

Abstract
Death-associated protein kinase (DAPK) is a death domain-containing serine/threonine kinase, and participates in various apoptotic paradigms. Here, we identify the extracellular signal-regulated kinase (ERK) as a DAPK-interacting protein. DAPK interacts with ERK through a docking sequence within its death domain and is a substrate of ERK. Phosphorylation of DAPK at Ser 735 by ERK increases the catalytic activity of DAPK both in vitro and in vivo. Conversely, DAPK promotes the cytoplasmic retention of ERK, thereby inhibiting ERK signaling in the nucleus. This reciprocal regulation between DAPK and ERK constitutes a positive feedback loop that ultimately promotes the apoptotic activity of DAPK. In a physiological apoptosis system where ERK–DAPK interplay is reinforced, downregulation of either ERK or DAPK suppresses such apoptosis. These results indicate that bidirectional signalings between DAPK and ERK may contribute to the apoptosis-promoting function of the death domain of DAPK.
Keywords: anoikis, DAPK, death domain, ERK
Introduction
Apoptosis is essential for normal development and maintenance of tissue homeostasis and is controlled by a complex interplay between pro- and antiapoptotic proteins. The death domain superfamily, composed of the death domain (DD), death effector domain and caspase recruitment domain family of proteins, has emerged as the pivotal mediator of protein–protein interactions required for the transmission and regulation of apoptotic signals (Weber and Vincenz, 2001). Death-associated protein kinase (DAPK), a DD-containing, calmodulin (CaM)-regulated serine/threonine kinase, exerts proapoptotic activity and functions as a positive mediator of apoptosis induced by a variety of stimuli, such as interferon-γ, Fas, TNF-α, TGF-β, ceramide and oncogenes c-myc and E2F (Deiss et al, 1995; Cohen et al, 1997, 1999; Inbal et al, 1997; Raveh et al, 2001; Jang et al, 2002; Pelled et al, 2002). DAPK displays a multidomain structure, comprising a kinase domain, a CaM-binding motif, eight ankyrin repeats, a cytoskeleton binding region and a DD, in which the kinase domain and DD are both important for its proapoptotic activity (Cohen et al, 1997, 1999). Emerging evidence reveals the role of DAPK in tumor suppression. DAPK expression is frequently lost in tumors due to hypermethylation of the DAPK gene (Cohen and Kimchi, 2001). Furthermore, DAPK is capable of suppressing oncogenic transformation induced by c-myc and E2F in vitro (Raveh et al, 2001), and blocking tumor metastasis in vivo (Inbal et al, 1997).
The mechanism underlying the proapoptotic function of DAPK has not been completely unraveled. Activation of a p53-mediated apoptotic pathway was found to account for its apoptosis inducibility (Raveh et al, 2001). Accordingly, several p53-deficient cell lines escape from apoptosis but undergo autophagic death in response to DAPK overexpression (Inbal et al, 2002). Recently, we demonstrated that DAPK inactivates integrin through an inside-out mechanism, which subsequently blocks matrix survival signal, thereby activating p53 and inducing an anoikis-type apoptosis (Wang et al, 2002). The catalytic activity of DAPK is absolutely required for all the biological functions of this kinase, including anoikis, whereas deletion of DD greatly impairs but not completely abolishes the death-promoting function of DAPK (Cohen et al, 1999; Cohen and Kimchi, 2001; Kuo et al, 2003). Notably, overexpression of DD alone protects cells from apoptosis induced by the full-length protein (Raveh et al, 2000), suggesting that protein–protein interactions through this domain could positively regulate the proapoptotic activity of DAPK. However, the interacting partner of the DD of DAPK has not been identified.
The extracellular signal-regulated kinase (ERK) family of mitogen-activated kinase (MAPK) is activated by mitogens through a pathway involving Ras, Raf and MEK (Cobb, 1999; Schaeffer and Weber, 1999; Chang and Karin, 2001). Once activated, ERK translocates from the cytoplasm to the nucleus, which is critical for the fulfillment of many cellular functions of ERK, such as gene transcription, cell proliferation and differentiation (Howe et al, 2002). Thus, subcellular localization of ERK is a significant factor in determining the biological responses of ERK pathway. ERK subcellular localization is regulated by interactions with various proteins. For example, MEK1 is responsible for both cytoplasmic retention of ERK prior to its activation and nuclear export after its dephosphorylation (Fukuda et al, 1997; Adachi et al, 2000). Furthermore, interactions with several cytoplasmic and nuclear proteins promote ERK cytoplasmic and nuclear retentions, respectively (Volmat and Pouyssegur, 2001). In addition, ERK nuclear translocation is also regulated by cell adhesion status (Danilkovitch et al, 2000; Aplin et al, 2001) through an integrin-dependent mechanism (Aplin et al, 2001).
The biological consequence of ERK activation in a given cell is determined in part by the cell-specific combination of downstream substrates. Substrate recognition by ERK and other MAPKs is achieved by interactions with conserved docking sequences in the substrates. These sequences, which are distinct from the phosphoacceptor residues, are responsible for increasing substrate phosphorylation efficiency and for providing specificity (Holland and Cooper, 1999; Sharrocks et al, 2000). One class of the docking sites, termed D-domain, is also found in MAPK upstream kinases, MAPK phosphatases and MAPK scaffolds (Sharrocks et al, 2000; Enslen and Davis, 2001). Mutagenesis studies reveal that a conserved domain (termed CD domain), which lies in the near C-terminal region outside of the catalytic domain of ERK, is required for binding D-domain-containing proteins (Tanoue et al, 2000). In addition, studies on the structures of p38 MAPK (Chang et al, 2002) and ERK2 (Lee et al, 2004) bound to D-domain peptides reveal a docking groove in these kinases responsible for interacting with the D-domain.
Here we report that DAPK interacts with ERK through a D-domain within its DD and is a substrate of ERK. Phosphorylation of DAPK by ERK increases the catalytic activity of DAPK. Conversely, DAPK promotes the cytoplasmic retention of ERK, thereby inhibiting ERK signaling in the nucleus. This reciprocal regulation between ERK and DAPK plays a positive and physiological role in apoptosis regulation. Together, our results suggest that bidirectional signaling between DAPK and ERK may be one mechanism that contributes to the apoptosis-promoting function of the DD of DAPK.
Results
DAPK interacts with ERK through the DD of DAPK
In a search for potential partner of the DAPK DD, we carried out a yeast two-hybrid screen of a human placenta cDNA library using the DD of DAPK as bait. Both ERK1 and ERK2 were recovered from this screen as strongly positive clones. The specificity of interaction between ERK and DAPK DD was verified by one-on-one transformation (Figure 1A). To examine this interaction further, we expressed the DD of DAPK as a GST fusion protein (GST-DD) and tested its ability to pull down in vitro-translated ERK1 and ERK2. GST-DD, but not GST, efficiently interacted with ERK1 and ERK2 (Figure 1B), suggesting direct associations of ERK1/2 with this DD. To determine if full-length DAPK interacts with ERK, 293T cells were transfected with Flag-tagged DAPK together with ERK1 or ERK2. Western blotting of the anti-Flag immunoprecipitates from lysates of transfected cells revealed co-precipitations of ERK1 and ERK2 with DAPK (Figure 1C). Furthermore, the interaction of endogenous DAPK with endogenous ERK in 293T cells (Figure 1D) and HeLa cells (data not shown) was detected by a reverse immunoprecipitation assay with the anti-ERK antibody followed by Western blotting with the anti-DAPK antibody. To examine whether the interaction of DAPK with ERK involves a region other than the DD of DAPK, we generated a series of DAPK deletion mutants fused with GST. Pull-down analysis revealed that only the DD segment was capable of interacting with ERK (Figure 2A). Accordingly, deletion of DD almost completely abolished the interaction between DAPK and ERK, as determined by co-immunoprecipitation assays (Figure 2B). In contrast, the kinase activity of DAPK is not required for ERK interaction, as both DAPK active (ΔCaM) and kinase-dead (K42A) mutants could still bind ERK (Figure 2B). Together, our results identify ERK as a DAPK-interacting protein, and the DD of DAPK is both sufficient and necessary for this interaction.
