

Abstract
Infection with the fish-borne liver fluke Opisthorchis viverrini is classified by the International Agency for Research on Cancer as a Group 1 carcinogen: definitely carcinogenic in humans. Cofactors likely contribute to bile duct cancer (cholangiocarcinoma) caused by this infection. Here we review recent findings that address the role of liver fluke associated H. pylori in hepatobiliary disease and malignancy. We hypothesize that co-infection by O. viverrini and the bacillus Helicobacter pylori is central of liver fluke infection associated cholangiocarcinoma.
Keywords: Liver fluke, Opisthorchis viverrini, Helicobacter spp, Co-infection, pathogenesis, hepatobiliary diseases, cholangiocarcinoma
Graphical Abstract
1. Introduction
Foodborne trematodiasis caused by infection with the opisthorchiid liver flukes Opisthorchis viverrini, O. felineus and Clonorchis sinensis remains a major public health problem in East Asia and Eastern Europe where >40 million people are infected [1, 2]. O. viverrini is endemic in Thailand, Lao People’s Democratic Republic (Lao PDR), Vietnam and Cambodia with over 10 million people are infected [1]. Humans acquire the infection by eating raw or undercooked fish harboring infective stage metacercariae (reviewed in [3]). Upon ingestion, the metacercariae excyst in the duodenum and juvenile flukes migrate into the biliary tree. In the bile ducts, the parasites mature over four weeks into adult flukes. Parasites eggs are shed in the fecal stream to the environment where the eggs are ingested by freshwater snails of the genus Bithynia. The parasite undergoes transformations and multiplications within the snail, culminating in the release of cercariae that seek out and penetrate the skin of a freshwater cyprinid fish, completing the cycle. Human infection causes several hepatobiliary abnormalities, including cholangitis, obstructive jaundice, hepatomegaly, periductal fibrosis, cholecystitis and cholelithiasis (see [3]). Both experimental and epidemiological evidence strongly implicates liver fluke infection in the etiology of one of the primary liver cancer subtypes – cholangiocarcinoma (CCA), a fatal bile duct cancer [1, 4, 5]. Khon Kaen province in north-eastern Thailand where the O. viverrini liver fluke is endemic has reported the highest incidence of CCA in the world, >100 cases per 100,000 [6]. However, additional risk factors for hepatobiliary diseases and CCA have been documented including primary sclerosing cholangitis (see [7]), inflammatory bowel disease [8], metabolic syndromes [9], hepatitis virus [10], fluke infection-associated oxysterols [11], and infection with Helicobacter spp. [12]. The last is attracting increasing research interest [13].
2. Helicobacter spp. and extragastric diseases
Infection with Helicobacter pylori, a Gram-negative bacillus is the first bacterial infection known to be an etiological agent of gastric diseases including gastric adenocarcinoma [14–17]. Virulence factors of H. pylori including cytotoxin-associated gene A (CagA) and vacuolating cytotoxin A (VacA) contribute to the pathogenesis of H. pylori-associated disease [18, 19]. Although chronic H. pylori infection is associated with the stomach possible association with several extragastric complications including hepatobiliary and pancreatic diseases have been proposed [20, 21]. There is strong evidence that H. pylori seropositivity and biliary tract cancer with overall OR 5.47 and, specifically, for extrahepatic (OR 7.01) and intrahepatic cancer (OR 10.67) but not for hepatocellular carcinoma in the Alpha-Tocopherol, Beta-Carotene Cancer Prevention (ATBC) cohort [22]. For liver fibrosis, prevalence of cagA H. pylori was directly proportional to severity of liver disease and was more positive in advanced stages of fibrosis (28.2%) compared to early stages (5.9%) in HCV-related chronic hepatitis and cirrhosis [23]. The mechanism by which H. pylori induces liver fibrosis may involve increased cytokines, i.e. TGF-β1 and oxidative stress induced pro-inflammatory signaling pathways in hepatic stellate cell line (HSC) [24, 25]. Other species of Helicobacter, specifically H. hepaticus and H. bilis also are implicated in hepatobiliary disease [12, 13, 22].
3. Associations among Helicobacter, opisthorchiasis and cholangiocarcinoma
H. pylori has been reported to be involved in a case series of hepatobiliary diseases in opisthorchiasis endemic Thailand [26]. We first systematically reported an association between H. pylori, specifically cagA positive-H. pylori and CCA but not hepatolithiasis or normal controls in patients from Northeast Thailand, a region endemic for opisthorchiasis [26, 27]. CCA cases with H. pylori infection exhibit higher portal inflammation and biliary cell proliferation as determined by PCNA immunohistochemistry [26]. Molecular mechanisms integral to H. pylori induced hepatobiliary diseases have also been described [28–30]. Since CCA is strongly associated with opisthorchiasis in endemic areas, as noted, O. viverrini may have an integral though still cryptic relationship with H. pylori. Indeed, we have been aware of this relationship for some time, and recently reported an association between O. viverrini and Helicobacter spp. in a hamster model [31]. The liver fluke infected hamsters showed significantly higher H. pylori and H. bilis than control, non-liver fluke-infected hamsters. In addition, H. pylori can be detected in the gut epithelium of O. viverrini and hence we have concluded that the liver fluke represents a reservoir of H. pylori within the biliary system [31]. Similar findings have been seen in humans infected with O. viverrini. The higher the liver fluke infection intensity, as determined by O. viverrini eggs per gram of feces, the greater the fecal numbers of H. pylori. Moreover, we also demonstrated that cagA positive H. pylori associated with increased risk of periductal fibrosis as determined by ultrasonography in opisthorchiasis (Deenonpoe et al., manuscript submitted). Given our pioneering research in the pathogenesis of liver fluke induced pathology and CCA and over 20 years research experience in this field [2, 3, 11, 29, 32, 33], we hypothesize that the liver fluke/H. pylori co-infection is the central player in biliary disease manifestations including CCA in opisthorchiasis in northeastern Thailand. However, the underlying mechanisms by which H. pylori associates with the liver fluke, opisthorchiasis and CCA remain to be established.
