

Abstract
Rationale:
Lipoid congenital adrenal hyperplasia (LCAH) is extremely rare, but is the most fatal form of congenital adrenal hyperplasia resulting from mutations in the steroidogenic acute regulatory protein (STAR) gene. LCAH arises from severe defects in the conversion of cholesterol to pregnenolone, the precursor of all steroids.
Patient concerns:
A case was reported that an 11-month-old Chinese girl who presented with a sex development disorder and hyponatremia. The clinical and genetic tests were carried out to confirm the diagnosis. The genogram of this case was also explored and analyzed. The girl presented with hyponatremia, decreased cortisol level, elevated adrenocorticotropic hormone level and female vulva despite a 46, XY karyotype. Enlarged adrenal glands and testicular-like tissue in the bilateral inguinal regions were detected with abdominal ultrasound. She was suspected of having LCAH, and definitive diagnosis was made after Sanger sequencing detected a homozygous frameshift variant c.707_708delins CTT (p.Lys236Thrfs∗47) on exon 6 of the STAR gene.
Diagnoses:
LCAH.
Interventions:
She was prescribed hydrocortisone 10 to 12 mg/m2 and 9a- fludrocortisone 100 mg/d.
Outcomes:
Her skin hyperpigmentation and vomiting disappeared, and she had normal growth and development without adrenal crisis attacks. Her hormone and electrolyte levels remained normal, except for a persistently elevated ACTH level throughout 2 years of follow-up. At follow-up for 2 years, the patient is now 104.5 cm tall and weighs 23.3 kg at the age of 4 years old. Her plasma sodium and potassium concentration were normal. Her ACTH level is still elevated (1176 pg/mL). Her baseline sex hormone levels are testosterone <0.1 ng/dL and progesterone <0.08 ng/dL. The level of PRA (1.06 ng/mL per h) is within normal range.
Lessons:
This mutation was in accordance with previously reported gene mutations. The patient's parents were nonconsanguineous; her parents, paternal grandfather, and maternal grandmother were all found to be carriers of a STAR gene mutation. This 46 XY disorders of sex development case presented with adrenal insufficiency and female phenotype initially. The diagnosis was complicated depending on the clinical hormone workup. LCAH was confirmed by genetic tests and genogram of the family.
Keywords: congenital adrenal hyperplasia, lipoid congenital adrenal hyperplasia, STAR mutation
1. Introduction
Lipoid congenital adrenal hyperplasia (LCAH) is an autosomal recessive inherited disease.[1] It is the most fatal form of adrenal hyperplasia and is characterized by impaired adrenal and gonadal steroidogenesis. Mineralocorticoid, glucocorticoid, and sex steroids are all affected due to severe defects in the conversion of cholesterol to pregnenolone, the precursor of all steroids.[2] Severe insufficiency of adrenocortical hormone can result in an Addisonian crisis with salt loss. The lack of gonadal hormones leads to a disorder of sex development.[3] There are a wide variety of phenotypes for this disease. The nonclassical ones frequently present with atypical clinical manifestations, such as with severely disordered glucocorticoid hormone secretion but normal electrolytes and normal male genitalia.[4]
LCAH arises from defects in the transport of cholesterol to the mitochondrial inner membrane, which is physiologically mediated by steroidogenic acute regulatory protein (StAR), coded by the STAR gene.[5] StAR is normally expressed in the adrenal glands and gonads, which helps explain the pathogenesis of LCAH.[2] Loss-of-function mutations of the STAR gene lead to the absolute or partial inability of cholesterol to move to the mitochondrial inner membrane and participate in the biosynthesis of pregnenolone through interacting with the side chain cleavage enzyme P450scc.[1,6]
For the patients presenting with genitalia abnormality and Addison disease, several diagnoses should be considered such as 17 alpha hydroxylase deficiency (17OHD), 3β-hydroxysteroid dehydrogenase (3β-HSD) deficiency, cytochrome P450 oxidoreductase deficiency (PORD), and LCAH. It is crucial for the patients to identify the etiology early since all of them have similar symptoms. Herein, we report a Chinese girl with classic LCAH with a homozygous STAR mutation c.707_708delins CTT (p.Lys236Thrfs∗47) who presented to our hospital.