Figure 1.

Identification of ERK1/2 as DAPK-binding proteins. (A) Yeast strain L40 cotransformed with Gal- and LexA-based fusion constructs was assayed for β-gal activity or His3 phenotype (−His). (B) In vitro-translated, 35S-labeled ERK1 or ERK2 was incubated with either GST or GST-DD. The pull-down products and 5% amount of input ERK1/2 were analyzed by autoradiography (upper panel). The lower panel indicates the equal input of GST and GST-DD in pull-down reactions. (C) 293T cells were transfected with ERK1 or ERK2, together with Flag-DAPK (F-DAPK) or a control plasmid (−). Cell lysates were subjected to immunoprecipitations followed by Western blot with antibodies as indicated. The amounts of ERK1/2 in cell lysates are shown on the bottom. As the ERK antibody used in this study detects ERK2 more efficiently than ERK1, a strong ERK2 band, corresponding to the endogenous ERK2, was seen in the ERK1 transfectants. (D) DAPK and ERK interact endogenously. Lysates of 293T cells were used for immunoprecipitations with control IgG or anti-ERK and followed by immunoblot with anti-DAPK or anti-ERK.
Figure 2.

Mapping the ERK binding region in DAPK. (A) Lysates of 293T cells were incubated with GST-DAPK deletion mutants as illustrated in the upper panel. The pull-down products were assayed by Western blot with anti-ERK. The equal inputs of GST fusion proteins are shown on the bottom (input). (B) 293T cells transfected with various Flag-tagged DAPK constructs were subjected to immunoprecipitations and Western blot with antibodies as indicated. The amounts of ERK and Flag-DAPK proteins in lysates are shown on the bottom. (C) Alignment of the ERK docking sequences found in DAPK DD and several other ERK substrates. In the sequence of DAPK, the first amino acid listed is numbered. Conserved residues are highlighted. The feature of consensus D-domain sequence is indicated on the bottom; ϕ and X represent a hydrophobic amino acid and any amino acid, respectively. (D) GST fusion proteins immobilized on beads were incubated with equal amounts of 293 lysates. The pull-down products were analyzed by Western blot with anti-ERK2 antibody. The lower panel shows an equal input of the GST and GST fusion proteins in pull-down reactions.
Recruitment of ERK to its binding partners often requires conserved docking sequences (Holland and Cooper, 1999; Sharrocks et al, 2000; Enslen and Davis, 2001). Examination of the sequence of DAPK DD revealed the existence of such sequence (D-domain) (Figure 2C). To determine whether the integrity of D-domain is required for ERK–DAPK interaction, GST pull-down analysis was performed using wild-type or mutant GST-DD proteins. In GST-DDm1 and GST-DDm2 mutants, the consensus LXL and RR residues were replaced by AXA and AA, respectively, whereas GST-DDm1/2 carried mutations in both LXL and RR sequences. Compared to wild-type DAPK DD, mutation of either RR or LXL residues markedly reduced binding to ERK, and mutation of both sequences completely abrogated binding (Figure 2D). Thus, the ERK–DAPK interaction is mediated through a specific ERK docking sequence present in the DD of DAPK.
ERK stimulates the catalytic activity of DAPK
Having identified a physical interaction between DAPK and ERK, we next determined their functional relationships. Our previous studies revealed an elevation of DAPK catalytic activity in response to serum stimulation (Kuo et al, 2003). To test whether this effect involves ERK, 293T cells expressing Flag-DAPK were pretreated with the MEK inhibitor PD98059 followed by serum stimulation, and DAPK catalytic activity was assessed by its ability to phosphorylate the regulatory light chain of myosin II (MLC), a DAPK substrate both in vitro and in vivo (Kuo et al, 2003; Bialik et al, 2004). In vitro kinase assay using the Flag-DAPK immunoprecipitated from cell extract revealed that PD98059 blocked serum-induced activation of DAPK (Figure 3A). Similar results were obtained by coexpressing Flag-DAPK with myc-MKP3 (Figure 3A), a dual-specificity phosphatase specifically deactivating ERK1/2 (Muda et al, 1996). The steady-state kinetic parameters for DAPK isolated from differentially treated cells were determined by measuring the rate constant of phosphorylation at various concentrations of substrate. Although the Vmax and Kcat values were comparable at each condition, serum stimulation led to a two-fold decrease of the Km value, which was reversed by either PD98059 or MKP3 (Figure 3B). To demonstrate directly the induction of DAPK catalytic activity by ERK, 293T cells were cotransfected with Flag-DAPK and a constitutively active MEK1 mutant followed by serum starvation. Expression of this MEK1 mutant indeed induced an increase in the DAPK catalytic activity (Figure 3C and Supplementary Figure S1). However, this activation of DAPK was not observed with a DAPK mutant lacking its DD (DAPKΔDD) (Supplementary Figure S1). As DD mediates the interaction between DAPK and ERK, this result suggests that activation of DAPK by ERK requires their direct association.
Figure 3.

ERK upregulates DAPK catalytic activity. (A) Serum-starved 293T cells transfected with Flag-DAPK and/or MKP3 were pretreated with 50 μM PD98059 and/or stimulated with or without serum for 15 min. Lysates with equal amounts of proteins were subjected to immunoprecipitations followed by in vitro kinase assays with MLC as a substrate or by Western blot with anti-Flag. Cell lysates were also used for Western blot analysis to detect the expression of various proteins (bottom panels). (B) Double reciprocal (Lineweaver–Burk) plots for analyzing the kinetic parameters of MLC phosphorylation catalyzed by DAPK or DAPKS735D isolated from transfected 293T cells at various conditions. The initial velocities (V0) were measured using 1 μg DAPK at increasing substrate concentrations, and kinetic analysis was performed as described in Materials and methods. (C) 293T cells transfected with various constructs were serum-starved and then lysed. DAPK kinase activity was assayed as in (A). The expression of various proteins in cell lysates is shown in the bottom panels.
