

Abstract
Objective:
The aim of this study was to observe the counter-effect of carbon monoxide-releasing molecule-2 (CORM-2) on lipopolysaccharide (LPS)-suppressed thrombomodulin (TM) and endothelial protein C receptor (EPCR) expressions from human umbilical vein endothelial cell (HUVEC), and to reveal its mechanisms.
Methods:
HUVECs were divided into 5 treatment groups, wherein reagents were added simultaneously. TM and EPCR proteins of the cells and the culture medium levels of soluble TM, soluble EPCR, and matrix metalloproteinase-2 (MMP-2) were detected after administration, whereas mRNA levels of TM and EPCR, as well as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activity among groups, were also evaluated.
Results:
No significant difference was observed in any indicator between CORM-2 and sham groups. Addition of LPS produced drastic increase in MMP-2 expression, NF-κB activity, shedding of TM and EPCR (into the culture medium), as well as remarkable decrease in both mRNA and protein expressions of TM and EPCR, and cell viability. LPS + CORM-2 treatment significantly reduced the increase in MMP-2, NF-κB activity, and TM/EPCR shedding, whereas maintained both mRNA and protein levels of TM and EPCR, and preserved cell viability.
Conclusions:
CORM-2 protects HUVEC from LPS-induced injury, by way of suppressing NF-κB activity, which downregulates TM and EPCR mRNAs. It also decreases MMP-2 expression and prevents the shedding of TM and EPCR from the surface of endothelial cells, so as to preserve their protective effect.
Keywords: CORM-2, endothelial cell, endotoxin, EPCR, thrombomodulin
1. INTRODUCTION
Inflammatory reaction and coagulation disorder are 2 major pathological processes during sepsis, which are interconnected and mutually promotive with vascular endothelial cells acting as a bridge in between.[1] Studies show that protection of endothelial cells considerably improves the prognosis of sepsis. Thrombomodulin (TM) and endothelial protein C receptor (EPCR), both transmembrane protein receptors widely expressed on the surface of vascular endothelial cells, are major components in the protein-C system (PC) and important regulators for its activation. They not only improve coagulation status, but also inhibit inflammatory reaction.[2] It is believed that during sepsis, both proteins and mRNA are downregulated as the result of NF-κB activation by proinflammatory factors like lipopolysaccharides (LPS), tumor necrosis factor-α (TNF-α), and so on. Song et al and Sohn et al[3,4] found that activated NF-κB migrates into the nucleus and downregulates the gene expression of TM and EPCR, whereas suppression of NF-κB activity significantly increases their expression. However, under the influence of LPS and cytokines, the secretion and activation of matrix metalloproteinases (MMPs) are increased, which directly injure the endothelial cells by hydrolyzing their surface receptors and proteins, including TM and EPCR.[5] In fact, studies have demonstrated close relationship between increased MMP activity and the shedding of TM and EPCR.[6]
Carbon monoxide (CO) is a small gaseous molecule with anti-inflammatory and antimicrobial properties that is able to penetrate cell membranes.[7] Metal carbonyl compounds was commonly known as CO-releasing molecules or CORMs. It mainly generated from the decomposition of heme by heme oxygenase (HO) in the body. It is well established by many studies that both endogenous and exogenous CO suppresses inflammation, protects endothelial cells, and improves microcirculation during sepsis, but the mechanism of its function is not clearly defined.[8] The released CO in a controlled manner has been developed for therapeutic applications.[9]
However, the role of CORM-released CO in regulation of the systemic inflammation during sepsis has not been investigated yet. Therefore, in this study, we employed tricarbonyldichlororuthenium (II) dimer (carbon monoxide releasing molecule-2, CORM-2), one of the novel CO donors, to assess the effects and potential mechanisms of CORM-released CO in modulation of EPCR and TM. We established an in vitro cell model for sepsis, which induced an injury of human umbilical vein endothelial cells (HUVECs) stimulated with LPS. CORM-2 was used as the source of exogenous CO in this cell model of sepsis, and the effect of CORM-2 on the expression status of TM and EPCR was examined.
2. MATERIALS AND METHODS
2.1. Drugs and Reagent
CORM-2 and LPS (Escherichia coli 0128:B12) were purchased from Sigma (USA); the enzyme-linked immunosorbent assay (ELISA) kits for TM, EPCR, MMP-2 from Bluegene Biology Company (China); primary antibodies to TM and EPCR from Abcam (USA); RNA Kits from Takara (Japan); Cell-Counting Kit-8 (CCK-8) kits from Biyuntian Biology Company (China); electrophoretic mobility shift assay (EMSA) kits from Pierce (USA); cell culture medium, RPMI-1640 and fetal bovine serum (FBS) both from Gibco (USA). Umbilical cords were obtained from the Department of Obstetrics, The First Affiliated Hospital of Harbin Medical University. All procedures were approved in advance by the ethical committee of The First Affiliated Hospital of Harbin Medical University with proper patient consent on file.
