

Abstract
The APOBEC3 (A3) family of proteins are DNA cytidine deaminases that act as sentinels in the innate immune response against retroviral infections and are responsive to interferon. Recently, a few A3 genes were identified as potent enzymatic sources of mutations in several human cancers. Using human cancer cells and lymphocytes we show that under stress conditions and immune challenges all A3 genes are direct transcriptional targets of the tumor suppressor p53. While the expression of most A3 genes (including A3C and A3H) was stimulated by activation of p53, treatment with the DNA damaging agent doxorubicin or the p53 stabilizer Nutlin, led to repression of the A3B gene. Furthermore, p53 could enhance interferon type-I induction of A3 genes. Interestingly, overexpression of a group of tumor-associated p53 mutants in TP53-null cancer cells promoted A3B expression. These findings establish that the “guardian of the genome” role ascribed to p53 also extends to a unique component of the immune system–the A3 genes–thereby integrating human immune and chromosomal stress responses into an A3/p53 immune axis.
Keywords: cytidine deaminase, retrotransposons, innate immunity, mutant p53, cancer therapy
Introduction
The well-known tumor suppressor p53 is a transcriptional master regulator that regulates the expression of genes associated with a wide range of functions including cell cycle arrest, apoptosis and senescence in response to genotoxic and non-genotoxic stresses that challenge cellular genomic integrity (1). Following various stresses, the activated p53 protein is accumulated in the nucleus, where it binds to DNA as a tetramer at p53 response elements (p53REs) (1).
The commonly held view of p53 as a “guardian of the genome” has expanded greatly during the last few years to cover many biological processes (1–3), including the role of p53 in modulating the human immune system and antiviral defense (4–7). Previously, we found that activation of p53 by common anti-tumor agents in human primary and cancer cell lines directly alters expression of several members of the innate immune Toll-like receptor (TLR) gene family, which is involved in host defense against invading pathogens (8,9). This results in modulation of TLR downstream responses to cognate ligands (10,11).
p53 ChIP-seq approaches in combination with transcriptome analysis have revealed many new p53 regulated target genes. From our p53 cistrome studies p53 in human cancer (3) and human primary lymphocytes (unpublished data), we have identified new p53 targets involved in immune response signaling. Included are several members of the innate immune gene family APOBEC3 (A3; Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3).
The A3 genes, which consists of seven highly related DNA cytidine deaminases that are tandemly distributed on human chromosome 22, catalyze the deamination of cytidine to uracil (12). The A3 genes are a key component of the innate immune system in vertebrates that inhibit replication of a variety of retroviruses, endogenous retroelements and DNA viruses (13,14). Recently, the C-to-T hyper-mutagenesis genomic changes in multiple cancers have been attributed to A3A and A3B (15–17).
The A3 genes are generally viewed as constitutively expressed in immune related cells as well as in immune-associated cancer cells. In the context of innate immune responses to infection, the expression of several A3 genes is modulated by IFNs that may be cell type and IFN-type dependent (18,19). However, little is known about the transcription factors involved in the expression of the A3 genes in response to immune challenges and other environmental stressors.
Here, we describe a new role for p53: regulation of the A3 gene family in response to common anticancer drugs and direct activation of p53. Using both a panel of human cancer cell lines as well as primary immune cells obtained directly from human subjects we discovered that most A3 genes are transcriptionally responsive to p53. Furthermore, cancer-associated p53 mutants can dramatically impact the pattern of expression of the A3 genes, including novel responses of the cancer hypermutator A3B gene. In addition, p53 can participate and influence the IFN-induced transcriptional responses of several A3 family members. Overall, we provide the first evidence that p53 is a key node linking DNA damage, immune-induced responses and A3 gene expression in human cells.
Material and methods
Cell lines and treatments
Human cancer cell lines were cultured in RPMI-1640, McCoy’s 5A or DMEM supplemented with 10% of FBS and 100 units/ml penicillin/streptomycin as described elsewhere. More information about the cancer and primary human cells is provided in the Supplemental Material.
RNA isolation and qPCR
Total RNA was isolated with RNEasy kits (Qiagen). Quantification and purity of the samples was determined using Nano drop spectrophotometer (Thermo Fisher Scientific) and 1 μg total RNA was reverse transcribed using Transcriptor reverse transcriptase with random hexameric primers (Roche) following manufacturer’s recommendations. qPCR was performed following established procedures, primers, and Universal Primary Library System probes as described (19) using 7000 ABI sequence Detection System (Applied Biosystems). All reactions were done in triplicate and relative quantification values were calculated based on the 2-ΔΔCt method using expression from the housekeeping gene Tata Binding Protein (TBP). Primers and probes are described in Supplemental Table S6. Additional material and methods can be found in the supplemental material.
Statistical analysis
Analysis was performed using GraphPad Prism statistical software. Data are represented as mean ± standard deviations from at least three separate experiments. Two-tailed student’s t test was applied for comparisons of two groups. p values <0.05 were considered significant.
Results
Chromosomal stress and p53 activation alters A3 gene family expression
Recently, in our p53 ChIP-seq study that included associated gene expression analysis in osteosarcoma U2OS cells following p53 activation by Nutlin-3 (Nutlin) or Doxorubicin (DXR) we identified several new p53 target genes related to immune responses (3). Binding of p53 to the regulatory regions of the A3C and A3H members of the A3 gene family was associated with increased transcription based on Taqman analysis. Given that homology is high among the A3 family sequences (20), we hypothesized that p53 responsiveness may have been conserved among other members of the A3 family.
