

Abstract
Commonly upregulated in human cancers, the scaffolding protein NEDD9/HEF1 is a known regulator of mesenchymal migration and cancer cell plasticity. However, the functional role of NEDD9 as a regulator of different migration/invasion modes in the context of breast cancer metastasis is currently unknown. Here, it is reported that NEDD9 is necessary for both mesenchymal and amoeboid individual cell migration/invasion in triple-negative breast cancer (TNBC). NEDD9 deficiency results in acquisition of the amoeboid morphology, but severely limits all types of cell motility. Mechanistically, NEDD9 promotes mesenchymal migration via VAV2-dependent Rac1 activation, and depletion of VAV2 impairs the ability of NEDD9 to activate Rac1. Additionally, NEDD9 supports a mesenchymal phenotype through stimulating polymerization of actin via promoting CTTN phosphorylation in an AURKA-dependent manner. Interestingly, an increase in RhoA activity in NEDD9-depleted cells does not facilitate a switch to functional amoeboid motility, indicating a role of NEDD9 in the regulation of downstream RhoA signaling effectors. Simultaneous depletion of NEDD9 or inhibition of AURKA in combination with inhibition of the amoeboid driver ROCK results in an additional decrease in cancer cell migration/invasion. Finally, we confirmed that a dual targeting strategy is a viable and efficient therapeutic approach to hinder the metastasis of breast cancer in xenograft models, showcasing the important need for further clinical evaluation of this regimen in order to impede the spread of disease and improve patient survival.
Keywords: NEDD9, mesenchymal, amoeboid, breast cancer, metastasis
Introduction
Scaffolding protein NEDD9/HEF1, commonly overexpressed in many human cancers, is a critical regulator of cancer cell migration (1, 2). Cancer cells are capable of migrating either collectively or individually (3–5). In collective migration, cells retain most of their epithelial characteristics including cell-cell adhesions and invade as a sheet (6), thus enabling regional invasion and lymphatic-based metastasis. Alternatively, individual migration is distinguished by a loss of cell-cell adhesions and the ability to invade via two different methods known as mesenchymal and amoeboid (7–9), leading to blood-borne metastasis. Depending on extracellular matrix (ECM) composition and rigidity of the tumor microenvironment, the mesenchymal and amoeboid modes can be used interchangeably by the same cell population (3, 8). Initial nascent adhesions are highly present in amoeboid cells, consisting of integrin, talin, and non-phosphorylated paxillin. The recruitment of vinculin and FAK/Src complex and activation/phosphorylation of paxillin and FAK is indicative of focal adhesion (FA) maturation, which can culminate in formation of very elongated fibrillar adhesions (10), which are often found in mesenchymal cells.
Elevated expression of NEDD9 protein is required for individual mesenchymal migration of diverse tumor cells (1, 11, 12). Mesenchymal migration is characterized by an elongated cell morphology, multiple focal/3D adhesions, and the ability to degrade ECM by matrix metalloproteinases (MMPs) (7, 13), creating a path through the basement membrane/tissue. A master regulator of mesenchymal migration, Rac1 GTPase, is activated by a number of guanine nucleotide exchange factors (GEFs), including melanoma-specific DOCK3 (14), which is recruited/activated by NEDD9 (15). Rac1 activates the WAVE regulatory complex leading to polymerization of actin filaments (16). We have previously shown that NEDD9 also activates and stabilizes Aurora A kinase (AURKA) (17), which phosphorylates and activates histone deacetylase HDAC6 (18), leading to deacetylation and subsequent increased activity of the actin branching/stabilizing protein cortactin (CTTN) (19), which is necessary for lamellipodia and invadopodia formation - structures that define mesenchymal invasion.
Rac1-driven mesenchymal migration runs counter to the corresponding RhoA GTPase and ROCKII kinase-driven amoeboid pathway. Amoeboid migration is characterized by a rounder cell morphology, small/weak adhesions, and increased cell contractility (7, 20). This type of locomotion does not typically rely on matrix degradation, and instead slightly deforms the cell shape to squeeze through pre-existing gaps in the ECM (21). Because of this, amoeboid movement is more suited for invasion through less dense and more elastic matrices such as collagens. Mechanistically, RhoA binds and activates ROCKII which phosphorylates myosin II light chain (MLC2), thus increasing actomyosin contractility and amoeboid invasion. NEDD9 contributes to downregulation of the amoeboid pathway through binding/activating Src kinase, leading to phosphorylation/inhibition of ROCKII (11).
The impact of NEDD9 on collective tumor cell invasion has not been explored. However, collective migration of lymphocytes in NEDD9 knockout mice was shown to be significantly reduced (22), and neuronal tube formation in mouse and chick embryos was inhibited upon NEDD9 depletion (23). We have shown that a decrease in NEDD9 expression leads to significant deficiency in activation of MMPs (24, 25), suggesting a deficiency of these cells to create tunnels through the matrix.
Since individual tumor cells are capable of utilizing either one of these modes of movement, simultaneous targeting of both pathways may provide significant clinical benefit against metastatic cancers (14, 26). In this current study, we define the molecular mechanisms of NEDD9-dependent Rac1 activation and the efficacy of combined inhibition of both types of cell movement to hinder the metastasis of triple negative breast cancer (TNBC).
Materials and Methods
Cell lines, RNAs, and plasmids
TNBC cell lines MDA-MB-231LN (Caliper); Hs578T, HCC1143, BT549, HCC1395 (American Type Culture Collection); and SUM159 (gift from Dr. Boerner, Karmanos Cancer Institute) were authenticated and propagated for less than 6 months. si/sh/sgRNAs and plasmids used in this study are described in detail in the Supplementary Materials and Methods. The plasmids/RNAs were introduced via nucleofection (Amaxa) according to manufacturer’s recommendation or virus infection (293T packaging cells, LifeTechnologies) as previously described (24).
Recombinant protein production
Recombinant AURKA (19), wild type and N/C-term CTTN (27), and GST and GST-PBD proteins were produced as previously described (28).
Cell morphology analysis
104 cells were plated on 2D or in 3D 1.5mg/ml collagen (Advanced BioMatrix) or 4mg/ml Matrigel (Corning) as previously described (30). Cells were treated with 50 or 100μM Y-27632 (ApexBio) where indicated. Cells were brightfield imaged on a Leica DM/IL microscope. Cell elongation was measured as a function of cell length divided by cell width (ImageJ software/NIH).
Western blotting
Western blots were performed as previously described (24). Further details and a list of antibodies used can be found in the Supplemental Material and Methods.
Immunofluorescence
The edges of 18mm round glass coverslips (Fisher) were coated with a Super-HT-PAP-PEN hydrophobic slide marker and coated inside with 0.01% poly-L-lysine (Electron Microscopy Sciences). For 3D matrix embedding, 2.5×104 cells suspended in 200μl of 1.5mg/ml collagen-I or 4mg/ml Matrigel plug were loaded on the coverslips. Coverslips were placed into a 12-well plate and incubated in a 37°C humid chamber during polymerization. Full media was added, and the cells were allowed to acclimate to the matrix for 48 hours. Fixation and immunofluorescence staining was carried out as previously described (29). The list of used antibodies and quantification details are provided in the Supplemental Material and Methods.
Collagen gel contraction assay
1×106 cells per sample were suspended in 1.5mg/ml collagen and inserted into a well in a 12-well plate. Triplicate wells were included per cell line and a well with no cells was included as control. Once polymerized, media was placed on top of the collagen plug. After 24 hours, the collagen plugs were detached from the walls and bottom of the well. Gels were imaged (Syngene/G-Box) every 15 minutes for the first few hours followed by hourly images up to 24 hours. Gel area was measured by manual outlining of the collagen plugs and analyzed in ImageJ.
Radioactive kinase assays
2.5μCi of [γ-32P]-ATP (Perkin Elmer) suspended in kinase buffer (28) was added to recombinant AURKA and/or CTTN (25-50ng) protein and incubated for 30 minutes at 30°C. Samples were then subjected to gel electrophoresis and membrane transfer followed by X-ray film exposure for 2 hours to visualize phosphorylation and western blotting to visualize total protein level.
Actin polymerization assays
Actin polymerization assays were carried out according to manufacturer’s recommendation (Cytoskeleton). Recombinant AURKA (3mM), CTTN (75nM), Arp2/3 (50nM, Cytoskeleton) or WASP-VCA domain (5μM, Cytoskeleton) suspended in magnesium/ATP cocktail were added to 5μM pyrene-labeled G-actin in a 96-well plate and actin polymerization was measured based on the increase of fluorescence at excitation 365nm/emission 407nm every 30 seconds for 20 minutes at 30°C using a GENios plate-reader (Tecan).
