

Abstract
Reperfusion of ischemic myocardium is crucial for salvaging myocardial cells from ischemic cell death. However, reperfusion itself induces various deleterious effects on the ischemic myocardium. These effects, known collectively as reperfusion injury, comprise stunned myocardium, reperfusion-induced arrhythmia, microvascular reperfusion injury, and lethal reperfusion injury. No approach has proven successful in preventing any of these injuries in the clinical setting. My colleagues and I recently proposed a new postconditioning protocol, postconditioning with lactate-enriched blood (PCLeB), for the prevention of reperfusion injury. This new approach consists of intermittent reperfusion and timely coronary injections of lactated Ringer's solution, aiming to achieve controlled reperfusion with cellular oxygenation and minimal lactate washout from the cells. This approach appeared to be effective in preventing all types of reperfusion injury in patients with ST-segment elevation myocardial infarction (STEMI), and we have already reported excellent in-hospital outcomes of patients with STEMI treated using PCLeB. In this review article, I discuss a possible mechanism of reperfusion injury, which we believe to be valid and which we targeted using this new approach, and I report how the approach worked in preventing each type of reperfusion injury.
Abbreviation: CAG, coronary angiography; CK, creatine kinase; CRP, C-reactive protein; ECG, electrocardiography; MI, myocardial infarction; MPT, mitochondrial permeability transition; STEMI, ST-segment elevation myocardial infarction; PCI, percutaneous coronary intervention; PCLeB, postconditioning with lactate-enriched blood; PVC, premature ventricular contraction; TIMI, thrombolysis in myocardial infarction; VF, ventricular fibrillation; VT, ventricular tachycardia
Keywords: Lactate, No-reflow phenomenon, Postconditioning, Reperfusion arrhythmia, ST-segment elevation myocardial infarction, Stunning
Highlights
-
•
Postconditioning with lactate-enriched blood (PCLeB) targets hypercontracture.
-
•
PCLeB employs lactate as an inherent contractile activity blocker.
-
•
PCLeB appears to be effective in preventing all four types of reperfusion injury.
-
•
PCLeB may also suppress early inflammation after myocardial infarction.
-
•
PCLeB may improve outcomes of patients with acute myocardial infarction.
1. Introduction
Reperfusion of ischemic myocardium is crucial for salvaging myocardial cells from ischemic cell death. However, reperfusion itself induces various deleterious effects on the ischemic myocardium; this has become known as reperfusion injury. Originally, reperfusion injury was classified into four types of injury [1]: 1. stunned myocardium (stunning); 2. reperfusion-induced arrhythmia; 3. microvascular reperfusion injury (the no-reflow phenomenon or, more recently, microvascular obstruction); and 4. lethal reperfusion injury. No approach has proven successful in preventing any of these injuries in the clinical setting.
My colleagues and I recently reported a new modified postconditioning protocol, postconditioning with lactate-enriched blood (PCLeB), which appeared to be effective against all four types of reperfusion injury [2], [3], [4], [5], [6]. In this review article, I discuss a possible mechanism of reperfusion injury, which we believe to be valid and which we targeted using the new postconditioning protocol, and I report how this new approach worked in preventing each type of reperfusion injury.
2. Hypercontracture as a new target for the prevention of reperfusion injury
Hypercontracture develops within the reperfused ischemic myocardium and mechanically disrupts myocardial cell skeletons that ischemic insults have already made vulnerable to such strong forces. Consequently, contraction band necrosis ensues in the myocardium [7], [8]. This has been included among the mechanisms of lethal reperfusion injury [1], [9], [10], [11], [12]. However, hypercontracture has not been regarded as the primary cause or trigger, but only as one component of the cascade of events that eventually results in reperfusion injury.
Currently, the most popular mechanism for reperfusion injury involves mitochondrial disorders that result from a permeability transition [11], [12], [13]. During ischemia, the mitochondrial permeability transition (MPT) pore remains closed, but it opens within minutes of reperfusion [14], [15]. This transition is thought to initiate the irreversible process of reperfusion injury. Meanwhile, a well-developed hypercontracture (contraction band necrosis) is reportedly observed within 120 s of reperfusion of ischemic myocardium in animal experiments [16]. Although a causal relationship between hypercontracture and MPT has not been established, development of a mature hypercontracture appears to coincide chronologically with the onset of MPT. Although MPT may play an important role in the development of reperfusion injury, its role in hypercontracture development is rather questionable. We therefore targeted hypercontracture for the prevention of reperfusion injury, rather than MPT.
In a recent large-scale clinical trial, cyclosporine, a potent inhibitor of MPT, failed to improve the long-term outcomes of patients with ST-segment elevation myocardial infarction (STEMI) and failed to prevent left ventricular remodeling [17]. Thus, for an effective breakthrough, a new strategy for the prevention of reperfusion injury would appear to be essential.