4. Pathogenesis of H. pylori induced biliary diseases
Pathogenesis and disease outcomes following infection with H. pylori are mediated by a complex interplay between bacterial virulence factors, host, and environmental factors. Unfortunately, only a few studies describe the pathogenesis in H. pylori induced biliary diseases [26, 28, 30]. In brief, following entry of H. pylori into host tissue, four steps are critical for the bacterium to establish successful colonization, persistent infection, and disease: 1) survival in environment (the acidic stomach or the alkaline bile ducts); 2) movement toward epithelial cells by flagella-mediated motility; 3) attachment to host cells mediated by interactions between bacterial cell adhesins and host cell receptors; and 4) tissue damage following the release of toxins, specifically CagA and VacA [34, 35].
5. Survival of H. pylori in the bile
H. pylori and other species of Helicobacter can survive at the alkaline pH of the bile as they can be detected in bile sampled from biliary diseases including CCA [36, 37]. Most of the H. pylori and other bacteria in the bile are coccoid form that may reflect responses to bile acids [38]. In addition, biofilm formation by the bacteria in the bile seems facilitate survival in the environment within the human biliary tract [36]. Interestingly, an increased abundance of H. pylori virulence genes, i.e. cagA and vacA was observed in extrahepatic CCA compared to benign biliary diseases [37]. Similarly, significant higher frequencies of cagA-positive H. pylori have been reported during CCA than cholelithiasis and in bile from healthy individuals [26]. Biliary micro-environmental components such cholesterol may enhance the pathogenicity of H. pylori as it acquires host cholesterol for catabolism of lipopolysaccharide (LPS) for cell membranes [39]. In addition, cholesterol promotes growth of H. pylori in serum-free media [40]. Together, these findings provide support to the notion that H. pylori can be more virulent within the biliary environment.
6. Opisthorchis is a reservoir of H. pylori and host-bacterial interaction
We recently reported that O. viverrini was a reservoir of H. pylori (Figure 1) [31]. The intensity of infection with H. pylori within O. viverrini infected hamsters was significantly greater than that of uninfected hamsters (Figure 2). The H. pylori bacteria localized on the gut epithelium of fluke and survived in the adult liver flukes in vitro co-cultured with antibiotics for more than 30 days [31]. These findings indicate the establishment of H. pylori benefits from the support of the liver fluke gut. As the pH in the secretions from the gut of O. viverrini is approximately pH 5–6, compared to the pH 7–8 of the bile, perhaps the liver fluke protects the H. pylori bacilli from the otherwise inimical environment of the bile. Beyond survival in environmental milieu and pH of the gut of the liver fluke within the lumen of the bile duct, establishment of infection by H. pylori requires adhesion and colonization of the gut epithelium by H. pylori to protect the bacilli from displacement from the gut by forces such as those generated by peristalsis and emptying similar to those described in the mammalian stomach. Specific interaction between the bacterial adhesins and the host (fluke) gut epithelium receptors is needed.
Figure 1.

Identification of Helicobacter pylori in the gut of Opisthorchis viverrini. (A) Cluster of H. pylori-like (short arrow) stained with the Warthin-Starry method shown as dark brown curved rod-like bacteria in the lumen. (B) Specific detection of H. pylori with anti-H. pylori antibody immunostained as a dark brown color (short arrow). Original magnification, x400. (From Deenonpoe et al (2015) Asian Pac J Cancer Prev 18, 1751, with permission.)
Figure 2.

Quantification of Helicobacter pylori in colorectal feces of hamsters using quantitative RT-PCR. Total bacterial cell counts among the following 5 groups of hamsters are compared; 1. Control (Group 1), 2. Control+ ABx (Group 2), 3. Infected hamster (Group 3), 4. Infected hamster + ABx (Group 4), and 5. Infected hamster + ABx+ PZQ (Group 5). * Significant differences among groups, P ≤ 0.001. (From Deenonpoe et al (2015) Asian Pac J Cancer Prev 18, 1751, with permission.)