2.2. HUVEC Culture
The umbilical cords were perfused and digested using a digestive solution containing 0.125% pancreatin, 0.01% ethtylediaminetetraacetic acid (EDTA). Isolated HUVECs were cultured in regular cell incubator under 5% CO2 at 37°C. When the adhering cells reached confluence, passage by trypsin digestion was performed. After 3 to 5 passages, a monolayer of polygonal cells with the “cobble stone” appearance was formed and cells were identified as endothelial cells by immunohistochemical staining for factor VIII-associated antigen. Cells with a viability >95% by trypan blue were seeded into 6-well plates at a density of 105 cells/mL and divided into 5 groups: control (negative control), LPS (positive control), CORM-2, CORM-2 + LPS, and inactive carbon monoxide-releasing molecule-2 (iCORM-2) + LPS. The control group received only equal volume of dimethyl sulfoxide (DMSO), whereas other groups received LPS (10 μg/mL), CORM-2 (100 μmol/L), iCORM-2 (100 μmol/L), which resolved in DMSO, or their combinations accordingly. Supernatant was collected at 8, 16, and 24 hours for the detection of respective targets.
2.3. CCK-8 assay
Repeat all experiment groups on 96-well plates with same cell density relating to the culture area. Each group occupied 18 wells, 6 for each time point, and duplicates were made to secure accuracy. The culture medium was replaced after 24-hour incubation, and respective concentrations of CORM-2, iCORM-2, and LPS were added accordingly. At 8, 16, and 24 hours, medium in assigned wells was changed into 100 μL medium without serum, and 10 μl CCK-8 reagent was added. Cells were incubated for another 2 hours, before light absorbance (A value) was determined under 450-nm wavelength while using 655 nm as a reference.[10]
2.4. ELISA
Medium on the 6-well plates was sampled at 8, 16, and 24 hours, and soluble thrombomodulin (sTM), soluble endothelial protein C receptor (sEPCR), and MMP-2 protein concentrations were determinate by ELISA assay following the instructions of the manufacturer.
2.5. Western Blot (WB)
Cells were collected by trypsinization 8 hours after treatment and lysed by established method. Protein concentration was estimated by Coomassie blue, and 40 μg protein from each well was loaded onto 10% SDS-Polyacrylamide Gel (Biyuntian Biology Company, China) for electrophoresis under 80 V for 2 hours, and then transferred onto nitrocellulose membranes under 120 V for 3 hours. Membrane was then blocked with 5% skimmed milk at 4°C overnight, before primary rabbit anti-human monoclonal antibodies (1:1000) were added and incubated at room temperature for 2 hours. Membranes were then washed and incubated with horseradish peroxidase-conjugated secondary antibodies for another 1 hour (Goat anti-rabbit 1:2000; Zhongshan Jinqiao, Beijing, China), before developed under ultraviolet light and analyzed by an image-analyzing system.
2.6. Reverse transcription polymerase chain reaction (RT-PCR)
Cells were collected by trypsinization 8 hours after treatment, and RT-PCR was performed following manufacturer instructions. Total RNA was extracted, quantified by spectrophotometer, and reverse-transcribed into cDNA using following primers:
TM:
ss-5’-CGAGTGCCACTGCATCCCATA-3’,
as-5’-GCAGATGAAACCGTCGTCC-3’;
EPCR:
ss-5’-CAGGTGGACGGCGATGTT-3’
as-5’-CTTCCTGCCCCAACTCCATTATC-3’;
GAPDH:
ss-5’-CGCTGAGTACGTGGAG-3’,
as-5’-GAGGAGTGGGTGTCGCTGTT-3’.
PCR protocol for TM was: melting (95°C, 3 minutes), then 30 cycles of denaturation (94°C, 30 seconds), annealing (58°C, 30 seconds), and polymerization (72°C, 30 seconds), ending with an additional extension (72°C, 5 minutes). EPCR and GAPDH were annealed at 54.2°C and 60°C, respectively. PCR product was stained with ethidium promide, electrophoresed on 2% agar gel, and sized against the standard marker. The relative richness of the mRNA was calculated from its ReinhoitZahl value against internal control using a gel image analyzing system.