Cell kymography
Kymography analysis was carried out as previously described (19). In brief, cells were plated on glass bottom dishes (Fisher Scientific) 48 hours after nucleofection with mCherry-LifeAct. Cells were then treated overnight with 100nM AURKA inhibitor MLN8237 (Selleckchem) and live-imaged the next day every 45 seconds for 60 minutes. Kymograms were compiled from a perpendicular 1 pixel-width line from each frame on the same spot of the cell membrane.
Active Rac1 pulldown assays
Cells were nucleofected with the indicated plasmids/siRNAs and allowed to recover overnight, followed by suspension of 1.5×105 cells in 450μl of 1.5mg/ml collagen-I or 4mg/ml Matrigel. In indicated assays, cells were also treated overnight with 4μM EHop-016 (ApexBio). After 24 hours, cells were lysed for 10 minutes on ice with 1X Rac1 lysis buffer as previously described (30). Lysates were spun down at max speed for 12 minutes at 4°C and supernatants were added to Glutathione-4FastFlow sepharose (GEHealthcare) loaded with GST or GST-PBD protein, rotating at 4°C for at least 1 hour. Sepharose was then washed three times with lysis buffer, boiled in Laemmli buffer and subjected to gel electrophoresis and western blotting.
Active Rac1 and RhoA G-LISA assays
G-LISA activation assays for Rac1 and RhoA were performed as recommended by the manufacturer’s protocol (Cytoskeleton).
Immunoprecipitations of GEF proteins
pcDNA3.1-mRFP-NEDD9 and Rac1 GEF-expressing plasmids were co-transfected into 293T cells using TurboFect (ThermoFisher) per manufacturer protocol. After 24 hours, cells were lysed with PTY lysis buffer and processed as previously described (28). Lysates were precleared with protein-A/G-sepharose (GEHealthcare) for 1 hour at 4°C. Mouse IgG control/or anti-NEDD9 (2G9) protein-A/G sepharose, or anti-HA/anti-FLAG EZview-Red antibody (Sigma) were added to precleared lysates for 12 hours rotating at 4°C. Beads were then washed three times with PTY buffer. To elute proteins from the beads, 10% ammonia hydroxide was added for 15 seconds followed by speed-vacuum centrifuge to remove ammonia hydroxide. Protein was then resuspended in Laemmli buffer for gel electrophoresis and western blotting. The IP of CTTN is detailed in the Supplemental Materials and Methods.
Live-cell imaging of individual cell invasion
Cells treated with si/sgRNAs against NEDD9 or RhoA and/or overexpressing CA-RhoA and/or treated with 50μM Y-27632 or 100nM MLN8237 for 24h were re-suspended in 1.5mg/ml collagen and placed into 3D-Chemotaxis μ-slides (Ibidi) according to manufacturer protocol. Fetal bovine serum was added to the left media reservoir to create a chemoattractant gradient. When used, Y-27632 and/or MLN8237 were also added to both reservoirs to keep the drugs present during the assay. Cells were imaged every 15 minutes via DIC 10X objective on a Nikon Sweptfield/Eclipse/TE2000-E confocal microscope for 23 hours. Time lapse images were imported into ImageJ software and combined to form video files. Quantification methods are detailed in the Supplemental Materials and Methods.
Live-cell imaging of collective cell invasion
1×105 cells in full medium were plated on top of 4-5mg/ml Matrigel-coated delta-T dishes (Bioptechs). After 1 hour, the medium was removed and the cell layer was covered with more Matrigel. After polymerizing for 30 minutes at 37°C, media was re-added. Live-cell imaging was conducted every 10 minutes for 24 hours at 37°C via Nikon Sweptfield/Eclipse/TE2000-E confocal microscope (20X objective). Time lapse images were imported into ImageJ software and combined to form video files. Quantification methods are detailed in the Supplemental Materials and Methods.
Fluorescent imaging of collective cell invasion sandwich assay
Cells were pretreated overnight with 50μM Y-27632 or 100nM MLN8237, and 3×104 cells were plated in 8-well chamber slides (ThermoFisher) coated with 150μl 4mg/ml Matrigel. After 1 hour, the medium was removed and the cells covered with another 200μl Matrigel, followed by full media after Matrigel polymerization. Cells were live imaged in 5μm Z-steps away from the cell seeding position at 72 hours using a Nikon Sweptfield/Eclipse/TE2000-E confocal microscope. Invasion was quantified by total cell fluorescence intensity at the furthest distance cells reached (100μm). 3D invasion boxes were constructed using the Volume-Viewer plugin for ImageJ to show cell fluorescence signal throughout the Matrigel from the seeding position.
Mouse xenograft study
Animals were housed in the WVU Animal Facility under pathogen-free conditions according to an approved Institutional Animal Care and Use Committee protocol. 1×106 MDA-MB-231-luc2 cells expressing doxycycline-inducible pTRIPZ-RFP-shControl or shNEDD9 (25) were suspended in 4mg/ml Matrigel (Trevigen) and injected into the mammary fat pad of 6–8-weeks old immunodeficient female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory). Mice with mammary tumors (150–200mm3) were pre-treated in neoadjuvant settings with 10mg/kg AURKA inhibitor MLN8237 via oral gavage for 1 week (4 days on, 3 days off) as previously described to temporarily halt metastasis, but continue mammary tumor growth (31). shRNA expression in tumor cells was induced via introduction of doxycycline-containing food (Bio-Serv) in combination with vehicle or 10mg/kg Y-27632 which was administered by oral gavage for 3 weeks (4 days on, 3 days off). Tumor growth and dissemination was evaluated weekly through in vivo whole-body bioluminescence imaging (BLI) using an IVIS/Lumina-II system as previously described (17). After 3 weeks, lungs, mammary tumors, and blood were collected for analysis. Paraffin-embedded lung sections were stained by hematoxylin and eosin (H&E) and analyzed for the number of metastases per lung area by a pathologist as previously described (17). Primary tumors were analyzed for NEDD9 expression by western blot. Ten mice per both shRNA groups (further separated into 5 per drug group for a total of 20 mice) were used based on statistical analysis.
Quantification of circulating tumor cells
Submandibular mouse blood samples were collected into EDTA-coated tubes on ice to prevent clotting. Erythrocytes were lysed and removed from blood via incubation with RBC lysis buffer (eBioscience) according to manufacturer’s protocol. Cells were fixed in 2% paraformaldehyde for 10 minutes, followed by centrifugation at 500g for 5 minutes at 4°C, and resuspended in 1% BSA/PBS. Flow cytometry (FACS) was performed using a BD Biosciences/LSR Fortessa to count RFP-positive circulating tumor cells in the blood samples. Final counts were normalized to initial primary tumor size at week zero.
Statistical analysis
One-way ANOVA or student’s t-test statistical analyses were performed with GraphPad Prism software where noted. p ≤ 0.05 was considered to be significant (*). Experimental values were reported as the mean ± SEM (standard error of mean).
Results
TNBC cell morphology changes upon NEDD9 and ROCK inhibition
Metastatic TNBC cell lines with primarily mesenchymal morphology (MDA-MB-231, BT549, HCC1143, HCC1395, Hs578T and SUM159) were used to assess the role of NEDD9 in switching cells from mesenchymal to amoeboid morphology. NEDD9 was depleted using previously characterized siRNAs (17, 32) (Fig.1A) in a SMARTpool, and the changes in individual cell morphology were monitored using live cell imaging microscopy when plated in 2D or suspended in 3D matrix (collagen I or Matrigel).
Fig. 1. TNBC cell morphology changes upon NEDD9 and ROCK inhibition.
