

Abstract
Chromosomal translocations that fuse the mixed lineage leukemia (MLL) gene with multiple partners typify acute leukemias of infancy as well as therapy-related leukemias. We utilized a conditional knockin strategy to bypass the embryonic lethality caused by MLL-CBP expression and to assess the immediate effects of induced MLL-CBP expression on hematopoiesis. Within days of activating MLL-CBP, the fusion protein selectively expanded granulocyte/macrophage progenitors (GMP) and enhanced their self-renewal/proliferation. MLL-CBP altered the gene expression program of GMP, upregulating a subset of genes including Hox a9. Inhibition of Hox a9 expression by RNA interference demonstrated that MLL-CBP required Hox a9 for its enhanced cell expansion. Following exposure to sublethal γ-irradiation or N-ethyl-N-nitrosourea (ENU), MLL-CBP mice developed myelomonocytic hyperplasia and progressed to fatal myeloproliferative disorders. These represented the spectrum of therapy-induced acute myelomonocytic leukemia/chronic myelomonocytic leukemia/myelodysplastic/myeloproliferative disorder similar to that seen in humans possessing the t(11;16). This model of MLL-CBP therapy-related myeloproliferative disease demonstrates the selectivity of this MLL fusion for GMP cells and its ability to initiate leukemogenesis in conjunction with cooperating mutations.
Keywords: granulocyte/macrophage progenitors, leukemogenesis, MLL-CBP, myeloproliferative disease
Introduction
Hematopoiesis, the highly regulated process that produces mature blood cells, depends on the coordinated proliferation, differentiation, and survival of hematopoietic stem cells (HSCs) and their committed progenitors. An additional level of regulation is imposed on the HSC, which is the only population capable of sustaining lifelong hematopoiesis by undergoing self-renewal (Spangrude et al, 1988). Distortion of any of these cellular programs is potentially oncogenic and might contribute to leukemia.
Leukemia-associated chromosomal translocations have revealed the identity of multiple transcriptional regulators that control normal hematopoiesis (Rabbitts, 1994). The mixed lineage leukemia (MLL) protein represents one such transcriptional regulator. Chromosomal rearrangements of the MLL gene typify acute leukemias of infancy as well as therapy-related leukemias (Hunger et al, 1993; Super et al, 1993; Felix et al, 1995; Behm et al, 1996). However, the critical stage of hematopoietic development targeted by each MLL fusion protein is unclear. The observation that leukemic cells bearing an MLL translocation often coexpress both myeloid and lymphoid markers raises the possibility that an MLL fusion selectively transforms HSC. If so, this HSC population would have the inherent self-renewal capacity that might be co-opted for leukemogenesis. Alternatively, the translocation event between MLL and a partner gene may affect a more committed population such as common myeloid progenitors (CMPs), granulocyte/macrophage progenitors (GMPs), or common lymphoid progenitors (CLPs). In this scenario, the MLL fusion protein might confer a self-renewal/proliferative capacity to these short-lived progenitors to initiate leukemogenesis. Finally, we require further models to define the downstream mechanism by which MLL fusion proteins disrupt the delicate balance of hematopoietic self-renewal/proliferation and differentiation to result in leukemogenesis.
MLL is involved in chromosomal translocations with more than 40 different cytogenetic loci. Of these, the t(11;16)(q23;p13) that fuses MLL to CBP (CREB binding protein) is unique because it is almost exclusively associated with therapy-related myeloproliferative diseases across the gamut from acute myelomonocytic leukemia (t-AMML), to chronic myelomonocytic leukemia (t-CMML) and myelodysplastic syndrome (t-MDS) (Rowley et al, 1997; Sobulo et al, 1997; Taki et al, 1997). This is in contrast to other MLL translocations, such as the t(9;11) and t(4;11), which usually occur in de novo acute leukemias. The high incidence of t-MDS among patients with the t(11;16) further distinguishes the MLL-CBP fusion from other therapy-related MLL rearrangements that are more restricted to t-AML or t-ALL with no preceding MDS.
The well-characterized biochemical roles of CBP provide an excellent opportunity to assess its functional contribution within the MLL-CBP fusion protein to leukemogenesis. CBP serves as an important coactivator for a diverse set of transcription factors (e.g. CREB, p53, GATA-1) (Kwok et al, 1994; Blobel et al, 1998; Van Orden et al, 1999). Coactivation depends on the ability of CBP to recruit components of the basal transcriptional machinery to promoters. It also depends on the intrinsic ability of CBP to acetylate both histones and transcription factors themselves (Bannister and Kouzarides, 1996; Gu and Roeder, 1997; Boyes et al, 1998). We hypothesize that one likely mechanism by which MLL-CBP contributes to malignant transformation may involve constitutive recruitment of CBP to MLL-regulated genes. Our finding that the activation domain of MLL normally interacts with CBP and utilizes CBP to activate transcription supports this hypothesis (Ernst et al, 2001). In one testable model, the MLL-CBP fusion would result in deregulated expression of MLL target genes that contribute to leukemogenesis.
Gene targeting experiments in mice suggest that MLL functions as a positive regulator of Hox gene expression (Yu et al, 1995, 1998). Hox genes are involved in normal hematopoiesis and leukemogenesis, making them attractive oncogenic targets for MLL fusion proteins. HOX genes are differentially expressed in hematopoietic lineages, suggesting that they may play a role in lineage commitment and maintenance (Shen et al, 1989; Magli et al, 1991, 1997). Furthermore, a number of studies have already implicated HOX genes in hematologic malignancies. Overexpression of Hox a7 and Hox a9 by proviral integration caused myeloid leukemia in BXH-2 mice (Nakamura et al, 1996; Afonja et al, 2000). In addition, the t(7;11), which fuses the Hox a9 gene with the nucleoporin gene NUP98, is implicated in a subset of acute myeloid leukemias (AMLs) (Borrow et al, 1996). Together, these findings warrant an assessment of HOX gene expression in models of MLL fusion leukemia.
In an attempt to assess the transformation capabilities of MLL-CBP, we generated an Mll-Cbp knockin allele by gene targeting. However, the standard Mll-Cbp knockin allele resulted in embryonic lethality of mice and precluded the use of this system to examine MLL-CBP in leukemogenesis (data not shown). To circumvent this limitation, here we generate mice with a conditional Mll-Cbp allele whose expression can be controlled during murine development. This allele was created by fusing the Cbp cDNA into exon 8 of Mll and inserting a transcriptional stop cassette flanked by loxP sequences just 5′ to the fusion site (Lasko et al, 1992; Higuchi et al, 2002). Subsequent excision of the stop cassette mediated by Cre recombinase then allowed us to assess directly the effect of MLL-CBP on hematopoiesis in adult mice. This conditional model of Mll-Cbp also has the advantage of more accurately mimicking the human t(11;16) event that occurs acutely in a somatic cell of hematopoietic origin rather than in the germ line. Consequently, we are able to examine the immediate effects of MLL-CBP expression on hematopoiesis, avoiding any compensatory event that might transpire in the setting of a germline knockin.
Results
Conditional Mll-Cbp knockin mice
We generated a knockin targeting construct for the conditional expression of a Cbp cDNA fused with exon 8 of the endogenous murine Mll gene. The construct was designed to recapitulate the most common type of MLL-CBP fusion found in human leukemias possessing a t(11;16) (Sobulo et al, 1997; Taki et al, 1997) (Figure 1A). A polyadenylation signal from simian virus 40 (SV40) was placed downstream of the Cbp translation stop codon, and a PGK-puromycin gene was also inserted to enable positive selection. In order to regulate expression of the fusion gene in a lineage- and temporal-specific manner, we inserted a transcriptional stop cassette flanked by loxP sites just 5′ to the site of Mll-Cbp fusion (Figure 1B). This cassette arrests transcription within its sequence, and prevented expression of full-length Mll-Cbp. Subsequent excision of the stop cassette, mediated by Cre recombinase, would enable expression of Mll-Cbp from the endogenous Mll promoter.
Figure 1.