3. PCLeB, a novel postconditioning protocol
In 2005, Staat et al. reported that four brief cycles of intermittent reperfusion applied immediately after reopening of the culprit coronary artery, a procedure called “postconditioning”, reduced the infarct size in patients with STEMI [18]. The beneficial effects of postconditioning were attributed to the delay in recovery from tissue acidosis produced during ischemia. However, recent larger clinical trials did not demonstrate any protective effects of postconditioning in patients with STEMI [19], [20]. To achieve more consistent results, my colleagues and I modified the original protocol of postconditioning to increase the delay in recovery from intracellular acidosis during the early reperfusion period [2]. Fig. 1 shows an overview of the protocol of PCLeB. In our postconditioning protocol, the duration of each brief reperfusion is prolonged from 10 s to 60 s in a stepwise manner. This approach sought to prevent rapid and abrupt washout of lactate during the very early phase of reperfusion. Lactated Ringer's solution (20–30 mL) containing 28 mM lactate is injected directly into the culprit coronary artery at the end of each brief reperfusion and the balloon is quickly inflated at the site of the lesion, so that the lactate is trapped inside the ischemic myocardium. Each brief ischemic period lasts 60 s. After 7 cycles of balloon inflation and deflation, full reperfusion is performed. This approach aims to achieve controlled reperfusion with cellular oxygenation and minimal lactate washout from the cells. Lactate accumulation is generally accepted to be responsible for intracellular acidosis during ischemia. Therefore, the delay in recovery from intracellular acidosis, achieved by simple intermittent reperfusion postconditioning, may be increased through this approach.
Fig. 1.

Overview of the protocol for postconditioning with lactate-enriched blood. The duration of each brief reperfusion was prolonged from 10 to 60 s in a stepwise manner. At the end of each brief reperfusion, lactate was supplied by injecting lactated Ringer's solution into the culprit coronary artery. Each brief ischemic period lasted 60 s. After 7 cycles of balloon inflation and deflation, full reperfusion was performed; subsequently, stenting was performed. LCA, left coronary artery; RCA, right coronary artery.
(Reprinted from Koyama et al. [3]).
More specifically, this method precisely targets the prevention of hypercontracture. My colleagues and I have previously reported that abrupt washout of lactate during the reperfusion following simulated ischemia induced contracture in guinea-pig myocytes. This occurred despite a substantial decrease in their intracellular Ca2 + concentrations, which were elevated during the simulated ischemia [21]. Based on these findings, we attempted to create a transition period between ischemia and reperfusion. This transition period allowed the elevated intracellular Ca2 + concentration to normalize safely, while suspending the restoration of vigorous myocardial contraction by keeping the tissue lactate concentrations high (maintaining tissue acidosis). This was the basic concept of our novel postconditioning protocol. To summarize our protocol, lactate was employed as an inherent contractile activity blocker with the aim of preventing the rapid recovery of contractile force immediately following reperfusion, which otherwise might result in the development of hypercontracture.
This idea was fueled by previous experimental studies. Garcia-Dorado et al. reported that inhibition of the contractile apparatus of ischemic myocardium using 2,3-butanedione monoxime during the first 30 min of reperfusion reduced the infarct size in pigs [22]. Buckberg and colleagues also showed that reperfusion for 20 to 30 min with hyperkalemic blood prevented contraction and reduced myocyte death during totally vented bypass in the canine heart [23], [24]. This approach is currently widely used in the field of open-heart surgery as a method of cardioprotection, and is called terminal warm blood cardioplegia. Thus, inhibition of myocardial contraction during the early reperfusion period may be an effective approach for cardioprotection.
4. Effects of PCLeB on microvascular reperfusion injury (the no-reflow phenomenon or microvascular obstruction)
Mechanical restoration of coronary blood flow by percutaneous coronary intervention (PCI) often results in tissue hypoperfusion in patients with STEMI; this is reperfusion injury in the microvasculature, known as the no-reflow phenomenon [25], [26], or more recently as microvascular obstruction [27], [28]. My colleagues and I reported that PCLeB was consistently followed by angiographically excellent microcirculation recovery [2], [3], [4], [5], [6]. The coronary angiography findings of a representative patient are presented in Fig. 2. This patient had a totally occluded obtuse marginal artery. After completion of PCI using PCLeB, coronary blood flow in the obtuse marginal artery appeared to be faster than that in the other coronary arteries. Furthermore, the washout of the contrast medium from the obtuse marginal artery appeared to be faster still, indicating that microcirculation recovery was apparently augmented. These angiographic observations have been confirmed by a more quantitative approach. We reported that the mean corrected Thrombolysis in Myocardial Infarction (TIMI) frame count of 55 consecutive STEMI patients treated using PCLeB was 20.1 ± 10.1 frames (normal value 21 frames) [5]. Among these patients, 30 so far have returned for follow-up coronary angiography and have shown no angiographic restenosis. In these 30 patients, the corrected TIMI frame counts immediately after PCI completion (19.1 ± 9.3 frames) were significantly smaller than those in the chronic phase (27.4 ± 12.2 frames, p < 0.0001). These results were in stark contrast to the no-reflow phenomenon, and the results obtained indicated more than its prevention. Because our approach did not include any pharmacological intervention, the consistently observed excellent recovery of microcirculation can be viewed as an emergence of what it truly is, unveiled by our approach. Reactive hyperemia caused by endogenous vasodilators, such as adenosine or nitric oxide, is known to be transiently observed during the early reperfusion period following ischemia. The reason for the transient nature of reactive hyperemia is unknown. However, the consistently observed excellent microcirculation recovery in our studies suggests that reactive hyperemia had been prematurely abolished by microvascular reperfusion injury that developed immediately after reperfusion. Successful attenuation of microvascular reperfusion injury by our approach might possibly unveil the true nature of reactive hyperemia, i.e., long-lasting reactive hyperemia, for the first time ever. Hypercontracture obliterates microcirculatory coronary flow by compressing the vessel walls from outside; this action has been incorporated into the mechanisms of the no-reflow phenomenon [29], [30]. The consistently observed excellent microcirculation recovery may indicate that our approach, targeting hypercontracture, has been successful.
