

Abstract
Calcium influx can promote neuronal differentiation and survival, at least in part by activating Ras and its downstream targets, including the Erk pathway. However, excessive calcium influx can initiate molecular signals leading to neuronal death during excitotoxicity or in neurodegenerative diseases. Here we describe a new signaling pathway associated with calcium influx that contributes to neuronal cell death in cerebellar neurons. Influx of calcium, mediated either by L-type voltage-sensitive calcium channels or glutamate receptors, is associated with the suppression of brain-derived neurotrophic factor (BDNF) activation of Ras and its effectors Erk and Akt. This is the result of enhanced association of the tyrosine phosphatase Shp-2 with TrkB receptors, which inhibits BDNF-induced TrkB autophosphorylation and activation. Deletion of the Shp2 gene in neuronal cultures reverses inhibition of TrkB function and increases neuronal survival after extended depolarization or glutamate treatment. These findings implicate Shp-2 in a feedback system initiated by calcium that negatively regulates neurotrophin signaling and sensitizes neurons to excitotoxicity.
Keywords: BDNF, calcium, excitotoxicity, neurons, Shp-2
Introduction
Two of the most important classes of modulators of neuronal function are the neurotrophin receptors and the voltage-sensitive and ligand-activated calcium channels. Both groups of proteins play essential roles in neuronal differentiation, survival and plasticity. For example, differentiation of PC12 cells, used as a model for neuronal differentiation, can be induced by both NGF (Greene and Tischler, 1976) and calcium influx (Rusanescu et al, 1995). Both signals can also promote neuronal survival in moderate concentrations (Finkbeiner and Greenberg, 1996; Cowan et al, 2001), but calcium has neurotoxic effects at higher concentrations. High calcium levels initiate apoptotic signals leading to neuronal death after glutamate excitotoxicity (Sattler and Tymianski, 2001), traumatic brain injury (Lee et al, 1999) or in neurodegenerative diseases (Mattson, 2000; LaFerla 2002). This process is thought to be largely mediated by activation of proteases such as calpain (Brorson et al, 1994) and caspases (Yuan and Yankner, 2000). Neurotrophins protect against apoptosis induced by stimuli such as serum deprivation or calcium channel blockers and may also inhibit the damaging effects of high calcium influx.
Calcium and neurotrophin signaling pathways interact positively with each other in several ways. Calcium influx after neuronal depolarization induces the transcription of the BDNF gene via CREB activation (West et al, 2001). Furthermore, neuronal activity results in brain-derived neurotrophic factor (BDNF) secretion by neurons (Goodman et al, 1996; Hartmann et al, 2001), generating a potential autocrine response that can affect synaptic activity and enhance neuronal survival. Depolarization also increases the recruitment of the BDNF receptor TrkB to the plasma membrane (Meyer-Franke et al, 1998), thereby enhancing both factors involved in BDNF/TrkB signaling. Reciprocally, BDNF treatment can potentiate synaptic transmission (Korte et al, 1995; Figurov et al, 1996; Patterson et al, 1996; Schuman, 1999) or directly induce neuronal depolarization (Kafitz et al, 1999) and is necessary in activity-dependent neuronal survival (Ghosh et al, 1994). These data suggest the existence of a synergistic interaction between calcium and neurotrophin signaling, as they share some intracellular signaling mechanisms to carry out their functions.
A common signal shared by neurotrophins and calcium is the activation of the Ras/Erk pathway (Rosen et al, 1994; Farnsworth et al, 1995; Kaplan and Miller, 2000). However, the timing of Erk activation after neurotrophin or calcium stimulation is different, with Trk-induced signaling lasting significantly longer. The neurotrophin–Trk complex is internalized within 10–15 min after treatment in vesicles that are retrogradely transported from nerve terminals, and the complex keeps signaling throughout its axonal transit, inducing Erk activity for several hours. Increases in intracellular calcium concentration through voltage-sensitive calcium channels (VSCCs) or from intracellular stores are transient events, due to ion channel inactivation, internal store depletion and buffering of excess calcium. This results in relatively brief Erk activation in PC12 cells that lasts up to 10–15 min. However, an extension of calcium influx does not result in an extended Erk activation, suggesting the existence of a calcium-dependent inhibitory feedback. For example, when calcium influx in PC12 cells is prolonged by using L-channel agonist S(−)BayK8644 (BayK), Erk tyrosine phosphorylation levels increase in amplitude but very little in duration (Rusanescu et al, 1995). Moreover, although glutamate receptors have slow and incomplete desensitization, they only generate a similar transient Erk activation.
Under normal physiological conditions, neurons are simultaneously subjected to combinations of growth factor gradients and ion channel activity. Most studies have addressed only one signal at a time when investigating the activation of the Ras/Erk pathway. In this paper, we analyzed the output signal leading to Ras, Erk and Akt activation in cerebellar neurons after convergent stimulation by BDNF and depolarization. We found that calcium influx generated by L-type VSCCs or glutamate inhibits subsequent BDNF activation of TrkB receptors through enhanced receptor association of the Shp-2 tyrosine phosphatase. Shp-2 is a ubiquitously expressed SH2-containing tyrosine phosphatase that can be both a positive or negative regulator of tyrosine kinase receptor signaling and is involved in nervous system development (Neel et al, 2003). We also show that suppressing Shp-2 function in neurons protects neurons from toxicity associated with excessive calcium influx, suggesting that Shp-2 activity sensitizes neurons to excitotoxicity by suppressing the protective effects of TrkB receptor activity.
Results
Neuronal depolarization causes a transient inhibition of BDNF-mediated Erk and Akt activation
Cultured mouse cerebellar granule neurons were used as a model system to investigate the potential role of calcium influx in the modulation of TrkB receptor function. Cells were treated with 30 ng/ml BDNF and 50 mM KCl, separately or combined, for various lengths of time and then Erk and Akt activation was observed using phospho-specific antibodies in immunoblots of cell lysates (Figure 1). When used individually, both stimuli activated Erk; however the time course was significantly different, as expected. BDNF caused a peak in Erk activity within 10 min, which slowly declined over a 60 min period (Figure 1A) and was maintained above baseline for at least 2 h (data not shown). KCl-induced activation peaked at 7 min but rapidly declined such that Erk levels were back to baseline by 12 min. Interestingly, there was a secondary rise in Erk activity, which peaked at 20 min (Figure 1B) and it too declined rapidly (data not shown). To simulate physiological conditions, cultured neurons were depolarized for various periods of time with 50 mM KCl before being treated with BDNF for 7 min (Figure 1C). Compared to BDNF stimulation alone, pretreatment of neurons with KCl for 7 min suppressed subsequent BDNF-induced phosphorylation of Erk by ∼75% (lanes 2 and 3). This inhibition occurred 14 min after the onset of depolarization, just when Erk phosphorylation by depolarization alone was also significantly reduced (see Figure 1B, lane 3). The major inhibitory effect of depolarization on BDNF signaling was transient however. At 12 min of KCl pretreatment, Erk phosphorylation was actually higher than the initial BDNF stimulation (lane 4), probably due to overlap of the two stimuli, but remained at ∼80% of original BDNF-induced signal thereafter (lanes 5 and 6).