There are several adhesins in H. pylori including blood-antigen binding protein A (BabA) and sialic acid-binding adhesin (SabA), neutrophil-activating protein (NAP), heat shock protein 60 (Hsp60), adherence-associated proteins (AlpA and AlpB), H. pylori outer membrane protein (HopZ), and lacdiNAc-binding adhesin (LabA) [35] that interact with host tissues and cells. BabA mediates binding of the bacteria to Lewis B antigens, Leb [41] and related terminal fucose residues found on blood group O (H antigen), A and B antigens [42]. Similar to BabA, the SabA has affinity for sialyl-Lex [43]. Toll-like receptor 4 (TLR4) is a known receptor for bacterial LPS [44]. The LabA adhesin specifically binds to GalNAcβ1-4GlcNAc motif, also known as N,N′-diacetyllactosediamine [lacdiNAc]), carried by MUC5AC mucins [45, 46]. However, orthologues of the human cell receptors and antigens have yet to be described from the epithelium of the gastrodermis of the gut of O. viverrini. We have begun laboratory investigation these aspects, with initial findings that support the role of liver fluke as a reservoir of H. pylori.
7. Helicobacter pylori – biliary epithelium interaction
Concerning the gastric epithelium, H. pylori colonizes on the mucosal layer, and adheres to the epithelium through interactions between bacterial adhesins with cellular receptors (as described above) [35], after which virulence factors stimulate cascades of inflammatory signalling, anti-apoptosis, cell proliferation and transformation pathways [18, 19]. Moreover, cagA-positive H. pylori has been shown to induce mutation of the gastric carcinoma cell line [47]. However, only a few reports have described the interaction between biliary epithelium and H. pylori. Our previous studies revealed that H. pylori induces multiple effects in CCA cell lines in vitro, including inflammation (IL-8 production), cellular proliferation and apoptosis [28, 30]. In addition, at a low multiplicity of infection (MOI=1), H. pylori induces pro-inflammatory cytokine production and proliferative responses in CCA cell lines. These findings suggest that the small numbers of H. pylori bacteria that reach the biliary epithelial cells suffice to promote inflammation and transformation within this niche. Hence, the findings provide support for the potential role of this carcinogenic microbe in the development of hepatobiliary disease [28]. In order to investigate this hypothesis, as well as links between cagPAI-positive H. pylori strains and CCA, we tested the ability of various H. pylori wild-type and isogenic cag mutant strains to adhere and induce pro-inflammatory responses in two CCA cell lines [29]. All of the strains adhere to both CCA cell lines without significant differences among the different virulence factors, and in addition, H. pylori wild-type bacteria stimulated significantly higher responses in all cell types compared with cagA−, cagL− or cagPAI− strains. Furthermore, H. pylori requires a functional type 4 secretion system (T4SS) for the activation of NF-κB, leading to the production of IL-8, in biliary tract epithelial cells [41]. Together, these several reports reveal that the cagPAI is critical for H. pylori–related pathogenesis in biliary epithelia, and thus provide a potential causal link for H. pylori in biliary tract disease including CCA.
8. Liver fluke enhances H. pylori colonization and adhesion to biliary epithelium?
Since O. viverrini has a blind gut (like all trematodes it does not have an anus), all ingested materials in the gut including bacteria are regurgitated out through the oral sucker. To date there are no reports on the link between O. viverrini infection and enhancement of H. pylori colonization. However, we have reported the up-regulation of TLR 4 but not other TLRs in normal cholangiocytes (H69) co-cultured with O. viverrini excretory-secretory products [48]. As TLR 4 is a known receptor for H. pylori, this result may imply the possible increased adhesion on biliary epithelium in O. viverrini infection. Other host receptors for H. pylori also may be involved in the colonization. Indeed, colonization of Helicobacter in mucosal layer needs its sheathed flagella to move across the mucus layer to the epithelium [35]. Given O. viverrini is rather large in size (up 1 cm in length in the human biliary tract) and can mechanically damage the mucous layer of the biliary epithelium, and thus facilitate bacterial adhesion.
9. Pathogenesis of liver fluke/Helicobacter induced pathology and carcinogenesis
Three main mechanisms are proposed to contribute to CCA through chronic infection with O. viverrini: mechanical damage to the biliary epithelia caused by the feeding activities of the parasites, immunopathology due to infection-related inflammation, and toxic effects of parasite ES. The interplay of these mechanisms aligns with current understanding of this malignancy, suggesting formation and progression relies on many interrelated factors creating a microenvironment that is conducive for malignant transformation [49]. However, these interplays may not explain or entirely explain the role of O. viverrini. Additionally, the influence and activities of H. pylori transported by O. viverrini into the biliary tract may be central to pathogenesis and carcinogenesis of liver fluke-associated CCA.
9.1. Mechanical injury
Mechanical injury caused by the liver flukes contributes to biliary damage. The suckers of the fluke attach to biliary epithelia, damage the bile ducts, even in early infection. As the flukes mature, the lesions enlarge and ulcerate. The ulcers provide a portal of egg entrapment, inducing circumoval granuloma during chronic opisthorchiasis that in turn leads to biliary periductal fibrosis [2]. Along with inflammation, the ulcer allows facilitates entry of for bile acids and other bile constituents, exposure to which can predispose to malignant transformation given these metabolites represent endogenous etiologic agents in gastrointestinal cancers [50]. In addition, migration of the liver flukes within the bile ducts and their grazing activity in mucin along the biliary epithelium ensures close contact between H. pylori and the cholangiocytes lining the bile ducts.
