

Key Clinical Message
We report a compound heterozygous mutation (c.135delC; c.423+2dupT) of MLC1 gene in a Chinese patient underlying infantile macrocephaly and neurological deterioration in early childhood. Brain MRI revealed diffusion abnormality in swollen white matter and a subcortical cyst. The cDNA sequencing analysis for the c.423+2dupT variant revealed skipping of exon 5.
Keywords: Compound heterozygous mutation, exon skipping, MLC1
Introduction
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a rare type of leukodystrophy. Patients develop macrocephaly during the first year of life. Thereafter, slow neurological deterioration with cerebellar ataxia and spasticity occurs. Magnetic resonance imaging (MRI) of the brain reveals diffuse signal abnormality, swelling of the cerebral white matter, and the presence of subcortical cysts in the anterior temporal regions 1, 2, 3, 4.
Three types of MLC that share similar clinical symptoms are listed in OMIM: MLC1 (#604004), MLC2A (#613925), and MLC2B (#613926). MLC1 is caused by mutations of the MLC1 gene on chromosome 22q13.33 in approximately 75% of MLC cases, with an autosomal recessive mode of inheritance. In approximately 20% of patients, the disease is explained by mutations in the hepatocyte cell adhesion molecule precursor gene (HEPACAM) on chromosome 11q24.2 5. HEPACAM‐associated MLC can be inherited in an autosomal recessive form (classified as MLC2A) or in a less severe autosomal dominant form of “transient” MLC, with or without developmental disability and/or autism (MLC2B) 6.
Currently, nearly 90 mutations of MLC1 have been reported worldwide. Mutations are distributed throughout the entire coding region and include all different types: missense and nonsense mutations (53%), splice‐site mutations (20%), deletions (20%), and insertions (7%) [HGMD http://www.hgmd.cf.ac.uk/ac/all.php]. However, the genotype‐phenotype correlation has not been described 7, 8.
Clinical presentations of MLC1 gene mutations are common; however, functional studies are mostly restricted to computational predictions only. Thus, deviation in the knowledge of mutations arises inevitably.
In this study, we present a compound heterozygous mutation in the MLC1 gene in an affected child with moderate clinical symptoms from Hunan, China. A compound heterozygous mutation (c.135delC; c.423+2dupT) was identified in the MLC1 gene. One is the novel deletion, which induces protein truncation with a premature stop codon. The second variation c.423+2dupT has been reported previously 9; however, we present a functional study demonstrating exon 5 skipping for the first time.
Patient and Methods
The proband was a 9‐year‐old Chinese boy born to healthy parents. Clinically, he was diagnosed with megalencephalic leukoencephalopathy. After an uneventful delivery, macrocephaly was noted during his first year of life. The physical examination data at his ten‐month follow‐up showed a large head circumference of 57 cm (45.4 ± 1.3 cm is the normal range of the WTO head circumference by age in boys) (http://www.who.int/childgrowth/standards/en/). Several years later, the boy presented symptoms of mild developmental disability. Weak language ability and poor grades were reported during primary school. Epilepsy or spasticity had not occurred, but ataxia was noted with unsteady walking and poor coordination ability. At 7 years, Encephalo‐CT revealed extensive density, a decrease of the cerebral white matter in each lobe of the bilateral cerebral hemisphere, and a slight expansion in the anterior cerebral longitudinal fissure and subarachnoid space. The brain MRI showed abnormal white matter and a subcortical cyst in the left temporal lobe (Fig. 1A).
Figure 1.