Fig. 2.

Coronary angiography (CAG) findings of a representative patient (an 81-year-old woman), before and after percutaneous coronary intervention (PCI).
Panel 0: CAG before PCI. The obtuse marginal artery was totally occluded (black arrow). Panel 1: The early phase of the final CAG view after PCI completion. The contrast medium reached the far end of the obtuse marginal artery (white arrow) while still only extending halfway through the left anterior descending artery and the main trunk of the left circumflex artery (black arrows). Panel 2: The mid phase of the final CAG view. Panel 3: The late phase of the final CAG view. The contrast medium still remained in both the left anterior descending and the main trunk of the left circumflex arteries (white arrows), while it had already washed out from the obtuse marginal artery.
5. Effects of PCLeB on reperfusion-induced arrhythmia
Reperfusion therapy for STEMI is frequently associated with serious ventricular arrhythmias, such as ventricular tachycardia (VT) or ventricular fibrillation (VF). This phenomenon has been described as reperfusion-induced arrhythmia [1]. My colleagues and I quite recently reported the absence of reperfusion-induced arrhythmia in 58 consecutive STEMI patients treated using PCLeB [6]. None of the 58 study patients experienced sustained or non-sustained VT or VF during the course of the reperfusion therapy. In that study, for a more analytical investigation of reperfusion-induced arrhythmia, the last 9 patients with non-inferior STEMI underwent Holter electrocardiography (ECG) during and after reperfusion. Because we noticed that some hemodynamic deterioration during reperfusion, such as bradycardia or hypotension, induced scattered premature ventricular contractions (PVCs) in patients with inferior STEMI, we included only patients with non-inferior STEMI in the Holter ECG analysis, thus eliminating the effects of hemodynamic deterioration on the emergence of ventricular arrhythmia.
Fig. 3 shows the PVC counts, which were determined from the Holter ECG recordings made during and after reperfusion in the 9 patients. The maximum PVC counts were observed several hours after reperfusion in most of the patients. The mean PVC count during the first hour of reperfusion was only 3.7 ± 3.6 counts/h. Multiple PVCs were not observed during the first hour of reperfusion in any of the 9 patients, thus providing additional support for the protective effects of PCLeB against reperfusion-induced arrhythmia. The absence of reperfusion-induced arrhythmia suggests that additional insult to the ischemic myocardium upon reperfusion was minimal in STEMI patients treated using PCLeB.
Fig. 3.

PVC counts during and after reperfusion in the patients with non-inferior STEMI. The counts were determined from the Holter electrocardiographic recordings. The patients' data are shown individually, using a different color for each patient.
Pt, patient; PVC, premature ventricular contraction; STEMI, ST-segment elevation myocardial infarction.
(Reprinted from Akima et al. [6]).
6. Effects of PCLeB on myocardial stunning
Prolonged contractile dysfunction observed after reperfusion of the ischemic myocardium is defined as stunned myocardium [31]. In clinical settings, attenuation of stunning may not be easy to demonstrate. My colleagues and I reported a STEMI patient with muscle squeezing of the culprit coronary artery immediately after PCI completion using PCLeB (Fig. 4) [32]. This indicates prompt recovery of myocardial contraction, specifically in the epicardial portion of the reperfused myocardium. Moreover, we reported excellent in-hospital outcomes in 55 consecutive STEMI patients treated using PCLeB [5]. None of the 55 study patients died or required continuation of oral diuretic or inotropic therapy for heart failure on discharge. In fact, pulmonary congestion was observed on chest radiographs in 13 patients during the early days of their hospital stay. Among these 13 patients, only 3 required temporary use of a diuretic, while the other 10 patients required no specific heart failure therapy for remission. These results suggest prompt recovery of myocardial contraction after reperfusion therapy using PCLeB, without substantial myocardial stunning.
Fig. 4.