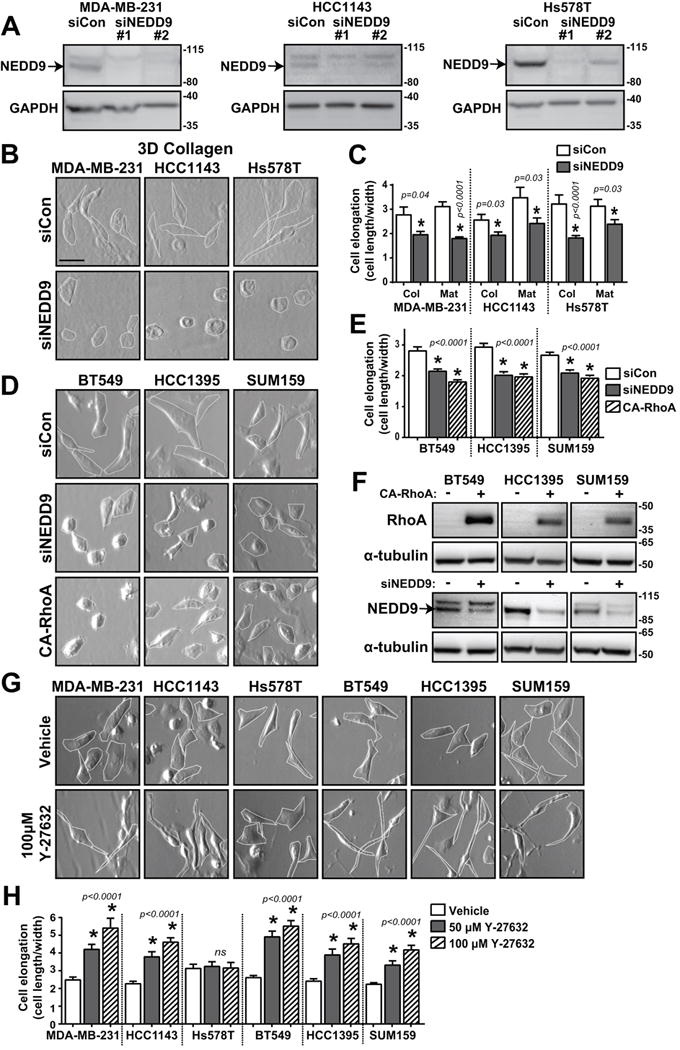
(A) Western blot analysis of NEDD9 expression in MDA-MB-231, HCC1143, and Hs578T treated with multiple siRNAs. (B) Brightfield images of MDA-MB-231, HCC1143, and Hs578T cells treated with siCon or siNEDD9 in 3D collagen I and (C) cell elongation quantified as cell length/width (40 cells/group). (D) Brightfield images of BT549, HCC1395, and SUM159 cells expressing siCon, siNEDD9, or CA-RhoA, (E) cell elongation quantified as cell length/width (100 cells/group), and (F) western blots of NEDD9 knockdown and CA-RhoA expression. (G) Brightfield images of TNBC cells, vehicle or Y-27632 treatment and (H) cell elongation quantified as cell length/width (40 cells/group). ns, not significant; *p<0.05, student’s t-test vs control.
In 2D, control (siCon) cells have well-defined elongated morphology, while NEDD9-depleted (siNEDD9) cells show up to a 40% decrease in cell elongation (Fig.S1A–B) reflected by a decrease in the ratio of cell length over cell width. In 3D matrix, the pore size is a critical determinant of cell morphology and migration type (20). The average pore size of the collagen I matrix used was about 15 μm2 (Fig.S1C–D), which supports both amoeboid and mesenchymal movement (33). Alternatively, the tightly-packed fibers that make up Matrigel are not conducive for amoeboid migration (<1 μm) and are permissive primarily for matrix-degrading mesenchymal movement. Interestingly, when seeded in either of these 3D scaffolds, siNEDD9 cells show acquisition of rounded amoeboid morphology and up to a 50% decrease in cell elongation (Fig.1B–C), which is similar to 2D results.
Knockout of NEDD9 using sgRNAs and CRISPR/Cas9 technology led to similar changes in morphology with less variability in cell elongation and showing an elongation ratio closer to 1, indicating that they are more circular with similar cell length and width (Fig.S1E–F). This phenotype was similar to cells overexpressing constitutively active RhoA GTPase (Fig.1D–F), suggesting RhoA activity may be high in NEDD9 deficient cells. Importantly, almost all TNBC cells demonstrated significant plasticity in their ability to not only be able to switch to amoeboid morphology, but also be able to increase mesenchymal morphology, which was confirmed as control cells became further elongated upon application of the well-characterized Rho kinase inhibitor, Y-27632 (Fig.1G–H).
NEDD9 depletion leads to increased myosin light chain phosphorylation but decreased contractility
Next, we tested if the acquisition of amoeboid morphology by NEDD9-deficient TNBC cells is accompanied by a switch to amoeboid migration, which could potentially promote intra/extravasation during metastasis of breast cancer. Actomyosin contractility and force-mediated remodeling of the matrix is a signature of the amoeboid type of cell migration (34). A key driver of actomyosin contractility is the Rho kinase ROCK, which phosphorylates myosin II light chain (MLC2) (35). In both collagen and Matrigel, NEDD9 deficiency resulted in up to a threefold increase in pMLC2 (Fig.2A–D, Fig.S1J), which co-localized with cortical actin around the cell membrane.
Fig. 2. NEDD9 depletion increases phosphorylation of MLC2 and decreases phosphorylation of FAK and paxillin.
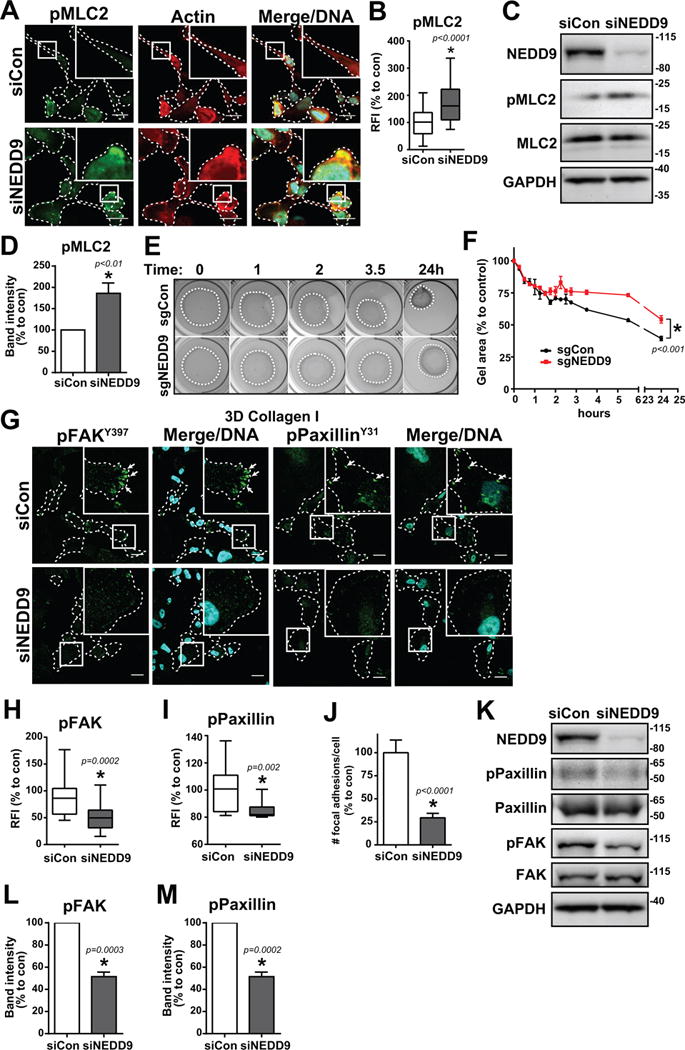
(A) MDA-MB-231 siCon and siNEDD9 cells in collagen I stained for pMLC2, actin, and Hoechst DNA dye. Scale bar, 20 μm. (B) Box and whisker plot of pMLC2 relative fluorescence intensity (RFI), n = 3, 20–30 cells/group. Intensity was normalized to the cell area. (C) Western blot and (D) quantification of MLC2 phosphorylation, n = 3. (E) Collagen gel contraction assay of MDA-MB-231 CRISPR sgCon/sgNEDD9 cells over 24 hours and (F) quantification of collagen gel area, n = 3. (G) MDA-MB-231 siCon and siNEDD9 cells in collagen I stained for pFAKY397 or pPaxillinY31 and Hoechst DNA dye. Scale bar, 20 μm. (H) Box and whisker plot of pFAKY397 fluorescence and (I) pPaxillinY31 fluorescence, n = 3, at least 20 cells/group. (J) Quantification of # of focal adhesions per cell, n = 3, at least 20 cells/group. (K) Western blot and quantification of (L) FAK and (M) paxillin phosphorylation. n = 3. *p<0.05, student’s t-test vs control.
Interestingly, despite this increase in pMLC2 in NEDD9-depleted cells, there was a significant decrease in the ability of the collective cell population to contract floating collagen gels (Fig.2E–F), indicating that morphologically amoeboid NEDD9-depleted cells may not possess all of the mechanistic properties of amoeboid cells. The inability of NEDD9-deficient cells to translate the increased pMLC2 signaling to matrix deformation/contraction may be indicative of impaired adhesion or actin dynamics connecting the internal and external forces of the cell.
