is presented in Table 1. The age at presentation varied from 3 months to 7 years. The chief complaint differed in each case. Case 1 was unable to walk or stand alone by 17 months of age despite reaching other developmental milestones. Case 2 was noted to have early morning awakenings for feeding and an increased appetite. She had episodes of morning fatigue and a decreased energy that improved following feeding. This pattern likely represented recurrent hypoglycemia. Case 3 was incidentally found to have hepatomegaly on physical examination. Case 4 was being evaluated by an endocrinologist because of her short stature and was noted to have a firm liver edge 3 cm below the costal margin.
Table 1
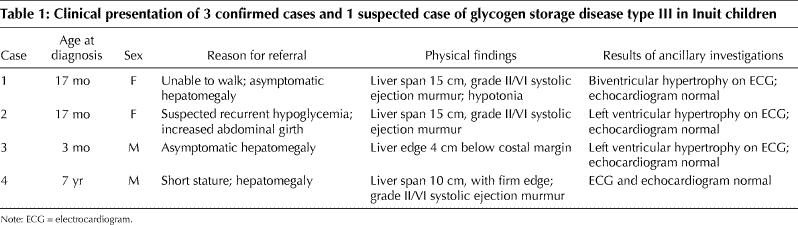
Hepatomegaly, found in all cases, ranged from mild to moderate. Systolic ejection murmurs were heard in 3 of the cases. Electrocardiogram findings consistent with ventricular hypertrophy were noted in 3 cases, but all 4 had normal echocardiograms. Only 1 case (case 4) was associated with short stature. The serum biochemistry profiles of the 4 children on presentation are summarized in Table 2. All had elevated liver transaminase levels, although of varying severity. The creatinine kinase (CK) level was elevated in cases 1 and 3 on presentation; the level in case 2 was elevated at the last follow-up visit.
Table 2
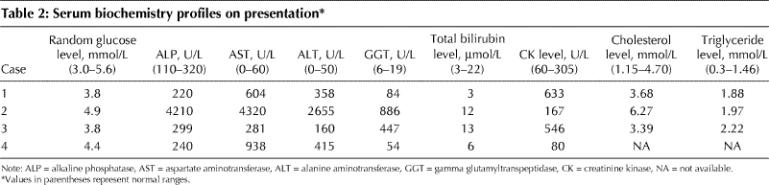
Random blood glucose levels were well within normal limits in all of the cases. When children with hepatomegaly who have normal random blood glucose levels are suspected of having GSD III, they should undergo a fasting challenge to determine the duration until hypoglycemia and response to glucagon. If GSD III is present, the children usually cannot fast for more than a few hours and will often have an increased serum glucose level after glucagon administration. The latter feature helps to distinguish GSD III from GSD I. However, with prolonged fasting, GSD III patients lose their ability to respond to glucagon. Formal fasting challenges confirmed hypoglycemia after 3–3.5 hours in 3 of the 4 cases. The results of the fasting challenges and liver biopsies are presented in Table 3. The glycogen-laden hepatocytes observed on biopsy in all 4 cases establishes the diagnosis of GSD.
Table 3
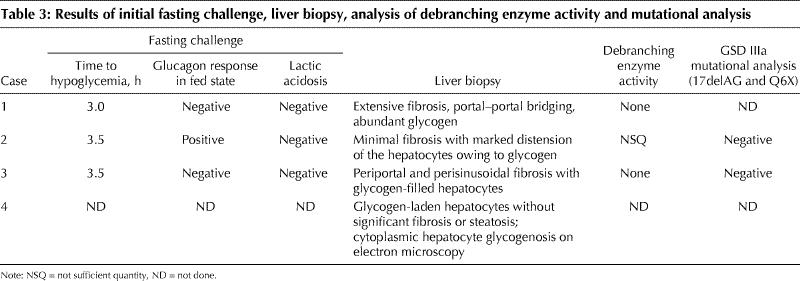
Absent debranching enzyme activity in the biopsy specimens in cases 1 and 3 confirmed the diagnosis of GSD III. The response to glucagon after a short fast observed in case 2 is typical of GSD III. The elevated CK level in cases 1 through 3 is most compatible with GSD IIIa. Genetic testing for the 2 common mutations that account for 90% of GSD IIIb (17delAG and Q6X) was negative in the 2 cases in which it was performed. There was no enzymatic analysis of case 4's biopsy specimen to determine the type of GSD, but the clinical findings and available investigations were consistent with GSD type III, VI or IX variant.