DAPK is a substrate of ERK
Next, we investigated the mechanism by which ERK induces DAPK activity. As this effect presumably requires a direct interaction of the two proteins, one possibility is that phosphorylation of DAPK by ERK enhances the kinase activity of DAPK. Notably, DAPK possesses a serine-containing peptide (PPSP, at amino acids 733–736) that resembles the reported consensus site PXS/TP for phosphorylation by ERK, and this peptide is conserved in mouse DAPK (Jin et al, 2001). To test the possible phosphorylation of DAPK by ERK, we performed in vitro kinase assays using purified and active ERK2 as the enzyme and various recombinant DAPK proteins purified from baculovirus as the substrates. To distinguish DAPK phosphorylation by ERK from DAPK autophosphorylation, the kinase-dead K42A mutant was used. As shown in Figure 4A, the K42A mutant was phosphorylated by ERK2 in a dose-dependent manner. This phosphorylation, however, was greatly impaired in the K42A/S735A double mutant. This result not only identifies DAPK as a direct substrate of ERK but also indicates Ser 735 as the principal site for phosphorylation by ERK. The time-course and stoichiometry analysis of phosphorylation for DAPK by ERK2 indicates that up to 73% of the K42A mutant was phosphorylated by ERK2 in this kinase assay (Supplementary Figure S2). To demonstrate DAPK as an in vivo substrate of ERK, we generated an antiserum to a phosphopeptide corresponding to the region around Ser 735 of DAPK, and used it to detect phosphorylation of DAPK at Ser 735 in vivo. As shown in Figure 4B, DAPK or S735A mutant expressed in quiescent cells showed little reactivity to the anti-phospho-DAPK antibody. Activation of ERK signaling by introducing the active MEK1 significantly enhanced the recognition of wild-type DAPK, but not S735A, by this antibody, suggesting a role of ERK in phosphorylating DAPK at Ser 735 in vivo. Furthermore, the level of phosphorylated DAPK was elevated by serum stimulation, whereas either PD98059 or MKP3 was capable of blocking serum-induced DAPK phosphorylation at Ser 735 (Figure 4C). Together, these data indicate that DAPK is a substrate of ERK both in vitro and in vivo.
Figure 4.
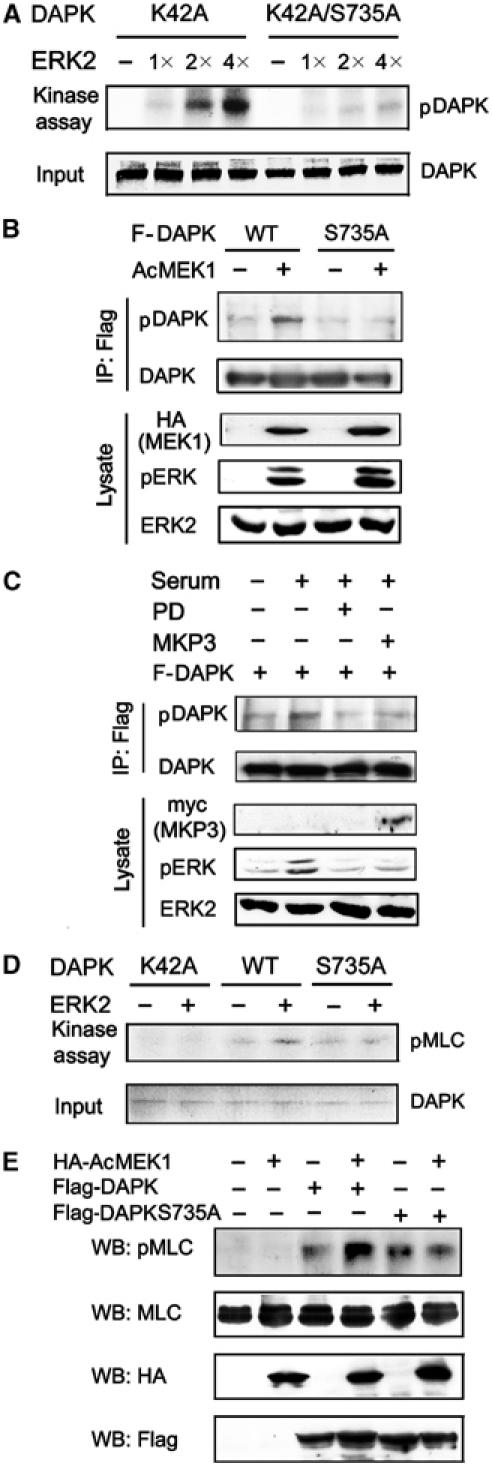
ERK activates DAPK kinase activity through a direct phosphorylation at Ser 735. (A) DAPK proteins as indicated were purified from baculovirus and used as substrates in the in vitro kinase reactions with increasing amounts of purified and active ERK2. The reactions were analyzed by autoradiography (upper) or Coomassie blue staining (lower). (B, C) 293T cells transfected with plasmids as indicated were serum-starved (B), or pretreated with or without PD98059 (PD) and stimulated with or without serum for 15 min (C). Cell lysates were subjected to immunoprecipitations with anti-Flag followed by immunoblot with anti-DAPK or anti-phospho-DAPK(S735). The expression levels of various proteins in cell lysates are shown on the bottom. (D) In vitro phosphorylation of DAPK by ERK enhances the kinase activity of DAPK. Purified DAPK or its mutants immobilized on beads were subjected to in vitro kinase reactions with or without active ERK2 as described in Materials and methods. The phosphorylated and unphosphorylated DAPK or its mutants were assayed for their kinase activities using MLC as a substrate. [γ-32P]ATP was included only in the second kinase reaction. The final reaction products were analyzed by autoradiography (upper) or Coomassie blue staining (lower). (E) 293 cells transfected with plasmids as indicated were serum-starved and lysed. Cell lysates with equal amounts of proteins were used for Western blot with antibodies as indicated.
Phosphorylation of DAPK by ERK stimulates the kinase activity of DAPK
To assess the functional consequence of DAPK phosphorylation by ERK, we compared the kinase activity of ERK-phosphorylated DAPK to that of unphosphorylated DAPK. To this end, purified DAPK or its mutants was introduced into a phosphorylation reaction in the presence or absence of purified and active ERK2. After incubation, these DAPK proteins were subjected to the second phosphorylation reaction for testing their capabilities to phosphorylate MLC in vitro. As shown in Figure 4D, the ERK-phosphorylated DAPK displayed an increased activity in MLC phosphorylation, compared to the unphosphorylated DAPK. Mutation of the Ser 735 residue of DAPK to Ala (S735A), however, abrogated this activation by ERK. The observed MLC phosphorylation was specific to DAPK, as the K42A mutant, either phosphorylated or unphosphorylated by ERK, did not result in MLC phosphorylation. To ascertain that the induction of DAPK activity by ERK indeed requires ERK phosphorylation of DAPK at Ser 735, the S735A mutant was expressed in 293T cells in the presence or absence of active MEK1, and the kinase activity of this mutant was determined by in vitro kinase assays. In contrast to the wild-type DAPK, the activity of S735A mutant could no longer be upregulated in response to ERK activation (Figure 3C). Furthermore, a phosphorylation-mimicking mutant S735D displayed a higher basal activity than the wild-type protein, and could not be further activated by active MEK1 (Figure 3C). Steady-state kinetic analysis revealed that the phosphorylation-mimicking mutation led to an ∼3.5-fold decrease of the Km value without significantly affecting the Vmax and Kcat values (Figure 3B). Thus, both ERK activation and S735D mutation resulted in a similar mode of alterations in DAPK kinetic parameters, which provides additional evidence for Ser 735 phosphorylation as the prime mechanism of ERK-induced DAPK activation. Finally, we investigated whether ERK phosphorylation of DAPK at Ser 735 stimulates DAPK catalytic activity in vivo. Antibody specifically recognizing MLC phosphorylated at Thr 18 and Ser 19 (Ratcliffe et al, 1999) was used to monitor DAPK activity in vivo, as MLC has been the only known in vivo substrate of DAPK (Kuo et al, 2003; Bialik et al, 2004). As expected, expression of DAPK in quiescent cells increased the level of phosphorylated MLC. This induction of phosphorylated MLC was further augmented by introducing the active MEK1 mutant, demonstrating the ability of ERK to promote DAPK kinase activity in vivo. However, this enhancement of DAPK in vivo activity by ERK was not observed with the S735A mutant, even though in the absence of active MEK1, this mutant induced a comparable level of phospho-MLC to the wild-type protein (Figure 4E). In conclusion, these data provide substantial evidence that phosphorylation of DAPK at Ser 735 by ERK promotes the catalytic activity of DAPK.