NEDD9 depletion leads to a decrease in mature focal adhesion dynamics
Mature, large focal adhesions have been associated with the mesenchymal phenotype, whereas a decrease in size and maturity has been linked to amoeboid morphology (36, 37). Staining for phospho-FAK (pFAKY397) or phospho-paxillin (pPaxillinY31) has been commonly used to label mature focal adhesions (37, 38). Immunofluorescence analysis shows up to a twofold decrease in the number of adhesions positive for pFAKY397 and pPaxillinY31 upon NEDD9 depletion (Fig.2G–J,S1G–J), indicating a critical role of NEDD9 in adhesion maturation. The decrease in FAK and paxillin phosphorylation was further confirmed by western blot in multiple breast cancer cell lines (Fig.2K–M,S2). NEDD9 deficiency trended toward a decrease in the number of adhesions containing the more mature/stable focal adhesion marker vinculin (39), which would be concurrent with amoeboid phenotype, however the length of many of these adhesions was increased compared to control, suggesting the stabilization of fibrillar adhesions (Fig.S3). Nevertheless, NEDD9-depleted cells also had an increase in the total number of small adhesions that contained talin (Fig.S4), which is one of the first proteins recruited to focal contacts/adhesions (39). This indicates that there is an increase in small, weak nascent adhesions and a decrease in mature adhesions, which is concurrent with amoeboid phenotype (40). This may suggest that NEDD9 is not needed for adhesion initiation but is required for fibrillary adhesion disassembly. Such duality may potentially affect both mesenchymal and amoeboid adhesion dynamics.
NEDD9 regulates AURKA-driven phosphorylation of CTTN and stability of actin filaments
Amoeboid morphology is characterized by a decrease in filamentous actin polymerization while the mesenchymal elongated shape relies on an increase in filamentous actin (7). Previously, we have shown that NEDD9 binds and activates AURKA and the actin stabilizing protein CTTN, which results in an increase in actin polymerization and stabilization of lamellar protrusions and invadopodia (19). Here we show that AURKA directly phosphorylates CTTN (Fig.3A) at the (C-terminus 350–546aa) (Fig.3B,S5). To assess the impact of this phosphorylation on actin dynamics, an in vitro actin polymerization assay was carried out using recombinant AURKA and CTTN proteins. Incubation of AURKA with CTTN resulted in a significant increase in the actin polymerization rate (Fig.3C) compared to CTTN without AURKA. AURKA alone or in combination with Arp2/3 and WASP did not significantly affect actin polymerization, indicating that CTTN is the major substrate of AURKA in this assay. Moreover, kymography analysis of mCherry-LifeAct-expressing cells shows that the inhibition of AURKA results in a complete blockade of lamellar protrusions (Fig.3D–F), indicating that AURKA has a key role in cellular actin dynamics.
Fig. 3. NEDD9 regulates AURKA-driven phosphorylation of CTTN and stability of actin filaments.
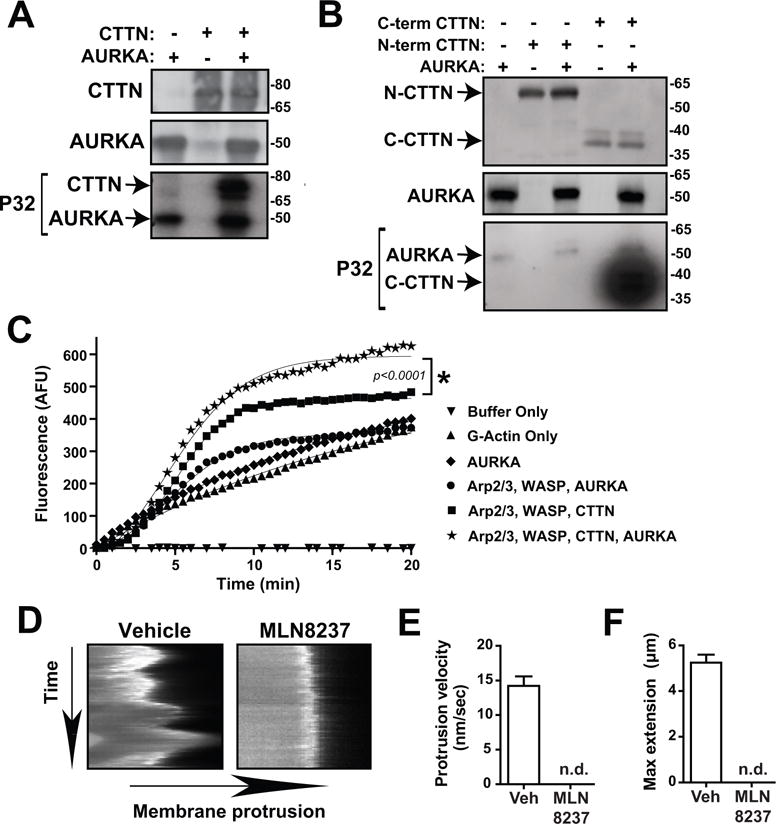
(A) Radioactive in vitro kinase assay of recombinant AURKA and full length WT CTTN proteins using radiolabeled P32-ATP, n = 3. WB shows total protein (top and middle) while autoradiograph shows P32-phosphorylated protein (bottom). (B) Radioactive in vitro kinase assay of recombinant AURKA and N-term (1-350AA) and C-term (350-546AA) CTTN proteins using radiolabeled P32-ATP, n = 3. WB shows total protein (top and middle) while autoradiograph shows P32-phosphorylated protein (bottom, 24 hour exposure). (C) Pyrene-actin polymerization assay of AURKA and CTTN, n = 4. Curves were fit with Boltzmann sigmoidal analysis, *p<0.05. (D) Kymographs of cell membrane of MDA-MB-231 cells treated with Vehicle or MLN8237 and quantifications of (E) protrusion velocity and (F) maximum membrane extension. n.d., not detected.
NEDD9 depletion decreases Rac1 activity in breast cancer cells
Rac1 GTPase has been shown to be one of the key drivers of mesenchymal migration/morphology. To measure Rac1 activity in cells grown in either 3D collagen or Matrigel, GST-tagged PBD (PAK p21 binding domain) protein was used to specifically pull down Rac1-GTP. Cells overexpressing dominant negative Rac1 (DN) or constitutively active Rac1 (CA) were used as controls (Fig.S6A). NEDD9 depletion resulted in a significant decrease in the amount of active Rac1 in cells grown in both scaffolds (Fig.4A–B). This was consistent when repeated in two other mesenchymal breast cancer cell lines as well, Hs578T and HCC1143 (Fig.S6B–C). As a complementary approach, Rac1 G-LISA assays (41) were used to evaluate the effect of NEDD9 depletion. Similarly to the active Rac1 pulldown assay, Rac1 G-LISA shows a decrease in the amount of active Rac1 (Fig.4B, Fig.S6D). Additionally, the RhoA G-LISA shows an increase in the amount of active RhoA, which is concurrent with amoeboid phenotype (Fig.4C).
Fig. 4. NEDD9 drives Rac1 activity through interaction with GEF VAV2.