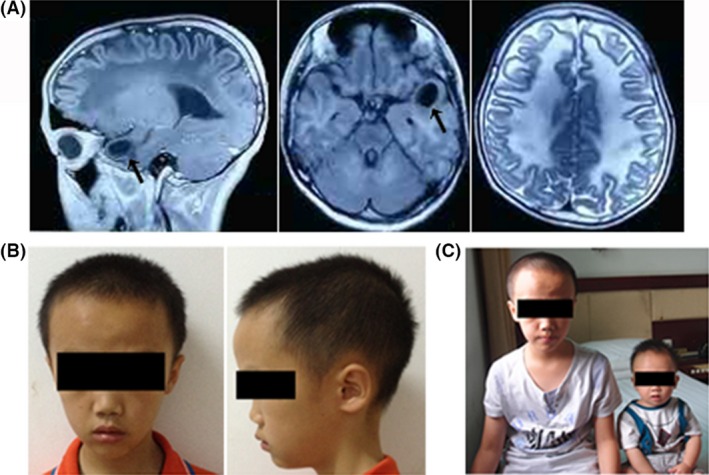
(A) Brain MRI of the proband. The image shows a subcortical cyst in the left temporal lobe (arrow) and a diffuse lesion in the white matter. (B) The patient's photographs at the age of nine. The head circumference was large. (C) Proband (left) and his healthy brother (right). The baby was about ten months old. No abnormity was found.
This case study was approved by the institutional research ethics committee of the Reproductive and Genetic Hospital of Citic‐Xiangya. Written informed consent for the research and for the publication of the patients’ anonymous details and images was obtained from the parents.
For mutation analysis, genomic DNA was extracted from peripheral blood of the affected son and the two parents. Genomic DNA and total RNA were extracted following the manufacturer's instructions (Qiagen, Germany). The coding region of MLC1 (GenBankNM_015166) was amplified by polymerase chain reaction (PCR) using the primers listed in Table 1. The mRNA expression of MLC1 was examined by transcription‐polymerase chain reaction (RT‐PCR) using the following primers: forward (5′‐CCAGGAGGAACGCCAATGTG‐3′) and reverse (5′‐TCAGGACCCGAGCAGGAAAT‐3′). Both PCR and RT‐PCR were performed in a 25‐μL reaction containing 50 ng template, 12.5 μL Gold Taq Green Master Mix (Promega, Madison, Wisconsin, USA), and 0.5 μmol/L each of the primers. The reactions were performed under the following conditions: 95°C for 1.5 min, 35 cycles of 94°C for 40 sec, 60°C for 40 sec, and 72°C for 40 sec, followed by a final extension at 72°C for 5 min. Amplimers were purified using the Wizard SV Gel and PCR Clean‐Up System (Promega, Madison, Wisconsin, USA), followed by direct sequencing on an Applied Biosystems 3130 Genetic Analyzer (Applied Biosystems, Hitachinaka, Ibaraki, Japan). The results of the mutant samples were validated by reuploading.
Table 1.
Primer sequences used for MLC1 amplification and sequencing
| Fragment | Oligonucleotide primers | Annealing temperature(°C) | Length(bp) |
|---|---|---|---|
| Exon 2 | 5′‐CTCAGAGTGGCCAAAGCC‐3′ | 60 | 328 |
| 5′‐ACCAGAGGGACCAGATGC‐3′ | |||
| Exon 3 | 5′‐CAGAAGTTGAAGGGTCAGGG‐3′ | 57 | 561 |
| 5′‐GAAGTTTCACTCTCATTGCCC‐3′ | |||
| Exon 4/5 | 5′‐GCTCATGGGATTCCGGTT‐3′ | 60 | 548 |
| 5′‐ TGTGGGTGTCAGGCGTCT‐3′ | |||
| Exon 6 | 5′‐ GGTGGCGTGAGAAAGGCG‐3′ | 60 | 222 |
| 5′‐CCCACCTCGCTCACCCTG‐3′ | |||
| Exon 7 | 5′‐ AGTGCTGAGTCCCTGTGC‐3′ | 60 | 363 |
| 5′‐GCAGTAACAAACTCCCCC‐3′ | |||
| Exon 8 | 5′‐ GGTGGGTGTGTCCTATGG‐3′ | 60 | 652 |
| 5′‐ GGTGACTCTCTGTCTGAA‐3′ | |||
| Exon 9 | 5′‐ TACCCCTGCTTCCCTGCG‐3′ | 60 | 396 |
| 5′‐ ACCCCACCTTCCTCATTG‐3′ | |||
| Exon 10 | 5′‐ GAACCAGCTTGGGACTAT‐3′ | 55 | 275 |
| 5′‐ GGGGGGCTCTGAAATAAA‐3′ | |||
| Exon 11 | 5′‐ GCTCACACCTCCTTCCGC‐3′ | 60 | 278 |
| 5′‐ CCCACCCCACAGGCTTCT‐3′ | |||
| Exon 12 | 5′‐ GCAGGCGTTTCTGGGACA‐3′ | 60 | 355 |
| 5′‐ GCTCAGGGCGATTAGGGG‐3′ |
Results
Sequencing of the MLC1 gene from the patient identified two heterozygous mutations, c.135delC, p.Cys46Alafs*12 and c.423+2dupT, p.Pro109_Ile142del (skipping exon 5). Chromatograms are shown. The two mutations were confirmed in his parents by Sanger sequencing (Fig. 2A–G).