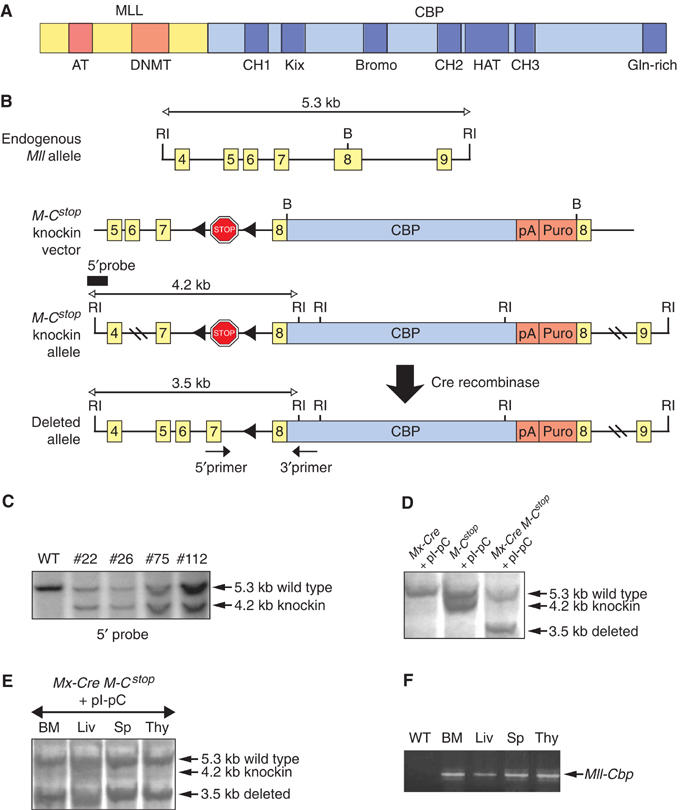
Generation of mice with a conditional Mll-Cbp knockin allele. (A) The most common MLL-CBP fusion protein associated with the human t(11;16) and its recapitulation in the Mx-Cre Mll-CbpSTOP mice. AT: AT hooks; DNMT: DNA methyltransferase homology domain; CH1, 2, 3: zinc-finger domains 1, 2, 3; Bromo: bromodomain; HAT: histone acetyltransferase domain; Gln-rich: glutamine-rich domain. (B) The endogenous Mll allele, the targeting vector, the predicted Mll-CbpSTOP (M-Cstop) knockin allele resulting from homologous recombination, and the activated Mll-Cbp allele following deletion of the stop cassette by Cre recombinase. (RI) EcoRI; (B) BamHI. (C) Southern blot analysis of EcoRI-digested genomic DNA from wild-type and targeted (ES #22, 26, 75, 112) embryonic stem (ES) cells, probed with a 5′ external probe. (D) Southern blot analysis of EcoRI-digested genomic DNA from the BM of Mx-Cre Mll-CbpSTOP mice and control littermates. Excision of the STOP cassette is indicated by the presence of the 3.5-kb band. (E) Southern blot analysis showing deletion of the STOP cassette in different tissues of Mx-Cre Mll-CbpSTOP mice following pI-pC treatments. BM: bone marrow; Liv: liver; Sp: spleen; Thy: thymus. (F) RT–PCR analysis confirming the presence of the Mll-Cbp transcript in different tissues of Mx-Cre Mll-CbpSTOP mice following pI-pC treatments. WT: Mx-Cre BM.
Embryonic stem cells possessing the correct, homologous recombination of the targeting construct were verified by Southern blot and used to achieve germline transmission of the knockin allele (Figure 1C). We always tested mice heterozygous for this conditional knockin allele, Mll-CbpSTOP, which were born at the expected Mendelian frequency and exhibited no obvious abnormalities. RT–PCR analysis confirmed the absence of Mll-Cbp expression in multiple tissues (data not shown). In order to be able to activate expression the Mll-CbpSTOP fusion allele in adult mice, we introduced the Mx-Cre transgene in which Cre recombinase expression can be activated by induction of interferon through administration of polyinosinic-polycytidylic acid (pI-pC). Mx-Cre Mll-CbpSTOP mice (4 weeks old) and control littermates received three intraperitoneal injections of pI-pC over a 6-day period. To assess the in vivo efficiency of Cre-mediated excision, we determined the level of deletion of the stop cassette in various hematopoietic tissues by Southern blot. High levels of Cre-mediated excision were achieved in the bone marrow (BM), liver, spleen, and thymus (Figure 1D and E). Furthermore, RT–PCR analysis confirmed expression of the Mll-Cbp fusion gene in those tissues following pI-pC treatment (Figure 1F).
MLL-CBP induced myeloid hyperplasia
To study the immediate effects of MLL-CBP, we monitored myeloid and lymphoid hematopoiesis in Mx-Cre Mll-CbpSTOP mice following the pI-pC treatments. There was no significant difference in total white blood cell count or differential counts between the Mx-Cre Mll-CbpSTOP mice and treated control littermates. We also performed flow cytometric analysis (FACS) of BM cells, splenocytes, and thymocytes, and noted no significant differences in the lymphoid compartments. However, we consistently observed an increase in Mac-1+/Gr-1+ myeloid cells in Mx-Cre Mll-CbpSTOP BM. This myelomonocytic expansion was evident as early as 4–10 days following the last pI-pC treatment and was still present 4 weeks following activation of the Mll-Cbp allele (Figure 2A and B). Histological examination of the BM revealed hypercellularity with maturation of the myelomonocytic cell series (data not shown). The onset of myeloid hyperplasia within days of activating MLL-CBP expression indicates that MLL-CBP alone is sufficient to induce myeloid hyperplasia.
Figure 2.
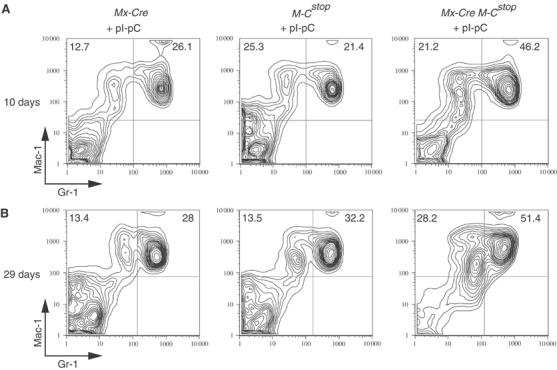
MLL-CBP induced myeloid hyperplasia. (A) We analyzed BM cells isolated from Mx-Cre Mll-CbpSTOP mice and control littermates at 10 days following the last pI-pC treatment. A representative FACS profile of BM cells stained with Mac-1 and Gr-1 antibodies is shown. (B) A representative FACS profile of BM cells at 29 days following the last pI-pC treatment.
MLL-CBP increases GMP cells
The myeloproliferation induced by MLL-CBP could reflect a direct effect of the fusion protein on HSCs and/or myeloid progenitors. To explore this, we assessed the frequency of cells that mark as HSCs and committed progenitors by FACS in Mx-Cre Mll-CbpSTOP mice at 3 weeks following pI-pC treatments. The percentages of HSCs, CMPs, and megakaryocyte/erythroid progenitors (MEPs) in the BM of Mx-Cre Mll-CbpSTOP mice were similar to that of control littermates lacking the MLL-CBP fusion. However, GMPs were substantially increased in Mx-Cre Mll-CbpSTOP mice (Figure 3A). Analysis of the total number of HSC, CMP, GMP, and MEP cell populations in the BM indicated that Mx-Cre Mll-CbpSTOP mice had an average three-fold increase in GMPs (Figure 3B). In contrast, the CLP compartment was decreased in the BM of Mx-Cre Mll-CbpSTOP mice (Figure 3A). Furthermore, the total number of CLP was reduced by about three-fold (Figure 3B). Together, these data indicate that MLL-CBP selectively expanded progenitor cells with GM differentiation potential, warranting exploration of the mechanism.
Figure 3.