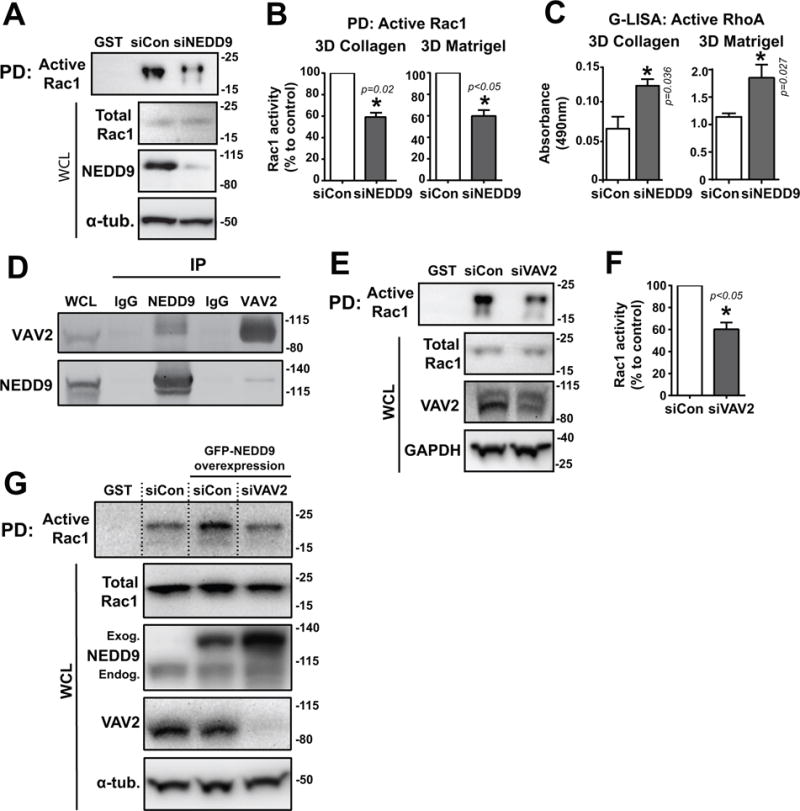
(A) Western blot of active-Rac1 pulldown (PD) from MDA-MB-231 siCon/siNEDD9 cells in 3D Matrigel using GST-PBD conjugated beads. Lane 1: GST-empty beads control. WCL, whole cell lysate. (B) Quantification of Rac1 activity in 3D collagen and Matrigel (active/total Rac1) by WB. (C) Quantification of RhoA activity in 3D collagen and Matrigel by G-LISA. (D) Co-immunoprecipitation of HA-VAV2 and RFP-NEDD9 through IP of each protein. IgG, mouse IgG control. (E) Western blot of active-Rac1 pulldown from MDA-MB-231 siCon/siVAV2 cells and (F) quantification of Rac1 activity. (G) Western blot of active-Rac1 pulldown from GFP-NEDD9 overexpressing MDA-MB-231 siCon/siVAV2 cells. Lane 1: GST-empty beads control. WCL, whole cell lysate. Exog, exogenous. Endog, endogenous. For all experiments: n = 3, *p<0.05, student’s t-test vs control.
NEDD9 drives Rac1 activity through interaction with GEF VAV2
Previously, it was shown in melanoma cells that NEDD9 activates Rac1 via interaction with the Rac1-GEF DOCK3 (15). Based on the breast cancer cell expression profiles of different GEFs and their downstream effectors, several potential Rac1 GEF candidates were examined for their ability to bind NEDD9 in co-immunoprecipitation experiments including DOCK4, DOCK180, VAV2, Tiam1, GEF-H1, ARHGEF2, ARHGEF4, DEF6, ECT2, and DBL (Supplementary Table 1). Some GEFs (such as DEF6, Fig.S6E) were co-immunoprecipitated with NEDD9 using anti-NEDD9 specific monoclonal (2G9) antibody, however only VAV2 was found to also reciprocally pull down NEDD9 (Fig.4D, Fig.S6F) when immunoprecipitating VAV2. To confirm that VAV2 is required for Rac1 activation in breast cancer cells, active Rac1 pulldowns were performed on cells treated with siCon or siRNA targeting VAV2 (Fig.4E). VAV2 depletion resulted in a 40–60% reduction in activity of Rac1 (Fig.4F), supporting the notion that VAV2 is a critical downstream effector of NEDD9-dependent Rac1 activation. Importantly, treatment of siCon cells with EHop-016 (42), a small molecular compound which inhibits Rac1 activation by VAV2 through interaction with Rac1’s VAV2-binding site, resulted in a reduction in Rac1 activity that was similar to the effect caused by siNEDD9 (Fig.S6G–H). This inhibition effect by EHop-016 was not found to be additive when combined with NEDD9 depletion. Additionally, an increase in Rac1 activity through NEDD9 overexpression was able to be blocked by VAV2 knockdown, further supporting that NEDD9-induced Rac1 activity is VAV2 dependent (Fig.4G).
Simultaneous targeting of NEDD9 and ROCK/RhoA hinders breast cancer cell motility/invasion in vitro
Since NEDD9 depletion supports the ROCK/RhoA pathway, knockdown of NEDD9 was combined with the ROCK-targeting compound Y-27632 to test for an additional therapeutic benefit in 3D invasion assays. NEDD9 depletion alone (via siRNA or sgRNA) led to an increase in amoeboid morphology and drastically decreased invasion (Fig.5A–E, Vid.S1; Fig.S7A–D, Vid.S2), similar to the overexpression of constitutively active RhoA (Fig.S7F–J, Vid.S3). Combination of Y-27632+siNEDD9 led to an increase in leading edge speed (extension/retraction), but decreased the cell body speed, thus rendering the elongated cell immobile (Fig.5C). The directionality and elongation of Y-27632-treated cells was significantly increased regardless of NEDD9 depletion (Fig.5D–E). As a complementary approach, RhoA depletion was performed and resulted in comparable trends (Fig.S8, Vid.S4). Effects on invasion were also evaluated using Hs578T and HCC1143 cells treated with siNEDD9, confirming similar results (Fig.S9, Vid.S5-6). Additionally, the AURKA inhibitor MLN8237 was tested as a substitute for siNEDD9 in combination treatments. Cell invasion distance was found to be decreased with MLN8237/Y-27632 treatment, and was similar to the efficacy of siNEDD9/Y-27632 treatment (Fig.S10A–B, Vid.S7). This suggests that MLN8237/Y-27632 could potentially be a viable combination regimen as well.
Fig. 5. Simultaneous targeting of NEDD9 and ROCK/RhoA hinders breast cancer cell motility/invasion in vitro.
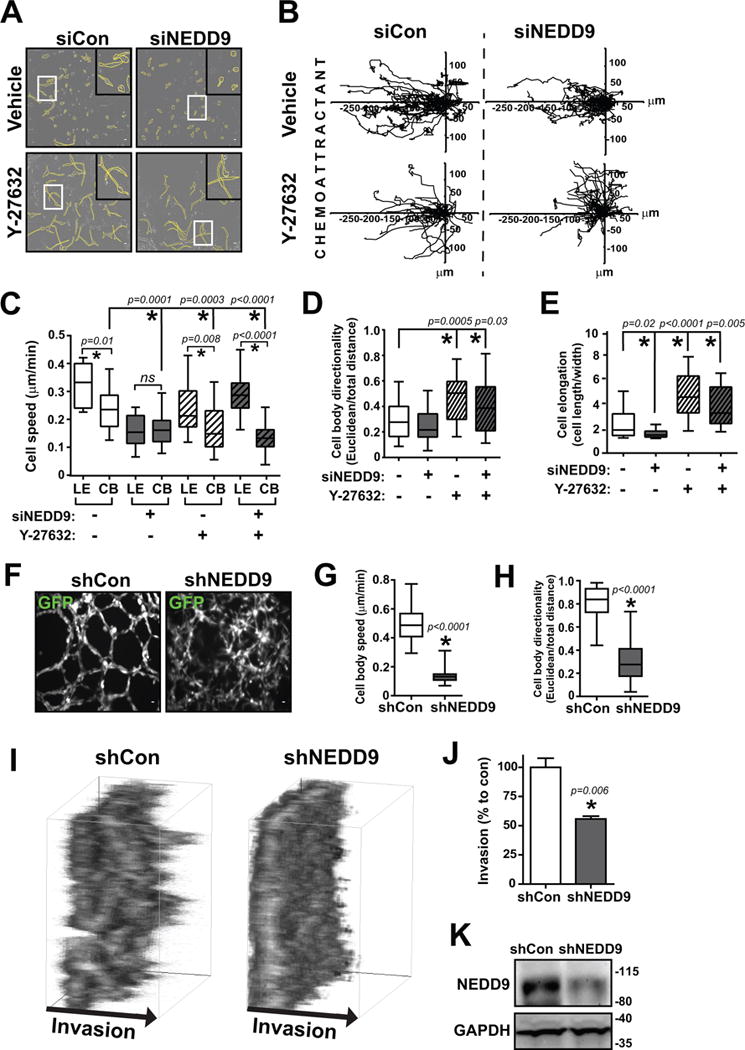
(A) Brightfield outlined cell morphology images (~12 hours), (B) individual cell tracking movement plots toward FBS chemoattractant (left), box and whisker plots of (C) cell body (CB) and leading edge (LE) speed, (D) cell body directionality, and (E) cell elongation of MDA-MB-231 cells with siCon/siNEDD9 + Vehicle/Y-27632 treatment in 3D collagen Ibidi chemotactic invasion movies. Scale bar, 20 μm. n = 3, 30–40 cells per group. ns, not significant; *p<0.05, one-way ANOVA. (F) GFP immunofluorescence images and box and whisker plots of (G) cell body speed and (H) cell body directionality of MDA-MB-231 pGIPZ-GFP shControl and shNEDD9 cells after 24 hours being densely seeded in a 3D Matrigel sandwich assay. Scale bar: 20 μm. n = 3, at least 60 cells per group. *p<0.05, student’s t-test vs control. (I) 3D projection boxes and (J) collective invasion quantification of the amount of MDA-MB-231 pGIPZ-GFP shControl/shNEDD9 cells reaching 100 μm invasion distance after 72 hours being densely seeded in a 3D Matrigel sandwich assay. *p<0.05, student’s t-test vs control. (K) Western blot of shNEDD9 knockdown in collective invasion assay.