9.2. Metabolic products
Liver flukes release excretory/secretory products (ES) from the tegument and excretory openings into the bile, or culture medium in vitro, some of which are highly immunogenic [51, 52]. Recent findings revealed that O. viverrini ES products contained H. pylori [31]. These ES products, aside from inducing immune responses, may be toxic to or interact with the biliary epithelium [53]. Murine fibroblasts (NIH-3T3) co-cultured with O. viverrini (but physically separated from the worms in Transwell plates) proliferate compared to cells in media alone [54]. Human biliary cells also proliferate in the presence of ES-derived parasite growth factors, i.e. granulin [55] and anti-apoptotic proteins [56]. H. pylori itself can promote cell proliferation and transformation and anti-apoptosis as noted above. These data together demonstrate clearly that metabolic products of Opisthorchis viverrini, which includes H. pylori or its components, orchestrate the induction of cell proliferation and anti-apoptosis, confirming pioneering reports of hyperplasia of opisthorchiasis-associated biliary epithelial cells [57, 58].
9.3. Immunopathology
Host immune responses and immunopathological processes mediate hepatobiliary damage in opisthorchiasis [57–60]. We have implicated parasite-specific IL-6 in the pathogenesis of advanced periductal fibrosis in opisthorchiasis, with links to other hepatobiliary abnormalities, including CCA [61]. Sripa and Kaewkes [57] showed that inflammation around infected hamster bile ducts was a consequence of host cellular responses to antigens of O. viverrini. Marked infiltration of inflammatory cells in periductal sites of infected liver was associated with the presence of fluke antigens in the bile duct epithelium as detected by immunohistochemistry (IHC). Intense antigen staining was seen adjacent to the flukes. Small bile ducts, the secondary/third order ducts - where flukes do not occur because the diameter of the ducts is not sufficiently wide to accommodate parasites – also were positive for O. viverrini antigens and were markedly inflamed. By comparison, H. pylori also triggers pro-inflammatory cytokine responses by biliary cells [29] that can cause inflammation in infected hosts. O. viverrini ES products of the liver flukes, which may contain H. pylori components, induce IL-8 production that is unique cytokine for H. pylori in normal cholangiocytes [48]. Therefore, we predict that both O. viverrini and H. pylori synergistically induce pathologies through immunopathogenic process in the infected biliary tract.
10. Multi-factorial pathway from O. viverrini/H. pylori infection to CCA
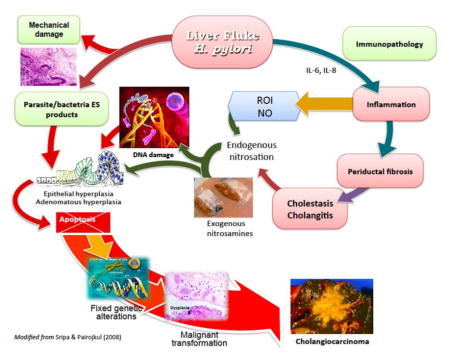
Figure 3 pictorially indicates the several mechanisms that have been proposed to explain how infection with O. viverrini provokes cholangiocarcinogenesis. The primary pathologic change, i.e. epithelial desquamation and ulcer, may be due to mechanical irritation caused by the liver fluke, H. pylori and/or the ES released by the liver flukes. However, as outlined above, immunopathological processes contribute to the longstanding hepatobiliary damage. With liver fluke and H. pylori co-infection, inflammation, periductal fibrosis and proliferative responses including epithelial hyperplasia, goblet cell metaplasia and adenomatous hyperplasia represent predisposing lesions that may enhance susceptibility of host cell chromosomal DNA to genotoxic lesions [4, 62]. N-nitroso compounds and their precursors occur at low levels in fermented food, including fermented fish, e.g., plara, which is ubiquitous in the diet of people in much of Thailand and neighboring Laos. Indeed, it has been argued that these compounds are requisite along with liver fluke infection as carcinogens leading to CCA in the inhabitants of these regions [63].
Figure 3. Hypothesized pathways of pathogenesis of opisthorchiasis/H. pylori-induced cholangiocarcinoma.

The liver fluke Opisthorchis viverrini/Helicobacter damages bile duct tissue via at least three distinct pathways: 1) mechanical damage to biliary epithelia caused by parasites sucking; 2) inflammation-induced immunopathology, particularly due to reactive oxygen intermediates (ROI) and nitric oxide (NO); and 3) direct effects of fluke/Helicobacter secreted proteins on biliary epithelia including cell proliferation induced by parasite-derived growth factors. These pathways converge, resulting in genetic lesions and unregulated proliferation. Damaged DNA/genes after successive replications become fixed, leading to malignant transformation of cholangiocytes. Adapted from [32].