Figure 1.
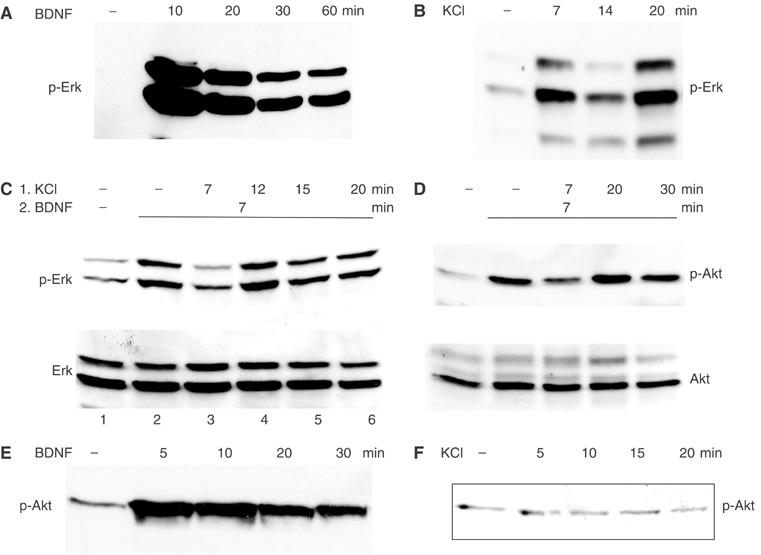
Time course of Erk and Akt phosphorylation by BDNF, depolarization or both. Cultured mouse cerebellar neurons were serum-starved for 3 h and then stimulated as shown, either with 30 ng/ml BDNF (A, E), 50 mM KCl (B, F) or with 50 mM KCl followed by addition of 30 ng/ml BDNF (C, D). The samples were analyzed by Western blot and stained with anti-p-Erk antibodies (A–C) or anti-p-Akt antibodies (D–F). Total protein levels for Erk (C) and Akt (D) are also shown.
Analysis of Akt Ser473 phosphorylation using the same protocol revealed a similar pattern: depolarization caused a transient inhibition of BDNF signaling, which was maximal after 7 min of KCl pretreatment (14 min total depolarization; Figure 1D). BDNF-induced Akt phosphorylation was restored to ∼80% of initial signal after 30 min of depolarization. BDNF alone induced a continuous Akt activation in the time frame analyzed (Figure 1E), whereas depolarization alone did not activate Akt (Figure 1F).
The activation of both Erk and Akt is dependent on Ras. Ras activation by BDNF, as assayed by affinity purification with the Ras-binding domain of Raf, was also suppressed by previous depolarization (Figure 2A), matching the timing of Erk and Akt inhibition. Again, Ras activation did not return completely to levels seen upon BDNF stimulation without KCl pretreatment (compare lanes 4–6 to lane 2). The timing of this inhibition is species-dependent, as in rat neurons maximum inhibition developed 30 min after the initial depolarization (data not shown).
Figure 2.
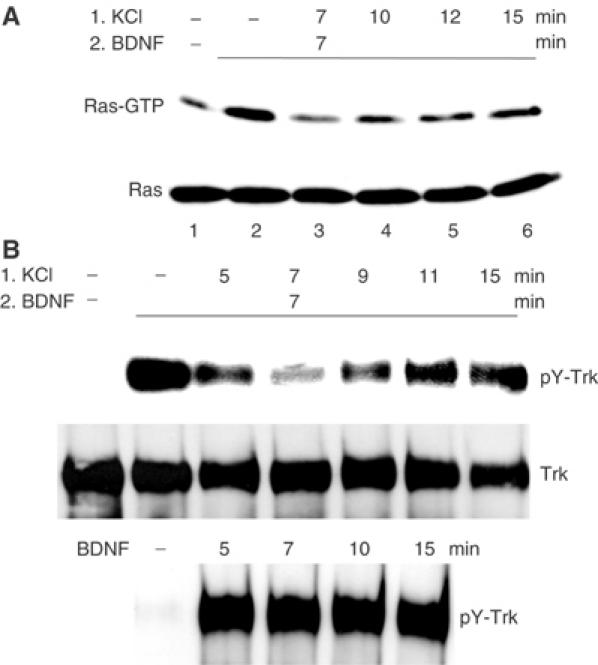
Time course of Ras and TrkB activation by BDNF in the presence of depolarization. Cultured mouse cerebellar neurons were serum-starved for 3 h and then treated with (1) KCl (50 mM) for various times followed by (2) addition of BDNF (30 ng/ml) for another 7 min. (A) Time course of Ras activation, measured as Ras-GTP levels. Total Ras levels from the same samples are shown below. (B) Time course of TrkB tyrosine phosphorylation and activation. Total Trk levels after reprobing the blot with anti-Trk antibodies are shown below. The lower panel shows Trk tyrosine phosphorylation by BDNF only. Maximum TrkB and Ras inhibition occurred at 14 min after calcium entry.
Depolarization suppresses BDNF-induced TrkB tyrosine phosphorylation
To identify the step affected by depolarization, the response of the BDNF receptor, TrkB, to the combination of depolarization/BDNF stimulation was assessed by probing TrkB immunoprecipitates for phosphotyrosine levels. As expected, TrkB tyrosine autophosphorylation was enhanced markedly by BDNF treatment alone (Figure 2B, lane 2). However, TrkB tyrosine phosphorylation was reduced by prior depolarization, and peak inhibition (lane 4, total of 14 min of depolarization) coincided with the time point of maximum Erk, Akt and Ras inhibition, suggesting that the wave of inhibition initiates at the level of the TrkB receptor and propagates along the entire pathway to Erk and Akt. Maximal suppression of BDNF-induced TrkB phosphorylation always occurred approximately 14 min after the start of depolarization in murine neurons (7 min depolarization followed by 7 min of depolarization/BDNF stimulation) and was not dependent on the duration of BDNF treatment, or whether BDNF was added before, simultaneously or after depolarization (data not shown). After this peak inhibition, TrkB tyrosine phosphorylation returned to 85–90% of the original BDNF-induced signal for at least 15 min. In contrast, BDNF stimulation on its own gave a steady signal through this period (Figure 2B, lower panel).
Depolarization of cerebellar neurons causes calcium influx through L-type VSCCs. Suppression of calcium influx through these channels by nimodipine (10 μM), a specific inhibitor of L-type VSCCs, restored TrkB phosphorylation after depolarization (Figure 3A). Conversely, an increase in calcium influx using the L-type channel agonist BayK (Rusanescu et al, 1995) enhanced the suppression of TrkB phosphorylation in both amplitude and duration, while BayK alone did not have any effect (Figure 3B). The incomplete recovery of TrkB phosphorylation after peak inhibition may be explained by the continuous low-level calcium influx during long-term depolarization (Di Virgilio et al, 1987). Due to calcium-dependent calcium channel inactivation, calcium influx is not significantly increased by longer KCl treatments, but is increased by stronger depolarization (higher KCl concentration). Consistent with this, we find that a 1 min pulse of 75 mM KCl followed by a 13 min chase with serum-free medium suppressed BDNF-induced TrkB phosphorylation more than 50 mM KCl treatment for the entire 14 min (Figure 3C, compare lanes 3 and 5).