Because of the redundancy in the regulatory and coding regions for A3 genes, there have been inconsistent conclusions about the transcriptional regulation of the A3 genes as well as the quantification of A3 expression by common approaches (microarray and qPCR) (19). To validate our previous findings and explore the extent to which p53 regulates the expression of other A3 members, we measured expression of all A3 genes in U2OS cells following exposure to Nutlin and DXR using the Universal Primary Library System technology. (This technology, which is more robust than the general SybrGreen primers or Applied Biosystems Taqman primers/probes assays for evaluating A3 mRNA changes (19), allows highly related sequences to be distinguished.) Activation of p53 by these agents led to time-dependent upregulation of several A3 genes (from ~2.5 to 13 fold), as shown in Fig. 1A. The A3B gene differed in that both drug treatments repressed it. In addition to A3A and A3D, we confirmed upregulation of A3C and A3H as well as the p53 target gene CDKN1A (p21) used as a positive control. We also observed that DXR, but not Nutlin, increased A3G mRNA. Except for repression of A3B, a similar pattern of expression of A3 genes was observed in A549 lung cancer cells. (Supplemental Fig. S1A). The expression of A3A was not detected in A549 cells. Overall, we establish that expression of A3 genes can be regulated in response to genotoxic stress and p53 activation.
Figure 1. Induced expression of the A3 gene family by DNA stress and activation of the p53 pathway in human cancer cell lines.

(A) Expression of A3 genes in U2OS cells treated for 24h with DMSO (vehicle 0.1%), the p53 activating drug Nutlin (10 μM) and Doxorubicin (DXR, 1.5 μM). Changes in gene expression presented as fold-change compared to untreated cells (value of 1) were analyzed by real time-qPCR. p21 expression was a positive control for a p53 transcriptional target. “*” indicates p<0.05 compared to untreated cells. (B) Nutlin or (C) Doxorubicin are heat maps for expression of A3 genes after 24h treatment in human cancer cell lines that are p53 proficient or deficient. The heat map fold-change values compared to untreated cells and statistical analysis are available in Supplemental Table S3.
Given that induction of innate immune genes is relevant in disease and cancer treatments and to determine if the impact of Nutlin and DXR on A3 genes was general rather than cell type specific, we expanded the analysis for A3 responses to DNA damaging agents and p53 activation in a panel of cell lines harboring wild-type, null and mutated p53 examined (Supplemental Table S2). Presented as heat maps in Figs. 1B (Nutlin) and 1C (DXR) are the mRNA fold-changes for each A3 gene and cell line (See Supplemental Table S3 for raw data). Expression of A3A was mainly observed in cell lines of immune origin (LCL35, GM12878, THP1, Jurkat and RAJI). The expression of the A3 genes was clearly induced by drug treatments only in WT p53 cells, where there was consistent induction of A3C and A3H as well as p21 among the cell lines. Cell type and agent-specific effects in mRNA changes were observed for A3A, A3D, A3F and A3G. As observed in U2OS and A549, A3B was repressed in all WT p53 cell lines examined in response to Nutlin and DXR. We also found that other chromosomal stressors, such as etoposide and ionizing radiation, altered the A3 genes expression profile in various cancer cell lines in a p53 dependent manner (Supplemental Figs. S1B–C). Although there are dramatic differences in the expression spectra for some A3 genes between cell lines, we establish that many genes of the A3 family are responsive to genotoxic stress and p53 activation in human cancer cells.
Since most of the A3 genes are expressed in immune cells, we examined their response to p53 induction in human primary lymphocytes. Relative levels of mRNA expression were determined in phytohemagglutinin (PHA) stimulated lymphocytes obtained from peripheral blood mononuclear cells freshly isolated from 11 healthy human volunteers. Despite variability among subjects, the overall A3 gene family expression profiles induced by Nutlin and DXR were like those in WT p53 cancer cell lines, as described in Supplemental Figs. 2A–B. The expression of the internal controls p21 and Mdm2 were also induced in all donors after Nutlin or DXR treatments. In agreement with previous reports (18,19), A3A expression was undetectable in T lymphocytes. A3B mRNA was not detected in all donors, but in those that it was observed (8 out of 11), both treatments induced repression of this gene. Pre-treatment of lymphocytes from 4 volunteers with the p53 inhibitor pifithrin-α (21) prior to Nutlin or DXR strongly altered expression, identifying a direct role for p53 in regulating Nutlin and DXR induced A3 gene expression (Supplemental Figs. S2C–D).
p53 directly modulates A3 gene family expression in human cells
A direct comparison between the A3 expression profiles for HCT116 p53+ and its isogenic version lacking p53 (HCT116 p53-) revealed that most of the changes in A3 genes were dependent on p53 (Figs. 1B and 1C). Few significant changes in expression were observed for the A3 genes after Nutlin or DXR in the null and mutant p53 cell lines. When changes were detected in the p53 mutant cell lines, they were typically opposite to the changes observed in WT p53 cell lines. An example is A3C, which was repressed in p53 mutant cells but significantly induced in WT p53 cells. However, the most dramatic example was found for the A3B response to DXR in that it was upregulated in several mutant cell lines but repressed in WT p53 cells. The p53-dependent gene expression changes in A3 genes were addressed by expressing RNAi p53 from a cassette that was integrated in MCF7 (22) or in U2OS (3), or using a lentiviral vector harboring small-hairpin RNAs that target p53 mRNA (termed p53sh-3755 and p53sh-3756) in A549 and LCL35 cells (Fig. 2 and Supplemental Fig. S3). As shown in Figs. 2A–C and Supplemental Fig. S3, the upregulation observed for A3 genes following DXR or Nutlin exposure in control cells (parental or scrambled) was completely lost or reduced in all p53 depleted cell lines regardless of tissue origin, confirming that these are p53 targets. The A3B induced repression was prevented by silencing of p53 expression.