Figure 2.
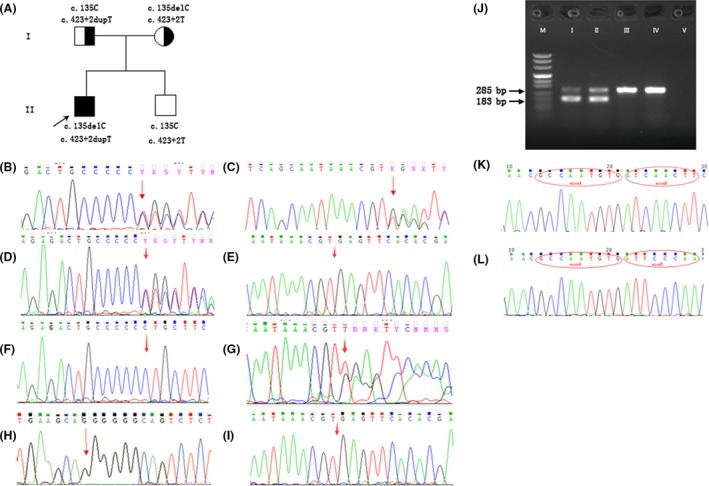
Genetic analysis. (A) Pedigree chart. The proband was noted with compound heterozygous mutations. The father and mother both carried heterozygous mutations. The prenatally diagnosed boy was free of mutations. (B, C) Sequence of the proband's sample. A: Arrow pointing to the heterozygous mutation c.135delC; B: heterozygous mutation c.423+2dupT. (D, E) Sequences of the two mutation regions in the mother. (F, G) Sequences of the two mutation regions in the father. (H, I) gDNA sequence of amniotic fluid. H: Arrow pointing to the normal c.135C; I: arrow showing the position of normal c.423+2T. (J) Electrophoretogram of cDNA amplification. “M” is the DNA marker; “I”, “II”, “III”, “IV,” and “V” indicate the proband, the father, the mother, normal, and blank control, respectively. There were two separable bands in the samples of the proband and the father. The top band was 285 bp in size; the lower one was 183 bp. The mother's RT‐PCR product was equal to the normal control. (K, L) cDNA sequence of the proband. K: The smaller product was 183 bp, and red ovals at the bases of MLC1 mark exon 4 and exon 6. L: Product of normal length, the bases in the red ovals belong to exon 4 and exon 5 of MLC1.
The region 302–586 bp of MLC1 cDNA was amplified. A 285‐bp normal band and a shorter band appeared in both of the proband and his father. The sequencing of cDNA amplifiers showed that the shorter product corresponded to the deletion of exon 5 in the MLC1 gene (Fig. 2J–L). Indirectly, mRNA from the allele with C.423+2dupT exhibited skipping of exon 5.
In a new pregnancy of the mother, a prenatal diagnosis was carried out, and no mutations at either site of the MLC1 gene were detected in the fetus (Fig. 2H and I). The next year, a healthy baby was born as a full‐term normal delivery. The physical indicators of the baby were normal, including the head circumference, but no MRI examination was performed.
Discussion
MLC1 is a genetic puerile leukoencephalopathy rarely found in China. Here, we describe an affected boy from a Chinese family. The patient had the classical symptoms of infantile macrocephaly, including motor developmental delay, mild ataxia, and intellectual disability (Fig. 1B). Two variations, c.135delC, p.Cys46Alafs*12 and c.423+2dupT, p.Pro109_Ile142del (skipping exon 5), detected in the MLC1 gene from the patient were highly likely to be pathogenic mutations. First, either mutation could result in a protein truncation or structure defect. Additionally, both mutations segregated concomitantly with the pathological phenotype of the patient and his parents. Additional evidence of the causative effect of the identified mutation in the affected sibling was that an unaffected pregnancy (16th gestational week) was negative (Fig. 1C).