2. Case presentation
An 11-month-old baby girl presented to the outpatient clinic at Peking Union Medical College Hospital with skin hyperpigmentation and vomiting for 2 weeks. The child was born at full-term by Cesarean section and was the second child of the nonconsanguineous parents. The patient's older sibling was phenotypically female, but died in infancy without a clear diagnosis. Although the karyotype was 46, XY, the patient was raised as female gender since she had female external genitalia. In addition, hyponatremia and an elevated adrenocorticotropic hormone (ACTH) level were detected by tests.
The blood pressure at clinic was 80/50 mm Hg. Laboratory analysis revealed that the baby had a mildly elevated cholesterol level of 5.72 mmol/L (normal: 2.85–5.70 mmol/L), but a normal triglyceride level. Her plasma sodium level was 131.7 mmol/L (normal: 135–145 mmol/L) with potassium concentration of 6.61 mmol/L (normal: 3.5–5.5 mmol/L). ACTH was >1250 pg/mL (0–46 pg/mL) with a morning cortisol of 1.23 μg/dL (normal: 4–22 μg/dL). Baseline sex hormone tests revealed luteinizing hormone <0.10 U/L, follicle stimulating hormone 1.78 U/L, testosterone 2.50 ng/dL, progesterone 0.44 ng/dL, estradiol 22.99 pg/mL, and dehydroepiandrosterone (DHEA) sulfate <0.1 μg/dL. Adrenal computed tomography (CT) scan revealed enlarged adrenal glands. Neither ovaries nor uterus could be identified with pelvic ultrasound. However, testicular-like tissues were detected in the bilateral inguinal regions (left: 0.6 × 0.4 cm; right: 0.7 × 0.4 cm) by ultrasonography. Plasma renin activity (PRA) was 0.11 ng/mL per h (normal: 0.93–6.56 ng/mL per h) with a normal aldosterone level of 8.84 ng/dL (normal: 6.5–29.6 ng/dL). Given ambiguous genitalia, karyotype of 46, XY and decreased DHEA and testosterone levels, the diagnosis of 21-hydroxylase deficiency and 11 beta hydroxylase deficiency were excluded. Lacking hypertension and hypopotassemia, the clinical presentation of salt loss and decreased progesterone also did not support the diagnosis of 17OHD. Without an elevated progesterone level, cytochrome PORD was less considered. The differential diagnosis included 3β-HSD deficiency and LCAH. After the informed consent form was signed by the families and approved by the Institutional Review Board of Peking Union Medical College Hospital, further genetic analysis with gene sequencing indicated it is not defective HSD3B2 (related to 3β-HSD deficiency). However, a homozygous variant c.707_708delins CTT (p.Lys236Thrfs∗47) was found on exon 6 of the STAR gene. A truncated disease-causing protein was predicted with MutationTaster software. The variant mutation was found to cause a frameshift at codon 236 with a stop at codon 282. Both parents were found to be heterozygous for the variant (c.707_708delins CTT, p.Lys236Thrfs∗47) at the same locus of the STAR gene without phenotypic consequences. To further evaluate the family pedigree for mutation carriers, the patient's paternal and maternal grandparents also took part in the gene sequencing. The patient's paternal grandfather and maternal grandmother both carried the same mutant allele; her paternal grandmother and maternal grandfather were negative for the mutant allele. All the family members denied consanguinity and no subclinical phenotypes (such as mild amenorrhea in women) were recorded, and there were no abnormalities in physical and laboratory examinations. The results of the patient, her parents, and paternal grandfather and maternal grandmother are shown in Fig. 1. The genogram of LCAH in this family is presented in Fig. 2.
Figure 1.