The main findings at last follow-up of each case are presented in Table 4. The fasting tolerance of the 3 cases with hypoglycemia responded to frequent feeding and cornstarch supplementation and has improved with age. The course of the hepatomegaly has been variable. Significant portal hypertension has developed in case 1. Clinical evidence of muscular involvement did not progress in case 1 or develop in cases 2 and 3.
Table 4

Comments
We diagnosed 4 cases of GSD III in Inuit children from northern Quebec and the Baffin Region of eastern Nunavut in the last decade. According to 2001 census data,7 the Inuit population of these regions totals 20 265. This number of cases represents an inordinately high prevalence for this rare disease, which is estimated to have an incidence of 1 in 100 000 live births in North America.8 Of note, there was no consanguinity of the parents of these children, nor was there any known shared pedigree between the 4 cases, all of whom lived in different villages. Cases 1 through 3 came from villages in northern Quebec, bordering the eastern coast of Hudson Bay. From north to south, case 1 was separated from case 2 by about 100 km, and case 2 was separated from case 3 by about 400 km. Case 4 came from a village off the Melville Peninsula in eastern Nunavut, bordering on the western shores of Hudson Bay.
Three of the cases clearly fit the diagnosis of GSD IIIa on the basis of the clinical picture, liver biopsy findings and increased creatinine kinase levels. About 80% of GSD III cases in North America are of the type IIIa variant, in which the debranching enzyme is deficient in muscle as well as hepatocytes.2 This is comparable to the distribution of cases in other countries with an increased incidence of the disease, such as Japan. Interestingly, linkage studies tracing mitochondrial DNA sequences suggest that the circumpolar Inuit are originally of Asian progeny.9
Genetic confirmation of the diagnosis of GSD type III would require demonstration of a mutation in the gene coding for the debranching enzyme. Testing is available to identify the 2 mutations that account for over 90% of cases of GSD IIIb. However, the gene defects responsible for GSD IIIa are far too heterogeneous to perform genetic testing on a routine basis.10 Therefore, the condition is diagnosed on the basis of the clinical features, liver biopsy results confirming diminished debranching enzyme activity and ancillary testing. Debranching enzyme activity can now also be measured in blood or fibroblast specimens. (For a detailed review of GSD III see reference 11.)
The clinical presentation of case 1 was of particular interest because the girl had significant hypotonia and muscle weakness without any other detectable cause than the GSD. Clinical muscle involvement is usually minimal, if at all evident, in childhood but can become severe by the third or fourth decade of life.2 Accumulation of the incompletely degraded glycogen molecule, known as phosphorylase limit dextrin, can also occur in cardiac muscle with a variable spectrum of clinical significance.12 Abnormal findings on electrocardiography can occur early and subclinically,12,13 as in 3 of the 4 cases, and are suggestive of the type IIIa variant of GSD.
Case 4 came to our attention only when the patient was 7 years of age, when mild hepatomegaly was noted on evaluation for short stature. Liver biopsy findings were highly suggestive of glycogen storage disease; however, further testing would be required to determine exactly which type. Unfortunately, this patient was lost to follow-up, and no CK level was measured, but mild cases are part of the large spectrum of clinical presentations of GSD III. There has even been a case of GSD IIIa diagnosed in a 54-year-old woman only after evaluation of an elevated CK level.14
Not only is the presentation of GSD variable, so too is the prognosis. Hepatic involvement is often transient, with elevated transaminase levels and hepatomegaly resolving with age. However, progression to cirrhosis and end-stage liver disease has also been documented.4,5,6 This is certainly a concern in case 1, in whom evidence of portal hypertension has already been detected at 7 years of age.
These 4 cases represent a high prevalence of GSD III among the Inuit. The presentations and severity were quite varied, and it is possible that other cases in these communities have gone undiagnosed. Although the initial features may be subtle, the long-term complications can be serious and include impaired neurodevelopment (secondary to recurrent hypoglycemia), hepatic cirrhosis, cardiomyopathy and debilitating muscle weakness. It is important for health care providers caring for this population to consider and recognize the possible presentations of GSD III.