A patient (42-year-old man) with muscle squeezing of the culprit coronary artery immediately after the completion of percutaneous coronary intervention (PCI), performed 100 min after the onset of chest pain: peak creatine kinase (CK) level, 10,140 IU/L; time to peak CK release, 2.5 h. Coronary angiography (CAG) before and after PCI and resting thallium-201 scintigraphy before discharge are shown. Upper left panel: CAG prior to PCI. The proximal left anterior descending artery (LAD) is completely occluded. Upper middle panel: Diastolic phase of the final CAG view after completion of PCI. Upper right panel: The systolic phase of the same CAG view as shown in the upper middle panel. The mid-portion of the LAD shows segmental narrowing caused by muscle squeezing (arrow). Lower panel: The resting thallium-201 scintigraphy before discharge. The take-up of thallium by the myocardium of the LAD area is excellent, showing well-preserved myocardial viability, despite the high peak CK level achieved.
(Reprinted from Koyama et al. [32]).
7. Effects of PCLeB on lethal reperfusion injury
Reperfusion of ischemic myocardium is crucial for salvaging myocardial cells from ischemic cell death. However, reperfusion itself induces another type of myocardial cell death, called lethal reperfusion injury [1], [9], [10], [11], [12], the most critical kind of reperfusion injury in terms of patient prognosis. Although infarct-size reduction would be the most robust evidence for the successful prevention of lethal reperfusion injury, this has not yet been demonstrated in patients treated using PCLeB. However, my colleagues and I recently demonstrated that PCLeB abolished a biphasic relationship between the time to reperfusion and the time to peak creatine kinase (CK) release, which might be attributed to the consequences of lethal reperfusion injury [4].
It is well known that reperfusion therapy for patients with acute myocardial infarction (MI) induces an early peaking of blood levels of cardiac markers such as CK or cardiac troponin. This phenomenon has been explained as a washout mechanism induced by reperfusion therapy [33]. However, we were all aware that the front-loading of peak CK release was not uniform, but was observed to a variable degree, even in STEMI patients who were treated similarly with successful reperfusion therapy. My colleagues and I previously reported a biphasic relationship between the time to reperfusion and the time to peak CK release in 55 consecutive patients with acute MI who were undergoing successful PCI; the latest CK release occurred in patients treated approximately 7 h after MI onset, with progressively earlier CK release occurring in patients treated either earlier or later than 7 h after MI onset (Fig. 5a) [34]. This biphasic relationship might add some clarity to the diversity observed among MI patients treated with successful PCI, in terms of the early-peaking CK phenomenon. We claimed that this biphasic relationship could not be explained by the traditional CK washout mechanism, but could be explained if one considered the possibility that cell death caused by lethal reperfusion injury made a significant contribution to early CK release.
Fig. 5.
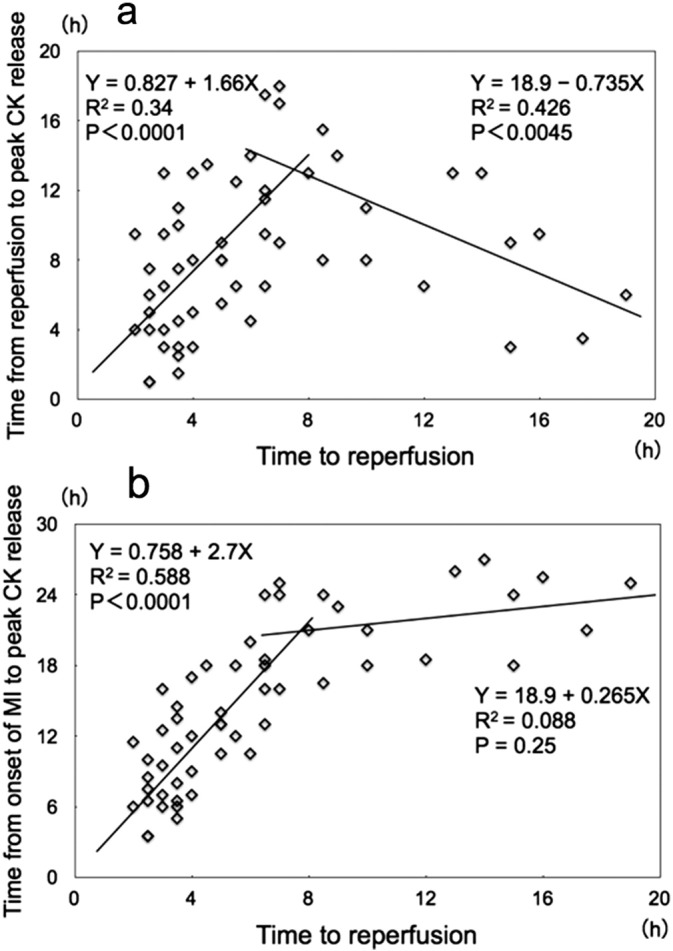
a) Relationship between the time to reperfusion and the time to peak serum creatine kinase (CK) value in 55 consecutive patients with acute myocardial infarction (MI) undergoing successful percutaneous coronary intervention. There was a positive linear correlation between the time to reperfusion and the time to peak serum CK release in cases where the time to reperfusion was within 7 h of myocardial infarction onset. After peaking at approximately 7 h, the time to peak serum CK levels declined linearly as the time to reperfusion increased. A negative linear correlation existed between time to reperfusion and time to peak serum CK when the time to reperfusion was longer than 7 h.
b) Relationship between the time to reperfusion and the time from the onset of MI to peak serum CK release. There was a positive linear correlation between the time to reperfusion and the time to peak serum CK in cases where the time to reperfusion was within 7 h of MI onset; once the time to reperfusion exceeded 7 h, the regression line became fairly flat, and the time to peak serum CK release was constant at approximately 24 h.