Figure 3.
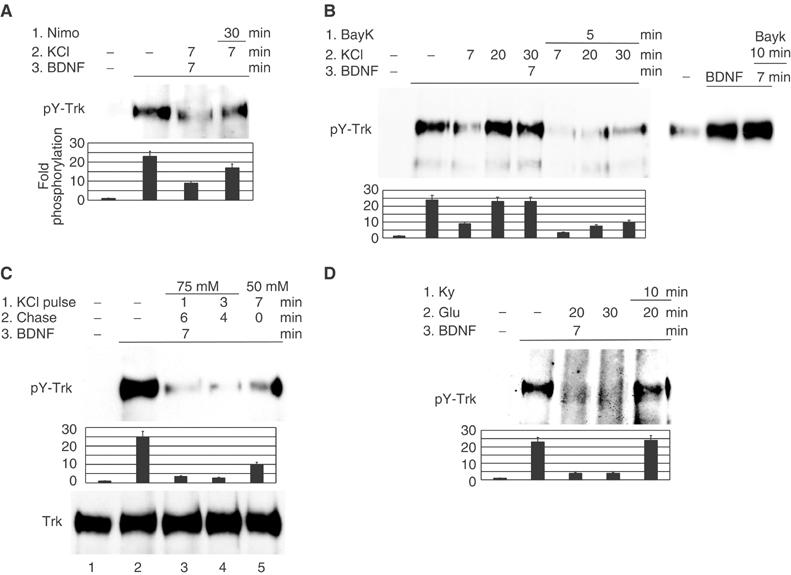
The amplitude and duration of calcium influx determine the level of TrkB dephosphorylation. Phosphotyrosine levels of TrkB immunoprecipitates are shown and quantified. (A) Calcium influx in neurons is necessary for TrkB dephosphorylation. Serum-starved neurons were treated in order with (1) nimodipine (10 μM) (Nimo), (2) KCl (50 mM) and (3) BDNF (30 ng/ml) for the times shown, each treatment being added to the previous ones. (B, C) Increased calcium influx enhances TrkB dephosphorylation. (B) Neurons were treated in order with (1) BayK (100 ng/ml) , (2) KCl (50 mM) and (3) BDNF (30 ng/ml) or with BayK followed by BDNF (right panel). (C) Neurons were depolarized for a 1 or 3 min pulse with 75 mM KCl or for 7 min with 50 mM KCl, and then the depolarizing medium was replaced with fresh serum-free medium for the time remaining up to a total of 7 min, followed by addition of 50 ng/ml BDNF for another 7 min. (D) Calcium influx induced by glutamate receptors generates a lasting inhibition of TrkB. Kynurenic acid (1 mM) (Ky), sodium glutamate (1 mM) (Glu) and BDNF (30 ng/ml) were added in order to cultured neurons, and then Trk immunoprecipitates were analyzed for tyrosine phosphorylation. The data are representative of results from at least three independent experiments.
Calcium influx through ligand-gated ion channels also suppressed TrkB activation by BDNF in neurons. Glutamate (1 mM) pretreatment of neurons suppressed TrkB tyrosine phosphorylation by BDNF (Figure 3D). This process was reversed by addition of 1 mM kynurenic acid, an inhibitor of the glycine-binding site of the NMDA receptor. In contrast to depolarization, glutamate-mediated dephosphorylation of TrkB was sustained, suggesting that the partial recovery of TrkB phosphorylation after depolarization was due to ion channel inactivation.
Two potential mechanisms by which calcium influx could lower BDNF-induced TrkB phosphorylation are a reduction in receptor availability on the cell surface or activation of a dephosphorylation mechanism. Therefore, we tested the possibility that neuronal depolarization reduces cell surface concentration of TrkB, thereby preventing BDNF binding. After BDNF or KCl treatment of neurons, cell surface TrkB molecules were labeled on ice using water-soluble sulfosuccinimidyl 6-(biotinamido) hexanoate (EZ-Link Sulfo-NHS-LC-Biotin), which binds to amino groups on extracellular protein domains. Neuron lysates were then incubated with agarose-bound avidin to collect the biotin-labeled protein fraction and the precipitates were immunoblotted with anti-Trk antibodies. In contrast with BDNF treatment, which caused Trk internalization within 30 min, depolarization in fact increased TrkB availability on neuronal surface (Meyer-Franke et al, 1998), which indicates that TrkB internalization was not the cause of its dephosphorylation (Figure 4A). In addition, the experiment in Figure 4B confirms that inhibition of TrkB phosphorylation took place 14 min after depolarization even as more TrkB receptors were present on cell surface (compare lanes 2 and 3). The transient nature of TrkB dephosphorylation and the fact that it was not caused by a change in TrkB localization suggested the alternate possibility that depolarization activates a tyrosine phosphatase that can dephosphorylate TrkB.
Figure 4.
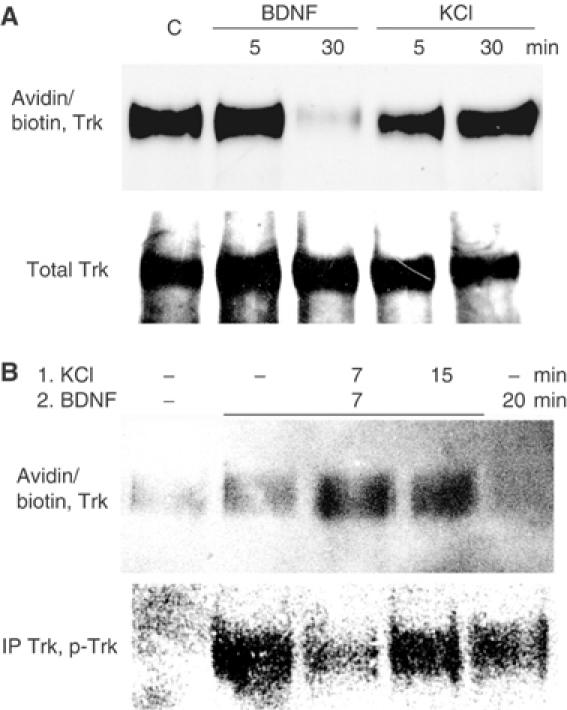
Calcium-induced TrkB dephosphorylation is not caused by receptor internalization. (A) Neuronal depolarization does not induce TrkB receptor internalization. After treatment with 30 ng/ml BDNF or depolarization with 50 mM KCl, neuronal surface proteins were labeled with water-soluble sulfosuccinimidyl 6-(biotinamido) hexanoate. Biotinylated proteins precipitated from cell lysates with agarose-immobilized avidin were then analyzed for Trk content. Duplicate samples were analyzed for total Trk levels. (B) The influence of depolarization on BDNF-induced TrkB internalization. Neurons, treated as shown, were labeled with sulfosuccinimidyl 6-(biotinamido) hexanoate and lysates were analyzed both for cell surface Trk levels and for Trk tyrosine phosphorylation. The results are representative of experiments performed three times.