Figure 2. p53 regulates expression of A3 genes in human cancer cells.
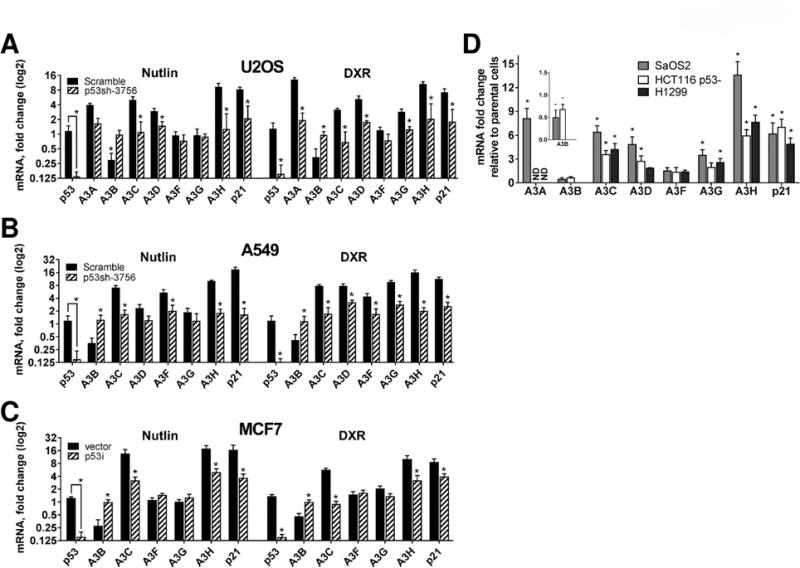
Changes in A3 mRNA levels were evaluated after 24h treatment with Nutlin (10μM) or DXR (1.5μM) in (A) U2OS, (B) A549 and (C) MCF7 cells with stably expressing scrambled shRNA, vector or p53 shRNAi (p53sh-3756 or p53i). Values are displayed as fold-changes (log2 scale) relative to their respective parental cell lines (value set to 1). (D) p53 null SaOS2, HCT116 p53- and H1299 cells were transfected either with empty vector or WT p53. A3 mRNA expression levels following transfection were examined by qPCR; p21 expression was a positive control. “*” corresponds to p<0.05 relative to untransfected cells (i.e., value of 1). “ND”, not detected. Calculations in SAOS2 cells for A3A and A3H after WT p53 transfection are approximations. See supplemental material for explanation.
To investigate further a direct connection between p53 and expression of A3 genes, WT p53 was overexpressed in the p53-null cancer cell lines SaOS2, HCT116 p53- and H1299. Although cell type differences were observed, the functional restoration of WT p53 expression resulted in ~2.5 to 14-fold upregulation of several A3 genes along with p21 mRNA used as a positive control (Fig. 2D). The A3F gene was the only family member that was not modified in any of the cell lines transfected. There was a lack of basal A3A or A3H expression in SaOS2 cells that appeared to be due to the absence of p53 since transfection of WT p53 resulted inexpression of both genes (Figs. 1B–C). Consistent with our previous observations, the restoration of functional p53 resulted in a significant inhibition of A3B gene expression in both transfected cell lines. Overall, these results confirm the direct participation of p53 in the regulation of A3 gene family members.
Activated p53 binds transcriptional regulatory regions of A3 genes
The p53 induced transcription of several A3 genes led us to investigate public p53 ChIP-seq datasets (summarized in Supplemental Table S4) for the impact of p53 across a broad range cell types and exposures. Included are the responses of normal diploid fibroblasts, embryonic stem cells, lymphoblasts as well as breast, colon, bone, and lung cancer cells. As shown in Fig. 3A, p53 can bind to the transcriptional regulatory regions of most of the A3 genes–A3A, A3B, A3C, and A3H genes–in response to the following stress conditions that activate p53: DXR, cisplatin, 5FU, actinomycin-D, ionizing radiation, Nutlin and RITA. To determine if A3 genes are direct transcriptional targets of p53 we screened for potential p53 REs in the promoter and regulatory regions of each A3 gene, from -5kb to +1.7 kb flanking the transcription start site (TSS) as well as the regions where ChIP-seq p53 binding peaks were observed. Using established guidelines for transcriptional functionality of potential p53REs (23) and p53Scan software (24), we identified several potential transactivation responsive p53REs located in the transcriptional regulatory regions of all A3 genes (Fig. 3A).
Figure 3. Activated p53 binds transcriptional regulatory region of A3 genes.