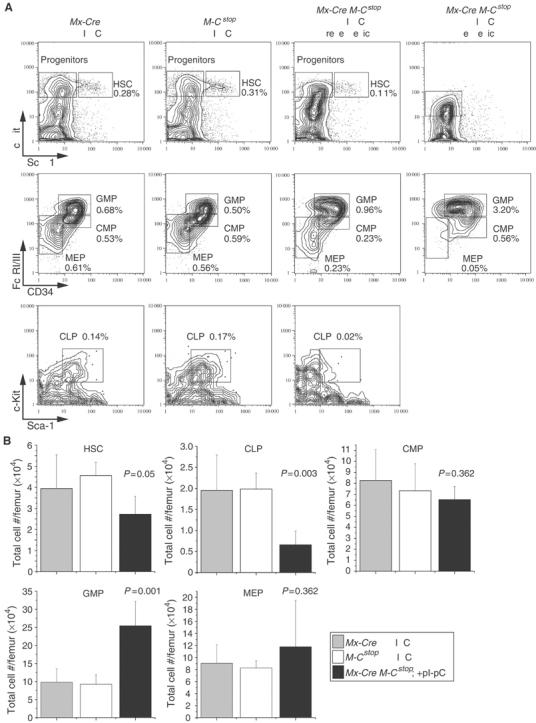
GMPs were substantially increased in Mx-Cre Mll-CbpSTOP mice. (A) Representative FACS-staining profiles of HSCs and progenitors from the BM of Mx-Cre Mll-CbpSTOP mice (3 weeks post-pI-pC treatments; gates are indicated in Materials and methods). Similar results were obtained from five Mx-Cre Mll-CbpSTOP mice and their control littermates. The FACS profile for a leukemic Mx-Cre Mll-CbpSTOP mouse is also shown for comparison and indicates an even greater expansion of GMP compared to the preleukemic phase of mice expressing MLL-CBP. (B) Bar graph showing the absolute numbers of HSC, CLP, CMP, GMP, and MEP cells in the BM of Mx-Cre Mll-CbpSTOP mice compared to controls. Values represent the average from five mice.
Enhanced replating efficiency of MLL-CBP-expressing HSC, GMP, and CMP
To determine which hematopoietic progenitor populations in Mx-Cre Mll-CbpSTOP BM express the Mll-Cbp fusion gene, we purified cells that mark as HSC, CLP, CMP, GMP, and MEP by their cell surface markers. Mll-Cbp transcript was detected in all of these progenitor populations by RT–PCR (Figure 4A). High levels of MLL-CBP expression were noted in CLPs although Mx-Cre Mll-CbpSTOP mice did not display detectable abnormalities in lymphopoiesis. The isolated expansion of GMP despite widespread expression of MLL-CBP argues for a lineage-specific effect of MLL-CBP on myelomonocytic progenitors.
Figure 4.
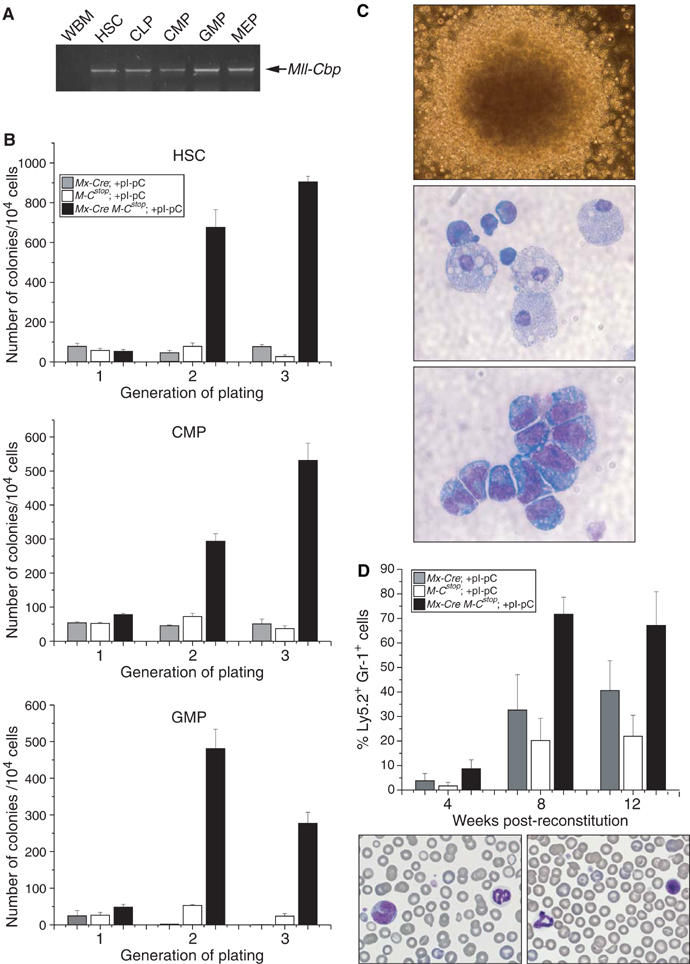
Enhanced replating efficiency of MLL-CBP-expressing GMP. (A) RT–PCR analysis for the Mll-Cbp transcript in HSC, CLP, CMP, GMP, and MEP cells. MLL-CBP is expressed in all of these cell populations. (B) Purified HSC, CMP, GMP cells were serially replated in methylcellulose culture. Bar graphs show the number of colonies obtained after each round of serial replating (the mean and s.d. of triplicate cultures are indicated). Similar results were obtained from two experiments. (C) Morphology of a typical tertiary colony derived from the MLL-CBP-expressing HSC, CMP, or GMP cultures. Cytospins of these colonies revealed the presence of myeloblasts and terminally differentiated macrophages. (D) Percent of donor-derived mature myeloid cells (% Ly5.2+Gr-1+) as assessed by FACS of peripheral blood (PB). Percentage values represent the average from 6–8 mice and representative PB smears from a recipient mouse that was reconstituted with MLL-CBP-expressing HSC, which show mature neutrophils, monocytes, and lymphocytes (magnification × 60).
To assess whether MLL-CBP might enhance the self-renewal/proliferative capacity of HSC or committed myeloid progenitors, we turned to an in vitro serial replating assay. Purified HSCs, GMPs or CMPs were plated in methylcellulose cultures under conditions optimal for the differentiation of multipotential progenitors. HSCs and progenitors derived from control littermates exhausted their proliferative capacity after a single round of replating and yielded very few secondary and tertiary colonies (Figure 4B). Instead, large mature macrophages dominated these plates, indicating that induction of terminal differentiation accompanied the loss of clonogenic potential. In contrast, HSC, GMP, and CMP cells expressing MLL-CBP demonstrated an enhanced proliferative potential continuing to generate colonies upon serial replating (Figure 4B). The MLL-CBP-expressing MEP cells did not demonstrate clonogenic activity in this assay (data not shown). The majority of the colonies initiated from HSC, GMP, and CMP populations were granulocyte–macrophage colonies consistent with MLL-CBP selectively affecting progenitors capable of giving rise to these differentiated cell types. Cytospin analysis of these colonies revealed the presence of myeloblasts as well as mature macrophages and/or mast cells, indicating that MLL-CBP does not completely block myelomonocytic differentiation (Figure 4C).
We utilized a long-term reconstitution assay to test whether MLL-CBP can enhance the self-renewal/proliferation of progenitors in vivo. To this end, we examined the ability of MLL-CBP-expressing HSC cells, the only preleukemic cell type with long-term repopulating abilities, to reconstitute the marrow compartment of lethally irradiated mice. Over a 3-month period, the Ly5.2 marked MLL-CBP cells increasingly comprised a much higher percentage of the Gr-1+ compartment in PB when compared to controls, which is consistent with our in vitro observation that MLL-CBP can enhance the self-renewal/proliferation of myeloid progenitors (Figure 4D).