(Reprinted from Koyama et al. [34]).
In Fig. 5b, the time from the onset of MI to peak serum CK release is plotted, rather than the time from the start of reperfusion, to make the relationship more comprehensible. We found a positive linear correlation between the time to reperfusion and the time from onset of MI to peak serum CK release, as long as the time from MI onset to the start of reperfusion was within 7 h. In contrast, when this time exceeded 7 h, the time to peak serum CK release was approximately constant at 24 h. Without reperfusion therapy, the time from the onset of MI to peak serum CK release is known to be approximately 24 h. This implies that reperfusion therapy performed 7 h or later after the onset of MI has no effect on the time from the onset of MI to peak serum CK release.
Fig. 6 shows a hypothetical schema of changes in the shape of the CK concentration time curve and the estimated contributions to the total CK concentration from both ischemic and reperfusion injury. This should make the observed biphasic relationship easier to understand. Reperfusion that starts relatively soon after the onset of MI is expected to lead to more reperfusion-related release of CK and less contribution by ischemic injury, because the myocardial cells have been exposed to oxygen deprivation for a shorter period of time. On the other hand, a later start of reperfusion leads to less reperfusion-induced release of CK and more ischemic injury, because the myocardial cells have been subjected to ischemic conditions for a longer period of time. Therefore, as the time from MI to reperfusion increases, the peak of the CK concentration time curve shifts toward the right, until it reaches the peak of CK release caused by ischemic injury, which is usually approximately 24 h after the onset of MI. Once these two peaks coincide, the CK concentration curve cannot be shifted any further to the right. Thus, if lethal reperfusion injury is involved in the mechanism of early peaking of CK release during reperfusion therapy, the observed biphasic relationship can be explained. This assumption will be valid if an attempt to prevent lethal reperfusion injury can abolish this biphasic relationship.
Fig. 6.

Hypothetical schema of changes in the shape of the creatine kinase (CK) concentration time curve, showing the estimated contributions to the total CK concentration by both ischemic injury and reperfusion injury. An early onset of reperfusion leads to a higher number of reperfusion-induced cell deaths than of cell deaths caused by ischemic injury, because the cells have been hypoxic for a shorter period of time. On the other hand, late-onset reperfusion leads to less reperfusion injury and more ischemic injury due to the prolonged hypoxic conditions experienced by the myocardial cells. Therefore, as the time to reperfusion increases, the peak of the CK concentration time curve shifts toward the right until it reaches the peak of CK release caused by ischemic injury, which is usually around 24 h after the onset of myocardial infarction.
(Reprinted from Koyama et al. [34]).
My colleagues and I recently reported that PCLeB abolished the aforementioned biphasic relationship between the time to reperfusion and the time to peak CK release [4]. Instead, we found a negative correlation between the two periods in 46 consecutive STEMI patients treated using PCLeB (Fig. 7a). This negative correlation revealed by PCLeB was thought to result from a combination of the effects of both attenuation of lethal reperfusion injury and enhanced CK washout caused by augmented coronary blood flow recovery. Fig. 7b shows the relationship between the time from reperfusion to peak CK release and the peak CK values in the same 46 patients. This should clarify the negative correlation in Fig. 7a. Because the washout mechanism works during the early reperfusion period, high peak CK values that peak early may be caused by this mechanism (as in the patient described in Fig. 4) [32]. On the other hand, relatively late and low CK peaks imply failure of the washout mechanism, possibly because only a small amount of CK remains to be washed out from the reperfused myocardium (Fig. 7b) [35]. Such cases were often seen in early reperfusion cases and were responsible for the plots in the upper left portion of the scatter plot (Fig. 7a), shifting the left shoulder of the linear regression line upward. Otherwise, the supposed enhanced CK washout mechanism appeared to induce consistent, early peaking of CK release, irrespective of the time to reperfusion (Fig. 7a). Thus, possible attenuation/prevention of lethal reperfusion injury by PCLeB abolished the biphasic relationship, and the early-peaking phenomenon of CK release might be exaggerated by the enhanced washout mechanism, allowing a simpler interpretation of the results.
Fig. 7.

a) Relationship between the time to reperfusion and the time from reperfusion to peak creatine kinase (CK) release; a linear, negative correlation is evident.
b) Relationship between the time from reperfusion to peak CK release and peak CK values; a clear, linear, negative correlation between these two variables, with peak CK values getting smaller as the time to peak CK release lengthened, is shown.
(Reprinted from Koyama et al. [4]).