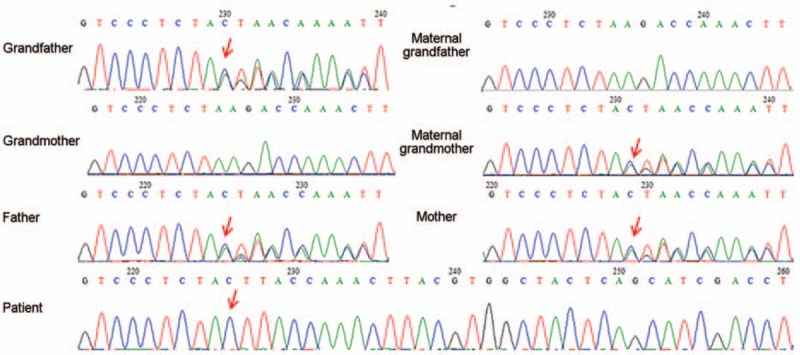
Sequence electropherograms showing the STAR gene mutation in an 11-month-old baby and her family members. Sequence analysis of the STAR gene revealed a homozygous frameshift for c.707_708delins CTT (p.Lys236Thrfs∗47). The paternal copy was inherited from the paternal grandfather. The same heterozygous mutation was found in the patient's mother and was inherited from the patient's maternal grandmother. The red arrows indicate the homozygous nucleotides of c.707_708delins CTT (p.Lys236Thrfs∗47) or the heterozygous mutation from the patient's family members.
Figure 2.
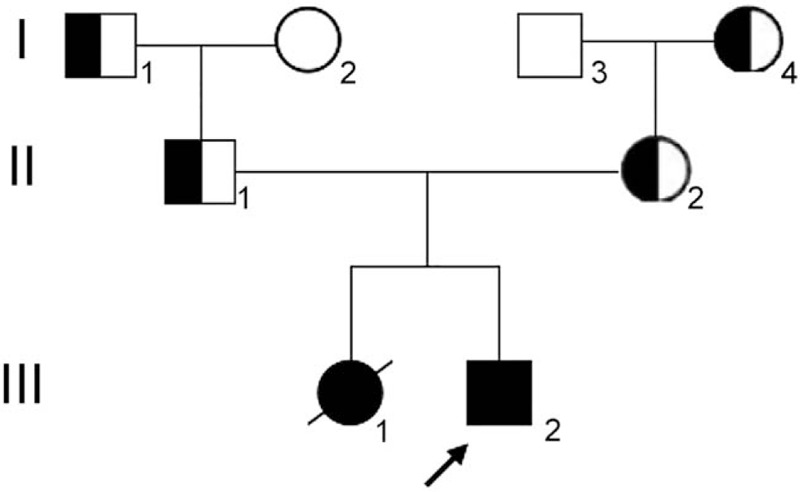
The pedigree of LCAH core family with STAR gene mutation.
The patient was prescribed hydrocortisone 10 to 12 mg/m2 and 9α-fludrocortisone 100 μg/d. Gradually, her skin hyperpigmentation and vomiting disappeared, and she had normal growth and development without adrenal crisis attacks. Her hormone and electrolyte levels remained normal, except for a persistently elevated ACTH level throughout 2 years of follow-up. At follow-up for 2 years, the patient is now 104.5 cm tall and weighs 23.3 kg at the age of 4 years old. Her plasma sodium level is 138 mmol/L with a normal potassium concentration of 4.8 mmol/L. Her ACTH level is still elevated (1176 pg/mL). Her baseline sex hormone levels are testosterone <0.1 ng/dL and progesterone <0.08 ng/dL. The level of PRA (1.06 ng/mL per h) is within normal range.
3. Discussion
In this case report, we presented the complete medical history, evaluation, and treatment of a patient ultimately diagnosed as LCAH with a STAR gene mutation. This patient manifested typical features of adrenal insufficiency, such as hyponatremia and skin hyperpigmentation, and had female external genitalia despite male chromosomal sex. Decreased serum cortisol resulted in a markedly elevated ACTH level. Critical symptoms or signs of adrenal insufficiency may manifest as soon as 2 months after birth.[7,8] This patient's diagnosis of LCAH was confirmed by the absence of steroid biosynthetic activity and resultant extremely low concentrations of all steroid hormones.[9] The differences between LCAH and 21-hydroxylase deficiency are easy to differentiate; patients with 21-hydroxylase deficiency are genetically 46, XX but are masculinized and have high levels of androgens. LCAH may be confused with 3β-HSD deficiency due to similar manifestations: ambiguous genitalia in males, Addisonian crisis, and abnormal hormone and electrolyte levels. LCAH can be distinguished from other various forms of congenital adrenal hyperplasia (CAH) by evaluating the concentrations of serum steroid metabolites. Elevated steroid metabolites can be found in CAH, but are not seen in LCAH.[10] For example, excessively high levels of 17-hydroxyprogesterone are present in 21-hydroxylase deficiency; high DHEA and 17-delta-5-hydroxypregnenolone are seen in 3β-HSD deficiency.