DAPK inhibits ERK nuclear translocation
Having demonstrated the activation of DAPK by ERK, we next explored the influence of DAPK on ERK function. Expression of DAPK, its active (ΔCaM) or dominant-negative (K42A) mutant in quiescent 293 cells (Supplementary Figure S3) or NIH3T3 cells (data not shown) did not lead to ERK activation, and did not affect MEK1-induced activation of ERK. However, DAPK attenuated MEK1-induced transcriptional activation of Elk-1 (Figure 5A) and phosphorylation of Elk-1 at Ser 383 (Figure 5B). As phosphorylation and activation of Elk-1 by ERK requires ERK nuclear translocation, we assessed whether DAPK impairs ERK translocation. In NIH3T3 cells stimulated with serum for 2 h, a significant portion of ERK was localized in the nucleus. However, in cells overexpressing DAPK, ERK was largely confined to the cytosol (Figure 5C). Indeed, in the population of cells overexpressing DAPK, only 33% displayed nuclear staining of ERK, which is in sharp contrast with the 82% observed in the population without overexpressing DAPK (Figure 5C, bottom panel). These results not only indicate that increased expression of DAPK impairs ERK nuclear translocation and nuclear signaling, but also establish a reverse direction of regulation between ERK and DAPK.
Figure 5.
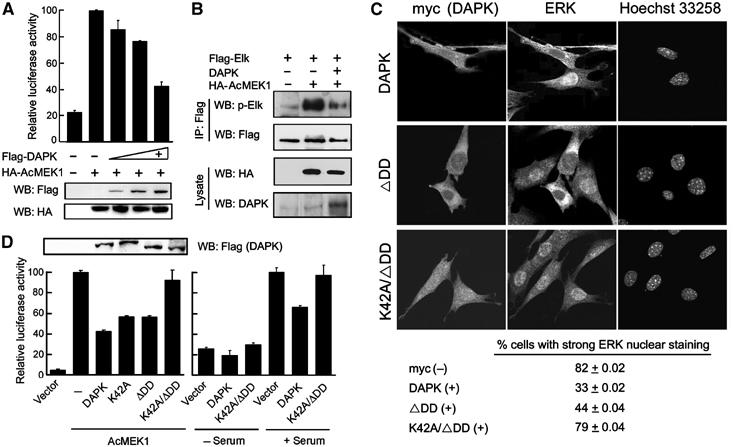
DAPK blocks ERK nuclear translocation and nuclear signaling. (A) DAPK blocks ERK signaling to Elk-1 activation. NIH3T3 cells were transfected with Gal4-Elk-1, Gal4-luciferase, pRK5β-Gal, and AcMEK1 (+) or a control vector (−), together with increasing amounts of Flag-DAPK or a control vector (−). Cells were serum-starved and lysed for luciferase and β-gal assays and Western blot analysis. (B) NIH3T3 cells transfected with plasmids as indicated were serum-starved. Cell lysates were subjected to immunoprecipitations and Western blot with antibodies as indicated. The levels of AcMEK1 or DAPK in cell lysates are shown on the bottom. (C) NIH3T3 cells transfected with myc-tagged DAPK or its mutants as indicated were serum-starved and then stimulated with serum for 2 h. Cells were triple stained with anti-myc, anti-ERK and Hoechst 33258, and visualized by confocal microscopy. The percent of cells showing stronger ERK nuclear staining than the cytoplasmic staining was quantitated and is listed on the bottom. The values shown are means±s.d. from three independent experiments, and at least 300 cells were counted for each population in each experiment. (D) NIH3T3 cells were cotransfected with Gal4-Elk-1, Gal4-luciferase, pRK5β-Gal, together with various DAPK constructs and/or AcMEK1. Transfectants were serum-starved or stimulated and then lysed for Elk-1 reporter assays as described in (A) and Western blot analysis (upper panel).
Next, we explored the mechanism by which DAPK interferes with ERK nuclear translocation. We reasoned that the tight association of ERK with cytoplasmically localized DAPK might prevent ERK nuclear transport. However, the noninteracting mutant DAPKΔDD could still inhibit serum-induced ERK nuclear translocation (Figure 5C) and downregulate MEK1-induced Elk-1 activation (Figure 5D, left panel), albeit with a lower efficiency, suggesting the existence of an additional mechanism. As cell adhesion is also a determining factor in the nuclear translocation of active ERK (Aplin et al, 2001), the antiadhesion function of DAPK, which requires its catalytic activity (Wang et al, 2002), may in part contribute to the ERK sequestration effect of DAPK. Indeed, the kinase-dead K42A mutant partially alleviated the inhibition of Elk-1 activation, and the K42A/ΔDD double mutant almost completely relieved this blockage (Figure 5D, left panel). Consistently, confocal analysis revealed that the K42A/ΔDD mutant could no longer sequester ERK in the cytoplasm in serum-stimulated cells (Figure 5C). Consequently, serum-induced Elk-1 activation was not affected by expression of K42A/ΔDD, in contrast to the effect of wild-type protein (Figure 5D, right panel). Together, these results suggest that DAPK-induced cytoplasmic retention of ERK involves both interaction-dependent and kinase activity-dependent mechanisms.