The mutation c.135delC (p.Cys46Alafs*12) had not been reported previously, nor was it present in the dbSNP or Human Gene Mutation Database. The analysis of biological information showed that this mutation could result in a change in the coding sequence after the 45th codon and create an in‐frame stop codon in exon 2, which probably led to a truncated protein of 56 residues or alternatively led to nonsense‐mediated decay of the mRNA (Fig. 3A).
Figure 3.
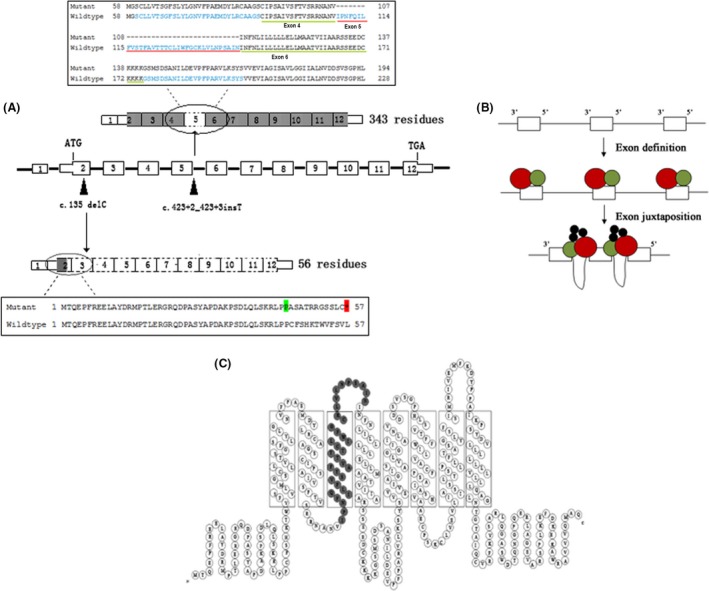
(A) Schematic representation of function patterns for the two mutations. Black arrowheads are mutation positions on MLC1 gDNA (middle). The mutation c.135delC creates an in‐frame stop codon in exon 2 and leads to a truncated protein of 56 residues (black box below). The mutation c.423+2dupT gives rise to an incomplete protein without exon 5 (black box above). Dotted boxes indicate the coding region that is not transcribed. (B) Sketch of “exon definition” with small exons and large introns. Splicing machinery (red and green) interacting with isolated exons during exon definition. Splicing factors (black) join the assembly during the exon juxtaposition following exon definition 11. (C) 2D model of the MLC1 protein 7. The gray parts show the deleted residues of exon 5.
The insertion or duplication mutation c.423_2dupT (p.Pro109_Ile142del) leading to exon 5 skipping had been presented previously; however, it had only been shown to create a new splicing site through in silico predictions 9. According to Leegwater's prediction, an in‐frame stop codon would arise among the 94‐bp sequence and lead to a truncated MLC1 gene product of 141 residues, with an addition of 26 unrelated residues at the C‐terminal end. However, in our study, cDNA analysis showed that the insertion apparently silenced the splicing acceptor site of exon 5 and gave rise to exon 5 skipping, while downstream exons after the fifth remained unaffected. The protein translated from the mutant MLC1 gene would contain 343 residues without the amino acids coded by exon 5, as shown in Figure 3A.
Our finding coincides with the exon‐oriented perspective of splice‐site pairing. This belongs to the “exon definition” of splice‐site recognition, including splice pairing across an exon for splicing with large introns and small exons 10. The “exon definition” model proposes that in pre‐mRNAs with large introns, the splicing machinery searches for a pair of closely spaced splice sites in exonic polarity (Fig. 3B). When such a pair is encountered, neighboring exons will be juxtaposed via interactions between the factors that recognize individual exons 11. Regarding the mutation (C.423+2dup) adjacent to exon 5, the exon is only 102 bp in length, while intron 5 is up to 2912 bp. This is the situation by which a short exon is separated by a long intron. When the extra T disabled the action of the wild‐type splice donor site of intron 5, the pairing of splice sites across exon 5 was inhibited, and the recognition of exon 5 failed. Rejection of the exon leads directly to exon 5 skipping.