MLL-CBP alters the genetic program of GMP
To elucidate the mechanism by which MLL-CBP enhanced the self-renewal/proliferation of GMP, we conducted gene expression profiling of purified GMP population. About 10 000 triple-sorted GMP cells were isolated from Mx-Cre Mll-CbpSTOP mice and littermate controls at 5 weeks following pI-pC treatments. Extracted RNA was amplified by two rounds of in vitro transcription, biotinylated, and applied to Affymetrix Mouse Expression Array 430A, which contains approximately 22 600 probe sets (data can be accessed at ArrayExpress, accession number E-MEXP-213). We assessed for genes differentially expressed in the Mx-Cre Mll-CbpSTOP sample as compared to controls that lacked the fusion gene (Figure 5). The classes of genes represented included signaling molecules, putative oncogenes, kinases, cell surface molecules, and other transcription factors. Collectively, these results indicate that MLL-CBP has a direct effect on the gene program of GMPs.
Figure 5.

MLL-CBP alters the genetic programs of GMP. The top 50 genes most closely associated with MLL-CBP expression—either upregulated (red) or downregulated (blue)—are shown. Each column represents a sample and each row a gene. Samples 1 and 2 represent Mx-cre GMP cells, samples 3 and 4 represent Mll-CbpSTOP GMP cells, and samples 5–7 represent Mx-Cre Mll-CbpSTOP GMP cells.
Hox a9 is required for maintaining MLL-CBP-mediated cell expansion
One of the most striking results of the microarray analysis was that Hox a9 is upregulated by MLL-CBP. Using real-time PCR analysis, we confirmed that MLL-CBP increased Hox a9 expression in GMP cells and the fold of induction was in close agreement with the microarray data. In addition, we found that the presence of MLL-CBP also upregulated Hox a9 expression in HSC, CLP, CMP, and MEP cells (Figure 6A). This result is consistent with recent data from several groups demonstrating an upregulation of HOX A9 expression in human MLL fusion-bearing leukemias (Armstrong et al, 2002; Yeoh et al, 2002; Debernardi et al, 2003).
Figure 6.
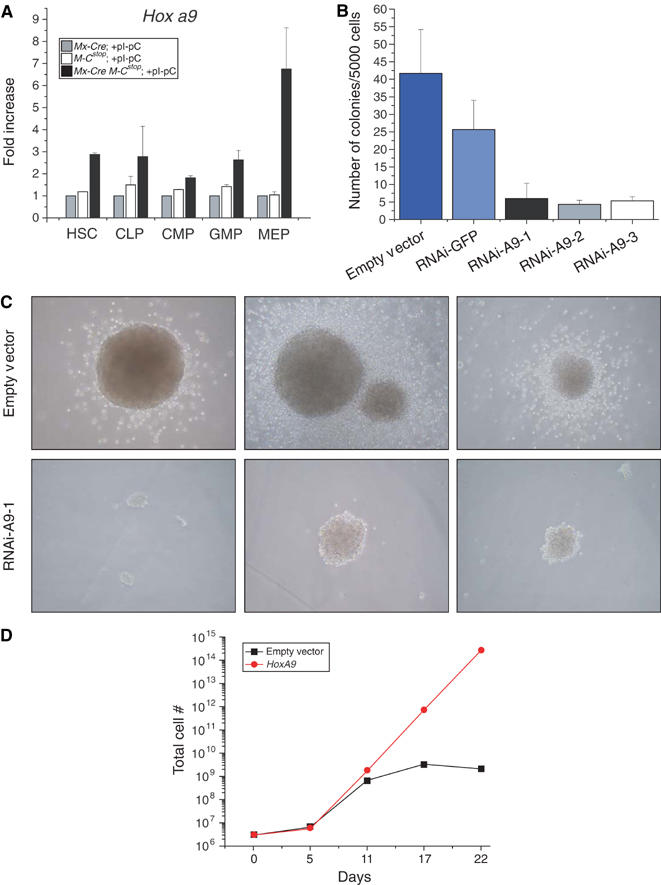
Hox a9 is required for mediating the effects of MLL-CBP. (A) MLL-CBP upregulated Hox a9 gene expression. As measured by real-time quantitative PCR, Hox a9 expression was increased in HSC, CLP, CMP, GMP, and MEP cells expressing MLL-CBP when compared to controls. Relative transcript levels were internally normalized to GAPDH levels. Values represent the average of at least two independent experiments performed in triplicate. (B) Effects of shRNA-HoxA9 constructs on BM cells expressing MLL-CBP. Three different shRNA constructs directed against Hox a9 were used (lanes 3–5). Empty retroviral vector and an shRNA construct directed against GFP were used as controls (lanes 1 and 2, respectively) (C) Effects of shRNA-Hox a9 on the size of the colony formation. (D) Effects of Hox A9 overexpression on the in vitro growth and replating efficiency of BM progenitors. Total cell numbers were counted at the specified days after the initial plating.
To test whether the high level of Hox a9 expression may be critical for MLL-CBP-mediated expansion of BM progenitors, we inhibited endogenous Hox a9 by RNA interference (RNAi). Three short-hairpin RNAs directed against Hox a9 (shRNA-A9) were constructed and the efficacy of these shRNAs in reducing Hox a9 expression was confirmed by quantitative PCR analysis (data not shown). To investigate the effects of reduced Hox a9 expression on MLL-CBP-mediated expansion of progenitors in vitro, we transduced BM cells with each of the three shRNA-A9 and subsequently plated the cells in methylcellulose culture. We observed that inhibition of Hox a9 expression significantly reduced the in vitro growth of MLL-CBP progenitors (Figure 6B and C). Each of the sh-RNA-A9 reduced the size as well as the number of colonies (Figure 6B and C). In contrast, a control shRNA directed against GFP did not substantially affect the colony-forming capacity of MLL-CBP-bearing BM cells. Taken together these data indicate that Hox a9 is a critical downstream target of MLL-CBP, required for maintaining the MLL-CBP mediated growth and expansion of progenitors.
We also tested the effects of Hox A9 overexpression on the replating efficiency of BM progenitors. We again utilized the in vitro serial replating assay as described previously. Wild-type BM cells were infected with a retrovirus that encoded for human Hox A9 and subsequently plated in methylcellulose culture. Whereas progenitors that were infected with empty control virus exhausted their growth potential by the third round of replating, cells that overexpressed Hox A9 continued to grow and generated numerous colonies after four rounds of replating (Figure 6D). In summary, these data are consistent with our hypothesis that Hox a9 plays an important role in MLL-CBP-mediated cell expansion and growth.
Mice expressing MLL-CBP develop ‘therapy-related' myeloproliferative diseases
A cohort of 11 pI-pC-treated Mx-Cre Mll-CbpSTOP mice were monitored over an 8-month period and none progressed to leukemia. Since human t(11;16)-bearing leukemias usually follow therapy, we reasoned that additional mutations might be required to complement MLL-CBP in leukemogenesis. To test this hypothesis, 4-week-old Mx-Cre Mll-CbpSTOP mice were treated with pI-pC to activate expression of the fusion gene and 1 week later were subjected to a single dose of sublethal γ-irradiation (4 Gy). We also treated a cohort of Mx-Cre Mll-CbpSTOP mice with a single injection of ENU (N-ethyl-N-nitrosourea) (100 g/kg body weight) following pI-pC treatment. Within 7 months, 60% (9/15) of the γ-irradiated (Figure 7A) and 73% (16/22) of the ENU-treated (Figure 7B) Mx-Cre Mll-CbpSTOP mice succumbed to a spectrum of myeloproliferative diseases resembling the therapy-induced t-AMML, t-CMML, and myelodysplastic/myeloproliferative disorder (t-MD/MPD) observed in humans possessing the t(11;16). In contrast, none of the γ-irradiated or ENU-treated control littermates that lacked MLL-CBP developed myeloid disease during the same period, indicating that MLL-CBP was required for the initiation of myeloid neoplasia. Among the affected Mx-Cre Mll-CbpSTOP mice, anemia, thrombocytopenia, leukocytosis, and splenomegaly were prominent (Table I). The increased WBC was predominantly mature monocytes and/or neutrophils with occasional immature forms present (Figure 7C and F). We never observed any incidence of lymphoid leukemia among the diseased mice.
Figure 7.