2.7. EMSA analysis of NF-κB activity
EMSA was performed using nonradioactive NF-κB EMSA kit (Viagene Biotech) and following the manufacturer's instructions and the reference.[11] And the sequence of NF-κB probe was 5‘-AGTTGAGGGGACTTTCC CAGGC-3‘ and the mutant probe was 5‘-AGTTGAGGCTACTTTCCCAGGC-3‘. Ten micrograms of crude nuclear protein was incubated for 20 minutes at room temperature in 15-μl binding reaction systems, which include: 1.5 μL of 10× binding buffer, ddH2O, and 1.5 μL poly (dI-dC) (1.0 μg/μL) to a final volume of 10 μL. Then 1 μL Bio-NF-κB probe or Bio-mutant-NF-κB probe (500fM) was added, and the reaction was incubated for 40 minutes at room temperature. Electrophoresis was carried out on a 6.5% nondenaturing polyacrylamide gel at 175 V in 0.25× Tris-borate-EDTA (TBE) (1× TBE is 89 mmol/L Tris-HCl, 89 mmol/L boric acid, and 5 mmol/L EDTA, pH 8.0) at 4°C for 1 hour. Gels were then transferred to the banding membrane at 394 mA in 0.5× TBE (1× TBE is 89 mmol/L Tris-HCl, 89 mmol/L boric acid, and 5 mmol/L EDTA, pH 8.0) at room temperature for 40 minutes. Membrane was then cross-linked under UV light for 10 minutes (Immobilization), blocked and labeled with Streptavidin-HRP. After washing and equilibration, pictures of the membrane were obtained utilizing an imaging apparatus (Alpha flurechemical).
2.8. Statistical analysis
Data are presented as mean ± standard deviation. The comparisons were conducted by analysis of variance. Multiple comparisons of mean were performed using the Bonfferoni procedure with type-I error adjustment and was used for evaluating statistical significance (SPSS 17.0, Chicago, IL). A value of P < .05 was considered significant.
3. Results
3.1. CORM-2 significantly promoted the cell viability of HUVEV stimulated with LPS
To examine the effects of CORM-2 in sepsis, we at first investigated that CORM-2 regulated cell viability of HUVEV after stimulated with LPS. The results indicated that there was a significant decrease (96.06% vs. 52.75%) (n = 3, P < .05) of cell viability of HUVEV treated by LPS compared with control group for 16 hours after LPS administration. When we treated HUVEV with LPS + CORM-2, and CORM-2 partially protects HUVEV from cell death induced by LPS, LPS + CORM-2 group has higher cell viability (64.77% vs. 52.75%) than the group treated with only LPS (Fig. 1) for 16 hours after LPS administration. CORM-2 was inhibited by iCORM in the LPS+CORM-2 group, the cell viability of HUVEV was significantly decreased in the LPS+iCORM-2 group compared to the LPS+CORM-2 group (54.33% vs.64.77%) (n = 3, P < .05). The LPS+ iCORM-2 group is similar as LPS group, and became worse with a prolonged cultivation. However, no obvious difference was found between control and CORM-2 groups. Those results indicate a protective effect of CORM-2 on the cell viability of LPS-induced HUVEC (Fig. 1).
Figure 1.

Cell viability determined by CCK-8 assay. LPS decreased cell viability in all groups compared with sham group. LPS, LPS + CORM, and LPS + iCORM groups had the most severe decrease. LPS + CORM group produced a lower cell viability than sham group (P < .05), but remained significantly higher than LPS and iCORM groups (P < .05). ∗P < .05, compared to sham group; +P < .05, compared to LPS group; #P < .05, compared to CORM group; &P < .05, compared to LPS + CORM group. CCK-8 = cell counting kit-8, CORM = carbon monoxide-releasing molecule, iCORM = inactive carbon monoxide-releasing molecule, LPS = lipopolysaccharides.