In fact, approximately 15% of mutations that cause genetic disease affect exon splicing 12, and the most frequent phenotype is exon skipping at a ratio of 51% 13. Because the splicing regulators and splice‐site signals are usually short and often degenerate, even the best computer programs are only 50% accurate in predicting actual splice sites 14, 15. Our work shows that for the genes whose transcripts are detectable by RT‐PCR in blood cells, cDNA anomalies should be screened for splicing results.
MLC1 is a plasma‐membrane protein containing eight transmembrane domains that is highly expressed in brain astrocytes (Fig. 3C). Although the exact function remains unclear, it has been demonstrated that MLC1 is related to the activation of volume‐regulated anion currents (VRAC) involved in the cellular osmotic response 16, 17. The mutations of MLC1 may reduce the regulatory volume of astrocytes, a key process in osmotic perturbation buffering in the central nervous system 18.
In our patient, the compound heterozygous alteration leads to premature termination and thus the nonavailability of normal protein leading to complete loss of function. The splicing machinery with the mutation c.423+2dupT, p.Pro109_Ile142del (skipping exon 5) may lead to a deficiency of the whole transmembrane domain 3 and the bonding pad (Fig. 3C). As no alternative transcript of the MLC1 gene has been reported, the combination of the two scenarios destroyed the protein function and promoted the start of the disease in our patient.
Several conclusions can be drawn from the identified results. (1) The identification of two mutations can assign the mutated gene to the disease; (2) a novel mutation c.135delC, p.Cys46Alafs*12 was identified for the MLC1 gene mutation group. Exon skipping but not an in‐frame stop codon was proven by cDNA sequencing for the reported mutation C.423+2dupT, p.Pro109_Ile142del (skipping exon 5); (3) our results may offer an experimental basis for further study of the gene splicing mechanism and gene therapy; (4) considering the timeliness and hardship of biopsy in gene diagnosis, RT‐PCR sequencing is an appropriate method for further functional verification of splicing mutations.
Authorship
CD: performed the experiments, collected the samples, analyzed the data, and wrote the manuscript. WH: designed the primers and collected the clinical materials. JD: analyzed the data. YT: offered financial support. GL and WL: devised and designed the experiments, provided administrative support, and approved the manuscript. WL: also collected the clinical materials and blood samples.
Conflict of Interest
The authors declare that no competing interests exist.
Acknowledgments
We sincerely thank the affected child and his family for their cooperation in this study. We are grateful to Dr. Ping Liang, Dr. Hsiao Chang Chan, and Dr. Kin Lam Fok for their careful revision of the text. This work was supported by a grant from the National Natural Science Foundation of China (No. 81471432).
Contributor Information
Guang‐Xiu Lu, Email: lugxdirector@aliyun.com.
Wen Li, Email: lwliwen@aliyun.com.