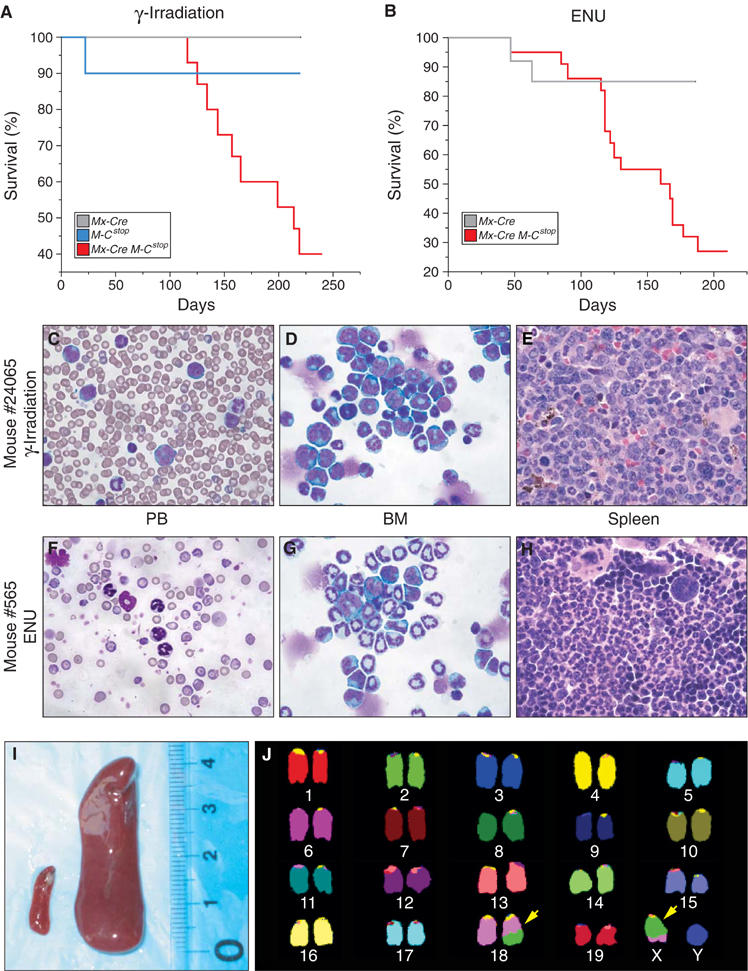
Mice expressing MLL-CBP progress to a fatal myeloid disease. (A) Kaplan–Meier survival plot for Mx-Cre Mll-CbpSTOP mice (n=15) that received a single dose of sublethal γ-irradiation. One Mll-CbpSTOP mouse (n=10) died from irradiation-induced BM failure. None of the Mx-Cre mice (n=11) developed disease. (B) Kaplan–Meier survival plot for ENU-treated Mx-Cre Mll-CbpSTOP mice (n=22). Two Mx-Cre mice (n=13) died from T-cell lymphomas. ENU-induced T-cell lymphoma in normal mice has been reported (Breuer et al, 1989). (C) Representative PB smear from an irradiated Mx-Cre Mll-CbpSTOP mouse #24065, which shows leukocytosis consisting predominantly of mature neutrophils and/or monocytes with immature forms present (mag × 60). (D) BM cytospin from the same animal demonstrated predominantly neutrophilic and monocytic cells of which immature forms/blasts were >20% (magnification × 60). (E) The spleen from the same animal showed an infiltrate of poorly–moderately differentiated myeloid cells (magnification × 60). (F) Representative PB smear from an ENU-treated Mx-Cre Mll-CbpSTOP mouse #565, which shows moderate neutrophilic leukocytosis. (G) BM cytospins from the same animal showed well-differentiated myelopoiesis without increased immature forms/blasts. (H) Spleen from the same animal showed marked red-pulp expansion with megakaryocytes, prominent erythropoiesis, and well-differentiated myelopoiesis. (I) Splenomegaly in a leukemic, irradiated Mx-Cre Mll-CbpSTOP mouse (right) compared to an age-matched wild-type control (left). (J) A representative spectral karyotype of leukemic cells from an irradiated Mx-Cre Mll-CbpSTOP animal. Tumor displays a t(18;X) as indicated by the arrows.
Table 1.
Analysis of MxCre Mll-CBPSTOP mice that received irradiation or ENU
| Mouse | No. of leukemic mice | Latency (median) | WBC × 103/μl (median) | RBC × 103/μl (median) | PLT × 103/μl (median) | Spleen wt mg (median) |
|---|---|---|---|---|---|---|
| MxCre Mll-CBPSTOP+Irrad | 9/15 | 116–219 (157) | 7–70 (41.1) | 2–9 (5.6) | 122–1261(463) | 320–2810 (1042) |
| MxCre Mll–CBPSTOP+ENU | 16/22 | 47–177 (126) | 9–51 (27.2) | 1–4 (2.2) | 193–1434 (698) | 450–1530 (1057) |
| Wild-type control | NA | 9 | 10 | 1048 | 100 | |
| NA=not applicable. |
However, the spectrum of myeloproliferative disease varied between the γ-irradiated and ENU-treated groups. The γ-irradiated mice demonstrated extensive disruption of the splenic architecture by poorly to moderately differentiated myeloid cells (Figure 7E) with leukemic infiltrates in the liver, lung, and lymph nodes (data not shown). BM cytospins showed a predominance of neutrophilic and monocytic cells with an increase of immature forms and a variable number of blast cells (Figure 7D). BM sections were also notable for the presence of pseudo-Gaucher cells, which are indicative of high cell turnover and often seen in human MD/MPD. Overall, the γ-irradiated Mx-Cre Mll-CbpSTOP mice represent a spectrum of myelomonocytic leukemias, from a more acute t-AMML (31% blasts) to others with more features of a chronic t-CMML. To assess whether neoplasms showed evidence of clonality, we performed spectral karyotypes on tumor specimens from γ-irradiated Mx-Cre Mll-CbpSTOP mice. Of the two tumor samples we analyzed, both contained a translocation. As seen in Figure 7J, all metaphase spreads from a representative leukemia displayed a t(18;X). These findings support the clonal origin of the tumor.
The typical myeloproliferative disease in ENU-treated mice characterized by leukocytosis with substantial anemia did not fulfill criteria for a nonlymphoid leukemia. BM cytospins and histologic sections revealed well-differentiated myelopoiesis without increased immature forms/blasts (Figure 7G). Spleens demonstrated marked expansion of red-pulp with megakaryocytes, prominent erythropoiesis, and well-differentiated myelopoiesis (Figure 7H). Liver and lymph node involvement were minimal. Given the cytopenias and leukocytosis, the ENU-induced disorder most closely resembles human disorders that would be classified as myelodysplastic/myeloproliferative disease (t-MD/MPD). Transduction of BM from γ-irradiated and ENU-treated mice did not transfer the disorder to lethally irradiated recipients, although transplantation per se is not the sole defining criterion for murine leukemias (Kogan et al, 2002).
FACS analysis of BM cells and splenocytes confirmed the expansion of myeloid cell types with a significant increase in either Mac-1+/Gr-1lo mature monocytes or Mac-1+/Gr-1+ immature myeloid cells and mature neutrophils (Figure 8). B and T cells were relatively deceased in the BM and spleen (Figure 7B). Overall, the pathology represents a spectrum of disease similar to human therapy-related myeloid neoplasms bearing the MLL-CBP fusion.
Figure 8.