3.2. CORM-2 dramatically decreased sEPCR, sTM, and MMP-2 expression of HUVEC induced by LPS
To further confirm the protection of CORM-2 in HUVEV after stimulated with LPS, we examined the regulation of CORM-2 in sEPCR, sTM, and MMP-2 expression of HUVEC induced by LPS. The results showed that no obvious difference was observed in sEPCR, sTM, and MMP-2 expression levels between control and CORM-2 groups; however, LPS significantly increased sEPCR, sTM, and MMP-2 expression (Fig. 2A-C) in HUVEV cells (control vs. LPS: sEPCR, 25.31 vs. 103.6 ng/mL; sTM, 1.29 vs. 7.81 ng/mL; MMP-2, 4.73 vs. 44.14 ng/mL) (n = 3 P < .05) for 16 hours after LPS administration. When CORM-2 was added, sEPCR, sTM, and MMP-2 expression levels in HUVEV induced by LPS was decreased compared with LPS group (LPS group vs. LPS + CORM-2: sEPCR, 103.6 vs. 64.77 ng/mL; sTM, 7.81 vs. 3.17 ng/mL; MMP-2, 44.14 vs. 19.41 ng/mL (n = 3, P < .05), which suggested that CORM-2 inhibited abilities of LPS-inducing sEPCR, sTM, and MMP-2 expression levels in HUVEV cells. More importantly, CORM-2 inhibitor significantly dismissed the inhibition of CORM-2 to LPS-inducing sEPCR, sTM and MMP-2 expression in HUVEV. Interestingly, sEPCR, sTM, and MMP-2 expression of HUVEV was gradually increased with prolonged LPS induction. LPS and iCORM groups had similar extent of increase with no statistical difference in between, whereas LPS + CORM-2 group maintained the lowest increase among groups. These data indicate sEPCR, sTM, and MMP-2 exist at low levels in normal cell culture supernatant. When LPS is added, the secretion of MMP-2 was increased with sEPCR, sTM expression levels. However, when CORM-2 is present in LPS + CORM-2 group, the secretion of MMP-2 is depressed with a simultaneously quenched increase in sEPCR, sTM levels, which indicate that CORM-2 reduces shedding of sEPCR and sTM by means of suppressing MMP-2 secretion in the process of LPS induction of HUVEC.
Figure 2.

(A–C) sEPCR (A), sTM (B), MMP-2 (C) levels in cell culture medium. Compared with sham group, after addition of LPS, sEPCR, sTM, MMP-2 levels significantly increased in all other groups (P < .05), among which LPS and LPS + iCORM groups produced highest increase, whereas their increase in LPS + CORM group was significantly suppressed (P < .05). CORM and SHAM groups had no significant difference. ∗P < .05, compared to sham group; +P < .05, compared to LPS group; #P < .05, compared to CORM group; &P < 0.05, compared to LPS + CORM group. CORM = carbon monoxide releasing molecule, iCORM = inactive carbon monoxide releasing molecule, LPS = lipopolysaccharides, MMP-2 = matrix metalloproteinase-2, sEPCR = soluble endothelial protein C receptor, sTM = soluble thrombomodulin.
3.3. CORM-2 maintained the expression of EPCR and TM in HUVEC after stimulated with LPS
We also examine whether CORM-2 regulates EPCR and TM mRNA and protein expression by qPCR and western blot. The results indicated that no difference was observed in EPCR and TM mRNA and protein expression between control and CORM-2 groups, but significant decrease in LPS, LPS + CORM-2, and LPS + iCORM-2 groups was observed (n = 3, P < .05).However, LPS and LPS plus iCORM-2 groups generated more significant decrease. There is no difference between LPS and LPS + iCORM-2 groups. LPS + CORM-2 group maintained much higher EPCR and TM mRNA and protein expression (n = 3, P < .05) (Fig. 3A-D). We also investigated the expression level of TM in the cell model. The results showed that no difference was observed in mRNA and protein expression of TM between control and CORM-2 groups, but significant decrease in all other groups was observed (n = 3, P < .05), among which LPS and iCORM-2 groups generated the most severe decrease but with no statistical difference in between. Compared with LPS group, LPS + CORM-2 group maintained much higher EPCR and TM protein expression (n = 3, P < .05) (Fig. 4A-D). Those results suggested that CORM-2 significantly maintained EPCR and TM mRNA and protein expression in HUVEV cells induced with LPS.
Figure 3.

(A–D) Epcr and EPCR protein levels were analyzed by RT-PCR and Western blot in HUVECs stimulated with LPS for 8 hours. (A) Epcr (315 bp) and Gapdh (576 bp) were analyzed by PCR; ratio of Epcr/Gapdh (Gapdh as internal control) was compared (B). Semiquantity of EPCR protein level also was examined and quantified by Western blot (C and D). 1. Sham group, 2. LPS group, 3. CORM-2 group, 4. CORM-2 + LPS group, 5. iCORM-2 + LPS group, Marker: DL2000, from bottom to top: 100, 250, 500, 750, 1000, 2000; (B,D) semiquantitative analyses of mRNA. ∗P < .05, compared to sham group; +P < .05, compared to LPS group; #P < .05, compared to CORM group; &P < .05, compared to LPS + CORM group. CORM = carbon monoxide-releasing molecule, EPCR = endothelial protein C receptor, HUVECs = human umbilical vein endothelial cells, iCORM = inactive carbon monoxide releasing molecule, LPS = lipopolysaccharides, RT-PCR = reverse transcription polymerase chain reaction.
Figure 4.