Calcium-induced TrkB inhibition is mediated by tyrosine phosphatase Shp-2
To search for a TrkB-associated tyrosine phosphatase, TrkB immunoprecipitates from neurons treated with the protocol described above were analyzed for phosphatase activity by in-gel tyrosine phosphatase assays. TrkB immunoprecipitates were run on a gel that contained radioactively labeled poly-(Glu4-Tyr-32P) (MW 44 000). After in-gel protein renaturation, a band (MW ∼70 kDa) of reduced radioactivity representing tyrosine phosphatase activity was detected (Figure 5A). The phosphatase activity of this protein peaked at 14 min of depolarization (7 min depolarization plus 7 min depolarization and BDNF), matching the maximal suppression of TrkB phosphorylation (compare with Figure 2B). This finding suggested that inhibition of TrkB tyrosine phosphorylation by calcium was the result of increased dephosphorylation by elevated tyrosine phosphatase activity associated with the receptor. The 70 kDa band matches the size of Shp-2, one of two SH2 domain-containing protein-tyrosine phosphatases. In fact, immunoprecipitation of Shp-2 from neuron lysates and subsequent in-gel phosphatase assay produced a band of activity of the same size as the band co-precipitated with TrkB (data not shown). Western blot analysis of duplicate lysates with antibodies to the C-terminus of Shp-2 also produced a band of same size as in the phosphatase assay (Figure 5A, right panel).
Figure 5.
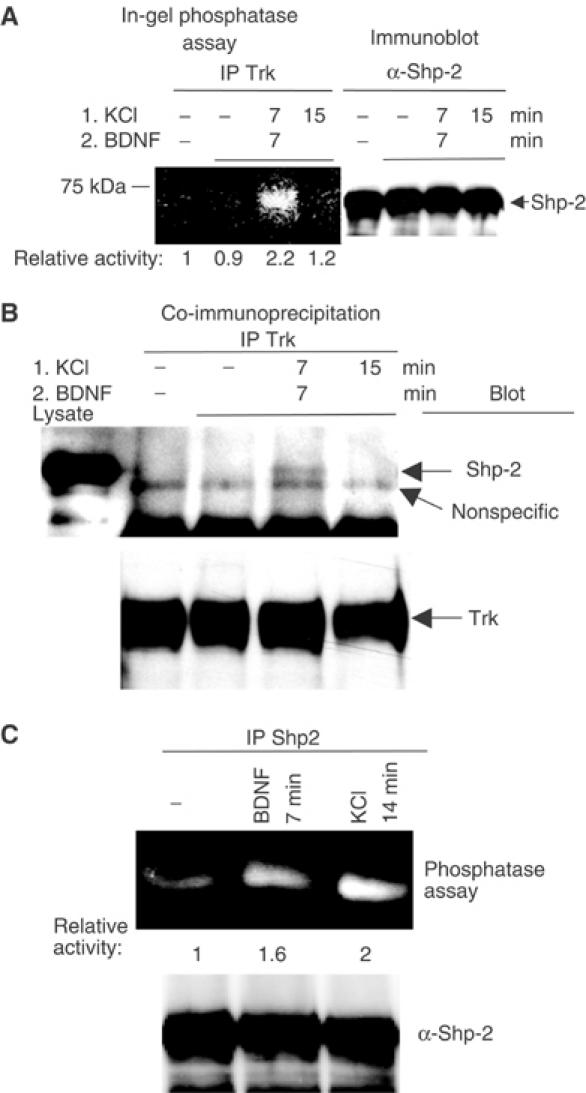
Implication of tyrosine phosphatase Shp-2 in TrkB dephosphorylation. (A) In-gel phosphatase assay of Trk immunoprecipitates. Trk immunoprecipitates from lysates of cerebellar neurons treated with 50 mM KCl and 30 ng/ml BDNF as shown were run on a gel containing 32P-labeled poly-(Glu4-Tyr) and subjected to renaturing conditions. Phosphatases present in the sample remove 32P and appear as lighter bands on a dark background. Right panel: a small fraction of each sample was used to assay total Shp-2 protein content. (B) Same experiment as in panel A, except that Trk immunoprecipitates were analyzed for the presence of Shp-2 by immunoblotting. The results are representative of experiments performed at least three times. (C) Phosphatase assay of Shp-2 immunoprecipitates after BDNF or 75 mM KCl treatment.
Increased TrkB-associated Shp-2 activity could be due to increased content of Shp-2 bound to TrkB or to higher Shp-2 catalytic activity or both. To test the first possibility, TrkB immunoprecipitates isolated from neuronal extracts after various stimuli were immunoblotted with Shp-2 antibodies (Figure 5B). An increase in Shp-2 protein was detected in TrkB immune complexes at the same time point that an increase in tyrosine phosphatase activity in TrkB immunoprecipitates was observed (compare to Figure 5A). Higher calcium influx in the presence of BayK increased Shp-2/TrkB association just before peak dephosphorylation (data not shown). In addition, Shp-2 catalytic activity was also increased by calcium influx alone to levels comparable to BDNF stimulation (Figure 5C). These findings suggest that an increase in Shp-2 association with TrkB as well as an increase in intrinsic Shp-2 activity is responsible at least in part for calcium-induced downregulation of the receptor's function.
Shp-2 deletion suppresses TrkB dephosphorylation
To confirm that Shp-2 participates in calcium-induced dephosphorylation of TrkB, experiments were performed in neurons with reduced levels of Shp-2. To generate such cells, the Cre/lox site-specific recombination system was used to delete the Shp-2 gene in cerebellar neurons from transgenic mice engineered to have the Shp-2 gene flanked by two loxP sites (floxed) (Figure 6A). The Cre DNA recombinase is a bacteriophage P1 protein that catalyzes recombination between two 34 bp loxP recognition sites. When the gene of interest is flanked by two directly repeated loxP sites, Cre expression promotes the excision of that gene (Sauer, 1998). This method avoids the severe developmental defects present in Shp-2 knockouts, and the modulatory effects of Shp-2 on TrkB activity can be studied directly and with less interference from other pathways. Cre recombinase delivered to neuronal cultures from floxed Shp-2 mice by adenovirus vector (AV) generated a four-fold reduction in Shp-2 protein levels, whereas control AV had no detectable effect (Figure 6B, lowest panel). In addition, depolarization-induced suppression of TrkB phosphorylation was partially inhibited, supporting a role for Shp-2 in TrkB downregulation. In contrast, neurons infected with control AV displayed normal calcium-induced inhibition of TrkB function (Figure 6B). As expected, reduction of Shp-2 levels also partially restored BDNF-induced Erk and Akt activation. In neurons with reduced Shp-2 expression, TrkB-associated phosphatase activity was also reduced (Figure 6C), confirming that Shp-2 is the main phosphatase associated with the receptor after calcium stimulation.
Figure 6.