8. Hypercontracture and an inverse relationship between ischemia duration and the severity of lethal reperfusion injury
More precisely, the aforementioned biphasic relationship between the time to reperfusion and the time to peak CK release can be explained if lethal reperfusion injury is involved in the mechanism of early peaking of CK release induced by reperfusion therapy, and if lethal reperfusion injury diminishes as the duration of the preceding ischemia increases. A longer ischemia duration reduces the number of viable cells; consequently, the number of cells subject to lethal reperfusion injury also decreases. This justification for the second of the above assumptions is superficially reasonable, but it may not be sufficient. If severe ischemic injury increases the number of cells susceptible to lethal reperfusion injury—a real possibility—the relationship between ischemia duration and the severity of lethal reperfusion injury may not be linear. Because reperfusion injury is based on something that happens inside the myocardium during ischemia, a shorter preparation period, i.e., a shorter duration of ischemia, is supposed to result in less reperfusion injury, while a longer ischemic period should result in more. However, the abovementioned biphasic relationship appears to indicate that this does not occur. If hypercontracture is hypothesized as the primary cause or trigger of the cascade of events that leads to lethal reperfusion injury, a more elegant explanation for the inverse relationship between the duration of ischemia and the severity of lethal reperfusion injury can be inferred.
The development of hypercontracture is recognized from necrosis of contraction bands in pathological specimens of reperfused ischemic myocardium [7]. Contraction band necrosis is found mainly in the marginal region of the reperfused ischemic myocardium, rather than in the center or the subendocardial side [8], [36]. The reason for this distribution has not been elucidated. However, the observation provides an important insight into the nature of hypercontracture; it appears to develop only in myocardial cells with mild to moderate ischemic injury. In severely damaged ischemic myocardium, i.e., myocardium in the ischemic center, hypercontracture does not seem to occur (Fig. 8). Accordingly, the longer the duration of the ischemia, the smaller the region of myocardium that develops hypercontracture. If hypercontracture triggers lethal reperfusion injury, this translates into an inverse linear relation between ischemia duration and the severity of lethal reperfusion injury.
Fig. 8.

Schema showing differences in myocardial ischemic injury caused by short-term ischemia and long-term ischemia, with each corresponding creatine kinase concentration time-curve image given below. The area of severe ischemic injury expands as the duration of ischemia increases. Hypercontracture appears to develop only in the myocardium with mild to moderate ischemic injury after reperfusion [8], [36]. Therefore, if hypercontracture is the primary cause or trigger of lethal reperfusion injury, a longer ischemia duration induces less lethal reperfusion injury.
Meanwhile, MPT does not seem able to explain the inverse relationship between the duration of ischemia and the severity of lethal reperfusion injury. The longer the ischemia lasts, the greater the severity of the mitochondrial disorders that may ensue. Thus, less mitochondrial disorder with longer ischemia appears unlikely.
9. Effects of PCLeB on early inflammation after MI
PCLeB may have some more beneficial effects on the reperfused ischemic myocardium. My colleagues and I reported less early inflammation in STEMI patients treated using PCLeB in our small matched case-control study [3]. Moreover, the peak C-reactive protein (CRP) level within 7 days of MI onset was only 3.50 ± 2.48 mg/dL in 55 consecutive STEMI patients treated using PCLeB [5]; this CRP level was substantially lower than those reported in the literature [37], [38]. A high peak CRP level early after MI has been demonstrated to be a predictor of poor outcomes in STEMI patients [37], [38]. A well-perfused tissue environment, along with less tissue inflammation, in STEMI patients treated using PCLeB can be expected to improve the healing process within the reperfused ischemic myocardium. Consequently, attenuation of left ventricular remodeling and resultant improvement of long-term outcomes can also be expected.
10. Conclusions
As described above, PCLeB appears to be effective in preventing all four types of reperfusion injury. Although the study populations were small and each finding might not be very robust, the apparent effectiveness of PCLeB over all types of reperfusion injury may indicate that the approach is right to the point. We have already reported the excellent in-hospital outcomes of STEMI patients treated using PCLeB. In addition, the augmented microcirculation recovery and lower level of inflammation after MI, consistently achieved by PCLeB, may also be expected to improve the long-term outcomes of STEMI patients. Because Lactated Ringer's solution has no pharmacological side effects, it is quite unlikely that adverse effects of this new approach could offset its beneficial effects in terms of both short- and long-term outcomes. Considering the current lack of any specific strategy for preventing reperfusion injury, the general use of this new approach may be justified without waiting for the advent of robust evidence—of course, only if patients are well informed about reperfusion injury, the serious adverse effects of conventional reperfusion therapy, and the details of this new therapy for the injury. I hope that lactated Ringer's solution will become “a solution” to the long-standing battle against myocardial reperfusion injury in the near future.
Conflicts of interest statement
The author has no conflicts of interest to declare.
Footnotes
Conflicts of interest: The author has no relationships with industry that are relevant to this work.