11. Conclusion
Infection with Opisthorchis viverrini is a cogent risk for cholangiocarcinoma. Thailand has the highest rates of both opisthorchiasis and cholangiocarcinoma in the world. Moreover, in this location, the prevalence and geographical range of carriage of Helicobacter pylori parallels those of opisthorchiasis and cholangiocarcinoma. Our review on recent literature reveals that the liver fluke is a reservoir of Helicobacter spp. and addresses the role of liver fluke associated H. pylori in hepatobiliary disease and malignancy. We hypothesize that co-infection by O. viverrini and H. pylori is central of liver fluke infection associated cholangiocarcinoma. Researches on several aspects of the two carcinogenic pathogens co-infection are currently carried out in our laboratory.
Highlights.
Opisthorchis/Helicobacter co-infection is central to opisthorchiasis
Opisthorchis is a reservoir of H. pylori
H. pylori may play key role in pathogenesis of opisthorchiasis-induced cholangiocarcinoma
Acknowledgments
This work was supported by the National Health Security Office, Thailand, the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, through the Health Cluster (SHeP-GMS), the Faculty of Medicine, Khon Kaen University, Thailand (award number I56110), and the Thailand Research Fund under the TRF Senior Research Scholar (RTA 5680006). The National Institute of Allergy and Infectious Diseases (NIAID), Tropical Medicine Research Center award number P50AI098639 and National Cancer Institute (NCI), National Institutes of Health (NIH) award CA164719 also provided support. RD acknowledges support as a PhD research scholar by the Commission on Higher Education, Thailand, under the program Strategic Scholarships for Frontier Research Network for the Joint PhD Program Thai Doctoral Degree, BS acknowledges support from Thailand Research Fund Senior Research Scholar. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders including TRF, NIAID, NCI, the NIH or other funders.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum; IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; Lyon. 7–14 June 1994; 1994. pp. 1–241. [PMC free article] [PubMed] [Google Scholar]
- 2.Sripa B. Pathobiology of opisthorchiasis: an update. Acta Trop. 2003;88(3):209–20. doi: 10.1016/j.actatropica.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Sripa B, Kaewkes S, Sithithaworn P, Mairiang E, Laha T, Smout M, Pairojkul C, Bhudhisawasdi V, Tesana S, Thinkamrop B, Bethony JM, Loukas A, Brindley PJ. Liver fluke induces cholangiocarcinoma. PLoS Med. 2007;4(7):e201. doi: 10.1371/journal.pmed.0040201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thamavit W, Bhamarapravati N, Sahaphong S, Vajrasthira S, Angsubhakorn S. Effects of dimethylnitrosamine on induction of cholangiocarcinoma in Opisthorchis viverrini-infected Syrian golden hamsters. Cancer Res. 1978;38(12):4634–9. [PubMed] [Google Scholar]
- 5.Thamavit W, Pairojkul C, Tiwawech D, Shirai T, Ito N. Strong promoting effect of Opisthorchis viverrini infection on dimethylnitrosamine-initiated hamster liver. Cancer Lett. 1994;78(1–3):121–5. doi: 10.1016/0304-3835(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 6.Cancer incidence in five continents. IARC Sci Publ. 2002;VIII(155):1–781. [PubMed] [Google Scholar]
- 7.Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. 2013;145(6):1215–29. doi: 10.1053/j.gastro.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huai JP, Ding J, Ye XH, Chen YP. Inflammatory bowel disease and risk of cholangiocarcinoma: evidence from a meta-analysis of population-based studies. Asian Pac J Cancer Prev. 2014;15(8):3477–82. doi: 10.7314/apjcp.2014.15.8.3477. [DOI] [PubMed] [Google Scholar]
- 9.Wu Q, He XD, Yu L, Liu W, Tao LY. The metabolic syndrome and risk factors for biliary tract cancer: a case-control study in China. Asian Pac J Cancer Prev. 2012;13(5):1963–9. doi: 10.7314/apjcp.2012.13.5.1963. [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto K, Onoyama T, Kawata S, Takeda Y, Harada K, Ikebuchi Y, Ueki M, Miura N, Yashima K, Koda M, Sakamoto T, Endo M, Horie Y, Murawaki Y. Hepatitis B and C virus infection is a risk factor for the development of cholangiocarcinoma. Intern Med. 2014;53(7):651–4. doi: 10.2169/internalmedicine.53.1410. [DOI] [PubMed] [Google Scholar]
- 11.Brindley PJ, da Costa JM, Sripa B. Why does infection with some helminths cause cancer? Trends Cancer. 2015;1(3):174–182. doi: 10.1016/j.trecan.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou D, Wang JD, Weng MZ, Zhang Y, Wang XF, Gong W, Quan ZW. Infections of Helicobacter spp. in the biliary system are associated with biliary tract cancer: a meta-analysis. Eur J Gastroenterol Hepatol. 2013;25(4):447–54. doi: 10.1097/MEG.0b013e32835c0362. [DOI] [PubMed] [Google Scholar]