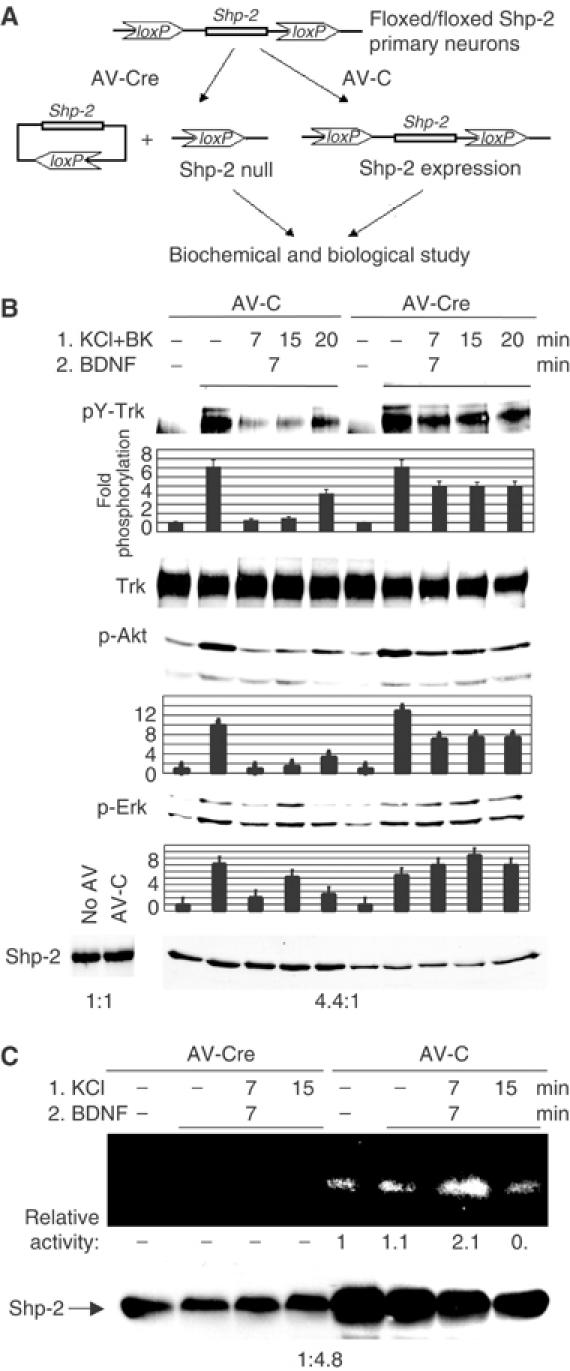
Reduction of Shp-2 protein level rescues TrkB signaling after calcium-induced inhibition. (A) Shp-2 expression in neurons is reduced with the Cre/loxP recombination system. The Cre DNA recombinase catalyzes recombination between two directly repeated loxP sites on either side of the gene and promotes the excision of Shp-2 gene. Neuron cultures from inducible (floxed) Shp-2 mice were infected with control AV (AV-C) or AV expressing the Cre protein (AV-Cre). (B) Reduction in Shp-2 protein levels rescues TrkB tyrosine phosphorylation and its downstream signaling. Shp-2 level in neurons was decreased 4.4-fold (averaged over lanes 1–5 versus 6–10) by AV-Cre infection relative to AV-C (last panel). Neurons were then treated with KCl (50 mM) plus BayK (BK) and BDNF (30 ng/ml) as shown, and Trk tyrosine phosphorylation was analyzed and quantified. Phospho-Akt and phospho-Erk levels were also quantified in the same samples. Data shown are representative of three independent experiments. (C) Phosphatase assay of Trk immunoprecipitates as in Figure 5A, using AV-infected neurons from Shp-2-Cre/lox mice. Shp-2 protein levels are shown below.
Shp-2 deletion increases neuronal survival after calcium influx
Deregulation of calcium homeostasis is thought to be the main cause of neuronal death in glutamate excitotoxicity and neurodegenerative diseases. Our results suggested that Shp-2 may participate in calcium-induced excitotoxicity by inhibiting BDNF-dependent survival signals that include Akt activation (Dudek et al, 1997). Therefore, we tested whether restoration of TrkB signaling in neurons by reducing Shp-2 protein levels promotes neuronal survival after intense neuronal stimulation. Cultures of cerebellar neurons from floxed Shp-2 mice, infected with either control or Cre AV for 3 days, were stimulated with 50 mM KCl, 20 mM KCl plus BayK or with 5 μM sodium glutamate in the presence of BDNF for 18 h (Figure 7). In neurons infected with control AV, approximately 50% cell death occurred after glutamate treatment for 18 h. In contrast, neurons with reduced Shp-2 levels were partially protected by BDNF against calcium excitotoxicity, displaying 77% survival rate, a 54% increase relative to controls with normal Shp-2 levels. Similar increases in survival rate occurred in low-Shp-2 neurons after depolarization with 20 mM KCl in the presence of BayK or with 50 mM KCl. This value likely underestimated the influence of Shp-2 since its expression was not completely reduced in these preparations of neurons. Survival rates after KCl or glutamate treatments were generally lower in virus-infected compared to uninfected neurons, which can survive up to 3 days after these treatments.
Figure 7.
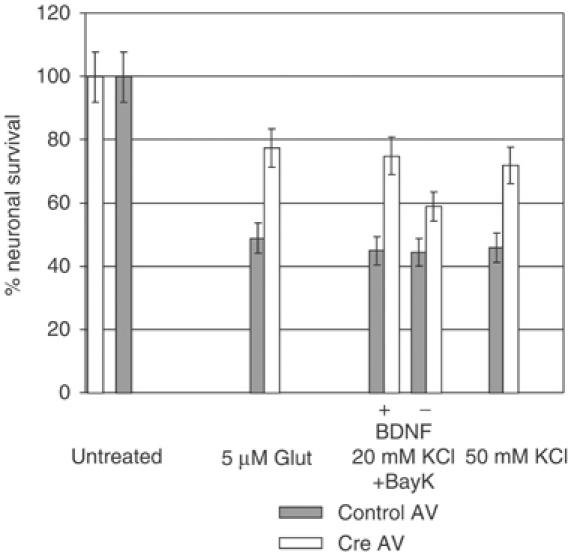
Reduction of Shp-2 protein levels promotes neuronal survival after excitotoxicity. Neuronal survival after excitotoxicity induced by 5 μM glutamate, 20 mM KCl plus 10 nM BayK or 50 mM KCl, each in the presence of 10 ng/ml BDNF, or 20 mM KCl plus BayK in the absence of BDNF. Survival rates are shown as a percentage of AV-infected, untreated neurons and represent an average of nine experiments. s.e.m.<±6.5%, P<0.001.
Discussion
We have shown that depolarization or glutamate treatment of cerebellar granule neurons inhibits subsequent or concomitant activation of TrkB receptors by BDNF. This negative crosstalk was detected as a suppression of both BDNF-induced tyrosine autophosphorylation of receptors and activation of signaling molecules that function downstream of these receptors, such as Ras, Erk and Akt. Calcium entry through either VSCCs or glutamate receptors appears to be the main event that induces the observed inhibition of TrkB signaling; however, the contribution of indirect events triggered by depolarization or glutamate, such as the release of other growth factors or neurotransmitters, cannot be excluded. The amplitude and duration of the peak inhibition were proportional to the intensity of depolarization, which determines the magnitude and duration of calcium influx (Di Virgilio et al, 1987). Thus, while neuronal depolarization generated a rather transient peak inhibition of TrkB activation, glutamate treatment resulted in a prolonged peak of inhibition, which was probably due to the slower desensitization of glutamate receptors. Moreover, prolonged peak inhibition of TrkB signaling was also observed when the open time of L-type VSCCs was extended by the addition of agonist BayK. In all cases, however, residual inhibition of Trk signaling persisted for at least 40 min.