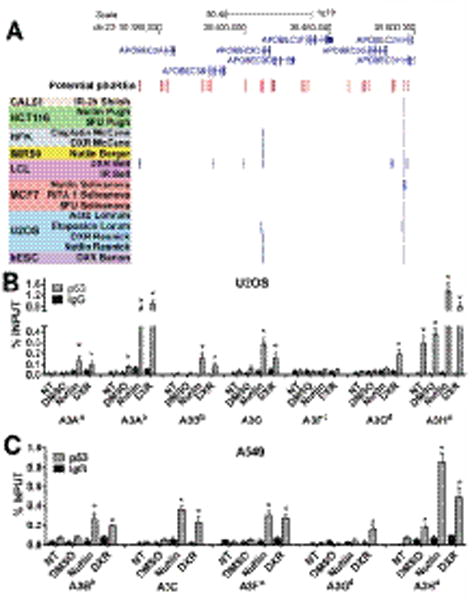
(A) Binding peak analysis of public p53ChIPseq datasets for human primary and cancer cells treated with DNA damaging agents and p53 activating drugs. Depicted is the region of human chromosome 22 containing the seven A3 genes. Red bars indicate relative position of potential p53 response elements (RE) identified by p53Scan software (24). Blue bars indicate p53 binding regions identified in the public p53ChIP-seq studies (see Supplemental Table S4). Validation of p53 binding to transcriptional regulatory regions of A3 genes containing putative p5REs in Nutlin (10μM) or DXR (1.5μM) is described in (B) U2OS and (C) A549 cells. p53 occupancy, determined by ChIP-PCR, is presented as % of total input DNA. Binding to the p21 promoter is used as a positive control. The superscript in A3 genes corresponds to p53RE sequences described in Supplemental Table S1. IgG serves as a negative control. Presented are means and standard deviations from 3 independent experiments. “*” indicates p<0.05 when compared to no treatment (“NT”) samples.
Overall, these results support a broad role for p53 in the A3 gene family regulation and suggest that the p53 binding regions containing the putative p53REs may regulate the expression of A3 genes. The top predicted functional p53REs are shown in Supplemental Table S1. A complete list of p53REs associated with A3 genes is presented in Supplemental Table S5. We note that for some of the putative p53REs the sequences were 95–100% conserved between several A3 genes, as found for p53REs of the A3D, A3F and A3G genes, which is consistent with the evolutionary duplication of the A3 genes. Although potential p53REs in A3B, A3D, A3F and A3G were identified around their TSS, none exhibited binding in reported p53 ChIP-seq studies. However, for the case of A3B, an intronic region containing a p53RE was bound by p53. The p53RE associated with A3F was previously reported as a p53 bound region in a ChIP-PET assay (25). Since a lack of binding in previous reports might be due to relatedness of the A3 genes, we investigated p53 binding to specific sites using a ChIP-PCR analysis that targets each of the putative REs. (Due to high sequence conservation, no reliable primers were identified to analyze some of the top predicted functional p53REs for A3D, A3F and A3G.) With this approach we confirmed that activated p53 can directly bind to the p53RE sequences identified in silico and/or previously found to bind in vivo for several A3 genes after Nutlin or DXR treatment in U2OS and A459 cells, as described in Figs. 3B and 3C and Supplemental Fig. S4. Binding of p53 to the promoter region of its target gene p21 was used as positive control while a random region of the GAPDH promoter was used as negative control (Supplemental Fig. S4). Collectively, these results demonstrate that most p53RE sites considered as associated with p53 induced expression of the A3 family members can be directly bound by p53 and strongly indicate that in response to chromosomal stresses p53 is a major transcription factor regulating the expression of A3 genes.
Tumor-associated p53 mutants can promote A3B expression
As described above, all the A3 genes were upregulated in WT p53 cancer cell lines in response to p53 activation except A3B, which was repressed. However, in cell lines with mutant p53 alleles, A3B was generally upregulated. As shown in Fig. 4A, transient expression in SaOS2 p53 null cells of the cancer-associated, hotspot p53 mutants R175H, G245S, R248Q, R273H and R280K resulted in upregulation of the A3B gene with little effect on expression of A3C, A3H or the internal control p21. The results were similar when the same p53 mutants were transiently overexpressed in HCT116 p53− cells, confirming that mutant p53 induces expression of A3B in human cancer cells (Fig. 4B). Interestingly, change-of-spectrum mutants A138Y, H178Y and R337H (10,26) that retain different levels of WT p53 transactivation change the expression profiles of A3 genes, including A3B. Altogether, these results imply that tumor-associated p53 mutants can influence the expression of several A3 genes, especially increasing the A3B mRNA levels.
Figure 4. Expression of mutant p53 in cancer cells leads to A3B upregulation.