NEDD9 is required for collective migration in 3D matrix
To test the impact of NEDD9 depletion on collective tumor cell invasion, cells were seeded in high density while sandwiched in between two Matrigel layers and time lapse microscopy was performed. Control cells invaded as strand-like collective streams (“highways”), similar to previous observations (9) (Fig.5F, Vid.S8). Formation of these streams is diminished following NEDD9 knockdown, which suggests the cells have minimal matrix degradation and/or rearrangement capacity. This deficiency in path-generating activity is required for leading cells during collective invasion. Additionally, cell speed and directionality were both significantly decreased in NEDD9-depleted cells (Fig.5G–H), which corroborates with a significant reduction in their invasion distance (Fig.5I–K). Furthermore, inhibition of AURKA yielded a similar decrease in collective cell invasion compared to NEDD9 depletion (Fig.S10C–D), again suggesting that AURKA could be a viable therapeutic target.
Combination of NEDD9 knockdown and Y-27632 treatment hinders breast cancer cell invasion and metastasis in vivo
MDA-MB-231-luc2 pTRIPZ-RFP-shControl or shNEDD9 cells were injected into the mammary fat pad of NSG mice. After 2.5 weeks, mice were treated with the AURKA inhibitor MLN8237 (4 days on, 3 days off) to prevent pulmonary metastases outgrowth without any effect on the mammary tumor growth as we previously reported (17). 72 hours post-MLN8237 pretreatment, mice with tumors were subjected to doxycycline-induced shCon or shNEDD9 expression and administration of Y-27632 compound for 3 weeks (Fig.6A). Primary tumor growth was measured weekly and was significantly reduced in shNEDD9/Y-27632 combination-treated animals compared to vehicle (Fig.6B–C). At the end of the study, NEDD9 depletion was confirmed in the mammary tumors by western blot (Fig.6D–E). To measure the metastatic ability of the tumor cells, terminal blood samples were collected and analyzed after euthanization for circulating tumor cells (CTCs) using flow cytometry. Nearly a 50% reduction in the number of CTCs was documented with shNEDD9 induction, which decreased even further with addition of Y-27632 (Fig.6F). A significant abatement in the number of metastases per lung area was seen with NEDD9 knockdown, and an even more pronounced inhibition was seen in combination with Y-27632 (Fig.6G), supporting our hypothesis that dual targeting of both the mesenchymal and amoeboid pathways via NEDD9 and ROCK/RhoA can be a promising therapy against breast cancer metastasis (Fig.6H).
Fig. 6. Simultaneous targeting of NEDD9 and ROCK hinders breast cancer cell invasion and metastasis in vivo.
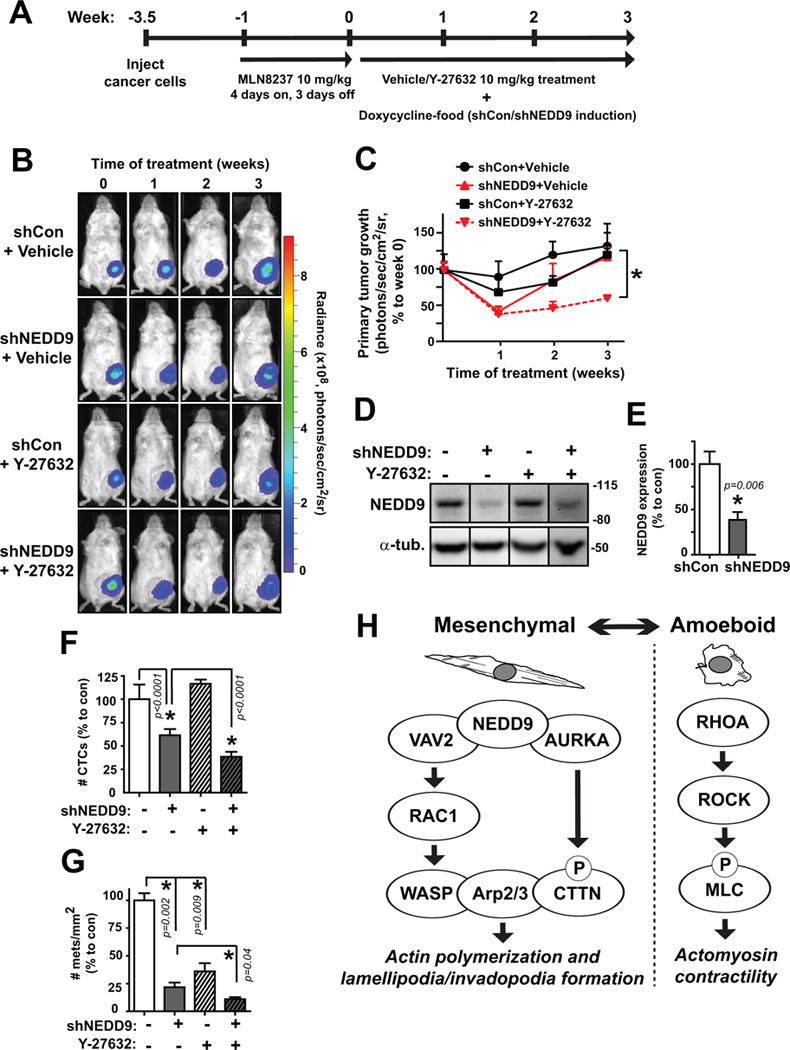
(A) MDA-MB-231-luc2 TZ RFP-shCon and RFP-shNEDD9 cells injected into the mammary fat pad of NSG mice were grown for ~3.5 weeks before starting treatment (time 0) for 3 weeks with 10 mg/kg Y-27632 and shRNA induction. (B) Weekly images of tumor cells in vivo via bioluminescence signal which was (C) quantified at the primary tumor site. (D) Western blot and (E) quantification of NEDD9 knockdown in final primary tumors. (F) Quantification of number of RFP-positive circulating tumor cells in mouse blood by FACS analysis. (G) Quantification of number of visible metastases per area of lung tissue. (H) Proposed schematic of mesenchymal and amoeboid pathways in breast cancer. n = 5 mice/group. *p<0.05, one-way ANOVA.
Discussion
NEDD9 protein is found consistently upregulated in TNBCs and highly correlates with invasive/metastatic spread (1, 17). Our findings provide a mechanistic explanation for NEDD9-driven invasion processes and strongly advocates for the combination of anti-NEDD9/AURKA and anti-ROCK targeting compounds to inhibit these movement signaling cascades. In this study, we report that deficiency in NEDD9 signaling itself leads to inhibition of key aspects of both mesenchymal and amoeboid migration in TNBC cells, resulting in substantial hindrance on cell invasion and metastasis. Similar to results in melanoma (14), NEDD9 deficiency in TNBC cells results in rounded/amoeboid morphology along with a decrease in the total number of mature (pFAK/pPaxillin positive) adhesions and an increase in the number of nascent adhesions (40). However, these cells also saw an increase in long fibrillar adhesions (Fig.S3D). These findings suggest that NEDD9 is also required for the disassembly of fibrillar adhesions similar to vinculin (43), which regulates the recruitment and release of focal adhesion proteins in a force-dependent manner (44). The role of NEDD9/HEF1 as a sensor of altered adhesion states has been previously reported (45). A decrease in disassembly could be due to matrix metalloprotease inhibition (46), as previously reported by our group (24). Hindered disassembly of FAs would be in disagreement with a previously noted increase in adhesion turnover of NEDD9-KO or mutant NEDD9 (Y189A) expressing fibroblasts in 2D cultures (47, 48), however the structure, composition, and dynamics of adhesions in 2D fibroblasts may be different from 3D tumor cells.
NEDD9 deficiency led to an increase in myosin-phosphorylated amoeboid-like cells, but this increase in pMLC2 did not translate to increased collective-cell contractility or invasion in 3D matrix. This disconnect may suggest a potential de-coupling between the actin filaments and myosin motors during cell body contraction (49). Alternatively, the inability of NEDD9-deficient cells to disassemble adhesions as we have suggested would limit amoeboid movement, which is in agreement with our previous reports on integrin dynamics (32).