ERK promotes the anoikis inducibility of DAPK
The inhibition of ERK nuclear translocation by DAPK, combined with the upregulation of DAPK kinase activity by ERK, establishes a bidirectional regulatory circuit between the two proteins. We hypothesize that this ERK–DAPK interplay would constitute a positive feedback loop to promote the function of DAPK via two mechanisms. First, blockage of ERK nuclear translocation would increase the cytoplasmic level of active ERK, thereby further potentiating the catalytic activity and subsequently apoptotic effect of DAPK. Second, although ERK signaling is generally considered to be antiapoptotic, DAPK might interfere with ERK survival signal by blocking the phosphorylation of its nuclear targets. In these regards, increased expression of DAPK would stimulate the formation of this feedback loop, which might ultimately switch the function of ERK signaling from prosurvival to proapoptotic. To test this model, we investigated the capability of ERK to promote the apoptosis induced by increased expression of DAPK. As our previous study revealed that DAPK functions as an anoikis inducer by blocking cell adhesion (Wang et al, 2002), we first examined the effect of ERK signaling on the antiadhesion function of DAPK. NIH3T3 cells, which are sensitive to the antiadhesion and proapoptotic activities of DAPK (Wang et al, 2002), were used for expressing various DAPK proteins and MEK1 (Figure 6A). Expression of wild-type DAPK or S735A in quiescent NIH3T3 cells resulted in a similar reduction in cell adhesion on fibronectin (Figure 6B). In cells expressing DAPK, but not S735A, activation of ERK by introducing the active MEK1 further potentiated this antiadhesion function. A similar result was observed in 293T cells (data not shown). As inhibition of cell adhesion by DAPK was found to trigger the induction of p53 and apoptosis (Wang et al, 2002), we next tested the effect of ERK on these activities of DAPK. Indeed, ERK activation significantly enhanced the p53 inducibility of DAPK, but displayed little effect on that of the S735A mutant (Figure 6C). As a consequence, the apoptosis-promoting activity of DAPK, but not the S735A mutant, was markedly potentiated in response to ERK activation (Figure 6D). Thus, in cells with elevated expression of DAPK, ERK signaling stimulates, rather than inhibits, the anoikis inducibility of DAPK. If phosphorylation of DAPK at Ser 735 by ERK does contribute in part to DAPK-mediated apoptosis, the phosphorylation-mimicking S735D mutant would be expected to display higher apoptosis inducibility than the wild-type protein in cells without receiving active MEK1. Indeed, when overexpressed in quiescent NIH3T3 cells, the S735D mutant exhibited significantly higher proapoptotic potency than the wild-type protein (Figure 6E), consistent with its higher catalytic activity.
Figure 6.

ERK activation stimulates the anoikis inducibility of DAPK. (A) NIH3T3 cells transfected with GFP together with various DAPK and AcMEK1 constructs were assayed by Western blot with antibodies as indicated. (B) Cells as in (A) were serum-starved and then assayed for their adhesion on fibronectin as described in Materials and methods. (C) NIH3T3 cells were cotransfected with p53-TA-luc and pRK5-βgal in the presence or absence of various DAPK constructs and AcMEK1, and then subjected to luciferase assays. (D, E) NIH3T3 cells transfected with various constructs were serum-starved and assayed for apoptosis using Cell Death-Detection ELISA. In (E), the expression levels of endogenous and exogenous DAPK are shown on the bottom. (F) DAPK or its mutants was introduced into MCF10A cells via retroviral-mediated gene transfer (bottom panels). Cells were seeded on plates coated with 25 ng/μl fibronectin and cultured in EGF-deprived medium for 16 h. Apoptosis was assayed as in (D).
Notably, because fibroblasts undergo anoikis only when serum is deprived (Almeida et al, 2000), in the experiments with NIH3T3 cells described above, the influence of ERK on the anoikis inducibility of DAPK was assessed in the absence of serum and ERK activation was achieved by ectopic expression of the active MEK1. To explore the role of ERK–DAPK interplay in DAPK-induced anoikis under physiological conditions, we utilized a human mammary epithelial cell line, MCF10A, in which DAPK-induced anoikis is readily detected in the presence of serum (Wang et al, 2002). To assess the anoikis inducibility of DAPK, cells were plated on fibronectin and incubated in medium with serum but without the EGF supplement, as the presence of a high concentration of EGF attenuates anoikis (Wang et al, 2002). Consistent with our previous study (Wang et al, 2002), expression of DAPK led to an induction of apoptosis in this system. Importantly, deletion of DD and mutation of the ERK phosphorylation site caused a similar reduction in this apoptotic effect (Figure 6F), suggesting that ERK–DAPK interplay contributes to a major part of the functional mechanism of the DD of DAPK.
Enhanced DAPK–ERK interaction promotes apoptosis
Having demonstrated that ERK–DAPK interplay promotes the apoptotic effect of DAPK in cells overexpressing DAPK, we next examined the physiological relevance of this finding. In particular, we investigated the role of endogenous ERK–DAPK interaction in apoptosis induced by a physiological stimulus. The phorbol-12-myristate-13-acetate (PMA)-treated human erythroblastic cell line D2, a cytokine-independent variant of TF-1 cell, was chosen for the following reasons. First, this cell line contained a high level of endogenous DAPK, compared with many other human cell lines of various origins (Supplementary Figure S4). Second, similar to the parental TF-1 cells, D2 cells can undergo distinct cell fates in response to PMA. When cultured in serum-free medium and exposed to PMA, D2 cells rapidly become attached and remain alive. However, if cell attachment is blocked by plating onto hydrogel-coated dishes, PMA treatment leads to massive apoptosis (Lai et al, 2002, 2003). Interestingly, ERK nuclear translocation induced by PMA was only observed in the adherent but not suspended population (Lai et al, 2002; Figure 7A), showing a tight correlation between ERK cytoplasmic retention and apoptosis induction. As DAPK was mainly localized in the cytoplasmic compartment in both adherent and suspended cells (Figure 7A), we suspected a more efficient ERK–DAPK complex formation in the suspended than adherent population. Indeed, immunoprecipitation analysis revealed a higher level of endogenous DAPK co-precipitated with endogenous ERK in the detached cells than adherent cells (Figure 7B). Furthermore, PMA induced a higher DAPK catalytic activity in detached than adherent cells (Figure 7C, left panel). This higher DAPK activity in suspended cells is most likely due to an enhanced DAPK–ERK complex formation, as inhibition of ERK activation by U0126 significantly suppressed PMA-induced DAPK activation in suspended cells (Figure 7C, right panel). To determine whether the DAPK–ERK interplay in detached cells plays any role in the cytoplasmic retention of ERK, we utilized RNA interference technology to knock down endogenous DAPK. DAPK expression was significantly reduced in cells treated with DAPK-specific siRNA1 and siRNA2, whereas the control siRNA did not affect DAPK expression (Figure 7D). Under this circumstance, the nonadherent population receiving either DAPK siRNA, but not control siRNA, displayed a partial restoration of ERK nuclear distribution in response to PMA. As expected, DAPK siRNA did not affect ERK subcellular localization in the attached population (Figure 7E). Together, these results demonstrate that both endogenous ERK–DAPK interaction and their reciprocal regulation are selectively enhanced in the suspended population. Next, we investigated whether this enhanced interplay between ERK and DAPK could contribute at least in part to the apoptotic fate of suspended population. As either blockage of ERK activation or downregulation of DAPK expression would interfere with the reciprocal regulation of the two proteins, we tested the effect of U0126 and DAPK siRNAs on the apoptosis of PMA-treated D2 cells. As shown in Figure 7F, U0126 greatly suppressed PMA-induced caspase-3 activity, a hallmark of apoptosis, in the suspended cells without affecting the survival of adherent cells. Furthermore, both DAPK siRNAs, but not the control siRNA, efficiently blocked PMA-induced apoptosis in the detached cells, whereas adherent population was not affected by siRNA treatment (Figure 7G). These findings not only indicate that the enhanced ERK–DAPK interplay and their reciprocal regulation play a crucial role in determining the apoptotic fate of D2 cells, but also highlight a physiological role of the ERK–DAPK regulatory circuit in promoting apoptosis.
Figure 7.