- 13.Mateos-Munoz B, Perez-de-la-Serna J, Ruiz-de-Leon A, Serrano-Falcon B, Casabona-Frances S, Velasco-Cerrudo A, Rey-Diaz-Rubio E. Enterohepatic Helicobacter other than Helicobacter pylori. Rev Esp Enferm Dig. 2013;105(8):477–84. doi: 10.4321/s1130-01082013000800006. [DOI] [PubMed] [Google Scholar]
- 14.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1(8390):1311–5. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 15.Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet. 1983;1(8336):1273–5. [PubMed] [Google Scholar]
- 16.Forman D. Helicobacter pylori infection: a novel risk factor in the etiology of gastric cancer. J Natl Cancer Inst. 1991;83(23):1702–3. doi: 10.1093/jnci/83.23.1702. [DOI] [PubMed] [Google Scholar]
- 17.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991;325(16):1127–31. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 18.Cid TP, Fernandez MC, Benito Martinez S, Jones NL. Pathogenesis of Helicobacter pylori infection. Helicobacter. 2013;18(Suppl 1):12–7. doi: 10.1111/hel.12076. [DOI] [PubMed] [Google Scholar]
- 19.Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe. 2014;15(3):306–16. doi: 10.1016/j.chom.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 20.Waluga M, Kukla M, Zorniak M, Bacik A, Kotulski R. From the stomach to other organs: Helicobacter pylori and the liver. World J Hepatol. 2015;7(18):2136–46. doi: 10.4254/wjh.v7.i18.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabelo-Goncalves EM, Roesler BM, Zeitune JM. Extragastric manifestations of Helicobacter pylori infection: Possible role of bacterium in liver and pancreas diseases. World J Hepatol. 2015;7(30):2968–79. doi: 10.4254/wjh.v7.i30.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy G, Michel A, Taylor PR, Albanes D, Weinstein SJ, Virtamo J, Parisi D, Snyder K, Butt J, McGlynn KA, Koshiol J, Pawlita M, Lai GY, Abnet CC, Dawsey SM, Freedman ND. Association of seropositivity to Helicobacter species and biliary tract cancer in the ATBC study. Hepatology. 2014;60(6):1963–71. doi: 10.1002/hep.27193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esmat G, El-Bendary M, Zakarya S, Ela MA, Zalata K. Role of Helicobacter pylori in patients with HCV-related chronic hepatitis and cirrhosis with or without hepatocellular carcinoma: possible association with disease progression. J Viral Hepat. 2012;19(7):473–9. doi: 10.1111/j.1365-2893.2011.01567.x. [DOI] [PubMed] [Google Scholar]
- 24.Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, Gabele E, Rusyn I, Yamashina S, Froh M, Adachi Y, Iimuro Y, Bradford BU, Smutney OM, Connor HD, Mason RP, Goyert SM, Peters JM, Gonzalez FJ, Samulski RJ, Thurman RG. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med. 2001;31(12):1544–9. doi: 10.1016/s0891-5849(01)00748-1. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Trevijano ER, Iraburu MJ, Fontana L, Dominguez-Rosales JA, Auster A, Covarrubias-Pinedo A, Rojkind M. Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology. 1999;29(3):960–70. doi: 10.1002/hep.510290346. [DOI] [PubMed] [Google Scholar]
- 26.Boonyanugomol W, Chomvarin C, Sripa B, Bhudhisawasdi V, Khuntikeo N, Hahnvajanawong C, Chamsuwan A. Helicobacter pylori in Thai patients with cholangiocarcinoma and its association with biliary inflammation and proliferation. HPB (Oxford) 2012;14(3):177–84. doi: 10.1111/j.1477-2574.2011.00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boonyanugomol W, Chomvarin C, Sripa B, Chau-In S, Pugkhem A, Namwat W, Wongboot W, Khampoosa B. Molecular analysis of Helicobacter pylori virulent-associated genes in hepatobiliary patients. HPB (Oxford) 2012;14(11):754–63. doi: 10.1111/j.1477-2574.2012.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boonyanugomol W, Chomvarin C, Baik SC, Song JY, Hahnvajanawong C, Kim KM, Cho MJ, Lee WK, Kang HL, Rhee KH, Sripa B. Role of cagA-positive Helicobacter pylori on cell proliferation, apoptosis, and inflammation in biliary cells. Dig Dis Sci. 2011;56(6):1682–92. doi: 10.1007/s10620-010-1512-y. [DOI] [PubMed] [Google Scholar]
- 29.Boonyanugomol W, Chomvarin C, Hahnvajanawong C, Sripa B, Kaparakis-Liaskos M, Ferrero RL. Helicobacter pylori cag pathogenicity island (cagPAI) involved in bacterial internalization and IL-8 induced responses via NOD1- and MyD88-dependent mechanisms in human biliary epithelial cells. PLoS One. 2013;8(10):e77358. doi: 10.1371/journal.pone.0077358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boonyanugomol W, Chomvarin C, Song JY, Kim KM, Kim JM, Cho MJ, Lee WK, Kang HL, Rhee KH, Sripa B, Hahnvajanawong C, Baik SC. Effects of Helicobacter pylori gamma-glutamyltranspeptidase on apoptosis and inflammation in human biliary cells. Dig Dis Sci. 2012;57(10):2615–24. doi: 10.1007/s10620-012-2216-2. [DOI] [PubMed] [Google Scholar]