Consistent with past findings, we found that NEDD9 deficiency resulted in over a 40% decrease in active Rac1, suggesting that NEDD9 is critical for Rac1 activation to occur in TNBC cells. We discovered that an interaction between NEDD9 and the Rac1-GEF protein VAV2 is required for Rac1 activation. Similar to NEDD9 depletion, loss of VAV2 yields a comparable decrease in Rac1 activity. Furthermore, the small molecule compound EHop-016, which efficiently blocks the interaction of Rac1 with VAV2 (42), did not cause an additive effect on Rac1 inhibition when given to siNEDD9 cells, suggesting that NEDD9 and VAV2 share the same pathway to Rac1 activation. The potential clinical application of this compound has yet to be determined, however another VAV-family targeting drug, the purine analogue azathioprine, was recently found to inhibit pancreatic cancer metastasis (50). Additionally, overexpression of NEDD9 increased Rac1 activity, which was abrogated upon depletion of VAV2, supporting that NEDD9’s influence on Rac1 activation is VAV2-dependent. Selecting NEDD9 as a therapeutic target could potentially be more beneficial than targeting VAV2 due to its upstream placement in the mesenchymal pathway and the multiple downstream branches that it links to including VAV2/Rac1 and AURKA/CTTN. The effect of AURKA inhibition on actin dynamics in TNBCs and its ability to regulate filamentous actin polymerization via phosphorylation of CTTN suggests a potential mechanistic explanation for the mesenchymal to amoeboid morphology changes observed upon NEDD9 depletion and provides additional venues to explore CTTN/AURKA signaling in TNBC metastasis.
In collagen invasion assays, both NEDD9-deficient and MLN8237-treated cells underwent a drastic reduction in cell invasion. It was anticipated that amoeboid cells could potentially move faster with a more erratic cell trajectory. While a small decrease in directionality was seen upon NEDD9 depletion, it was not significant. Inhibition of ROCK or RhoA in NEDD9-deficient TNBC cells reverted them back to an elongated cell shape, but not their original migration proficiency. These combination-treated cells also had a significant increase in cell directionality - this is likely due to their significant increase in cell length which was even higher than control cells.
The substantially impeded movement of combination-treated cells could be related to the large difference in speed observed between the CB and LE. LE speed remains high, continuing to protrude and retract, but the cell is incompetent to degrade matrix and/or make anchor points, due to NEDD9 deficiency. Meanwhile, the cell rear cannot efficiently contract due to inhibition of ROCK/RhoA, leaving the CB more immobile. This combination therefore results in a severe gap between a stagnant, immotile CB along with a quickly protruding/retracting, yet functionally impaired LE. These results support the notion that simultaneous inhibition of both the NEDD9/AURKA and ROCK/RhoA pathways could be an efficient anti-migratory/metastasis treatment option. These conclusions were further supported by our findings in an in vivo breast cancer xenograft mouse model, demonstrating a significant reduction in the number of circulating tumor cells and pulmonary metastases of mice that received combination treatment. These findings are also in agreement with previously published reports of NEDD9 depletion showing a decrease in the invasion and dissemination of diverse cancer types in vivo (1, 2).
In conclusion, while NEDD9-deficient cells may have many hallmarks of amoeboid phenotype, we have shown that amoeboid movement appears defective in some respects such as decreased cell contractility. Nonetheless, depletion of RhoA or addition of ROCK inhibitor to NEDD9-deficient cells provided additional benefit in further hindrance of cell invasion/metastasis. Through concurrent targeting of these pathways, this approach merits further evaluation as a clinical option to augment breast cancer patient survival.
Supplementary Material
Implications.
This study provides new insight into the therapeutic benefit of combining NEDD9 depletion with ROCK inhibition to reduce tumor cell dissemination and discovers a new regulatory role of NEDD9 in the modulation of VAV2-dependent activation of Rac1 and actin polymerization.
Acknowledgments
The authors would like to thank and acknowledge the WVU Shared Research Facilities and the MBRCC and HSC core facilities including the WVU Microscope Imaging Facility (supported by the MBRCC and NIH grants P20 RR016440, P30 RR032138/GM103488 and P20 RR016477) and the WVU Flow Cytometry Core Facility (supported by the NIH equipment grant number S10OD016165 and the Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of NIH under grant numbers P30GM103488 (CoBRE) and P20GM103434 (INBRE)).
Financial support: This research was supported by grants CA148671 (E.N.P) and KG100539 (E.N.P) from the NIH/NCI and Susan G. Komen for the Cure foundation, and in part by a NIH/NCRR 5 P20 RR016440-09 (L.F.G). The WVU HSC Core Facilities were supported by the NIH grants P30-GM103488, S10-RR026378, S10-RR020866, S10-OD016165, and GM103434.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Disclaimers: This manuscript contains original work only and has not been published or submitted elsewhere. All of the authors have directly participated in the planning, execution and/or analysis of this study and have approved the submitted version of this manuscript.
References
- 1.Kong C, Wang C, Wang L, Ma M, Niu C, Sun X, Du J, Dong Z, Zhu S, Lu J, Huang B. NEDD9 is a positive regulator of epithelial-mesenchymal transition and promotes invasion in aggressive breast cancer. PLoS One. 2011;6(7):e22666. doi: 10.1371/journal.pone.0022666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim M, Gans JD, Nogueira C, Wang A, Paik JH, Feng B, Brennan C, Hahn WC, Cordon-Cardo C, Wagner SN, Flotte TJ, Duncan LM, Granter SR, Chin L. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125(7):1269–81. doi: 10.1016/j.cell.2006.06.008. Epub 2006/07/04. doi:S0092-8674(06)00718-5 [pii] 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188(1):11–9. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3(5):362–74. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 5.Haeger A, Wolf K, Zegers MM, Friedl P. Collective cell migration: guidance principles and hierarchies. Trends Cell Biol. 2015 doi: 10.1016/j.tcb.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res. 2010;8(5):629–42. doi: 10.1158/1541-7786.MCR-10-0139. [DOI] [PubMed] [Google Scholar]
- 7.Pankova K, Rosel D, Novotny M, Brabek J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell Mol Life Sci. 2010;67(1):63–71. doi: 10.1007/s00018-009-0132-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf K, Friedl P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 2011;21(12):736–44. doi: 10.1016/j.tcb.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Brocker EB, Friedl P. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. 2003;160(2):267–77. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Case LB, Baird MA, Shtengel G, Campbell SL, Hess HF, Davidson MW, Waterman CM. Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions. Nature cell biology. 2015;17(7):880–92. doi: 10.1038/ncb3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn J, Sanz-Moreno V, Marshall CJ. The metastasis gene NEDD9 product acts through integrin beta3 and Src to promote mesenchymal motility and inhibit amoeboid motility. J Cell Sci. 125(Pt 7):1814–26. doi: 10.1242/jcs.101444. [DOI] [PubMed] [Google Scholar]
- 12.Bradbury P, Mahmassani M, Zhong J, Turner K, Paul A, Verrills NM, O’Neill GM. PP2A phosphatase suppresses function of the mesenchymal invasion regulator NEDD9. Biochim Biophys Acta. 1823(2):290–7. doi: 10.1016/j.bbamcr.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 13.Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal. 2010;8:23. doi: 10.1186/1478-811X-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135(3):510–23. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 15.Ahn J, Sanz-Moreno V, Marshall CJ. The metastasis gene NEDD9 product acts through integrin beta3 and Src to promote mesenchymal motility and inhibit amoeboid motility. J Cell Sci. 2012;125(Pt 7):1814–26. doi: 10.1242/jcs.101444. [DOI] [PubMed] [Google Scholar]