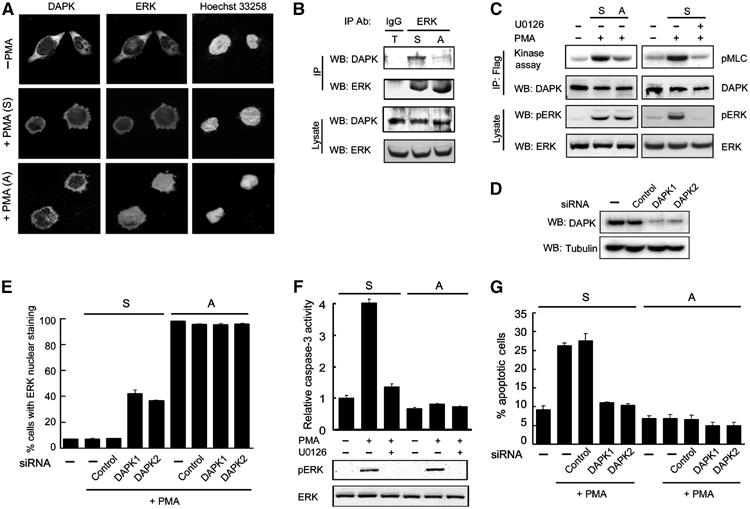
DAPK and ERK are both required for apoptotic death of cells with elevated ERK–DAPK interaction. (A) D2 cells were seeded on culture dishes treated with (+PMA (A)) or without (−PMA) PMA in serum-free condition for 3 h, or seeded on hydrogel-coated dishes followed by PMA treatment (+PMA (S)). Cells were triple stained by anti-DAPK, anti-ERK and Hoechst 33258, and visualized by confocal microscopy. (B) PMA-treated attached (A) and suspended (S) D2 cells as described in (A) and 1:1 mixture of both populations (T) were used for immunoprecipitation followed by Western blot analyses with antibodies as indicated. ERK and DAPK expressions in each population are shown on the bottom. (C) PMA induces a higher DAPK activity in suspended cells. Flag-DAPK-transfected D2 cells plated as in (A) were pretreated with or without 20 μM U0126, and then exposed to PMA for 3 h. DAPK catalytic activity was assayed as in Figure 3. The levels of ERK and pERK in lysates are shown on the bottom. (D) D2 cells were transfected with or without (−) DAPK siRNA or control siRNA, together with a CD2 marker plasmid. Transfected cells were isolated as described in Materials and methods and lysed for Western blot analysis. (E) D2 cells transfected with or without various siRNAs together with GFP were cultured and treated as in (A). The percentage of GFP-positive cells with ERK nuclear staining was quantitated. (F) Suspended or attached D2 cells were pretreated with 20 μM U0126 and then exposed to PMA or DMSO (−) for 6 h. Cells were harvested for analyzing the activity of caspase-3 (upper panel) or the status of ERK activation (bottom panels). (G) Knockdown of endogenous DAPK protects against PMA-induced apoptosis in suspended cells. Suspended or attached D2 cells transfected as in (E) were treated with or without PMA for 6 h. Cells were assayed for apoptosis with Annexin V staining as described in Materials and methods.
Discussion
The DD of DAPK positively regulates the proapoptotic function of this kinase (Cohen et al, 1999; Cohen and Kimchi, 2001). In this report, we have not only identified an interacting partner but also unraveled the functional mechanism of the DAPK DD. We found that DAPK binds ERK both in vitro and in vivo and this interaction is mediated by a docking sequence in the DD of DAPK. As illustrated in Figure 8, this DAPK–ERK interplay promotes the proapoptotic function of DAPK through two mechanisms. First, ERK functions as an upstream activating kinase for DAPK by phosphorylating DAPK at Ser 735. Second, DAPK promotes the cytoplasmic retention of ERK, which in one way further potentiates the phosphorylation and activation of the cytoplasmically localized DAPK and in another way could possibly impair ERK survival signals (see below). This reciprocal regulation between ERK and DAPK constitutes a positive feedback loop, thereby promoting the apoptotic effect of DAPK. Several lines of evidence support this conclusion. First, ERK, which is generally considered to be antiapoptotic, is capable of stimulating the proapoptotic function of DAPK in cells overexpressing DAPK. Second, this ERK–DAPK synergism is blocked by mutating a single residue in DAPK that prevents ERK phosphorylation. Third, deletion of DD and mutation of the ERK phosphorylation site cause a similar reduction in the apoptotic inducibility of DAPK. Finally and most importantly, in a PMA-induced apoptosis system in which the ERK–DAPK interaction and their reciprocal regulation are selectively stimulated, downregulation of either endogenous ERK or endogenous DAPK blocks such apoptosis, thus providing physiological evidence for a crucial role of ERK–DAPK regulatory circuit in promoting the apoptotic effect of DAPK. We conclude that recruitment of ERK and subsequent establishment of the ERK–DAPK two-way regulation circuit accounts for at least one mechanism by which the DAPK DD promotes apoptosis. In this regard, the DD of DAPK is capable of regulating apoptosis through a heterotypic mode of protein–protein interactions.
Figure 8.

Scheme depicting the promotion of apoptosis through a feedback regulatory circuit formed by DAPK–ERK interplay (see text).
The identification of the proapoptotic DAPK as a downstream target of ERK seems to conflict with the well-documented antiapoptotic effect of ERK signaling initiated by numerous stimuli (Ballif and Blenis, 2001). However, our demonstration that DAPK prevents ERK nuclear translocation without affecting its activation indicates that DAPK could alter the signaling outputs of ERK and may play an inhibitory role in its survival function. One well-characterized ERK survival pathway involves its phosphorylation and activation of the ribosomal S6 kinase (RSK) family kinases. Activated RSKs in turn phosphorylate the proapoptotic BAD and the prosurvival CREB, thereby inactivating and stimulating their functions, respectively (Ballif and Blenis, 2001). Although RSKs are largely cytosolic, the MSK subgroup of RSK family is constitutively located in the nucleus (Deak et al, 1998), thus requiring nuclear ERK for its activation. Therefore, sequestration of ERK in the cytoplasm could at least partially impair ERK survival signal. Accordingly, the antiapoptotic effect of ERK is abrogated in cells cultured in suspension, where ERK nuclear translocation is greatly impaired (Danilkovitch et al, 2000). In addition, cytoplasmic sequestration of ERK was previously found to correlate with the apoptotic inducibility of PMA in TF-1 and D2 cells (Lai et al, 2002). In this study, we further demonstrate that such cytoplasmic retention of ERK results in an elevated ERK–DAPK complex formation, which represents a key mechanism in determining the apoptotic fate of D2 cells. Notably, Rho-kinase and its induction of MLC phosphorylation were found to play a crucial role in restricting ERK in the cytoplasm in D2 cells (Lai et al, 2002, 2003). Consistently, DAPK, which similarly phosphorylates MLC, also contributes in part to the cytoplasmic distribution of ERK.