Despite its rarity, it is well established that STAR gene mutations lead to LCAH.[3,10] Presently, more than 40 STAR mutations have been identified[11,12] and 190 patients diagnosed with LCAH[2,6] have been reported and described. Despite the low worldwide prevalence, the LCAH cases have been identified mainly in Japanese, Korean, and Palestinian descent, but are rarely reported elsewhere.[13] Huang et al first reported the LCAH and STAR mutations identified in 10 Chinese children,[13] while there were no statistics on prevalence and incidence among Chinese population. Physiologically, StAR provides cholesterol substrate for steroid biosynthesis by trafficking it to P450scc on the mitochondrial inner membrane, where the first step of steroid synthesis is catalyzed.[14] The STAR gene is a 7.5 kb gene with 7 exons, but 40% of the StAR is encoded by exons 5 to 7; this is consistent with the fact that most reported mutations resulting in LCAH were found in exons 5 to 7 of the STAR gene.[2] The STAR gene is translated to a 37-kDa preprotein with 285 amino acids. The N-terminal mitochondrial import sequence (62 amino acids) of this preprotein is clipped off to form the 30-kDa mature StAR.[15] Cleavage of 62 residues from the N-terminus (N-62) does not significantly lower steroidogenic activity, but import and processing are impaired.[15] Twenty-eight biologically active C-terminal amino acids (C-28) are encoded by exons 5 to 7 on the outside of mitochondria; they play a crucial role in steroid biosynthesis, which was proved to lead to an inactive protein in vitro.[15] The StAR is characterized by a highly conserved amino acid sequence (from amino acids 67 to 280), which binds cholesterol and serves as a transporting module called StAR-related lipid transfer domain. This domain folds into a hydrophobic cavity formed by 9 twisted antiparallel β-sheets and 4α-helices for sterol binding and ligand accommodation.[11,13,16] The homozygous frameshift variant c.707_708delins CTT (p.Lys236Thrfs∗47) on exon 6 found in our patient was predicted to be located upstream of C-28, whose biological activity has been illustrated.[15] So this loss-of-function mutation resulted in severely impaired steroid biosynthesis of StAR protein, which makes it an inactive protein. Gene sequencing analyses revealed that the patient's parents, paternal grandfather, and maternal grandmother were all carriers of this mutation and were heterozygous for the variant of c.707_708delins CTT (p.Lys236Thrfs∗47). Consequently, our patient was homozygous for this disease-causing mutation. In a study that included 10 Chinese children diagnosed as LCAH, this variant accounted for 15% of the total mutant alleles with 1 parent being a carrier.[13] In addition to the 3 reported heterozygous variants, this is the first reported case of the homozygous STAR gene variant c.707_708delins CTT (p.Lys236Thrfs∗47) in a Chinese patient. The genetic result seen in this patient was in accordance with those seen at other medical centers in China, reflecting the reliability of the sequencing procedure used. In addition, our patient's genetic makeup supports the accuracy of the previous results. On the other hand, our patient's mode of inheritance was in accordance with that of an autosomal recessive-inherited disease. The patient had a detected homozygous gene mutation, and was found to be carrier of the mutant alleles without themselves having clinical manifestations. This report is special in that the patient's homozygous mutation has never been previously recorded as resulting from inheriting 1 affected allele from each heterozygous parent in a nonconsanguineous marriage. Given its rarity, the homozygous mutation is extremely rare in a nonconsanguineous marriage and it is the first reported homozygous STAR mutant among Chinese, which could accumulate the clinical experience in LCAH and enrich the records on STAR mutations. We were able to construct a rare pedigree analysis of this variant of the STAR gene with known information of the gene mutation in the patient, her parents, and her grandparents.
The c.707_708delins CTT (p.Lys236Thrfs∗47) mutation may be associated with a classic phenotype of LCAH because this patient presented with adrenal insufficiency and disordered sex development. In conclusion, a karyotype, STAR gene sequencing, and analysis of the activity and crystal structures, as well as 3-dimensional homology protein modeling of the StAR, may contribute to more precise diagnosis and treatment for patients with LCAH. The function of this gene requires further verification.
Acknowledgments
The authors thank the patient and her family. The authors also express our gratitude to all of the co-workers participating in this research.