- 16.Ammer AG, Weed SA. Cortactin branches out: roles in regulating protrusive actin dynamics. Cell Motil Cytoskeleton. 2008;65(9):687–707. doi: 10.1002/cm.20296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ice RJ, McLaughlin SL, Livengood RH, Culp MV, Eddy ER, Ivanov AV, Pugacheva EN. NEDD9 depletion destabilizes Aurora A kinase and heightens the efficacy of Aurora A inhibitors: implications for treatment of metastatic solid tumors. Cancer Res. 2013;73(10):3168–80. doi: 10.1158/0008-5472.CAN-12-4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129(7):1351–63. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozyreva VK, McLaughlin SL, Livengood RH, Calkins RA, Kelley LC, Rajulapati A, Ice RJ, Smolkin MB, Weed SA, Pugacheva EN. NEDD9 regulates actin dynamics through cortactin deacetylation in an AURKA/HDAC6-dependent manner. Mol Cancer Res. 2014;12(5):681–93. doi: 10.1158/1541-7786.MCR-13-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol. 2013;201(7):1069–84. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedl P, Wolf K, Lammerding J. Nuclear mechanics during cell migration. Curr Opin Cell Biol. 2011;23(1):55–64. doi: 10.1016/j.ceb.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo S, Ichikawa M, Kurokawa M. Structure and function of cas-L and integrin-mediated signaling. Crit Rev Immunol. 2006;26(5):391–406. doi: 10.1615/critrevimmunol.v26.i5.20. Epub 2007/03/08. doi: 023606d26a204d18,78c1314826d867c1 [pii] [DOI] [PubMed] [Google Scholar]
- 23.Aquino JB, Lallemend F, Marmigere F, Adameyko II, Golemis EA, Ernfors P. The retinoic acid inducible Cas-family signaling protein Nedd9 regulates neural crest cell migration by modulating adhesion and actin dynamics. Neuroscience. 2009;162(4):1106–19. doi: 10.1016/j.neuroscience.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loskutov YV, Kozyulina PY, Kozyreva VK, Ice RJ, Jones BC, Roston TJ, Smolkin MB, Ivanov AV, Wysolmerski RB, Pugacheva EN. NEDD9/Arf6-dependent endocytic trafficking of matrix metalloproteinase 14: a novel mechanism for blocking mesenchymal cell invasion and metastasis of breast cancer. Oncogene. 2014 doi: 10.1038/onc.2014.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLaughlin SL, Ice RJ, Rajulapati A, Kozyulina PY, Livengood RH, Kozyreva VK, Loskutov YV, Culp MV, Weed SA, Ivanov AV, Pugacheva EN. NEDD9 depletion leads to MMP14 inactivation by TIMP2 and prevents invasion and metastasis. Mol Cancer Res. 2014;12(1):69–81. doi: 10.1158/1541-7786.MCR-13-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamazaki D, Kurisu S, Takenawa T. Involvement of Rac and Rho signaling in cancer cell motility in 3D substrates. Oncogene. 2009;28(13):1570–83. doi: 10.1038/onc.2009.2. [DOI] [PubMed] [Google Scholar]
- 27.Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT, Cooper JA. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol. 2001;11(5):370–4. doi: 10.1016/s0960-9822(01)00098-7. [DOI] [PubMed] [Google Scholar]
- 28.Pugacheva EN, Golemis EA. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat Cell Biol. 2005;7(10):937–46. doi: 10.1038/ncb1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castello-Cros R, Cukierman E. Stromagenesis during tumorigenesis: characterization of tumor-associated fibroblasts and stroma-derived 3D matrices. Methods Mol Biol. 2009;522:275–305. doi: 10.1007/978-1-59745-413-1_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wozniak MA, Keely PJ. Use of three-dimensional collagen gels to study mechanotransduction in T47D breast epithelial cells. Biol Proced Online. 2005;7:144–61. doi: 10.1251/bpo112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozyreva VK, Kiseleva AA, Ice RJ, Jones BC, Loskutov YV, Matalkah F, Smolkin MB, Marinak K, Livengood RH, Salkeni MA, Wen S, Hazard HW, Layne GP, Walsh CM, Cantrell PS, Kilby GW, Mahavadi S, Shah N, Pugacheva EN. Combination of Eribulin and Aurora A Inhibitor MLN8237 Prevents Metastatic Colonization and Induces Cytotoxic Autophagy in Breast Cancer. Molecular cancer therapeutics. 2016;15(8):1809–22. doi: 10.1158/1535-7163.MCT-15-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozyulina PY, Loskutov YV, Kozyreva VK, Rajulapati A, Ice RJ, Jones BC, Pugacheva EN. Prometastatic NEDD9 Regulates Individual Cell Migration via Caveolin-1-Dependent Trafficking of Integrins. Mol Cancer Res. 2015;13(3):423–38. doi: 10.1158/1541-7786.MCR-14-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Artym VV, Matsumoto K. Imaging cells in three-dimensional collagen matrix. Curr Protoc Cell Biol. 2010 doi: 10.1002/0471143030.cb1018s48. Chapter 10:Unit 10 8 1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaggioli C, Sahai E. Melanoma invasion - current knowledge and future directions. Pigment cell research / sponsored by the European Society for Pigment Cell Research and the International Pigment Cell Society. 2007;20(3):161–72. doi: 10.1111/j.1600-0749.2007.00378.x. [DOI] [PubMed] [Google Scholar]
- 35.Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS, Sahai E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr Biol. 2006;16(15):1515–23. doi: 10.1016/j.cub.2006.05.065. [DOI] [PubMed] [Google Scholar]
- 36.Carragher NO, Walker SM, Scott Carragher LA, Harris F, Sawyer TK, Brunton VG, Ozanne BW, Frame MC. Calpain 2 and Src dependence distinguishes mesenchymal and amoeboid modes of tumour cell invasion: a link to integrin function. Oncogene. 2006;25(42):5726–40. doi: 10.1038/sj.onc.1209582. [DOI] [PubMed] [Google Scholar]
- 37.Hager MH, Morley S, Bielenberg DR, Gao S, Morello M, Holcomb IN, Liu W, Mouneimne G, Demichelis F, Kim J, Solomon KR, Adam RM, Isaacs WB, Higgs HN, Vessella RL, Di Vizio D, Freeman MR. DIAPH3 governs the cellular transition to the amoeboid tumour phenotype. EMBO Mol Med. 2012;4(8):743–60. doi: 10.1002/emmm.201200242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294(5547):1708–12. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 39.Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692(2–3):103–19. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 40.Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harb Perspect Biol. 2011;3(9):a005074. doi: 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlegel N, Burger S, Golenhofen N, Walter U, Drenckhahn D, Waschke J. The role of VASP in regulation of cAMP- and Rac 1-mediated endothelial barrier stabilization. American journal of physiology. 2008;294(1):C178–88. doi: 10.1152/ajpcell.00273.2007. [DOI] [PubMed] [Google Scholar]
- 42.Montalvo-Ortiz BL, Castillo-Pichardo L, Hernandez E, Humphries-Bickley T, De la Mota-Peynado A, Cubano LA, Vlaar CP, Dharmawardhane S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. The Journal of biological chemistry. 2012;287(16):13228–38. doi: 10.1074/jbc.M111.334524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teckchandani A, Cooper JA. The ubiquitin-proteasome system regulates focal adhesions at the leading edge of migrating cells. eLife. 2016;5 doi: 10.7554/eLife.17440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carisey A, Tsang R, Greiner AM, Nijenhuis N, Heath N, Nazgiewicz A, Kemkemer R, Derby B, Spatz J, Ballestrem C. Vinculin regulates the recruitment and release of core focal adhesion proteins in a force-dependent manner. Curr Biol. 2013;23(4):271–81. doi: 10.1016/j.cub.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Neill GM, Golemis EA. Proteolysis of the docking protein HEF1 and implications for focal adhesion dynamics. Mol Cell Biol. 2001;21(15):5094–108. doi: 10.1128/MCB.21.15.5094-5108.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stehbens SJ, Paszek M, Pemble H, Ettinger A, Gierke S, Wittmann T. CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nature cell biology. 2014;16(6):561–73. doi: 10.1038/ncb2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong J, Baquiran JB, Bonakdar N, Lees J, Ching YW, Pugacheva E, Fabry B, O’Neill GM. NEDD9 stabilizes focal adhesions, increases binding to the extra-cellular matrix and differentially effects 2D versus 3D cell migration. PLoS One. 7(4):e35058. doi: 10.1371/journal.pone.0035058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zou Z, Yuan Z, Zhang Q, Long Z, Chen J, Tang Z, Zhu Y, Chen S, Xu J, Yan M, Wang J, Liu Q. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy. 2012;8(12):1798–810. doi: 10.4161/auto.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wollrab V, Thiagarajan R, Wald A, Kruse K, Riveline D. Still and rotating myosin clusters determine cytokinetic ring constriction. Nature communications. 2016;7:11860. doi: 10.1038/ncomms11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Razidlo GL, Magnine C, Sletten AC, Hurley RM, Almada LL, Fernandez-Zapico ME, Ji B, McNiven MA. Targeting Pancreatic Cancer Metastasis by Inhibition of Vav1, a Driver of Tumor Cell Invasion. Cancer research. 2015;75(14):2907–15. doi: 10.1158/0008-5472.CAN-14-3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.