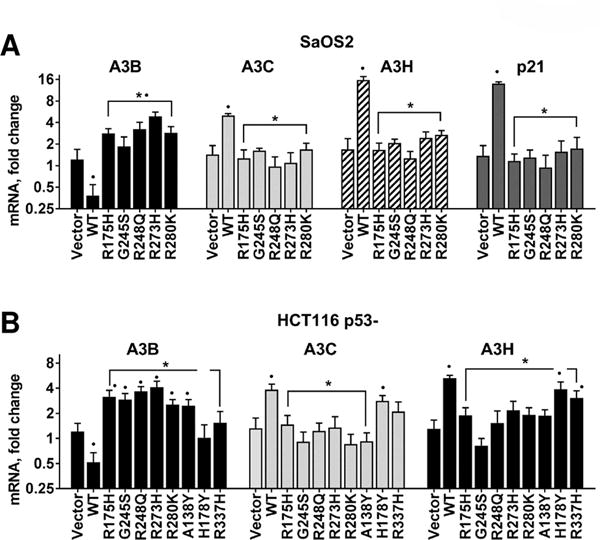
(A) p53 null SaOS2 and (B) HCT116 cells were transiently transfected with empty vector or p53 expression plasmids for WT or tumor-associated mutants. After 24h, mRNA levels for A3B were quantified by RT-qPCR. Expression of A3C, A3H and p21 were used as internal controls for genes upregulated by WT but not mutant p53. Presented are mRNA fold-changes relative to untransfected cells. For presentation purposes the Y-axis corresponding to mRNA fold-changes is presented in a log2 scale format. “*” indicates p<0.05 compared to expression induced by WT p53 transfection. “•” denotes p<0.05 compared to cells transfected with empty vector.
p53 modulates interferon induced expression of A3 genes in cancer cells
Consistent with its antiviral roles, several A3 genes are upregulated by various interferons (IFNs) (18,19). We, therefore, assessed if p53 can influence the expression of A3 genes in response to IFN type-I (IFN-I) in various cancer cell lines that differed in TP53 status.
As shown in the heat map of Fig. 5A, all A3 genes were modestly upregulated after 24h of IFN-I treatment in most cell lines regardless of p53 functional status, except for the substantial increase in A3G. To address whether p53 directly affects IFN-I mediated gene expression of A3 genes, U2OS cells with p53shRNAi or scramble shRNAi were treated with IFN-I. The IFN-I treatment stimulated the expression of all A3 genes (Fig. 5B), except A3B and A3C (Supplemental Fig. S5A) in a time dependent manner in U2OS scramble cells. Consistent with previous findings (27) there was a modest ~3-fold increase inTP53 mRNA levels after 24h of IFN-I treatment (Fig.5) in the scramble cells. While the absence of p53 due to p53shRNAi does not seem to have any impact on the early upregulation of the A3 genes by IFN-I challenge (1 to 6h), the presence of p53 was clearly required to maintain the upregulation of the A3A, -D, -F, -H and -G genes at 24h (Fig. 5B). Correspondingly, the protein levels of total p53 and phosphorylated Ser15 p53, a marker for activation, were increased (Fig. 5C) at 24 hr.
Figure 5. p53 modulates IFN induced expression of A3 genes.
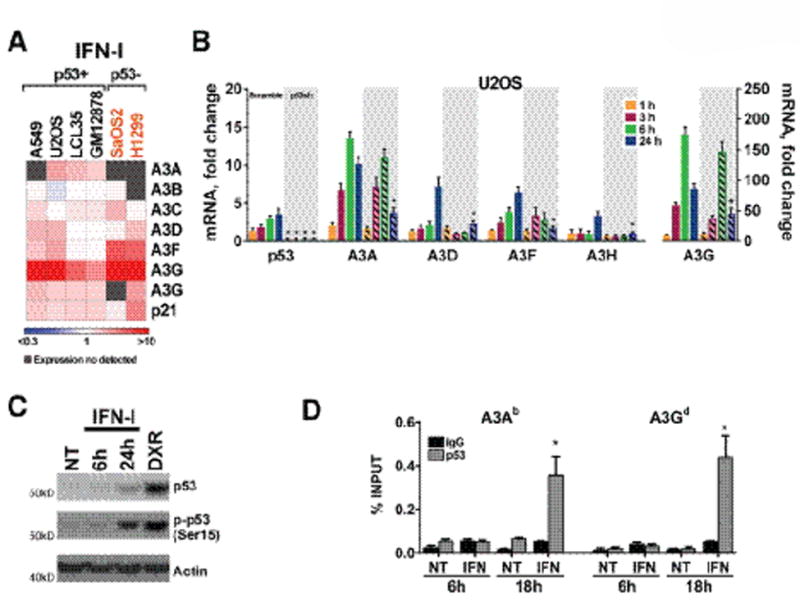
(A) IFN-I induced expression of A3 genes in human cancer cell lines with different p53 functional status. The heat map shows changes in A3 mRNA levels determined by qPCR after IFN-I treatment (500U, 24h). Fold-change values compared to untreated cells (value of 1) and statistical analyses are available in Supplemental Table S3. (B) TP53 depletion influences IFN induced responses of the A3 gene family expression. Expression of A3 genes, presented as fold-change relative to untreated parental cells, was evaluated in parental, scramble and TP53 shRNA U2OS cells after being challenged with 500U IFN-I during the indicated times. (C). Immunoblot of total p53 and phosphorylated Ser15 p53 used as activation maker in U2OS cells treated with IFN-I (500U) for 6 or 24h. The lysate from DXR-treated U2OS was used as positive control. Actin was used as loading control. (D) p53 occupancy for A3A and A3G transcriptional regulatory regions in U2OS cells treated for 6 or 18h with 500U of IFN-I. The A3 gene superscripts correspond to p53RE sequences described in Supplemental Table S1. p53 binding is expressed as % of total input DNA. IgG was a negative control. “*” indicates p<0.05 when compared to parental no treatment (“NT”) samples.