References
- 1.Kloner R.A. Does reperfusion injury exist in humans? J. Am. Coll. Cardiol. 1993;21:537–545. doi: 10.1016/0735-1097(93)90700-b. [DOI] [PubMed] [Google Scholar]
- 2.Koyama T., Shibata M., Moritani K. Ischemic postconditioning with lactate-enriched blood in patients with acute myocardial infarction. Cardiology. 2013;125:92–93. doi: 10.1159/000350595. [DOI] [PubMed] [Google Scholar]
- 3.Koyama T., Niikura H., Shibata M. Impact of ischemic postconditioning with lactate-enriched blood on early inflammation after myocardial infarction. IJC Metab. Endocr. 2014;2:30–34. [Google Scholar]
- 4.Koyama T., Niikura H., Shibata M. Possible creatine kinase washout mechanism revealed by postconditioning with lactate-enriched blood in patients experiencing ST-elevation myocardial infarctions. Int. J. Cardiol. 2014;177:492–493. doi: 10.1016/j.ijcard.2014.08.075. [DOI] [PubMed] [Google Scholar]
- 5.Koyama T., Munakata M., Akima T. Impact of postconditioning with lactate-enriched blood on in-hospital outcomes in patients with ST-segment elevation myocardial infarction. Int. J. Cardiol. 2016;220:146–148. doi: 10.1016/j.ijcard.2016.06.176. [DOI] [PubMed] [Google Scholar]
- 6.Akima T., Koyama T., Munakata M. Absence of reperfusion-induced arrhythmia in patients with ST-segment elevation myocardial infarction treated using postconditioning with lactate-enriched blood. Int. J. Cardiol. 2016;222:780–781. doi: 10.1016/j.ijcard.2016.08.145. [DOI] [PubMed] [Google Scholar]
- 7.Ganote C.E. Contraction band necrosis and irreversible myocardial injury. J. Mol. Cell. Cardiol. 1983;15:67–73. doi: 10.1016/0022-2828(83)90283-3. [DOI] [PubMed] [Google Scholar]
- 8.Jennings R.B. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ. Res. 2013;113:428–438. doi: 10.1161/CIRCRESAHA.113.300987. [DOI] [PubMed] [Google Scholar]
- 9.Hearse D.J., Bolli R. Reperfusion-induced injury manifestations, mechanisms, and clinical relevance. Trends Cardiovasc. Med. 1991;1:233–240. doi: 10.1016/1050-1738(91)90027-C. [DOI] [PubMed] [Google Scholar]
- 10.Piper H.M., García-Dorado D., Ovize M. A fresh look at reperfusion injury. Cardiovasc. Res. 1998;38:291–300. doi: 10.1016/s0008-6363(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 11.Yellon D.M., Hausenloy D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 12.Fröhlich G.M., Meier P., White S.K., Yellon D.M., Hausenloy D.J. Myocardial reperfusion injury: looking beyond primary PCI. Eur. Heart J. 2013;34:1714–1722. doi: 10.1093/eurheartj/eht090. [DOI] [PubMed] [Google Scholar]
- 13.Kloner R.A. Current state of clinical translation of cardioprotective agents for acute myocardial infarction. Circ. Res. 2013;113:451–463. doi: 10.1161/CIRCRESAHA.112.300627. [DOI] [PubMed] [Google Scholar]
- 14.Griffiths E.J., Halestrap A.P. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995;307(Pt 1):93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian T., Nieminen A.L., Herman B., Lemasters J.J. Mitochondrial permeability transition in pH-dependent reperfusion injury to rat hepatocytes. Am. J. Physiol. Cell Physiol. 1997;273:C1783–C1792. doi: 10.1152/ajpcell.1997.273.6.C1783. [DOI] [PubMed] [Google Scholar]
- 16.Kloner R.A., Ganote C.E., Whalen D.A., Jr., Jennings R.B. Effect of a transient period of ischemia on myocardial cells. II. Fine structure during the first few minutes of reflow. Am. J. Pathol. 1974;74:399–422. [PMC free article] [PubMed] [Google Scholar]
- 17.Cung T.T., Morel O., Cayla G. Cyclosporine before PCI in patients with acute myocardial infarction. N. Engl. J. Med. 2015;373:1021–1031. doi: 10.1056/NEJMoa1505489. [DOI] [PubMed] [Google Scholar]
- 18.Staat P., Rioufol G., Piot C. Postconditioning the human heart. Circulation. 2005;112:2143–2148. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- 19.Freixa X., Bellera N., Ortiz-Pérez J.T. Ischaemic postconditioning revisited: lack of effects on infarct size following primary percutaneous coronary intervention. Eur. Heart J. 2012;33:103–112. doi: 10.1093/eurheartj/ehr297. [DOI] [PubMed] [Google Scholar]