- 31.Deenonpoe R, Chomvarin C, Pairojkul C, Chamgramol Y, Loukas A, Brindley PJ, Sripa B. The carcinogenic liver fluke Opisthorchis viverrini is a reservoir for species of Helicobacter. Asian Pac J Cancer Prev. 2015;16(5):1751–8. doi: 10.7314/apjcp.2015.16.5.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sripa B, Pairojkul C. Cholangiocarcinoma: lessons from Thailand. Curr Opin Gastroenterol. 2008;24(3):349–56. doi: 10.1097/MOG.0b013e3282fbf9b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sripa B, Kanla P, Sinawat P, Haswell-Elkins MR. Opisthorchiasis-associated biliary stones: light and scanning electron microscopic study. World J Gastroenterol. 2004;10(22):3318–21. doi: 10.3748/wjg.v10.i22.3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keilberg D, Ottemann KM. How Helicobacter pylori senses, targets and interacts with the gastric epithelium. Environ Microbiol. 2016;18(3):791–806. doi: 10.1111/1462-2920.13222. [DOI] [PubMed] [Google Scholar]
- 35.Kao CY, Sheu BS, Wu JJ. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed J. 2016;39(1):14–23. doi: 10.1016/j.bj.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tajeddin E, Sherafat SJ, Majidi MR, Alebouyeh M, Alizadeh AH, Zali MR. Association of diverse bacterial communities in human bile samples with biliary tract disorders: a survey using culture and polymerase chain reaction-denaturing gradient gel electrophoresis methods. Eur J Clin Microbiol Infect Dis. 2016 doi: 10.1007/s10096-016-2669-x. [DOI] [PubMed] [Google Scholar]
- 37.Aviles-Jimenez F, Guitron A, Segura-Lopez F, Mendez-Tenorio A, Iwai S, Hernandez-Guerrero A, Torres J. Microbiota studies in the bile duct strongly suggest a role for Helicobacter pylori in extrahepatic cholangiocarcinoma. Clin Microbiol Infect. 2016;22(2):178 e11–22. doi: 10.1016/j.cmi.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 38.Nilius M, Strohle A, Bode G, Malfertheiner P. Coccoid like forms (CLF) of Helicobacter pylori. Enzyme activity and antigenicity. Zentralbl Bakteriol. 1993;280(1–2):259–72. doi: 10.1016/s0934-8840(11)80964-3. [DOI] [PubMed] [Google Scholar]
- 39.Hildebrandt E, McGee DJ. Helicobacter pylori lipopolysaccharide modification, Lewis antigen expression, and gastric colonization are cholesterol-dependent. BMC Microbiol. 2009;9:258. doi: 10.1186/1471-2180-9-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albertson N, Wenngren I, Sjostrom JE. Growth and survival of Helicobacter pylori in defined medium and susceptibility to Brij 78. J Clin Microbiol. 1998;36(5):1232–5. doi: 10.1128/jcm.36.5.1232-1235.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Boren T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279(5349):373–7. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 42.Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D, Roche N, Vikstrom S, Sjostrom R, Linden S, Backstrom A, Lundberg C, Arnqvist A, Mahdavi J, Nilsson UJ, Velapatino B, Gilman RH, Gerhard M, Alarcon T, Lopez-Brea M, Nakazawa T, Fox JG, Correa P, Dominguez-Bello MG, Perez-Perez GI, Blaser MJ, Normark S, Carlstedt I, Oscarson S, Teneberg S, Berg DE, Boren T. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305(5683):519–22. doi: 10.1126/science.1098801. [DOI] [PubMed] [Google Scholar]
- 43.Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, Altraja S, Wadstrom T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarstrom L, Boren T. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297(5581):573–8. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pachathundikandi SK, Tegtmeyer N, Backert S. Signal transduction of Helicobacter pylori during interaction with host cell protein receptors of epithelial and immune cells. Gut Microbes. 2013;4(6):454–74. doi: 10.4161/gmic.27001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kenny DT, Skoog EC, Linden SK, Struwe WB, Rudd PM, Karlsson NG. Presence of terminal N-acetylgalactosaminebeta1-4N-acetylglucosamine residues on O-linked oligosaccharides from gastric MUC5AC: involvement in Helicobacter pylori colonization? Glycobiology. 2012;22(8):1077–85. doi: 10.1093/glycob/cws076. [DOI] [PubMed] [Google Scholar]