Addendum
We have reason to believe that the diagnosis of GSD III among the Inuit, albeit rare, may be more common than previously suspected. The sibling of case 2 was recently found to have GSD III on the basis of clinical presentation and characteristic histologic findings on liver biopsy. We strongly suspect that another infant from the same community also had GSD III, since the autopsy performed after sudden death at age 3 months demonstrated significant hepatomegaly and abundant hepatic glycogen. Although a firm postmortem diagnosis is impossible, the circumstantial findings are consistent with those of the cases described here and are highly suggestive of GSD.
Footnotes
This article has been peer reviewed.
Contributors: Paul Zimakas was the principal author of the article and contributed to the article conception and design, and the data collection and analysis. Celia Rodd contributed to the conception and design, the data analysis and the critical revision of the article. Both authors approved the final version submitted to be published.
Competing interests: None declared.
Correspondence to: Paul James A. Zimakas, Department of Pediatric Endocrinology, Montreal Children's Hospital, 2300 Tupper St., Montreal QC H3H 1P3; fax 514 412-4264
References
- 1.Coleman RA, Winter HS, Wolf B, Gilchrist JM, Chen YT. Glycogen Storage Disease type III (glycogen debranching enzyme deficiency): correlation of biochemical defects with myopathy and cardiomyopathy. Ann Intern Med 1992; 116:896-900. [DOI] [PubMed]
- 2.Wolfsdorf JI, Holm IA, Weinstein DA. Glycogen storage diseases: phenotypic, genetic, and biochemical characteristics, and therapy. Endocrinol Metab Clin North Am 1999; 28(4):801-23. [DOI] [PubMed]
- 3.Coleman RA, Winter HS, Wolf B, Chen YT. Glycogen debranching enzyme deficiency: long-term study of serum enzyme activities and clinical features. J Inherit Metab Dis 1992;15: 869-81. [DOI] [PubMed]
- 4.Haagsma EB, Smit GP, Niezen-Koning KE, Gouw AS, Meerman L, Slooff MJ. Type IIIb glycogen storage disease associated with end-stage cirrhosis and hepatocellular carcinoma. The Liver Transplant Group. Hepatology 1997; 25 (3):537-40. [DOI] [PubMed]
- 5.Siciliano M, De Candia E, Ballarin S, Vecchio FM, Servidei S, Annese R, et al. Hepatocellular carcinoma complicating liver cirrhosis in type IIIa glycogen storage disease. J Clin Gastroenterol 2000;31(1):80-2. [DOI] [PubMed]
- 6.Matern D, Starzl TE, Arnaout W, Barnard J, Bynon JS, Dhawan A, et al. Liver transplantation for glycogen storage disease types I, III, and IV. Eur J Pediatr 1999;158(Suppl 2):S43-8. [DOI] [PMC free article] [PubMed]
- 7.Statistics Canada. 2001 Aboriginal population profile. Ottawa: Census Division, Statistics Canada; 2001.
- 8.Parvari R, Moses S, Shen J, Hershkovitz E, Lerner A, Chen YT. A single-base deletion in the 3'-coding region of glycogen-debranching enzyme is prevalent in glycogen storage disease type IIIA in a population of North African Jewish patients. Eur J Hum Genet 1997;5(5):266-70. [PubMed]
- 9.Bonatto SL, Salzano FM. A single and early migration for the peopling of the Americas supported by mitochondrial DNA sequence data. Proc Natl Acad Sci U S A 1997;94(5):1866-71. [DOI] [PMC free article] [PubMed]
- 10.Shen JJ, Chen YT. Molecular characterization of glycogen storage disease type III. Curr Mol Med 2002;2(2):167-75. [DOI] [PubMed]
- 11.Scriver CR, Beaudet AL, Sly WS, Valle DL, editors. The metabolic and molecular basis of inherited disease. 7th ed. Vol 1. New York: McGraw Hill; 2001. p. 949-50.
- 12.Moses SW, Wanderman KL, Myroz A, Frydman M. Cardiac involvement in glycogen storage disease type III. Eur J Pediatr 1989;148(8):764-6. [DOI] [PubMed]
- 13.Lee PJ, Deanfield JE, Burch M, Baig K, McKenna WJ, Leonard JV. Comparison of the functional significance of left ventricular hypertrophy in hypertrophic cardiomyopathy and glycogenosis type III. Am J Cardiol 1997; 79 (6): 834-8. [DOI] [PubMed]
- 14.Yang BZ, Stewart C, Ding JH, Chen YT. Type III glycogen storage disease: an adult case with mild disease but complete absence of debrancher protein. Neuromuscul Disord 1991;1(3):173-6. [DOI] [PubMed]