- 20.Limalanathan S., Andersen G.Ø., Kløw N.E., Abdelnoor M., Hoffmann P., Eritsland J. Effect of ischemic postconditioning on infarct size in patients with ST-elevation myocardial infarction treated by primary PCI results of the POSTEMI (POstconditioning in ST-Elevation Myocardial Infarction) randomized trial. J. Am. Heart Assoc. 2014;3 doi: 10.1161/JAHA.113.000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koyama T., Temma K., Akera T. Reperfusion-induced contracture develops with a decreasing [Ca2+]i in single heart cells. Am. J. Physiol. Heart Circ. Physiol. 1991;261:H1115–H1122. doi: 10.1152/ajpheart.1991.261.4.H1115. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Dorado D., Théroux P., Duran J.M. Selective inhibition of the contractile apparatus. A new approach to modification of infarct size, infarct composition, and infarct geometry during coronary artery occlusion and reperfusion. Circulation. 1992;85:1160–1174. doi: 10.1161/01.cir.85.3.1160. [DOI] [PubMed] [Google Scholar]
- 23.Langer G.A., Buckberg G.D., Tillisch J.H. Cardiac ischemia. Part II—reperfusion and treatment. West. J. Med. 1987;147:54–61. [PMC free article] [PubMed] [Google Scholar]
- 24.Okamoto F., Allen B.S., Buckberg G.D. Regional blood cardioplegic reperfusion during total vented bypass without thoracotomy: a new concept. J. Thorac. Cardiovasc. Surg. 1986;92(3 Pt 2):553–563. [PubMed] [Google Scholar]
- 25.Kloner R.A., Ganote C.E., Jennings R.B. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J. Clin. Invest. 1974;54:1496–1508. doi: 10.1172/JCI107898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niccoli G., Burzotta F., Galiuto L., Crea F. Myocardial no-reflow in humans. J. Am. Coll. Cardiol. 2009;54:281–292. doi: 10.1016/j.jacc.2009.03.054. [DOI] [PubMed] [Google Scholar]
- 27.Wu K.C., Zerhouni E.A., Judd R.M. Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation. 1998;97:765–772. doi: 10.1161/01.cir.97.8.765. [DOI] [PubMed] [Google Scholar]
- 28.Bekkers S.C., Yazdani S.K., Virmani R., Waltenberger J. Microvascular obstruction: underlying pathophysiology and clinical diagnosis. J. Am. Coll. Cardiol. 2010;55:1649–1660. doi: 10.1016/j.jacc.2009.12.037. [DOI] [PubMed] [Google Scholar]
- 29.Lee B.Y., Wilson G.J., Domenech R.J., MacGregor D.C. Relative roles of edema versus contracture in the myocardial postischemic “no-reflow” phenomenon. J. Surg. Res. 1980;29:50–61. doi: 10.1016/0022-4804(80)90008-6. [DOI] [PubMed] [Google Scholar]
- 30.Humphrey S.M., Gavin J.B. Reversal of the no reflow phenomenon in globally ischemic rat hearts by ventricular dilation. Pathology. 1985;17:437–442. doi: 10.3109/00313028509105497. [DOI] [PubMed] [Google Scholar]
- 31.Braunwald E., Kloner R.A. The stunned myocardium; prolonged, postischemic ventricular dysfunction. Circulation. 1982;66:1146–1149. doi: 10.1161/01.cir.66.6.1146. [DOI] [PubMed] [Google Scholar]
- 32.Koyama T., Kageyama T., Munakata M. Muscle squeezing of the culprit coronary artery in a patient with ST-elevation myocardial infarction after postconditioning with lactate-enriched blood. Int. J. Cardiol. 2015;182:77–78. doi: 10.1016/j.ijcard.2014.12.128. [DOI] [PubMed] [Google Scholar]
- 33.Apple F.S. Diagnostic use of CK-MM and CK-MB isoforms for detecting myocardial infarction. Clin. Lab. Med. 1989;9:643–654. [PubMed] [Google Scholar]
- 34.Koyama T., Shimada M., Baba A., Kosugi R., Akaishi M. Effects of early reperfusion on creatine kinase release in patients with acute myocardial infarction: implications for reperfusion injury. Int. J. Cardiol. 2012;155:335–337. doi: 10.1016/j.ijcard.2011.12.049. [DOI] [PubMed] [Google Scholar]
- 35.Munakata M., Koyama T., Akima T., Kanki H., Ishikawa S. Minimum ischemia reperfusion injury in a STEMI patient treated using postconditioning with lactate-enriched blood. Int. J. Cardiol. 2016;202:282–284. doi: 10.1016/j.ijcard.2015.08.131. [DOI] [PubMed] [Google Scholar]
- 36.Shintani-Ishida K., Unuma K., Yoshida K. Ischemia enhances translocation of connexin43 and gap junction intercellular communication, thereby propagating contraction band necrosis after reperfusion. Circ. J. 2009;73:1661–1668. doi: 10.1253/circj.cj-09-0079. [DOI] [PubMed] [Google Scholar]
- 37.Anzai T., Yoshikawa T., Shiraki H. C-reactive protein as a predictor of infarct expansion and cardiac rupture after a first Q-wave acute myocardial infarction. Circulation. 1997;96:778–784. doi: 10.1161/01.cir.96.3.778. [DOI] [PubMed] [Google Scholar]
- 38.Suleiman M., Khatib R., Agmon Y. Early inflammation and risk of long-term development of heart failure and mortality in survivors of acute myocardial infarction predictive role of C-reactive protein. J. Am. Coll. Cardiol. 2006;47:962–968. doi: 10.1016/j.jacc.2005.10.055. [DOI] [PubMed] [Google Scholar]