(A–D) Tm and TM protein levels were analyzed quantified by RT-PCR and Western blot in HUVECs stimulated with LPS for 8 hours. (A) Tm (227 bp) and Gapdh (576 bp) were analyzed by PCR; ratio of Epcr/Gapdh (Gapdh as internal control) was compared (B). 1. Sham group, 2. LPS group, 3. CORM-2 group, 4. CORM-2 + LPS group, 5. iCORM-2 + LPS group, Marker:DL2000, from bottom to top: 100, 250, 500, 750, 1000, 2000; B,D semi-quantitative analyses of mRNA. ∗P < .05, compared to sham group; +P < .05, compared to LPS group; #P < .05, compared to CORM group; &P < .05, compared to LPS + CORM group. Evaluation of tissue TM expression in the LPS-induced and CORM-2 pretreated groups by (C) Western blotting and (D) semiquantitative analyses of Western blots. ∗P < .05, compared to sham group; +P < .05, compared to LPS group; #P < .05, compared to CORM group; &P < .05, compared to LPS + CORM group. CORM = carbon monoxide-releasing molecule, iCORM = inactive carbon monoxide releasing molecule, HUVECs = human umbilical vein endothelial cells, LPS = lipopolysaccharides, RT-PCR = reverse transcription polymerase chain reaction, TM = thrombomodulin.
3.4. CORM-2 stabilized NF-κB activity of HUVEC after stimulated with LPS
Recently, it was reported that CO liberated by CORM-2 attenuated leukocytes sequestration in the liver and lung tissues of thermally injured mice by interfering with NF-κB activation suppressing endothelial cells proadhesive phenotype.[12] Therefore, we further investigated whether CORM-2 regulates NF-κB activity. The results indicated that no difference was observed in NF-κB activity between control and CORM-2 groups, but significant increase in all other groups was observed (n = 3, P < .05), among which LPS and iCORM groups generated the most dramatic increase but with no statistical difference in between. Compared with LPS group, LPS + CORM-2 group maintained much lower NF-κB activity (n = 3, P < .05). (Fig. 5A-B).
Figure 5.
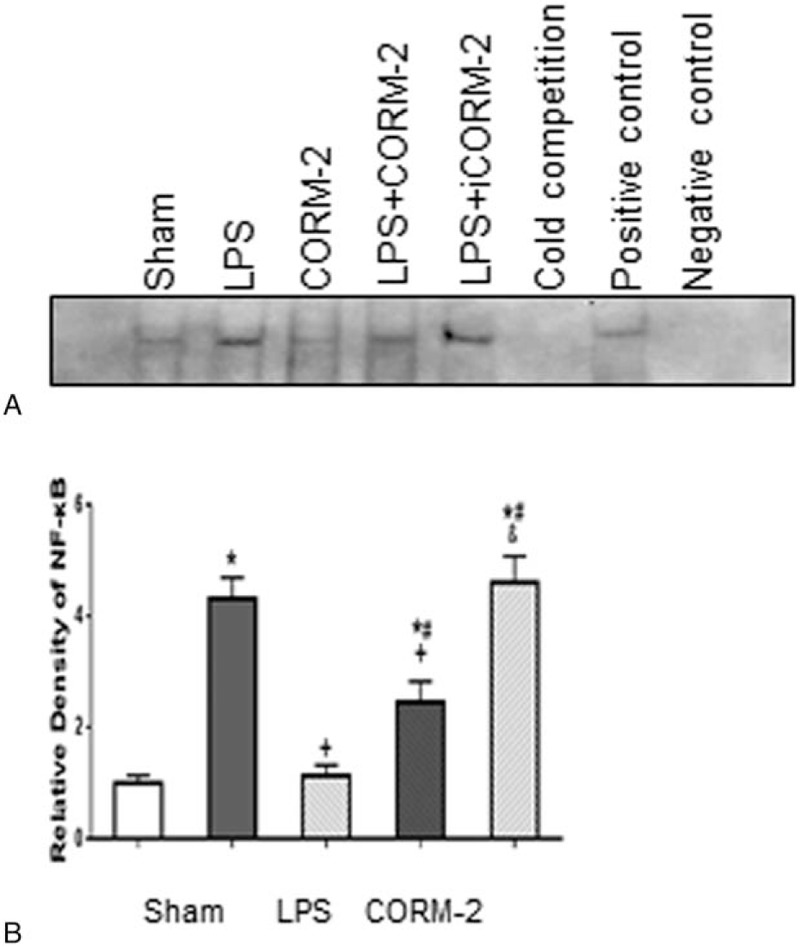
(A and B). NF-κB activity among groups 8 hours into LPS induction by EMSA. 1. Sham group, 2. LPS group, 3. CORM-2 group, 4. CORM-2 + LPS group, 5. iCORM-2 + LPS group, 6. Cold competition group, 7. Positive control, 8. Negative control. ∗P < 0.05, compared to sham group; +P < .05, compared to LPS group; #P < 0.05, compared to CORM group; &P < .05, compared to LPS + CORM group. CORM-2 = carbon monoxide-releasing molecule-2, EMSA = electrophoretic mobility shift assay, iCORM-2 = inactive carbon monoxide releasing molecule-2, LPS = lipopolysaccharides.