References
- 1. van der Knaap, M. S. , Barth P. G., Stroink H., van Nieuwenhuizen O., Arts W. F., Hoogenraad F., et al. 1995. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann. Neurol. 37:324–334. [DOI] [PubMed] [Google Scholar]
- 2. Leegwater, P. A. , Boor P. K., Yuan B. Q., van der Steen J., Visser A., Konst A. A., et al. 2002. Identification of novel mutations in MLC1 responsible for megalencephalic leukoencephalopathy with subcortical cysts. Hum. Genet. 110:279–283. [DOI] [PubMed] [Google Scholar]
- 3. Lopez‐Hernandez, T. , Sirisi S., Capdevila‐Nortes X., Montolio M., Fernandez‐Duenas V., Scheper G. C., et al. 2011. Molecular mechanisms of MLC1 and GLIALCAM mutations in megalencephalic leukoencephalopathy with subcortical cysts. Hum. Mol. Genet. 20:3266–3277. [DOI] [PubMed] [Google Scholar]
- 4. Mancini, C. , Vaula G., Scalzitti L., Cavalieri S., Bertini E., Aiello C., et al. 2012. Megalencephalic leukoencephalopathy with subcortical cysts type 1 (MLC1) due to a homozygous deep intronic splicing mutation (c.895‐226T>G) abrogated In vitro using an antisense morpholino oligonucleotide. Neurogenetics 13:205–214. [DOI] [PubMed] [Google Scholar]
- 5. Lopez‐Hernandez, T. , Ridder M. C., Montolio M., Capdevila‐Nortes X., Polder E., Sirisi S., et al. 2011. Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am. J. Hum. Genet. 88:422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Teijido, O. , Casaroli‐Marano R., Kharkovets T., Aguado F., Zorzano A., Palacín M., et al. 2007. Expression patterns of MLC1 protein in the central and peripheral nervous systems. Neurobiol. Dis. 26:532–545. [DOI] [PubMed] [Google Scholar]
- 7. Ilja Boor, P. K. , de Groot K., Mejaski‐Bosnjak V., Brenner C., van der Knaap M. S., Scheper G. C., et al. 2006. Megalencephalic leukoencephalopathy with subcortical cysts: an update and extended mutation analysis of MLC1 . Hum. Mutat. 27:505–512. [DOI] [PubMed] [Google Scholar]
- 8. Brignone, M. S. , Lanciotti A., Camerini S., De Nuccio C., Petrucci T. C., Visentin S., et al. 2015. MLC1 protein: a likely link between leukodystrophies and brain channelopathies. Front. Cell. Neurosci. 9:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leegwater, P. A. , Yuan B. Q., van der Steen J., Mulders J., Konst A. A., Boor P. K., et al. 2001. Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am. J. Hum. Genet. 68:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robberson, B. L. , Cote G. J., and Berget S. M.. 1990. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell. Biol. 10:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berget, S. M. 1995. Exon recognition in vertebrate splicing. J. Biol. Chem. 270:2411–2414. [DOI] [PubMed] [Google Scholar]
- 12. Krawczak, M. , Reiss J., and Cooper D. N.. 1992. The mutational spectrum of single base‐pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum. Genet. 90:41–54. [DOI] [PubMed] [Google Scholar]
- 13. Yang, X. , Bani M., Lu S., Rowan S., Ben‐David Y., and Chabot B.. 1994. The A1 and A1B proteins of heterogeneous nuclear ribonucleoparticles modulate 5′ splice site selection in vivo. Proc. Natl Acad. Sci. USA 91:6924–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cooper, T. A. , and Mattox W.. 1997. The regulation of splice‐site selection, and its role in human disease. Am. J. Hum. Genet. 61:259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith, C. W. , and Valcarcel J.. 2000. Alternative pre‐mRNA splicing: the logic of combinatorial control. Trends Biochem. Sci. 25:381–388. [DOI] [PubMed] [Google Scholar]
- 16. Lanciotti, A. , Brignone M. S., Molinari P., Visentin S., de Nuccio C., Macchia G., et al. 2012. Megalencephalic leukoencephalopathy with subcortical cysts protein 1 functionally cooperates with the TRPV4 cation channel to activate the response of astrocytes to osmotic stress: dysregulation by pathological mutations. Hum. Mol. Genet. 21:2166–2180. [DOI] [PubMed] [Google Scholar]
- 17. Teijido, O. , Martinez A., Pusch M., Zorzano A., Soriano E., del Rio J. A., et al. 2004. Localization and functional analyses of the MLC1 protein involved in megalencephalic leukoencephalopathy with subcortical cysts. Hum. Mol. Genet. 13:2581–2594. [DOI] [PubMed] [Google Scholar]
- 18. Sirisi, S. , Folgueira M., Lopez‐Hernandez T., Minieri L., Perez‐Rius C., Gaitan‐Penas H., et al. 2014. Megalencephalic leukoencephalopathy with subcortical cysts protein 1 regulates glial surface localization of GLIALCAM from fish to humans. Hum. Mol. Genet. 23:5069–5086. [DOI] [PubMed] [Google Scholar]