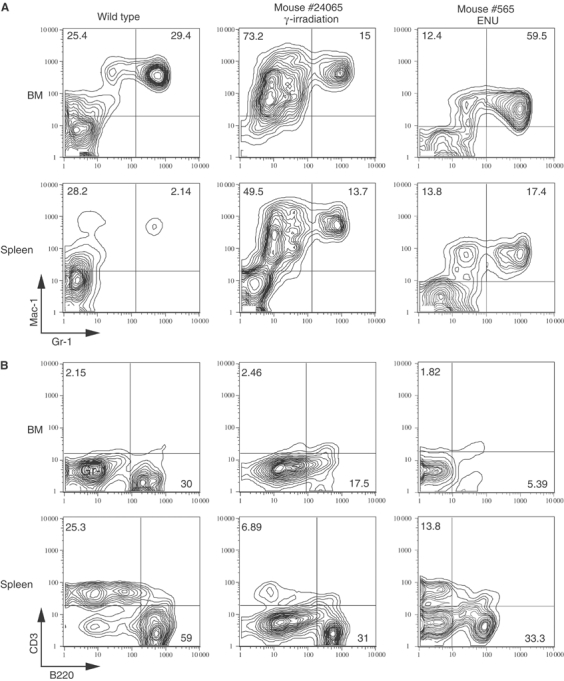
FACS analysis of BM and spleen cells from representative diseased Mx-Cre Mll-CbpSTOP mice. Single-cell suspensions were prepared from the BM and spleens of diseased Mx-Cre Mll-CbpSTOP mice and stained with a combination of Mac-1 and Gr-1 antibodies (A) or B220 and CD3 antibodies (B). The staining antibodies are indicated adjacent to each axis.
We examined hematopoietic progenitor populations in the BM of leukemic Mx-Cre Mll-CbpSTOP mice. A dramatic increase in GMP cells was noted in the BM of these diseased mice when compared to γ-irradiated control littermates and the preleukemic phase of mice expressing MLL-CBP (Figure 3A). Overall, these data indicate that MLL-CBP has its greatest effect on the GMP population, which is also the most prominently expanded progenitor population in the resultant leukemia.
Discussion
In the present study, we utilized a conditional knockin strategy to generate mice in which expression of MLL-CBP is temporally regulated. This model allowed us to bypass the embryonic lethality caused by a standard Mll-Cbp knockin and to assess the immediate effects of this fusion protein on hematopoiesis. Our successful modeling of the t(11;16)(q23;p13) revealed several important aspects of MLL-CBP leukemogenesis and therapy-related myeloproliferative disease.
MLL-CBP selectively expanded GMP in the BM and enhanced the self-renewal/proliferation of this progenitor population in vitro. We demonstrate that populations of sorted stem cells and progenitors that lacked MLL-CBP exhausted their clonogenic capacity after a single round of plating in methylcellulose, whereas cells expressing MLL-CBP continued to generate colonies after serial replating. This observation was confirmed in vivo when we found that MLL-CBP-expressing HSC and progenitors had a greater capacity to repopulate the marrow compartment of lethally irradiated recipients. Furthermore, in cultures initiated from MLL-CBP-expressing HSCs, CMPs, or GMPs, we consistently observed an outgrowth of granulocyte/macrophage colonies, indicating that the fusion protein had its greatest effect on the myelomonocytic lineage in vitro and in vivo. The presence of mature granulocytes and macrophages in these cultures indicates that MLL-CBP does not completely block terminal differentiation. This observation recapitulates a notable feature of human t(11;16) leukemias in which MLL-CBP was found in mature clonal cells with differentiation capacity (Rowley et al, 1997). Finally, in leukemic Mx-Cre Mll-CbpSTOP mice, the HSCs were almost undetectable, while the GMP compartment is even more dramatically expanded. This suggests that MLL-CBP-mediated leukemogenesis is likely to be initiated from the GMP population. In comparison, a previous report that retrovirally introduced MLL-GAS7 fusion into BM appeared to transform selectively a multipotent hematopoietic progenitor (MPP), the progeny of HSC with multilineage potential (So et al, 2003). Consistent with this, MLL-GAS7 induced an acute biphenotypic leukemia in mice. In contrast, MLL-CBP appeared to more selectively target the GMP, a progenitor population committed to myelomonocytic differentiation, and subsequently induce myelomonocytic leukemia in mice. MLL-AF9 knockin mice also exhibited a myeloproliferation before progressing to AML in adult animals (Corral et al, 1996; Dobson et al, 1999). It is interesting to note that the total number of CLP is actually decreased significantly in the Mx-Cre Mll-CbpSTOP mice despite robust MLL-CBP expression in this progenitor population. This argues that myelomonocytic progenitors may be particularly susceptible to the transforming effects of MLL-CBP, whereas this fusion may have deleterious effects on lymphoid progenitors. In concert, these results suggest that specific MLL fusion proteins target distinct stages of hematopoietic development and dictate the specific phenotype of the leukemia.
In another report, Lavau et al (2000) have generated a mouse model utilizing a truncated version of MLL-CBP in a retroviral transduction approach. In contrast to our model, the delivery of this fusion protein by retrovirus blocked myeloid differentiation and induced AML in mice. Several factors may account for the differences. In the conditional knockin system, the endogenous Mll promoter regulates transcription of the Mll-Cbp allele and its induction more closely mimics the lineage- and temporal-specific expression of the MLL-CBP fusion following a human chromosomal translocation. Furthermore, the Mll-Cbp knockin recapitulates the normal loss of one wild-type copy of Mll as a consequence of the chromosomal translocation. In contrast, expression of MLL-CBP directed by the retroviral long terminal repeat may vary and would not confer Mll haploinsufficiency. Together, these factors could have imparted biological differences.
To our knowledge, our study is the first to analyze the critical gene expression profiles of a phenotypically and functionally defined progenitor population that express an Mll fusion gene. Prior studies are usually based on composite profiles of crude BM that may not reflect the biology of the target cell of transformation or allow for analysis of early transformation events (Kumar et al, 2004). We showed that MLL-CBP altered the gene program of GMPs even before the development of frank leukemia. Several of the genes induced by MLL-CBP have been implicated in malignant transformation in human and mouse models. For example, GMP cells expressing MLL-CBP showed upregulation of CD14, which was previously shown to be induced by MLL-AF9 in vitro (Caslini et al, 2000; Munoz et al, 2003). MLL-CBP also induced Evi 2 expression. Disruption of this gene by proviral integration has been implicated in causing myeloid leukemias in BXH-2 mice, suggesting that this gene may contribute to transformation (Buchberg et al, 1990).
An attractive candidate for an MLL-CBP-induced relevant target is Hoxa9. This result is consistent with recent data from several groups demonstrating elevated HOX A9 expression in human MLL fusion-bearing leukemias (Armstrong et al, 2002; Yeoh et al, 2002; Debernardi et al, 2003). However, recent experiments examining the role of Hox a9 in leukemogenesis mediated by MLL fusion proteins came to mixed conclusions. When Hox a9-deficient BM cells were tranduced with a retrovirus expressing MLL-ENL, they failed to develop leukemia upon transplantation, suggesting a requirement for Hox a9 in the initiation of MLL-ENL-mediated leukemogenesis (Ayton and Cleary, 2003). In contrast, when Mll-AF9 knockin mice were mated with Hox a9 knockout mice, they developed leukemias at similar rates in the presence or the absence of Hox a9. The dichotomy in outcome of these well-performed studies might reflect a difference in specific Hox genes utilized by MLL-ENL versus MLL-AF9. Alternatively, this may represent a difference in somatic versus germline experimental models. In our approach, we asked whether Hox a9 is required for maintaining a transformed phenotype, cell growth, and expansion of premalignant BM progenitors expressing MLL-CBP. We found that inhibiting Hox a9 expression reduced the capacity of MLL-CBP-expressing cells to continually grow and expand in vitro. Furthermore, we also observed that overexpression of Hox a9 in wild-type BM progenitors significantly enhanced the replating efficiency of these cells. Together, these results suggest a molecular hierarchy in which the effects of MLL-CBP on hematopoiesis and leukemogenesis are mediated at least in part through sustained Hox a9 expression.
Notably, only the GMP population was significantly expanded in the presence of MLL-CBP even though Hox a9 was upregulated in HSC and all of the progenitor populations tested. One possible explanation is that GMPs are particularly sensitive to changes in Hox gene expression. Support for this hypothesis comes from the observation that overexpression of Hox a9 is predominantly associated with myeloid malignancies in human and mouse models (Nakamura et al, 1996; Kroon et al, 1998). Genes downstream of Hox a9 may help mediate the effects of MLL-CBP on myelomonocytic progenitors. Very few downstream targets of the Hox genes have been definitively established, although two recent studies describe Hox A9 upregulated genes following its overexpression in transformed cell lines (Dorsam et al, 2004; Ghannam et al, 2004). Further analysis of gene expression profile in systems such as the conditional MLL-CBP mouse model should help identify the complete gene programs required for leukemogenesis.