Our mutagenesis study suggests that DAPK restricts ERK nuclear transport through both interaction-dependent and kinase activity-dependent mechanisms. Thus, elevation of either DAPK expression or catalytic activity would not only by itself increase the proapoptotic signaling of DAPK, but also promote ERK cytoplasmic sequestration, thereby reinforcing the ERK–DAPK regulatory circuit, and consequently leading to the amplification of this proapoptotic signal. Importantly, upregulation of DAPK expression and/or catalytic activity has been reported to occur during apoptosis induced by a number of stimuli, such as ceramide, TGF-β, and oncogenes E2F and c-myc (Raveh et al, 2001; Jang et al, 2002; Pelled et al, 2002). Conceivably, in these circumstances, the ERK–DAPK two-way regulation circuit would facilitate the amplification of apoptotic signals and might thus play a significant role in these apoptotic paradigms. On the contrary, in normal cells without receiving apoptotic stimuli or overexpressing DAPK, ERK–DAPK interplay does not lead to apoptosis. Perhaps in cells with a low expression level or catalytic activity of DAPK, the ERK–DAPK regulatory circuit could not generate sufficient proapoptotic signal to overcome the prosurvival signals elicited by ERK and other antiapoptotic proteins. Furthermore, as ERK undergoes nuclear translocation upon activation, only a small portion of active ERK retaining in the cytoplasm is capable of activating DAPK via their interaction, thus limiting the apoptotic signal elicited by the ERK–DAPK circuit. However, under certain physiological conditions in which ERK cytoplasmic retention is promoted (e.g., the suspended D2 cells receiving PMA), the elevated ERK–DAPK interaction and their reciprocal regulation indeed contribute to apoptotic death of such cells. It would be interesting to determine whether upregulation of ERK–DAPK interaction contributes to other ERK-mediated apoptosis.
Materials and methods
Yeast two-hybrid screen
The DNA fragment encoding the DAPK DD was cloned to BTM116 to produce the LexA-DD for screening a human placenta cDNA library fused to the Gal4 activation domain. Yeast two-hybrid screen was performed as described (Vojtek and Hollenberg, 1995) and a total of 5 × 107 transformants were screened. Positive clones were selected on plates lacking uracil, histidine, tryptophan, lysine and leucine, followed by β-galactosidase (β-gal) assays and then verified by one-on-one transformation assays.
Antibodies
The polyclonal antibodies to DAPK (Jang et al, 2002) and phosphorylated MLC (Ratcliffe et al, 1999) were described previously. The phospho-DAPK(S735) antibody was produced by LTK Biolab Inc. (Taiwan) using the phosphopeptide TNSSRFPPpSPLASKPTV (corresponding to residues 727–742 of DAPK) to immunize rabbit. To enrich antibodies that specifically recognize phosphorylated DAPK, the antiserum was diluted and incubated with unphosphorylated, bacterially expressed DAPK immobilized on the PVDF membrane. The unbound antibodies were collected and used for Western blot analysis.
GST fusion proteins and pull-down analysis
Various segments of DAPK cDNA were cloned to pGEX-4T vector to generate GST fusion proteins. Production and purification of the GST fusion proteins were performed as described (Tsai et al, 2000). For pull-down analysis, equal amounts of GST fusion proteins immobilized on beads were incubated at 4°C for 2 h with cell lysates or various 35S-labeled proteins generated by in vitro transcription and translation (TNT transcription–translation system, Promega) in GST binding buffer containing 50 mM Hepes (pH 7.5), 1% NP-40, 50 mM NaCl, 10 mM EDTA, 20% glycerol, 1 mM PMSF, 1 μg/ml aprotonin and 1 μg/ml leupeptin. The beads were washed, and proteins bound on beads were analyzed by autoradiography or Western blot.
Phosphorylation of DAPK in vitro
A 4 μg portion of DAPK protein purified from baculoviral expression system was incubated at 25°C for 15 min in the presence or absence of purified and active ERK2 (0.05–0.2 μg; UBI) in 30 μl of kinase buffer containing 20 mM MOPS (pH 7.2), 25 mM β-glycerol phosphate, 5 mM EGTA, 1 mM DTT, 0.5 mM Na3VO5, 5 mM NaF, 20 μM cold ATP and 5 μCi [γ-32P]ATP. To determine the stoichiometry of phosphorylation of DAPK42A by ERK2, 0.4 μg of DAPK42A immobilized on protein G beads was incubated with 0.1 μg of active ERK2 in 30 μl of kinase reaction mixture described above.
DAPK kinase assay
DAPK activity was assayed as described (Kuo et al, 2003). Briefly, various DAPK proteins immunoprecipitated by anti-Flag antibody were incubated with MLC kinase buffer containing 50 mM Hepes (pH 7.5), 8 mM MgCl2, 2 mM MnCl2, 0.5 mM CaCl2, 50 μM ATP, 10 μCi [γ-32P]ATP, 0.1 mg/ml bovine serum albumin (BSA), 1 μM bovine CaM (Sigma) and 3 μg GST-MLC (Kuo et al, 2003) at 25°C for 10 min (enzyme concentration was adjusted to ensure linear kinetics at this time). For measuring steady-state kinetic parameters, 1 μg of DAPK was used to catalyze increasing amounts of GST-MLC (0.7–8.4 μM) in 25 μl of kinase reaction mixture described above.
Adhesion, p53 reporter and apoptosis assays
Cells cotransfected with various DAPK constructs, active MEK1 and GFP at a ratio of 5:5:1 were assayed for adhesion as described (Wang et al, 2002). Briefly, quiescent cells were allowed to adhere on plates coated with 10 ng/ml of fibronectin for 30 min at 37°C. To estimate the reference value for 100% attachment, cells were seeded on plates precoated with 20 ng/ml of fibronectin and incubated for 3–4 h at 37°C. After incubation, GFP-positive cells bound onto the experimental and reference plates were counted to determine the percent of adhesion. For p53 reporter and apoptosis assays, quiescent cells were plated on fibronectin and cultured in serum-free medium for another 6 h. Both attached and detached cells were harvested and combined for determining p53 reporter activity or apoptotic cells by Cell Death-Detection ELISA as described (Wang et al, 2002). For assaying apoptosis in D2 cells, cells were transfected with siRNA and GFP construct at a ratio of 5:1. At 12 h after transfection, cells were reseeded on regular or hydrogel-coated dishes and treated with 32 nM of PMA for 6 h. Cells were stained by phycoerythrin-conjugated Annexin V (BD Biosciences). The percentage of GFP-positive cells with Annexin V staining was analyzed by flow cytometry. Alternatively, D2 cells treated with various reagents were assayed for caspase-3 activity as described (Jang et al, 2002).
RNA interference
DAPK siRNAs and control siRNA were purchased from Dharmacon RNA Technologies. The siRNA was transfected into D2 cells together with a CD2 marker plasmid. Transfected cells were isolated at 48 h after transfection by the CELLection CD2 kit (DNYNAL Biotech).
Supplementary Material
Supplementary Material
Acknowledgments
We thank Andrew Sharrocks (The University of Manchester, UK), James Staddon (University College London, UK) and Hsiu-Ming Shih (National Health Research Institute, Taiwan) for reagents, Rik Derynck (UCSF, USA) for critical reading of the manuscript, Wei Ku for technical assistance and Jin-Mei Lai and Hsiao-Hui Lee for instructions on D2 cell experiments. This work was supported by NSC Frontier Grant NSC92-2321-B-002-017.