- 46.Rossez Y, Gosset P, Boneca IG, Magalhaes A, Ecobichon C, Reis CA, Cieniewski-Bernard C, Joncquel Chevalier Curt M, Leonard R, Maes E, Sperandio B, Slomianny C, Sansonetti PJ, Michalski JC, Robbe-Masselot C. The lacdiNAc-specific adhesin LabA mediates adhesion of Helicobacter pylori to human gastric mucosa. J Infect Dis. 2014;210(8):1286–95. doi: 10.1093/infdis/jiu239. [DOI] [PubMed] [Google Scholar]
- 47.Yao Y, Tao H, Park DI, Sepulveda JL, Sepulveda AR. Demonstration and characterization of mutations induced by Helicobacter pylori organisms in gastric epithelial cells. Helicobacter. 2006;11(4):272–86. doi: 10.1111/j.1523-5378.2006.00408.x. [DOI] [PubMed] [Google Scholar]
- 48.Ninlawan K, O’Hara SP, Splinter PL, Yongvanit P, Kaewkes S, Surapaitoon A, LaRusso NF, Sripa B. Opisthorchis viverrini excretory/secretory products induce toll-like receptor 4 upregulation and production of interleukin 6 and 8 in cholangiocyte. Parasitol Int. 2010;59(4):616–21. doi: 10.1016/j.parint.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1(1):46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernstein H, Bernstein C, Payne CM, Dvorak K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J Gastroenterol. 2009;15(27):3329–40. doi: 10.3748/wjg.15.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sripa B, Kaewkes S. Relationship between parasite-specific antibody responses and intensity of Opisthorchis viverrini infection in hamsters. Parasite Immunol. 2000;22(3):139–45. doi: 10.1046/j.1365-3024.2000.00286.x. [DOI] [PubMed] [Google Scholar]
- 52.Wongratanacheewin S, Bunnag D, Vaeusorn N, Sirisinha S. Characterization of humoral immune response in the serum and bile of patients with opisthorchiasis and its application in immunodiagnosis. Am J Trop Med Hyg. 1988;38(2):356–62. doi: 10.4269/ajtmh.1988.38.356. [DOI] [PubMed] [Google Scholar]
- 53.Harinasuta T, Riganti M, Bunnag D. Opisthorchis viverrini infection: pathogenesis and clinical features. Arzneimittelforschung. 1984;34(9B):1167–9. [PubMed] [Google Scholar]
- 54.Thuwajit C, Thuwajit P, Kaewkes S, Sripa B, Uchida K, Miwa M, Wongkham S. Increased cell proliferation of mouse fibroblast NIH-3T3 in vitro induced by excretory/secretory product(s) from Opisthorchis viverrini. Parasitology. 2004;129(Pt 4):455–64. doi: 10.1017/s0031182004005815. [DOI] [PubMed] [Google Scholar]
- 55.Smout MJ, Laha T, Mulvenna J, Sripa B, Suttiprapa S, Jones A, Brindley PJ, Loukas A. A granulin-like growth factor secreted by the carcinogenic liver fluke, Opisthorchis viverrini, promotes proliferation of host cells. PLoS Pathog. 2009;5(10):e1000611. doi: 10.1371/journal.ppat.1000611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matchimakul P, Rinaldi G, Suttiprapa S, Mann VH, Popratiloff A, Laha T, Pimenta RN, Cochran CJ, Kaewkes S, Sripa B, Brindley PJ. Apoptosis of cholangiocytes modulated by thioredoxin of carcinogenic liver fluke. Int J Biochem Cell Biol. 2015;65:72–80. doi: 10.1016/j.biocel.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sripa B, Kaewkes S. Localisation of parasite antigens and inflammatory responses in experimental opisthorchiasis. Int J Parasitol. 2000;30(6):735–40. doi: 10.1016/s0020-7519(00)00054-0. [DOI] [PubMed] [Google Scholar]
- 58.Bhamarapravati N, Thammavit W, Vajrasthira S. Liver changes in hamsters infected with a liver fluke of man, Opisthorchis viverrini. Am J Trop Med Hyg. 1978;27(4):787–94. doi: 10.4269/ajtmh.1978.27.787. [DOI] [PubMed] [Google Scholar]
- 59.Haswell-Elkins MR, Sithithaworn P, Mairiang E, Elkins DB, Wongratanacheewin S, Kaewkes S, Mairiang P. Immune responsiveness and parasite-specific antibody levels in human hepatobiliary disease associated with Opisthorchis viverrini infection. Clin Exp Immunol. 1991;84(2):213–8. doi: 10.1111/j.1365-2249.1991.tb08151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Flavell DJ, Flavell SU. Opisthorchis viverrini: pathogenesis of infection in immunodeprived hamsters. Parasite Immunol. 1986;8(5):455–66. doi: 10.1111/j.1365-3024.1986.tb00861.x. [DOI] [PubMed] [Google Scholar]
- 61.Sripa B, Mairiang E, Thinkhamrop B, Laha T, Kaewkes S, Sithithaworn P, Tessana S, Loukas A, Brindley PJ, Bethony JM. Advanced periductal fibrosis from infection with the carcinogenic human liver fluke Opisthorchis viverrini correlates with elevated levels of interleukin-6. Hepatology. 2009;50(4):1273–81. doi: 10.1002/hep.23134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim YI. Liver carcinoma and liver fluke infection. Arzneimittelforschung. 1984;34(9B):1121–6. [PubMed] [Google Scholar]
- 63.Migasena P, Reaunsuwan W, Changbumrung S. Nitrates and nitrites in local Thai preserved protein foods. J Med Assoc Thai. 1980;63(9):500–5. [PubMed] [Google Scholar]