4. Discussion
In this study, we observed that LPS decreased the expressions of EPCR and TM by both suppressing their production and increasing their shedding on the surface of HUVEC. We demonstrated, for the first time, the quenching effect of CORM-2 upon LPS stimulation of HUVEC, which could be stemmed from inhibition of NF-κB activity.
The progressing understanding on sepsis helps us gradually realize that the culprit of this pathological process is not limited to the pathogen and its toxin per se, but also involves excessively amplified inflammatory reaction, immunological irregularity, and coagulation disorders, which maximizes into disseminated intravascular coagulation (DIC). Coagulation disorders and DIC were originally considered terminal complications of sepsis; however, recent studies have confirmed that it runs through the entire pathological process and exerts decisive effect upon the prognosis.[13]
PC is an important coagulation-regulating mechanism composed of protein-C, TM, EPCR, protein-S, and protein-C inhibitor, among which protein-C plays a critical role. Clinically, Abraham et al[14] found that administration of recombinant activated protein C to septic patients could improve the survival rate. TM and EPCR, mainly expressed on the surface of vascular endothelial cells, could accelerate PC activation by thousands of times. Sepsis injures the endothelial cells, and the productions of TM and EPCR are suppressed, but the levels of sTM and sEPCR could increase as a result of shedding from vascular cell surface, which interferes with the activation of the PC system leading to coagulation disorders. It has been reported that there was a close association between the prognosis of septic patients and their plasma TM and EPCR levels. Thrombomodulin concentration independently predicted the development of DIC, multiple organ dysfunction syndrome, and mortality during intensive care unit stay. And the plasma levels of soluble EPCR in initially nonseptic critically ill patients appear elevated in the subjects who will subsequently become septic,[15] whereas Yamakawa et al[16] found that exogenous recombinant TM could improve organ function in septic patients and decrease their mortality. Others found that the decrease of TM and EPCR was related to many factors. On one hand, those multiple factors downregulate TM and EPCR mRNAs, and reduce their synthesis. On the other hand, excessive secretion of proteinases increase shedding of TM and EPCR from endothelial cells both contribute to their reduced expression.[17,18] For example, TNF-α inhibits the transcription of TM and EPCR in endothelial cells through the NF-κB pathway, as well as promotes the secretion of matrix metalloproteinases, mainly MMP-2, which hydrolyses cellsurface membrane receptors resulting in shedding of TM and EPCR.[19] Decreased TM and EPCR on the surface of endothelial cells suppress the activation of the PC system and perturb the anticoagulation system predisposing to blood coagulation disorders, formation of microthrombosis, microcirculatory deficiency, and eventually multiorgan failure.[20] In our study, compared with control group, addition of LPS not only increased NF-κB activity and suppressed the mRNA levels of TM and EPCR in all experimental groups, but also increased MMP2 production, which as mentioned above deteriorated the shedding of TM and EPCR.
HO/CO system is an important signaling pathway in the body. Multiple in vitro and in vivo studies have revealed that it could inhibit the release of inflammatory factors, maintain vascular tension, optimize blood coagulation, and protect the endothelial cells by its antioxidant and antiapoptotic effects. It is postulated that all these protective effects are mediated by the carbon monoxide generated in the process of HO-catalyzed heme oxidation.[21] Previous studies have proved that HO/CO system regulates vascular tension through iNOS system and inhibits platelet aggregation by regulating cGMP.[22,23] In vitro and in vivo studies show that exogenous CO could downregulate the expression of tissue factor and Plasminogen activator inhibitor-1 by inhibiting the activity of NF-κB in septic mice and LPS-stimulated HUVECs.[24,25]
All in all, CO improves coagulation status and inhibits microthrombosis via multiple pathways; however, to the authors’ knowledge, the relationship between HO/CO and PC systems is still lack of investigation, which may shed a new light on the study of sepsis. Current experiment has shown that the expression of CO was increased in sepsis, and administration of HO-1 agonist or exogenous CO can inhibit the endotoxin-induced releasing of interleukin-1β and TNF-α through MAPK and other pathways.[26] It is also demonstrated by both in vitro and in vivo experiments that exogenous CO can downregulate the activity of NF-κB, in septic mice and LPS-stimulated HUVECs, as well as alleviate LPS- or hyperoxia-induced apoptosis of endothelial cells through P38 pathway.[27] Megías et al[28] observed that CORM-2 can inhibit the expression of endotoxin-induced MMPs. In our study, we also observed that after administration of LPS, the expression of NF-κB nucleoprotein in endothelial cells was increased, whereas TM and EPCR mRNA levels decreased simultaneously. On the contrary, MMP2 expression was increased, which hydrolyzed and promoted the shedding of TM and EPCR from the HUVECs. All these factors resulted in a suppressed expression of TM and EPCR in endothelial cells. Our study also revealed that CORM-2 per se had no significant effect on HUVEC, but in the LPS-stimulated HUVECs, CORM-2 not only suppressed the activation of NF-κB and maintained the mRNA levels of TM and EPCR, but also inhibited the expression of MMP2 so that reduced the shedding of EPCR and TM, as manifested by lower levels of both proteins in the cultural media. These facts indicate CORM-2 exerts protective effect upon LPS-induced HUVEC.