Our strategy of inducing MLL-CBP in adult mice recapitulated many aspects of the human myeloproliferative disease associated with the t(11;16). Conditional MLL-CBP mice demonstrate a stepwise progression in which MLL-CBP is the initiating event that generates a pool of myeloid progenitors with enhanced self-renewal/proliferative capacities. Subsequent exposure to sublethal γ-irradiation or ENU, which are known strategies to induce cooperating mutations, resulted in progression to fatal therapy-related myeloproliferative disorders. This complementation of MLL-CBP to induce full leukemic transformation is consistent with features of human t(11;16) leukemia that are almost exclusively therapy-related and are accompanied by other genomic abnormalities in addition to t(11;16) (Rowley et al, 1997). Conditional MLL-CBP mice provide a much-needed model of therapy-related myeloproliferative disease. Furthermore, the spectrum of disorders varies following γ-irradiation, which results in t-AMML and t-CMML, versus ENU, which resembles t-MD/MPD. Perhaps, this mouse model will provide an experimental framework to characterize the complementing mutations that cooperate with MLL-CBP and are responsible for each distinct myeloproliferative disease.
Materials and methods
Generation of conditional Mll-Cbp knockin mice
The Mll-CbpSTOP targeting vector was assembled in the plasmid vector Litmus 28 (NEB, Beverly, MA). A 1.8-kb SmaI–BamHI mouse genomic clone (a gift from Jay L Hess), which includes sequences from Mll intron 4 to the BamHI site of exon 8, was blunt-end ligated to a 6.5-kb fragment of murine Cbp cDNA (codon 266 to codon 2441, GenBank accession no. S66385). A 0.5-kb fragment containing the Simian virus 40 polyadenylation signal (SV40 pA) was added immediately downstream of the stop codon of the Cbp cDNA sequence. To enable positive selection, the PGK-puromycin (PGK-puro) resistance cassette was ligated to the 3′ end of SV40 pA. A 1.5-kb BamHI–BamHI mouse genomic clone containing the last 65 bp of Mll exon 8 and part of intron 8 was cloned downstream of the PGK-puro fragment. Finally, we inserted a 1.6-kb DNA fragment containing a transcription termination sequence flanked by loxP sequences (Lasko et al, 1992) (Invitrogen, Carlsbad, CA) into an engineered NotI site in Mll intron 7. The targeting construct was electroporated into RW4 ES cells (strain 129Sv/J) and clones were selected in puromycin. Four out of 136 clones had undergone homologous recombination. Those clones with normal karyotypes were injected into C57BL/6 blastocysts, and male chimeras were bred with C57BL/6 females to achieve germline transmission.
Mx-Cre Mll-CbpSTOP mice were generated by crossing Mll-CbpSTOP/+ males with female mice transgenic for Mx-Cre. Mx-Cre Mll-CbpSTOP mice (4 weeks old) and control littermates were injected intraperitoneally with 400 μg of pI-pC (Sigma, St Louis, MO) three times at 2-day intervals.
HSC of reconstitution assay
At 5 weeks following pI-pC treatments, purified HSCs were obtained from Mx-Cre Mll-CbpSTOP mice or control littermates. Subsequently, 500 HSCs (Ly5.2+) together with 500 ‘helper' HSCs (Ly5.1+) were injected intravenously into the tail veins of lethally irradiated C57BL/6 recipients (Ly5.1+). Hematopoietic reconstitution was followed in peripheral blood at 4, 8, and 12 weeks after the transplantation.
Short hairpin RNA and retroviral infection
The oligos containing the AgeI and EcoRI restriction sites and hairpin DNA were annealed and ligated into AgeI–EcoRI-digested pmko-Neo (provided by William Hanh). Efficiency of the shRNAs was confirmed by real-time PCR analysis of BM cells transduced with the shRNA constructs. The shRNA sequences are available upon request.
293T cells were transfected with retroviral vectors encoding the shRNAs. Retroviral supernatants were collected at 48 h post-transfection. BM cells were plated onto nontissue culture-treated 10-cm plates and exposed to the retroviral supernatant for 3 h at 37°C in the presence of 8 μg/ml polybrene (Sigma). Cells were centrifuged at 1900 r.p.m. for 90 min and infection was repeated twice. Infected cells were then plated in methylcellulose culture in conditions identical to the in vitro replating assay.
Hox A9 retrovirus and in vitro serial replating assay
Total BM was extracted from 8-week-old C57BL6 donors, and red blood cells were lysed using Puregene RBC lysis Solution (Gentra Systems). In total, 5 × 106 BM mononuclear cells were infected with either an MSCV/EGFP retrovirus that encoded the human Hox A9 gene (NM_002142) or the control MSCV/EGFP retrovirus that lacked Hox A9 sequences. Cells were incubated in IMDM supplemented with 20% FBS, SCF (20 ng/ml), IL-6 (10 ng/ml), IL-11 (10 ng/ml) and polybrene (7 μg/ml). After 16 h, cells were plated at 1 × 104 cells/ml in triplicate in MethoCult™ M3434 media supplemented with GM-CSF (10 ng/ml). Cells were replated every 5–6 days in the same media at 1 × 104 cells/ml (Supplementary data).
Supplementary Material
Supplementary Data
Acknowledgments
We thank J Opferman for helping with ES cell cultures and in discussions, A Ranger for FACS analysis assistance, S Wade for colony management, E Braverman for SKY analysis, and JS Boehm and G Hinkle for help with RNAi. This work is supported in part by NIH grant #CA68484. KA is supported by NIH grant #DK50654. JW is supported by a predoctoral fellowship from the Howard Hughes Medical Institute.