Similarly, p53 occupancy at the p53REs for A3A and A3G was greatly increased only at 18 h of IFN-I treatment (Fig. 5D), suggesting that activated p53 is needed for IFN-I- regulation of A3 genes at late times. The impact of p53 on IFN-I induced expression and p53 occupancy of several A3 genes was also observed in A549 cells (Supplemental Figs. S5B–C). Unlike for U2OS cells, the expression of the A3B gene in the A549 cells was initially upregulated after IFN-I at early time points (3 and 6h) when p53 was not bound in the A3B transcriptional regulatory region. Notably, A3B mRNA levels were returned to basal levels after 24h while p53 occupancy increased overall, consistent with a p53 repressive activity on this gene.
The role of p53 in modulating IFN-I induced expression of A3 genes was further confirmed in SaOS2 p53- cells transiently transfected with WT p53. Restoration of WT p53 enhanced the IFN-I induced gene expression of most of the A3 genes except for A3C (Supplemental Fig. S5D). There is a difference with overexpressed p53 compared with non-transfected cells, in that IFN-I treatment in parental cells failed to induce the A3A and A3H genes over the undetectable basal levels while in p53 transfected cells the mRNA levels of both genes along with other A3 genes were synergistically increased, except for A3B. Consistent with our other observations, A3B was repressed after p53 expression. Although IFN-I had no effect on its own, the IFN-I abolished the p53 induced repression of A3B, returning its expression to basal levels. Collectively, our results demonstrate a role for p53 in IFN mediated expression of A3 gene family members.
Discussion
The A3 cytosine deaminases are key innate immune effector proteins providing defense against a range of viruses and retro-elements through their ability to mutagenize viral DNA, restrict viral replication and silence retrotransposition (28,29). Since the A3 proteins can deaminate cellular DNA and contribute to genomic instability, a fine-tuned control of expression of the A3 genes and enzymatic activities is needed to avoid collateral damage to the host genome. However, little was known about the transcriptional control of the A3 genes at basal or stress conditions beyond regulation by type-I IFN stimulation, estrogen and recently by replication stress (18,19,30,31). Here, we show that p53 can be a major transcriptional regulator of all A3 genes in response to chromosomal stress. Consistent with these observations, depletion of p53, inhibition by pifithrin-α and expression of loss-of-function tumor-associated p53 mutants alters A3 expression. Also, all A3 genes had p53 binding near transcription start sites, many of which were bound by activated. We observed that although p53 binding to these novel p53REs was relatively lower when compared to the binding of p53 to the p21 target gene, the binding of p53 in the A3 regulatory regions was consistent in two cell lines observed, not only following activation with classical the p53 activator drugs DXR and Nutlin but also after IFN-I treatment. Furthermore, we found that numerous CHIP-seq datasets across cell lines, exposures and tissue types provide further evidence for direct regulation of the A3 family by p53. Although we attempted to relate mRNA to protein changes with commercially available antibodies, results for A3A proteins were inconsistent, presumably due to the polyclonal nature of the antibodies used and/or multiple non-specific signals. However, a strong correlation has been reported for A3F and A3G mRNA and protein expression using noncommercial antibodies (19).
We also found that p53 can influence expression of the A3 gene family in response to IFN immune challenges, suggesting p53 may act as a central mediator of global innate immune responses. IFNs are the major cytokines produced by the innate immune system in response to viral infections, and several viral infections trigger p53 (32). Included in the IFN-stimulated group of genes (33) are TP53 (27) and A3 genes. (18,19,27). p53 was found to only influence late IFN-mediated transcriptional responses of the A3 genes. We suggest that p53 can enhance the antiviral arm of the innate immune response via the upregulation of A3 genes as well in consort with the antiviral response itself.
Based on our observations, we propose that activated p53 can serve as an integrator of DNA damage and immune responses. This is consistent with the recent report in which drug-induced replication stress can promote A3 activation (31). The role of p53 was not assessed in this work; however, replication stress typically involves p53 activation (31). We liken the action of p53 to a two-edged sword that has the potential to both increase genome instability through actions on viruses and retrotransposons or to decrease genome stability through mutagenesis of chromosomal DNA by the A3 deaminase activity. Through increased expression of A3 genes, p53 might guard against retrotransposition of large repeats that are active in the genome (34–37). Wylie et al. (37) demonstrated that WT p53 through its interaction with components of the piRNA (piwi-interacting RNA) pathway, suppresses transposon mobility in normal cells while mutant p53 in cancer cells could not, resulting in the activation of LINE mobility in cancer cells. This is consistent with depletion of A3C leading to greater LINE retrotransposition activity in a cancer cell line (38).
Several studies have shown a correlation between aberrant expression of A3B mRNA in multiple tumors and a specific mutation signature (15,17,39). We have established that WT p53 represses A3B expression and that p53 hotspot tumor-associated mutants can lead to upregulation. These p53 hotspot mutants have lost the wild type ability to interact with p53 RE sequences, opening the possibility that upregulation of A3B induced by p53 mutants could be through gain-of-function activities that enhance transactivation activities of other transcription factors such as NF-κB (40).Consistent with this hypothesis, Maruyama and collaborators recently found that the classical NF-κB pathway is responsible for activation of A3B mRNA in cancer cells (41).