5. Conclusion
CO, as a gaseous signaling molecule, is a hot subject in present research. It shows significant pharmacological effects in many areas including sepsis and transplantation. Our study clearly demonstrated that CORM-2 has protective effect on LPS-stimulated HUVEC, via maintaining adequate expression of EPCR and TM in HUVECs and added now evidence to the mechanisms, by which HO/CO system improves blood coagulation status.
Footnotes
Abbreviations: aPC = activated protein C, CCK-8 = Cell Counting Kit-8, CORM-2 = carbon monoxide-releasing molecule-2, DMSO = dimethyl sulfoxide, ELISA = enzyme-linked immunosorbent assay, EMSA = electrophoretic mobility shift assay, EPCR = endothelial protein C receptor, HUVEC = human umbilical vein endothelial cell, iCORM-2 = inactive carbon monoxide-releasing molecule-2, LPS = lipopolysaccharides, MMP-2 = matrix metalloproteinase-2, PC = protein C, sEPCR = soluble endothelial protein C receptor, sTM = soluble thrombomodulin, TM = thrombomodulin, TNF-α = tumor necrosis factor-α.
This study was supported by grants from the National Natural Scientific Foundation of China (NO. 81171785), the Scientific Research Foundation of Heilongjiang Provincial Health Department (NO.2013019) and the Foundation of the First Affiliated Hospital of Harbin Medical University (NO. 2017L004).
XLM carried out cell culture and drafted the manuscript, DSF carried out EMSA analysis of NF-κB activity, MML and KK carried out the Western blot, SLY and NS carried out the ELISA assay, LJ carried out the CCK8 assay, CCN carried out the RT-PCR, SHP participated in the design of the study, LJ and YPL performed the statistical analysis, MYZ conceived of the study, and participated in its design and coordination. All authors read and approved the final manuscript.
XM and DF contributed equally to this work.
The authors report no conflicts of interest.
References
- [1].Schouten M, Wiersinga WJ, Levi M, et al. Inflammation and coagulation in sepsis. J Leukoc Biol 2008;83:536–45. [DOI] [PubMed] [Google Scholar]
- [2].Van de Wouwer M, Collen D, Conway EM. Thrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol 2004;24:1374–83. [DOI] [PubMed] [Google Scholar]
- [3].Song D, Ye X, Xu H, et al. Activation of endothelial intrinsic NF-{kappa}B pathway impairs protein C anticoagulation mechanism and promotes coagulation in endotoxemic mice. Blood 2009;114:2521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sohn RH, Deming CB, Johns DC, et al. Regulation of endothelial thrombomodulin expression by inflammatory cytokines is mediated by activation of nuclear factor-kappa B. Blood 2005;105:3910–7. [DOI] [PubMed] [Google Scholar]
- [5].Nakamura T, Ebihara I, Shimada N, et al. Modulation of plasma metalloproteinase-9 concentrations and peripheral blood monocyte mRNA levels in patients with septic shock: effect of fiber-immobilized polymyxin B treatment. Am J Med Sci 1998;316:355–60. [DOI] [PubMed] [Google Scholar]
- [6].Menschikowski M, Hagelgans A, Eisenhofer G, et al. Reducing agents induce thrombomodulin shedding in human endothelial cells. Thromb Res 2010;126:e88–93. [DOI] [PubMed] [Google Scholar]
- [7].Li L, Moore PK. An overview of the biological significance of endogenous gases: new roles for old molecules. Biochem Soc Trans 2007;35(Pt 5):1138–41. [DOI] [PubMed] [Google Scholar]
- [8].True AL, Olive M, Boehm M, et al. Heme oxygenase-1 deficiency accelerates formation of arterial thrombosis through oxidative damage to the endothelium, which is rescued by inhaled carbon monoxide. Circ Res 2007;101:893–901. [DOI] [PubMed] [Google Scholar]
- [9].Garcia-Gallego S, Bernardes GJ. Carbon-monoxide-releasing molecules for the delivery of therapeutic CO in vivo. Angew Chem Int Ed Engl 2014;53:9712–21. [DOI] [PubMed] [Google Scholar]