Results from a series of experiments support the idea that depolarization or glutamate treatment suppresses TrkB receptor function at least in part by promoting its dephosphorylation by the tyrosine phosphatase, Shp-2. First, we showed through in-gel phosphatase assays that depolarization induced an increase in receptor-associated tyrosine phosphatase activity, which migrated in gels similarly to Shp-2, concomitant with the peak of TrkB dephosphorylation. Immunoblotting confirmed that the activity was due to Shp-2 by revealing an increase in Shp-2 content in TrkB immunoprecipitates that coincided with peak TrkB inhibition. Shp-2 phosphatase activity was also increased by depolarization. Finally, receptor-associated tyrosine phosphatase activity was reduced and depolarization-induced suppression of TrkB activity was reversed when Shp-2 expression was blunted through knockout of its gene in cultured neurons. We did note however lingering suppression of BDNF-induced activation of Trk receptors that did not coincide with detectable associated Shp-2 protein, suggesting that a secondary effect of depolarization on Trk activity may be involved.
Shp-2 is a ubiquitously expressed tyrosine phosphatase that has been implicated in tyrosine kinase signaling (Freeman et al, 1992; Feng et al, 1993; Vogel et al, 1993) through binding of its two SH2 domains to tyrosine phosphorylated proteins, such as the PDGF receptors and FRS2/SNT (Ong et al, 1997; Hadari et al, 1998) and Gab (Gu et al, 1998; Takahashi-Tezuka et al, 1998) adaptor proteins. Experiments in multiple animal models show that Shp-2 plays a positive role in receptor tyrosine kinase signaling to the Ras/Erk signaling cascade (Milarski and Saltiel, 1994; Tang et al, 1995) through both its scaffold function and the protein's phosphatase activity, at least in part by activation of c-src (Neel et al, 2003; Zhang et al, 2004). Evidence has also accumulated supporting a negative role of Shp-2 in receptor tyrosine kinase signaling (Peraldi et al, 1994). For example, Shp-2 negatively regulates PI3K activation by dephosphorylating PI3K-binding sites on the adaptor Gab1 (Zhang et al, 2002), and reactive oxygen species reverse the inhibitory effect of Shp-2 on PDGF receptor phosphorylation upon PDGF stimulation of cells (Meng et al, 2002).
Less is known about Shp-2 signaling in neurons, despite the fact that the protein's expression is particularly high in this tissue. Both loss-of-function and gain-of-function mutations in Shp-2 have been found to affect dramatically nervous system development. Mutations that constitutively activate Shp-2 cause Noonan's syndrome, which is associated with mental retardation (Tartaglia et al, 2001). Alternatively, deletion of Shp-2 during neurogenesis results in embryonic lethality (unpublished observation). Similar to other cell types, evidence exists for a positive role for Shp-2 in coupling tyrosine kinase receptors, in this case TrkB, to downstream signaling events in both cortical (Araki et al, 2000) and mesencephalic neurons (Takai et al, 2002). How this occurs is poorly understood, although receptor-induced association of Shp-2 with adaptor proteins such as Shps-1/BIT (Ohnishi et al, 1999), but not TrkB receptor, has been detected. In contrast, we have found that Shp-2 can negatively regulate BDNF signaling. What makes our investigation distinct is that we detected a negative role for Shp-2 only in response to depolarization or glutamate, which were not investigated in other studies. This difference implies that calcium causes a change in Shp-2 function from a positive regulator to a negative regulator. Although we do not yet know the details of this mechanism, we did note that depolarization induced an association of Shp-2 with the TrkB receptor that stimulation of cells with BDNF did not. This suggests that calcium changes the nature of the complex within which Shp-2 functions, presumably making the TrkB receptor more accessible to the phosphatase activity of Shp-2 and thus enabling Shp-2 to regulate receptor function negatively.
The ∼7-fold reduction in TrkB tyrosine phosphorylation (Figure 3C) suggests that most, possibly all, TrkB phosphorylation sites are affected, including the site of Shp-2 binding. In combination with the transient nature of calcium influx, this may explain the brief Shp-2–TrkB association. However, after the initial drop, TrkB phosphorylation does not fully recover during long-term depolarization, consistent with a low calcium influx that continues to leak into neurons. Thus, suppression of BDNF signaling by calcium influx may have a significant long-term impact. At the same time, depolarization also triggers massive exocytosis that may initiate a sequence of indirect events, which can also contribute to the observed outcome of neuronal survival. In addition, Shp-2 activation may influence other substrates to affect long-term neuronal function.
One function for this negative feedback system from calcium to TrkB receptor function may be to modulate signaling specificity. While calcium and BDNF activate many of the same signaling pathways in neurons, some clear differences have been noted. For example, while BDNF activates Akt in primary cerebellar granule cells, depolarization does not. However, calcium influx promotes the synthesis (West et al, 2001) and secretion of BDNF (Goodman et al, 1996; Hartmann et al, 2001). Thus, the negative feedback system we identified could function to prevent an autocrine effect of BDNF release that could obscure this signaling distinction between calcium and neurotrophins signaling (Morrison and Mason, 1998). Instead, secreted BDNF would still be able to influence neighboring cells, which could be the true target of BDNF. Moreover, this negative feedback system appears to discriminate between signals from different neurotrophin receptors. For example, Shp-2 has been implicated in the signaling of TrkA (Goldsmith and Koizumi, 1997); however, in contrast to TrkB, TrkA tyrosine phosphorylation after NGF stimulation of PC12 cells was not inhibited by depolarization (data not shown). TrkA may instead be negatively regulated through Shp-1 (Marsh et al, 2003), a related SH2 domain-containing tyrosine phosphatase. Depolarization-mediated inhibition of TrkB but not TrkA is in agreement with the fact that striatal neuronal death in Huntington's disease was substantially prevented by NGF-producing stem cells, but not by BDNF secretion (Martinez-Serrano and Bjorklund, 1996).
Another consequence of this negative feedback system from calcium to TrkB receptor function may arise under pathological conditions of excess calcium entry, where Shp-2 may contribute to neuronal death, at least in part by inhibiting survival pathways activated by BDNF, such as Akt. Moderate calcium influx is necessary for neuronal survival and can to some extent enhance TrkB signaling (Goodman et al, 1996; Meyer-Franke et al, 1998; Hartmann et al, 2001; Du et al, 2003). However, high intracellular calcium concentration has been considered to be a common major cause of neuronal death in glutamate-induced excitotoxicity or in neurodegenerative diseases, although the specific molecular mechanisms involved in each condition are different (Brorson et al, 1994; Lee et al, 1999; Mattson, 2000; Yuan and Yankner, 2000; Sattler and Tymianski, 2001; LaFerla, 2002). This effect has been thought to be mainly the result of calpain activation by calcium and subsequent caspase activation and cleavage of structural proteins like spectrin (Buchman et al, 1998). However, our data suggest that activity-dependent association of Shp-2 with TrkB and downregulation of the receptor may also be involved. This notion is supported by our finding that cell death in primary cultures of cerebellar granule cells induced by glutamate, depolarization or L-type channel agonist BayK treatment was suppressed in neurons where the Shp-2 gene was deleted.