References
- Afonja O, Smith JE Jr, Cheng DM, Goldenberg AS, Amorosi E, Shimamoto T, Nakamura S, Ohyashiki K, Ohyashiki J, Toyama K, Takeshita K (2000) MEIS1 and HOXA7 genes in human acute myeloid leukemia. Leuk Res 24: 849–855 [DOI] [PubMed] [Google Scholar]
- Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ (2002) MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 30: 41–47 [DOI] [PubMed] [Google Scholar]
- Ayton PM, Cleary ML (2003) Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev 17: 2298–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T (1996) The CBP co-activator is a histone acetyltransferase. Nature 384: 641–643 [DOI] [PubMed] [Google Scholar]
- Behm FG, Raimondi SC, Frestedt JL, Liu Q, Crist WM, Downing JR, Rivera GK, Kersey JH, Pui CH (1996) Rearrangement of the MLL gene confers a poor prognosis in childhood acute lymphoblastic leukemia, regardless of presenting age. Blood 87: 2870–2877 [PubMed] [Google Scholar]
- Blobel GA, Nakajima T, Eckner R, Montminy M, Orkin SH (1998) CREB-binding protein cooperates with transcription factor GATA-1 and is required for erythroid differentiation. Proc Natl Acad Sci USA 95: 2061–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow J, Shearman AM, Stanton VP Jr, Becher R, Collins T, Williams AJ, Dube I, Katz F, Kwong YL, Morris C, Ohyashiki K, Toyama K, Rowley J, Housman DE (1996) The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat Genet 12: 159–167 [DOI] [PubMed] [Google Scholar]
- Boyes J, Byfield P, Nakatani Y, Ogryzko V (1998) Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 396: 594–598 [DOI] [PubMed] [Google Scholar]
- Breuer M, Slebos R, Verbeek S, van Lohuizen M, Wientjens E, Berns A (1989) Very high frequency of lymphoma induction by a chemical carcinogen in pim-1 transgenic mice. Nature 340: 61–63 [DOI] [PubMed] [Google Scholar]
- Buchberg AM, Bedigian HG, Jenkins NA, Copeland NG (1990) Evi-2, a common integration site involved in murine myeloid leukemogenesis. Mol Cell Biol 10: 4658–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caslini C, Alarcon AS, Hess JL, Tanaka R, Murti KG, Biondi A (2000) The amino terminus targets the mixed lineage leukemia (MLL) protein to the nucleolus, nuclear matrix and mitotic chromosomal scaffolds. Leukemia 14: 1898–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral J, Lavenir I, Impey H, Warren AJ, Forster A, Larson TA, Bell S, McKenzie AN, King G, Rabbitts TH (1996) An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell 85: 853–861 [DOI] [PubMed] [Google Scholar]
- Debernardi S, Lillington DM, Chaplin T, Tomlinson S, Amess J, Rohatiner A, Lister TA, Young BD (2003) Genome-wide analysis of acute myeloid leukemia with normal karyotype reveals a unique pattern of homeobox gene expression distinct from those with translocation-mediated fusion events. Genes Chromosomes Cancer 37: 149–158 [DOI] [PubMed] [Google Scholar]
- Dobson CL, Warren AJ, Pannell R, Forster A, Lavenir I, Corral J, Smith AJ, Rabbitts TH (1999) The mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J 18: 3564–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsam ST, Ferrell CM, Dorsam GP, Derynck MK, Vijapurkar U, Khodabakhsh D, Pau B, Bernstein H, Haqq CM, Largman C, Lawrence HJ (2004) The transcriptome of the leukemogenic homeoprotein HOXA9 in human hematopoietic cells. Blood 103: 1676–1684 [DOI] [PubMed] [Google Scholar]
- Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ (2001) MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol Cell Biol 21: 2249–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix CA, Hosler MR, Winick NJ, Masterson M, Wilson AE, Lange BJ (1995) ALL-1 gene rearrangements in DNA topoisomerase II inhibitor-related leukemia in children. Blood 85: 3250–3256 [PubMed] [Google Scholar]
- Ghannam G, Takeda A, Camarata T, Moore MA, Viale A, Yaseen NR (2004) The oncogene Nup98-HOXA9 induces gene transcription in myeloid cells. J Biol Chem 279: 866–875 [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder RG (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90: 595–606 [DOI] [PubMed] [Google Scholar]
- Higuchi M, O'Brien D, Kumaravelu P, Lenny N, Yeoh EJ, Downing JR (2002) Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell 1: 63–74 [DOI] [PubMed] [Google Scholar]
- Hunger SP, Tkachuk DC, Amylon MD, Link MP, Carroll AJ, Welborn JL, Willman CL, Cleary ML (1993) HRX involvement in de novo and secondary leukemias with diverse chromosome 11q23 abnormalities. Blood 81: 3197–3203 [PubMed] [Google Scholar]
- Kogan SC, Ward JM, Anver MR, Berman JJ, Brayton C, Cardiff RD, Carter JS, de Coronado S, Downing JR, Fredrickson TN, Haines DC, Harris AW, Harris NL, Hiai H, Jaffe ES, MacLennan IC, Pandolfi PP, Pattengale PK, Perkins AS, Simpson RM, Tuttle MS, Wong JF, Morse HC III (2002) Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood 100: 238–245 [DOI] [PubMed] [Google Scholar]
- Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G (1998) Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J 17: 3714–3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar AR, Hudson WA, Chen W, Nishiuchi R, Yao Q, Kersey JH (2004) Hoxa9 influences the phenotype but not the incidence of Mll-AF9 fusion gene leukemia. Blood 103: 1823–1828 [DOI] [PubMed] [Google Scholar]
- Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, Roberts SG, Green MR, Goodman RH (1994) Nuclear protein CBP is a coactivator for the transcription factor CREB [see comments]. Nature 370: 223–226 [DOI] [PubMed] [Google Scholar]
- Lasko M, Sauer B, Mosinger B, Lee EJ, Manning RW, Yu S-H, Mulder KL, Westphal H (1992) Targeted oncogene activation by site-specific recombination in transgenic mice. Proc Natl Acad Sci USA 89: 6232–6236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavau C, Du C, Thirman M, Zeleznik-Le N (2000) Chromatin-related properties of CBP fused to MLL generate a myelodysplastic-like syndrome that evolves into myeloid leukemia. EMBO J 19: 4655–4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magli MC, Barba P, Celetti A, De Vita G, Cillo C, Boncinelli E (1991) Coordinate regulation of HOX genes in human hematopoietic cells. Proc Natl Acad Sci USA 88: 6348–6352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magli MC, Largman C, Lawrence HJ (1997) Effects of HOX homeobox genes in blood cell differentiation. J Cell Physiol 173: 168–177 [DOI] [PubMed] [Google Scholar]
- Munoz L, Nomdedeu JF, Villamor N, Guardia R, Colomer D, Ribera JM, Torres JP, Berlanga JJ, Fernandez C, Llorente A, Queipo de Llano MP, Sanchez JM, Brunet S, Sierra J (2003) Acute myeloid leukemia with MLL rearrangements: clinicobiological features, prognostic impact and value of flow cytometry in the detection of residual leukemic cells. Leukemia 17: 76–82 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Largaespada DA, Shaughnessy JD Jr, Jenkins NA, Copeland NG (1996) Cooperative activation of Hoxa and Pbx1-related genes in murine myeloid leukaemias (see comments). Nat Genet 12: 149–153 [DOI] [PubMed] [Google Scholar]
- Rabbitts TH (1994) Chromosomal translocations in human cancer. Nature 372: 143–149 [DOI] [PubMed] [Google Scholar]
- Rowley JD, Reshmi S, Sobulo O, Musvee T, Anastasi J, Raimondi S, Schneider NR, Barredo JC, Cantu ES, Schlegelberger B, Behm F, Doggett NA, Borrow J, Zeleznik-Le N (1997) All patients with the T(11;16)(q23;p13.3) that involves MLL and CBP have treatment-related hematologic disorders. Blood 90: 535–541 [PubMed] [Google Scholar]
- Shen WF, Largman C, Lowney P, Corral JC, Detmer K, Hauser CA, Simonitch TA, Hack FM, Lawrence HJ (1989) Lineage-restricted expression of homeobox-containing genes in human hematopoietic cell lines. Proc Natl Acad Sci USA 86: 8536–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- So CW, Karsunky H, Passegue E, Cozzio A, Weissman IL, Cleary ML (2003) MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell 3: 161–171 [DOI] [PubMed] [Google Scholar]
- Sobulo OM, Borrow J, Tomek R, Reshmi S, Harden A, Schlegelberger B, Housman D, Doggett NA, Rowley JD, Zeleznik-Le NJ (1997) MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13.3). Proc Natl Acad Sci USA 94: 8732–8737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangrude GJ, Heimfeld S, Weissman IL (1988) Purification and characterization of mouse hematopoietic stem cells. Science 241: 58–62 [DOI] [PubMed] [Google Scholar]
- Super HJ, McCabe NR, Thirman MJ, Larson RA, Le Beau MM, Pedersen-Bjergaard J, Philip P, Diaz MO, Rowley JD (1993) Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood 82: 3705–3711 [PubMed] [Google Scholar]
- Taki T, Sako M, Tsuchida M, Hayashi Y (1997) The t(11;16)(q23;p13) translocation in myelodysplastic syndrome fuses the MLL gene to the CBP gene. Blood 89: 3945–3950 [PubMed] [Google Scholar]
- Van Orden K, Giebler HA, Lemasson I, Gonzales M, Nyborg JK (1999) Binding of p53 to the KIX domain of CREB binding protein. A potential link to human T-cell leukemia virus, type I-associated leukemogenesis. J Biol Chem 274: 26321–26328 [DOI] [PubMed] [Google Scholar]
- Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, Behm FG, Raimondi SC, Relling MV, Patel A, Cheng C, Campana D, Wilkins D, Zhou X, Li J, Liu H, Pui CH, Evans WE, Naeve C, Wong L, Downing JR (2002) Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 1: 133–143 [DOI] [PubMed] [Google Scholar]
- Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ (1998) MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc Natl Acad Sci USA 95: 10632–10636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ (1995) Altered Hox expression and segmental identity in Mll-mutant mice. Nature 378: 505–508 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data