A recent study found that A3B mRNA expression and enzymatic activity were upregulated following transfection of a high-risk HPV genome. This effect was abrogated by inactivation of viral E6, a protein that causes degradation of p53 (42). Notably, in breast tumor samples and derived cancer cell lines A3B upregulation correlates with inactivation of TP53, strongly suggesting that p53 loss and increased A3B could be a tumor-initiating event allowing cells to bypass DNA damage checkpoints triggered by A3B (15).
Recently, the A3A protein was found to be a mutator in human cancers (16,17) since there is a stronger A3A mutagenesis signature than that associated with A3B (16). Furthermore, A3A itself can generate DNA breaks and activate the DNA Damage Response (DDR) (43,44) for which p53 is a major downstream regulator and effector (45). In our study, both WT and p53 mutant could upregulate A3A in response to DNA damage. Moreover, since chronic inflammation is commonly related to cancer and since A3A expression correlates with inflammatory environments (such as INF-I), A3A-induced damage may contribute to somatic mutation selection in cancers (46). Both A3A and A3B are induced in bladder and breast cancer cells after bleomycin treatment (47). Consistent with our results, the induction of A3B is more robust in cell lines harboring mutant p53 than those with WT p53 (47).
Among the seven A3 genes, both A3C and A3H were consistently upregulated in a p53 dependent manner. However, beyond their immune sentinel functions (19), there is little information about potential mutagenicity towards host genomes. Elevated expression of A3C along with lowered expression of A3B in breast cancer patients correlate with improved clinical outcome (48), consistent with our results where p53 activation promotes expression of A3C and represses expression of A3B. A role for A3H in A3 cancer-associated mutagenesis has been indicated by an A3H-I haplotype and associated A3 mutation pattern in breast tumors lacking A3B expression (49).
Thus, it appears that p53 regulation of the A3 genes can have both antitumor and pro-tumor consequences. Our results suggest an A3/p53 immune axis where activation of WT p53 in normal cells in response to internally and externally induced lesions (including replication collapse) could also influence A3 expression (particularly A3A and A3B) and subsequent appearance of mutations that have been identified in cancers.
We propose that after cancer establishment, the p53+ or p53- status along with cancer therapy may influence the potential for subsequent A3A and A3B mutagenesis as part of tumor evolution and metastasis. Most cancer therapies involve agents such as ionizing radiation, DXR and etoposide that induce WT p53. As described, mutant and WT p53 proteins can strongly influence expression of A3A and A3B as well as other A3 genes. Even the lack of p53 can lead to upregulation of A3B. Given that over 50% of cancers (some >90%) have altered p53, we suggest that specific p53 mutation status should be considered in treatments. Along this line, it would be interesting to address p53 status and mutagenesis patterns in secondary tumors as well as cancers that arise in association with Li Fraumeni disease (50), which is due to a germinal defect in p53.
Overall, our results provide molecular insights into p53 direct control of immune-surveillance under stress conditions, integrating p53 tumor suppressor activities with innate immune stimuli. Further studies are needed to establish the functional impact of A3 transcriptional regulation by p53. For example, under conditions of viral infection how does the p53-A3 axis impact the host immune function and the likelihood of mutation in the host and the pathogen. The integration of immune response and DDR may have implications for understanding therapeutic approaches. Understanding the relationship could provide novel ways to incorporate cell-extrinsic and cell-intrinsic defenses against tumorigenesis by pharmacological activation of p53, thereby achieving more effective therapies against cancer and other diseases.
Supplementary Material
Implications.
activated p53 can integrate chromosomal stresses and immune responses through its influence on expression of APOBEC3 genes, which are key components of the innate immune system that also influence genomic stability.
Acknowledgments
We thank the following NIEHS core facilities: Molecular Genomics, Viral Vector, Flow Cytometry and Clinical Research Unit. We thank Drs. Carl Anderson, Stavros Garantziotis, Dmitry Gordenin and Shepherd Schurman for critical reviewing and comments. This work was supported by Intramural Research Program of NIH (NIEHS Z01-ES065079 [to MAR]).