These results show that Shp-2 is part of a tightly regulated signaling network that connects calcium and growth factor action and regulates the timing and amplitude of downstream signals that control neuronal survival. Thus, Shp-2 may constitute an interesting drug target for preventing neuron loss during pathological conditions associated with increased intracellular calcium concentrations.
Materials and methods
Cerebellar neuron cultures
Cerebelli extracted from 6- to 7-day-old mice were dissociated in a solution of 0.3 mg/ml papain (Worthington) in Basal Medium Eagle (BME, Gibco) containing 1 mM kynurenic acid. After 30 min, the tissue was triturated in BME containing 10% fetal bovine serum, 35 mM glucose, 1 mM glutamine and 0.5% penicillin/streptomycin solution (Gibco). Triturated neurons were diluted in the same medium and plated on poly-L-lysine/laminin-coated plates. The next day, KCl (15 mM) and arabinoside C (10 μM) were added to the culture medium. Neurons were used for experiments 4–7 days after plating.
Protein phosphorylation assays
Cultured neurons were starved in serum-free BME for 3–4 h. For depolarization experiments, standard BME was replaced with modified BME where NaCl was reduced to 86–41 mM and KCl was increased to 30–75 mM. BDNF (30 ng/ml) was added to the depolarizing medium, as indicated. In pulse–chase experiments, the depolarizing medium was replaced after 1–3 min with standard BME for a total of 7 min, after which BDNF was added for another 7 min. Neurons were then quickly lysed on ice in a buffer containing 1% NP-40, 150 mM NaCl, 10 mM NaF, 50 mM Tris pH 7.5, 1 mM sodium vanadate, 1 mM PMSF, 15 ng/ml aprotinin and 20 μM leupeptin (Sigma). The supernatants were run on Western blots or used for TrkB immunoprecipitation, and the blots stained with anti-phospho-Erk, anti-phospho-Akt or anti-phosphotyrosine 4G10 (Cell Signaling) to detect the phosphorylation/activation levels of Erk, Akt and TrkB, respectively. Total TrkB protein was detected with anti-Trk C-14 (Santa Cruz Biotechnology). Where indicated, blots were scanned and bands quantified using an Alpha Innotech system.
Ras assay
Active Ras-GTP was affinity purified from neuron lysates by incubation for 30 min with S-hexylglutathione/agarose-bound GST-Ras-binding domain of c-Raf and analyzed by Western blot along with total lysates from the same samples, and then stained with Ras antibodies (Transduction Labs).
Labeling of cell surface TrkB
Cultured neurons were treated as above with depolarization and/or BDNF and then washed on the dish twice with ice-cold PBS. The neurons were then incubated on ice for 30 min with a solution of 0.5 mg/ml sulfosuccinimidyl 6-(biotinamido) hexanoate (EZ Link Sulfo-NHS-LC-biotin, Pierce) in PBS. After removing the biotinylation reagent, the cells were washed three times with ice-cold PBS and then lysed in 1% NP-40. Biotinylated cell surface proteins were extracted from lysates by affinity binding to avidin immobilized on agarose, and then run on Western blot and immunoblotted for Trk. Alternatively, after biotinylation, cell lysates were divided in half, and one-half was used for extraction of biotinylated proteins as above, while the other half was analyzed for Trk tyrosine phosphorylation of Trk immunoprecipitates.
In-gel tyrosine phosphatase assay
Poly-(Glu4-Tyr) (Sigma) was labeled with 32P in the presence of v-Abl (NE Biolabs) and [γ-32P]ATP overnight at room temperature. The radioactive peptide was separated from excess [γ-32P]ATP by chromatography on a Sephadex G50 column eluted with 50 mM imidazole, pH 7.2. The purified peptide was mixed and polymerized in a 10% acrylamide gel on which Trk or Shp-2 immunoprecipitates were run. The gel was incubated in 50 mM Tris pH 8/20% 2-propanol for 1 h, in 0.3% β-mercaptoethanol for 1 h and in 0.3% β-mercaptoethanol/1 mM EDTA/6 M guanidine–HCl/50 mM Tris pH 8 for 2 h for protein denaturation. The gel was then stored overnight in renaturation buffer containing 0.3% β-mercaptoethanol, 1 mM EDTA, 0.04% Tween 20 and 50 mM Tris pH 7.5, dried and exposed on a film. A fraction of each lysate was used to analyze total Shp-2 protein content, for uniformity and to compare protein size.
Shp-2 deletion in neurons from floxed Shp-2 mice
Cerebellar neuron cultures were prepared from floxed Shp-2 transgenic mice as described (Zhang et al, 2004). At 2 days after plating, control AV or AV expressing the Cre recombinase were added to the culture medium at a concentration of 25 MOI. At 3.5 days after infection, neurons were serum-starved for 3 h and used in experiments as shown.
Neuronal survival assay
Cerebellar neurons from floxed Shp-2 mice were cultured on 24-well plates and infected with control or Cre AV as above. At 3 days postinfection, the culture medium, already containing 20 mM KCl, was partially removed and BME containing 75 mM KCl instead of NaCl was added to a final concentration of 20–50 mM KCl. BayK (10 nM) was added to the 20 mM KCl medium and neurons were depolarized in the presence of 10 ng/ml BDNF. Alternatively, 5 μM sodium glutamate was added in the presence of 10 ng/ml BDNF. After 18 h, healthy neurons (assessed by phase-contrast reflection and axon integrity) were counted and stained with 0.4% Trypan blue, and unstained neurons were recounted.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
This work was supported by grants to LAF from NIH, CA47391 and GRASP Digestive Disease Center (P30-DK34928 GRASP) and to BGN from NIH, CA491452 and DK 50693.