References
- Adachi M, Fukuda M, Nishida E (2000) Nuclear export of MAP kinase (ERK) involves a MAP kinase kinase (MEK)-dependent active transport mechanism. J Cell Biol 148: 849–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida EAC, Ilic D, Han Q, Hauck CR, Jin F, Kawakatsu H, Schlaepfer DD, Damsky CH (2000) Matrix survival signaling: from fibronectin via focal adhesion kinase to c-Jun NH2-terminal kinase. J Cell Biol 149: 741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aplin AE, Stewart SA, Assoian RK, Juliano RL (2001) Integrin-mediated adhesion regulates ERK nuclear translocation and phosphorylation of Elk-1. J Cell Biol 153: 273–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballif BA, Blenis J (2001) Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ 12: 397–408 [PubMed] [Google Scholar]
- Bialik S, Brenick AR, Kimchi A (2004) DAP-kinase-mediated morphological changes are localization dependent and involve myosin-II phosphorylation. Cell Death Differ 11: 631–644 [DOI] [PubMed] [Google Scholar]
- Chang CI, Xu BE, Akella R, Cobb MH, Goldsmith EJ (2002) Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol Cell 9: 1241–1249 [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M (2001) Mammalian MAP kinase signaling cascades. Nature 410: 37–40 [DOI] [PubMed] [Google Scholar]
- Cobb MH (1999) MAP kinase pathways. Prog Biophys Mol Biol 71: 479–500 [DOI] [PubMed] [Google Scholar]
- Cohen O, Kimchi A (2001) DAP-kinase: from functional gene cloning to establishment of its role in apoptosis and cancer. Cell Death Differ 8: 6–15 [DOI] [PubMed] [Google Scholar]
- Cohen O, Feinstein E, Kimchi A (1997) DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. EMBO J 16: 998–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen O, Inbal B, Kissil JL, Raveh T, Berissi H, Spivak-Kroizaman T, Feinstein E, Kimchi A (1999) DAP-kinase participates in TNF-alpha- and Fas-induced apoptosis and its function requires the death domain. J Cell Biol 146: 141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilkovitch A, Donley S, Skeel A, Leonard EJ (2000) Two independent signaling pathways mediate the antiapoptotic action of macrophage-stimulating protein on epithelial cells. Mol Cell Biol 20: 2218–2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq LM, Alessi DR (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J 17: 4426–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A (1995) Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev 9: 15–30 [DOI] [PubMed] [Google Scholar]
- Enslen H, Davis RJ (2001) Regulation of MAP kinases by docking domains. Biol Cell 93: 5–14 [DOI] [PubMed] [Google Scholar]
- Fukuda M, Gotoh I, Adachi M, Gotoh Y, Nishida E (1997) A novel regulatory mechanism in the mitogen-activated protein (MAP) kinase cascade. Role of nuclear export signal of MAP kinase kinase. J Biol Chem 272: 32642–32648 [DOI] [PubMed] [Google Scholar]
- Holland PM, Cooper JA (1999) Protein modification: docking sites for kinases. Curr Biol 9: R329–R331 [DOI] [PubMed] [Google Scholar]
- Howe AK, Aplin AE, Juliano RL (2002) Anchorage-dependent ERK signaling-mechanisms and consequences. Curr Opin Cell Biol 12: 30–35 [DOI] [PubMed] [Google Scholar]
- Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A (2002) DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol 157: 455–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbal B, Cohen O, Polak-Charcon S, Kopolovic J, Vadai E, Eisenbach L, Kimchi A (1997) DAP kinase links the control of apoptosis to metastasis. Nature 390: 180–184 [DOI] [PubMed] [Google Scholar]
- Jang CW, Chen CH, Chen CC, Chen JY, Su YH, Chen RH (2002) TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat Cell Biol 4: 51–58 [DOI] [PubMed] [Google Scholar]
- Jin Y, Blue EK, Dixon S, Hou L, Wysolmerski RB, Gallagher PJ (2001) Identification of a new form of death-associated protein kinase that promotes cell survival. J Biol Chem 276: 39667–39678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo JC, Lin JR, Staddon JM, Hosoya H, Chen RH (2003) Uncoordinated regulation of stress fibers and focal adhesions by DAP-kinase. J Cell Sci 116: 4477–4490 [DOI] [PubMed] [Google Scholar]
- Lai JM, Hsieh CL, Chang ZF (2003) Caspase activation during phorbol ester-induced apoptosis requires ROCK-dependent myosin-mediated contraction. J Cell Sci 116: 3491–3501 [DOI] [PubMed] [Google Scholar]
- Lai JM, Wu S, Huang DY, Chang ZF (2002) Cytosolic retention of phosphorylated extracellular signal-regulated kinase and a Rho-associated kinase-mediated signal impair expression of p21(Cip1/Waf1) in phorbol 12-myristate-13-acetate-induced apoptotic cells. Mol Cell Biol 22: 7581–7592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG (2004) Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell 14: 43–55 [DOI] [PubMed] [Google Scholar]
- Muda M, Boschert U, Dickinson R, Martinou JC, Martinou I, Camps M, Schlegel W, Arkinstall S (1996) MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J Biol Chem 271: 4319–4326 [DOI] [PubMed] [Google Scholar]
- Pelled D, Raveh T, Riebeling C, Fridkin M, Berissi H, Futerman AH, Kimchi A (2002) Death-associated protein (DAP) kinase plays a central role in ceramide-induced apoptosis in cultured hippocampal neurons. J Biol Chem 277: 1957–1961 [DOI] [PubMed] [Google Scholar]
- Ratcliffe MJ, Smales C, Staddon JM (1999) Dephosphorylation of the catenins p120 and p100 in endothelial cells in response to inflammatory stimuli. Biochem J 338: 471–478 [PMC free article] [PubMed] [Google Scholar]
- Raveh T, Berissi H, Eisenstein M, Spivak T, Kimchi A (2000) A functional genetic screen identifies regions at the C-terminal tail and death-domain of death-associated protein kinase that are critical for its proapoptotic activity. Proc Natl Acad Sci USA 97: 1572–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh T, Droguett G, Horwitz MS, DePinho RA, Kimchi A (2001) DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat Cell Biol 3: 1–7 [DOI] [PubMed] [Google Scholar]
- Schaeffer HJ, Weber MJ (1999) Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol 19: 2435–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrocks AD, Yang SH, Galanis A (2000) Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem Sci 25: 448–453 [DOI] [PubMed] [Google Scholar]
- Tanoue T, Adachi M, Moriguchi T, Nishida E (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol 2: 110–116 [DOI] [PubMed] [Google Scholar]
- Tsai YT, Su YH, Fang SS, Huang TN, Qiu Y, Jou YS, Shih HM, Kung HJ, Chen RH (2000) Etk, a Btk family tyrosine kinase, mediates cellular transformation by linking Src to STAT3 activation. Mol Cell Biol 20: 2043–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojtek AB, Hollenberg SM (1995) Ras–Raf interaction: two-hybrid analysis. Methods Enzymol 255: 331–342 [DOI] [PubMed] [Google Scholar]
- Volmat V, Pouyssegur J (2001) Spatiotemporal regulation of the p42/p44 MAPK pathway. Biol Cell 93: 71–79 [DOI] [PubMed] [Google Scholar]
- Wang WJ, Kuo JC, Yao CC, Chen RH (2002) DAP-kinase induces apoptosis by suppressing integrin activity and disrupting matrix survival signals. J Cell Biol 159: 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CH, Vincenz C (2001) The death domain superfamily: a tale of two interfaces? Trends Biochem Sci 26: 475–481 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material