Footnotes
The authors declare no potential conflicts of interest
References
- 1.Aubrey BJ, Strasser A, Kelly GL. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb Perspect Med. 2016;6(5) doi: 10.1101/cshperspect.a026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen MA, Andrysik Z, Dengler VL, Mellert HS, Guarnieri A, Freeman JA, et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife. 2014;3:e02200. doi: 10.7554/eLife.02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menendez D, Nguyen TA, Freudenberg JM, Mathew VJ, Anderson CW, Jothi R, et al. Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 2013;41(15):7286–301. doi: 10.1093/nar/gkt504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359–70. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127(7):1323–34. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–5. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 7.Munoz-Fontela C, Macip S, Martinez-Sobrido L, Brown L, Ashour J, Garcia-Sastre A, et al. Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med. 2008;205(8):1929–38. doi: 10.1084/jem.20080383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menendez D, Shatz M, Azzam K, Garantziotis S, Fessler MB, Resnick MA. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 2011;7(3):e1001360. doi: 10.1371/journal.pgen.1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shatz M, Menendez D, Resnick MA. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. 2012;72(16):3948–57. doi: 10.1158/0008-5472.CAN-11-4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menendez D, Lowe JM, Snipe J, Resnick MA. Ligand dependent restoration of human TLR3 signaling and death in 53 mutant cells. Oncotarget. 2016 doi: 10.18632/oncotarget.11210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shatz M, Shats I, Menendez D, Resnick MA. p53 amplifies Toll-like receptor 5 response in human primary and cancer cells through interaction with multiple signal transduction pathways. Oncotarget. 2015;6(19):16963–80. doi: 10.18632/oncotarget.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, et al. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79(3):285–96. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- 13.Harris RS, Dudley JP. APOBECs and virus restriction. Virology. 2015;479–480:131–45. doi: 10.1016/j.virol.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stavrou S, Ross SR. Journal of immunology. 10. Vol. 195. Baltimore, Md: 2015. APOBEC3 Proteins in Viral Immunity; pp. 4565–70. 1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45(9):977–83. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, Malc EP, et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet. 2015;47(9):1067–72. doi: 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45(9):970–6. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol. 2009;83(18):9474–85. doi: 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010;38(13):4274–84. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol Biol Evol. 2005;22(2):367–77. doi: 10.1093/molbev/msi026. [DOI] [PubMed] [Google Scholar]
- 21.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285(5434):1733–7. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 22.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296(5567):550–3. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 23.Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9(10):724–37. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 24.Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, et al. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 2008;36(11):3639–54. doi: 10.1093/nar/gkn232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124(1):207–19. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 26.Menendez D, Inga A, Resnick MA. Estrogen receptor acting in cis enhances WT and mutant p53 transactivation at canonical and noncanonical p53 target sequences. Proc Natl Acad Sci U S A. 2010;107(4):1500–5. doi: 10.1073/pnas.0909129107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424(6948):516–23. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- 28.Kinomoto M, Kanno T, Shimura M, Ishizaka Y, Kojima A, Kurata T, et al. All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 2007;35(9):2955–64. doi: 10.1093/nar/gkm181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koito A, Ikeda T. Intrinsic restriction activity by AID/APOBEC family of enzymes against the mobility of retroelements. Mob Genet Elements. 2011;1(3):197–202. doi: 10.4161/mge.1.3.17430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates AID transcription and function. J Exp Med. 2009;206(1):99–111. doi: 10.1084/jem.20080521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanu N, Cerone MA, Goh G, Zalmas LP, Bartkova J, Dietzen M, et al. DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer. Genome Biol. 2016;17(1):185. doi: 10.1186/s13059-016-1042-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato Y, Tsurumi T. Genome guardian p53 and viral infections. Rev Med Virol. 2013;23(4):213–20. doi: 10.1002/rmv.1738. [DOI] [PubMed] [Google Scholar]
- 33.Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1(6):519–25. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, et al. LINE-1 retrotransposition activity in human genomes. Cell. 2010;141(7):1159–70. doi: 10.1016/j.cell.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levine AJ, Ting DT, Greenbaum BD. P53 and the defenses against genome instability caused by transposons and repetitive elements. Bioessays. 2016 doi: 10.1002/bies.201600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, et al. Human l1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110(3):327–38. doi: 10.1016/s0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- 37.Wylie A, Jones AE, D’Brot A, Lu WJ, Kurtz P, Moran JV, et al. p53 genes function to restrain mobile elements. Genes Dev. 2016;30(1):64–77. doi: 10.1101/gad.266098.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muckenfuss H, Kaiser JK, Krebil E, Battenberg M, Schwer C, Cichutek K, et al. Sp1 and Sp3 regulate basal transcription of the human APOBEC3G gene. Nucleic Acids Res. 2007;35(11):3784–96. doi: 10.1093/nar/gkm340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, et al. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013;73(24):7222–31. doi: 10.1158/0008-5472.CAN-13-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25(3):304–17. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maruyama W, Shirakawa K, Matsui H, Matsumoto T, Yamazaki H, Sarca AD, et al. Classical NF-kappaB pathway is responsible for APOBEC3B expression in cancer cells. Biochem Biophys Res Commun. 2016 doi: 10.1016/j.bbrc.2016.08.148. [DOI] [PubMed] [Google Scholar]
- 42.Vieira VC, Leonard B, White EA, Starrett GJ, Temiz NA, Lorenz LD, et al. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio. 2014;5(6) doi: 10.1128/mBio.02234-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011;12(5):444–50. doi: 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Schmitt K, Guo K, Santiago ML, Stephens EB. Role of the single deaminase domain APOBEC3A in virus restriction, retrotransposition, DNA damage and cancer. The Journal of general virology. 2016;97(1):1–17. doi: 10.1099/jgv.0.000320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nik-Zainal S, Wedge DC, Alexandrov LB, Petljak M, Butler AP, Bolli N, et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat Genet. 2014;46(5):487–91. doi: 10.1038/ng.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Middlebrooks CD, Banday AR, Matsuda K, Udquim KI, Onabajo OO, Paquin A, et al. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat Genet. 2016 doi: 10.1038/ng.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Delahanty R, Guo X, Zheng W, Long J. Integrative genomic analysis reveals functional diversification of APOBEC gene family in breast cancer. Hum Genomics. 2015;9:34. doi: 10.1186/s40246-015-0056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Starrett GJ, Luengas EM, McCann JL, Ebrahimi D, Temiz NA, Love RP, et al. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat Commun. 2016;7:12918. doi: 10.1038/ncomms12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol. 2015;33(21):2345–52. doi: 10.1200/JCO.2014.59.5728. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.