References
- Araki T, Yamada M, Ohnishi H, Sano S, Uetsuki T, Hatanaka H (2000) Shp-2 specifically regulates several tyrosine-phosphorylated proteins in brain-derived neurotrophic factor signaling in cultured cerebral cortical neurons. J Neurochem 74: 659–668 [DOI] [PubMed] [Google Scholar]
- Brorson JR, Manzolillo PA, Miller RJ (1994) Ca2+ entry via AMPA/KA receptors and excitotoxicity in cultured cerebellar Purkinje cells. J Neurosci 14: 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman VL, Adu J, Pinon LG, Ninkina NN, Davies AM (1998) Persyn, a member of the synuclein family, influences neurofilament network integrity. Nat Neurosci 1: 101–103 [DOI] [PubMed] [Google Scholar]
- Cowan WM, Hamburger V, Levi-Montalcini R (2001) The path to the discovery of nerve growth factor. Annu Rev Neurosci 24: 551–600 [DOI] [PubMed] [Google Scholar]
- Di Virgilio F, Milani D, Leon A, Meldolesi J, Pozzan T (1987) Voltage-dependent activation and inactivation of calcium channels in PC12 cells. Correlation with neurotransmitter release. J Biol Chem 262: 9189–9195 [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275: 661–665 [DOI] [PubMed] [Google Scholar]
- Du J, Feng L, Zaitsev E, Je HS, Liu XW, Lu B (2003) Regulation of TrkB receptor tyrosine kinase and its internalization by neuronal activity and Ca2+ influx. J Cell Biol 163: 385–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA (1995) Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 376: 524–527 [DOI] [PubMed] [Google Scholar]
- Feng G.S., Hui C.C., Pawson T (1993) SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science 259: 1607–1611 [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B (1996) Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381: 706–709 [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Greenberg ME (1996) Ca(2+)-dependent routes to Ras: mechanisms for neuronal survival, differentiation, and plasticity? Neuron 16: 233–236 [DOI] [PubMed] [Google Scholar]
- Freeman RM Jr, Plutzky J, Neel BG (1992) Identification of a human src homology 2-containing protein-tyrosine-phosphatase: a putative homolog of Drosophila corkscrew. Proc Natl Acad Sci USA 89: 11239–11243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME (1994) Requirement for BDNF in activity-dependent survival of cortical neurons. Science 263: 1618–1623 [DOI] [PubMed] [Google Scholar]
- Goldsmith BA, Koizumi S (1997) Transient association of the phosphotyrosine phosphatase SHP-2 with TrkA is induced by nerve growth factor. J Neurochem 69: 1014–1019 [DOI] [PubMed] [Google Scholar]
- Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F (1996) Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci 7: 222–238 [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA 73: 2424–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Pratt JC, Burakoff SJ, Neel BG (1998) Cloning of p97/Gab2, the major SHP2-binding protein in hematopoietic cells, reveals a novel pathway for cytokine-induced gene activation. Mol Cell 2: 729–740 [DOI] [PubMed] [Google Scholar]
- Hadari YR, Kouhara H, Lax I, Schlessinger J (1998) Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol Cell Biol 18: 3966–3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V (2001) Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J 20: 5887–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafitz KW, Rose CR, Thoenen H, Konnerth A (1999) Neurotrophin-evoked rapid excitation through TrkB receptors. Nature 401: 918–921 [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD (2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381–391 [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T (1995) Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA 92: 8856–8860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci 3: 862–872 [DOI] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW (1999) The changing landscape of ischaemic brain injury mechanisms. Nature 399: A7–A14 [DOI] [PubMed] [Google Scholar]
- Marsh HN, Dubreuil CI, Quevedo C, Lee A, Majdan M, Walsh GS, Hausdorff S, Said FA, Zoueva O, Kozlowski M, Siminovitch K, Neel BG, Miller FD, Kaplan DR (2003) SHP-1 negatively regulates neuronal survival by functioning as a TrkA phosphatase. J Cell Biol 163: 999–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Serrano A, Bjorklund A (1996) Protection of the neostriatum against excitotoxic damage by neurotrophin-producing, genetically modified neural stem cells. J Neurosci 16: 4604–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP (2000) Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1: 120–129 [DOI] [PubMed] [Google Scholar]
- Meng TC, Fukada T, Tonks NK (2002) Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell 9: 387–399 [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG Jr, Reichardt LF, Barres BA (1998) Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 21: 681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milarski KL, Saltiel AR (1994) Expression of catalytically inactive Syp phosphatase in 3T3 cells blocks stimulation of mitogen-activated protein kinase by insulin. J Biol Chem 269: 21239–21243 [PubMed] [Google Scholar]
- Morrison ME, Mason CA (1998) Granule neuron regulation of Purkinje cell development: striking a balance between neurotrophin and glutamate signaling. J Neurosci 18: 3563–3573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel BG, Gu H, Pao L (2003) The ‘Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28: 284–293 [DOI] [PubMed] [Google Scholar]
- Ohnishi H, Yamada M, Kubota M, Hatanaka H, Sano S (1999) Tyrosine phosphorylation and association of BIT with SHP-2 induced by neurotrophins. J Neurochem 72: 1402–1408 [DOI] [PubMed] [Google Scholar]
- Ong SH, Lim YP, Low BC, Guy GR (1997) SHP2 associates directly with tyrosine phosphorylated p90 (SNT) protein in FGF-stimulated cells. Biochem Biophys Res Commun 238: 261–266 [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER (1996) Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16: 1137–1145 [DOI] [PubMed] [Google Scholar]
- Peraldi P, Zhao Z, Filloux C, Fischer EH, Van Obberghen E (1994) Protein-tyrosine-phosphatase 2C is phosphorylated and inhibited by 44-kDa mitogen-activated protein kinase. Proc Natl Acad Sci USA 91: 5002–5006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen LB, Ginty DD, Weber MJ, Greenberg ME (1994) Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron 12: 1207–1221 [DOI] [PubMed] [Google Scholar]
- Rusanescu G, Qi H, Thomas SM, Brugge JS, Halegoua S (1995) Calcium influx induces neurite growth through a Src–Ras signaling cassette. Neuron 15: 1415–1425 [DOI] [PubMed] [Google Scholar]
- Sattler R, Tymianski M (2001) Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol 24: 107–129 [DOI] [PubMed] [Google Scholar]
- Sauer B (1998) Inducible gene targeting in mice using the Cre/lox system. Methods 14: 381–392 [DOI] [PubMed] [Google Scholar]
- Schuman EM (1999) Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol 9: 105–109 [DOI] [PubMed] [Google Scholar]
- Takahashi-Tezuka M, Yoshida Y, Fukada T, Ohtani T, Yamanaka Y, Nishida K, Nakajima K, Hibi M, Hirano T (1998) Gab1 acts as an adapter molecule linking the cytokine receptor gp130 to ERK mitogen-activated protein kinase. Mol Cell Biol 18: 4109–4117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai S, Yamada M, Araki T, Koshimizu H, Nawa H, Hatanaka H (2002) Shp-2 positively regulates brain-derived neurotrophic factor-promoted survival of cultured ventral mesencephalic dopaminergic neurons through a brain immunoglobulin-like molecule with tyrosine-based activation motifs/Shp substrate-1. J Neurochem 82: 353–364 [DOI] [PubMed] [Google Scholar]
- Tang TL, Freeman RM Jr, O'Reilly AM, Neel BG, Sokol SY (1995) The SH2-containing protein-tyrosine phosphatase SH-PTP2 is required upstream of MAP kinase for early Xenopus development. Cell 80: 473–483 [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD (2001) Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 29: 465–468 [DOI] [PubMed] [Google Scholar]
- Vogel W, Lammers R, Huang J, Ullrich A (1993) Activation of a phosphotyrosine phosphatase by tyrosine phosphorylation. Science 259: 1611–1614 [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001) Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 98: 11024–11031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Yankner BA (2000) Apoptosis in the nervous system. Nature 407: 802–809 [DOI] [PubMed] [Google Scholar]
- Zhang SQ, Tsiaras WG, Araki T, Wen G, Minichiello L, Klein R, Neel BG (2002) Receptor-specific regulation of phosphatidylinositol 3′-kinase activation by the protein tyrosine phosphatase Shp2. Mol Cell Biol 22: 4062–4072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T, Luo J, Thompson JA, Schraven BL, Philips MR, Neel BG (2004) Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol Cell 13: 341–355 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2