Footnotes
Abbreviations: 17OHD = 17 alpha hydroxylase deficiency, 3β-HSD = 3β-hydroxysteroid dehydrogenase, ACTH = adrenocorticotropic hormone, CAH = congenital adrenal hyperplasia, CT = computed tomography, DHEA = dehydroepiandrosterone, LCAH = lipoid congenital adrenal hyperplasia, PRA = plasma renin activity, STAR = steroidogenic acute regulatory protein.
This study was supported by grants from the National Key Program of Clinical Science (WBYZ2011-873).
The authors have no conflicts of interest to disclose.
References
- [1].King SR, Bhangoo A, Stocco DM. Functional and physiological consequences of StAR deficiency: role in lipoid congenital adrenal hyperplasia. Endocr Dev 2011;20:47–53. [DOI] [PubMed] [Google Scholar]
- [2].Miller WL, Bose HS. Early steps in steroidogenesis: intracellular cholesterol trafficking. J Lipid Res 2011;52:2111–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bose HS, Sugawara T, Strauss JF, III, et al. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N Engl J Med 1996;335:1870–8. [DOI] [PubMed] [Google Scholar]
- [4].Baker BY, Lin L, Kim CJ, et al. Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J Clin Endocrinol Metab 2006;91:4781–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bens S, Mohn A, Yuksel B, et al. Congenital lipoid adrenal hyperplasia: functional characterization of three novel mutations in the STAR gene. J Clin Endocrinol Metab 2010;95:1301–8. [DOI] [PubMed] [Google Scholar]
- [6].Kaur J, Casas L, Bose HS. Lipoid congenital adrenal hyperplasia due to STAR mutations in a Caucasian patient. Endocrinol Diabetes Metab Case Rep 2016;2016:150119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gassner HL, Toppari J, Quinteiro Gonzalez S, et al. Near-miss apparent SIDS from adrenal crisis. J Pediatr 2004;145:178–83. [DOI] [PubMed] [Google Scholar]
- [8].Fujieda K, Tajima T, Nakae J, et al. Spontaneous puberty in 46,XX subjects with congenital lipoid adrenal hyperplasia. Ovarian steroidogenesis is spared to some extent despite inactivating mutations in the steroidogenic acute regulatory protein (StAR) gene. J Clin Invest 1997;99:1265–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hauffa BP, Miller WL, Grumbach MM, et al. Congenital adrenal hyperplasia due to deficient cholesterol side-chain cleavage activity (20, 22-desmolase) in a patient treated for 18 years. Clin Endocrinol (Oxf) 1985;23:481–93. [DOI] [PubMed] [Google Scholar]
- [10].White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2000;21:245–91. [DOI] [PubMed] [Google Scholar]
- [11].Yuksel B, Kulle AE, Gurbuz F, et al. The novel mutation p.Trp147Arg of the steroidogenic acute regulatory protein causes classic lipoid congenital adrenal hyperplasia with adrenal insufficiency and 46,XY disorder of sex development. Horm Res Paediatr 2013;80:163–9. [DOI] [PubMed] [Google Scholar]
- [12].Camats N, Pandey AV, Fernández-Cancio M, et al. STAR splicing mutations cause the severe phenotype of lipoid congenital adrenal hyperplasia: insights from a novel splice mutation and review of reported cases. Clin Endocrinol (Oxf) 2014;80:191–9. [DOI] [PubMed] [Google Scholar]
- [13].Huang Z, Ye J, Han L, et al. Identification of five novel STAR variants in ten Chinese patients with congenital lipoid adrenal hyperplasia. Steroids 2016;108:85–91. [DOI] [PubMed] [Google Scholar]
- [14].Lin D, Sugawara T, Strauss JF, III, et al. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science 1995;267:1828–31. [DOI] [PubMed] [Google Scholar]
- [15].Arakane F, Sugawara T, Nishino H, et al. Steroidogenic acute regulatory protein (StAR) retains activity in the absence of its mitochondrial import sequence: implications for the mechanism of StAR action. Proc Natl Acad Sci USA 1996;93:13731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tsujishita Y, Hurley JH. Structure and lipid transport mechanism of a StAR-related domain. Nat Struct Biol 2000;7:408–14. [DOI] [PubMed] [Google Scholar]