- [10].Stone MK, Kolling GL, Lindner MH. Obrig TG. p38 mitogen-activated protein kinase mediates lipopolysaccharide and tumor necrosis factor alpha induction of shiga toxin 2 sensitivity in human umbilical vein endothelial cells. Infect Immun 2008;76:1115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wong C, Wang C, Ip W, et al. Role of p38 MAPK and NF-kB for chemokine release in coculture of human eosinophils and bronchial epithelial cells. Clin Exp Immunol 2005;139:90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sun B, Sun Z, Jin Q, et al. CO-releasing molecules (CORM-2)-liberated CO attenuates leukocytes infiltration in the renal tissue of thermally injured mice. Int J Biol Sci 2008;4:176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Levi M. The coagulant response in sepsis and inflammation. Hamostaseologie 2010;30:10. [PubMed] [Google Scholar]
- [14].Abraham E, Laterre PF, Garg R, et al. Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med 2005;353:1332–41. [DOI] [PubMed] [Google Scholar]
- [15].Lin SM, Wang YM, Lin HC, et al. Serum thrombomodulin level relates to the clinical course of disseminated intravascular coagulation, multiorgan dysfunction syndrome, and mortality in patients with sepsis. Crit Care Med 2008;36:683–9. [DOI] [PubMed] [Google Scholar]
- [16].Yamakawa K, Fujimi S, Mohri T, et al. Treatment effects of recombinant human soluble thrombomodulin in patients with severe sepsis: a historical control study. Critical Care 2011;15:R123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Esmon CT. New mechanisms for vascular control of inflammation mediated by natural anticoagulant proteins. Journal of Experimental Medicine 2002;196:561–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Laszik ZG, Zhou XJ, Ferrell GL, et al. Down-regulation of endothelial expression of endothelial cell protein C receptor and thrombomodulin in coronary atherosclerosis. AmJ Pathol 2001;159:797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gu JM, Katsuura Y, Ferrell GL, et al. Endotoxin and thrombin elevate rodent endothelial cell protein C receptor mRNA levels and increase receptor shedding in vivo. Blood 2000;95:1687–93. [PubMed] [Google Scholar]
- [20].Castellino F, Ploplis V. The protein C pathway and pathologic processes. J Thromb Haemost 2009;7:140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hoetzel A, Dolinay T, Schmidt R, et al. Carbon monoxide in sepsis. Antioxid Redox Signal 2007;9:2013–26. [DOI] [PubMed] [Google Scholar]
- [22].Furuichi M, Yokozuka M, Takemori K, et al. The reciprocal relationship between heme oxygenase and nitric oxide synthase in the organs of lipopolysaccharide-treated rodents. Biomed Res 2009;30:235–43. [DOI] [PubMed] [Google Scholar]
- [23].Johnson FK, Johnson RA. Carbon monoxide promotes endothelium-dependent constriction of isolated gracilis muscle arterioles. Am J Physiol Regul Integr Comp Physiol 2003;285:R536–41. [DOI] [PubMed] [Google Scholar]
- [24].Fujita T, Toda K, Karimova A, et al. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med 2001;7:598–604. [DOI] [PubMed] [Google Scholar]
- [25].Sun B, Shi G, Zhang P, et al. Inhibitive effect of exogenous carbon monoxide-releasing molecules 2 on tissue factor expression in sepsis. Zhonghua Shao Shang Za Zhi 2009;25:111–4. [PubMed] [Google Scholar]
- [26].Otterbein LE, Bach FH, Alam J, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 2000;6:422–8. [DOI] [PubMed] [Google Scholar]
- [27].Wang X, Wang Y, Kim HP, et al. Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation. J Biol Chem 2007;282:1718–26. [DOI] [PubMed] [Google Scholar]
- [28].Megias J, Busserolles J, Alcaraz M. The carbon monoxide-releasing molecule CORM-2 inhibits the inflammatory response induced by cytokines in Caco-2 cells. Br J Pharmacol 2007;150:977–86. [DOI] [PMC free article] [PubMed] [Google Scholar]