

Abstract
Some of the restarting events of stalled replication forks lead to sister chromatid exchange (SCE) as a result of homologous recombination (HR) repair with crossing over. The rate of SCE is elevated by the loss of BLM helicase or by a defect in translesion synthesis (TLS). We found that spontaneous SCE levels were elevated ∼2-fold in chicken DT40 cells deficient in Fanconi anemia (FA) gene FANCC. To investigate the mechanism of the elevated SCE, we deleted FANCC in cells lacking Rad51 paralog XRCC3, TLS factor RAD18, or BLM. The increased SCE in fancc cells required Xrcc3, whereas the fancc/rad18 double mutant exhibited higher SCE than either single mutant. Unexpectedly, SCE in the fancc/blm mutant was similar to that in blm cells, indicating functional linkage between FANCC and BLM. Furthermore, MMC-induced formation of GFP-BLM nuclear foci was severely compromised in both human and chicken fancc or fancd2 cells. Our cell survival data suggest that the FA proteins serve to facilitate HR, but not global TLS, during crosslink repair.
Keywords: Bloom syndrome, Fanconi anemia, homologous recombination, sister chromatid exchange, translesion synthesis
Introduction
The integrity of the mammalian genome is essential to suppress oncogenesis, and is maintained by an intricate network of molecules that guard against alterations from DNA damage (Rouse and Jackson, 2002). Fanconi anemia (FA) and Bloom syndrome (BS) are well-known examples of chromosomal instability disorders with an increased incidence of cancer. However, the clinical and cellular features of these diseases are quite distinct. FA is a hereditary disorder characterized by bone marrow failure, skeletal anomalies, and development of leukemia or squamous cell cancer (Grompe and D'Andrea, 2001; Joenje and Patel, 2001; D'Andrea and Grompe, 2003). FA cells are highly sensitive to killing by interstrand crosslinking (ICL) agents such as mitomycin C (MMC) and cisplatin, and display excessive chromosomal aberrations following MMC treatment (Sasaki and Tonomura, 1973; Sasaki, 1975). Complementation analysis indicates that at least 11 causative genes are involved (Levitus et al, 2003), and nine of them have been cloned (FANCA/B/C/D1/D2/E/F/G/L) (Joenje and Patel, 2001; D'Andrea and Grompe, 2003; Meetei et al, 2003a, 2004). Most of the encoded proteins (FANCA/B/C/E/F/G/L) form a nuclear core complex, which is responsible for monoubiquitination of FANCD2, a key event in the FA ‘pathway' (Garcia-Higuera et al, 2001). The exact biochemical function of FANCD2 remains unknown, but recent evidence indicates a role of FANCD2 in promoting homologous recombination (HR) (Yamamoto et al, 2005), which is crucial for maintaining genome integrity (Venkitaraman, 2004). Indeed, another FA protein, FANCD1/BRCA2 (Howlett et al, 2002), a tumor suppressor for familial breast cancer, is an important regulator of Rad51, the central molecule of HR (Venkitaraman, 2002; West, 2003).
BS is also a hereditary disorder characterized by immunodeficiency, small stature, and development of a variety of cancers (German, 1993). In contrast to FA, BS is caused by a defect in a single gene, BLM helicase (Ellis et al, 1995). The hallmark and a diagnostic feature of BS cells is ∼10-fold increase in sister chromatid exchange (SCE) levels (Chaganti et al, 1974). BLM is a member of the eukaryotic RecQ helicase family, which is thought to play multiple roles in DNA replication, recombination, or repair (Bachrati and Hickson, 2003; Hickson, 2003). For example, BLM is a part of BRCA1-containing mega-protein complex (Wang et al, 2000b), and colocalizes in subnuclear foci with Rad51(Wu et al, 2001). The RecQ family contains four more members (RecQ1, WRN, RecQ4, and RecQ5), and a defect in WRN or RecQ4 is known to cause the genome instability disorders, Werner syndrome (Yu et al, 1996) or Rothmund-Thomson syndrome (Kitao et al, 1999), respectively. Unexpectedly, BLM has been recently detected in the FA core complex (Meetei et al, 2003b), suggesting a potential functional link between FA and BLM proteins.
It is well accepted that replication forks often stall at various DNA lesions including endogenous base damage (Cox et al, 2000; Cox, 2001). At least two basic processes, translesion synthesis (TLS) and HR, function to ensure that replication can be resumed at stalled or broken forks. When single-strand breaks or blocking lesions are converted to double-strandbreaks or gaps, respectively, during passage of replication forks, these chromatid discontinuities may be repaired by HR occurring between daughter chromatids (Sonoda et al, 1999). In classical models, HR involves formation of double Holliday junctions, and their resolution may or may not result in crossing over. However, HR repair events in somatic cells primarily proceed without double Holliday junctions, hence without crossing over, through synthesis-dependent strand annealing pathway (Paques and Haber, 1999; Johnson and Jasin, 2000; Cromie et al, 2001). Thus, it is likely that only a fraction of restarting events lead to SCE, which is known to depend on Rad51, Rad54 (Sonoda et al, 1999; Dronkert et al, 2000), five Rad51 paralogs (Takata et al, 2000, 2001), and Nbs1 (Tauchi et al, 2002). Since TLS mediates lesion bypass by a group of specialized polymerases, dysfunctional TLS might lead to more SCE if the stalled fork breaks and requires HR to restart. Consistently, at least some of the TLS-deficient cells such as rad18, rev3, or dinB mutants display increased levels of SCE (Okada et al, 2002; Yamashita et al, 2002; Sonoda et al, 2003). In contrast, BLM may directly suppress SCEs by catalyzing the ‘dissolution' of the double Holliday junctions (Wu and Hickson, 2003).
In this study, we generated FANCC-deficient DT40 cells (hereafter referred to as fancc), and found that levels of spontaneous SCEs increased ∼2-fold compared to wild-type cells, similar to our fancd2 cells (Yamamoto et al, 2005). To investigate the basis of the elevated SCE, we made double mutants of FANCC with mutations in HR (XRCC3), TLS (RAD18), and BLM helicase. Our genetic analyses indicate functional linkage of FANCC with Xrcc3 or BLM but not with Rad18. Furthermore, crosslink damage-induced relocalization of BLM was defective in both human and chicken fancc or fancd2 cells. We propose that BLM, regulated by the FA pathway, functions in restarting stalled replication forks blocked by spontaneous lesions and ICLs.
Results
Disruption of chicken FANCC gene in DT40 cells
We obtained a cDNA clone containing full-length chicken FANCC by searching the chicken EST database (http://swallow.gsf.de/DT40/dt40Est.html). Chicken FANCC encodes a putative 559-amino-acid protein (DDBJ accession number AB176529) compared to 557 amino acids of human FANCC. The identity and similarity between two proteins are 45 and 59%, respectively. There are no domains or motifs suggestive of biochemical function in either protein.
Based on the sequence of the cDNA, we PCR-amplified a genomic sequence of chicken FANCC and designed a targeting vector (Figure 1A). A single transfection with the vector abrogated the band in Southern blot analysis (Figure 1B). This is not surprising, given the localization of FANCC on human chromosome 9 and the extensive synteny between human chromosome 9 and chicken Z sex chromosome (Nanda et al, 1999). DT40 was derived from a female chicken, which is hemizygous in terms of the Z chromosome. Another FA gene, FANCG, also lies on human chromosome 9, and we were able to disrupt FANCG in DT40 by single transfection (Yamamoto et al, 2003). RT–PCR analysis confirmed the FANCC gene disruption (Figure 1C) that is expected to delete one exon. Nucleotide sequencing revealed that the faint, shorter transcript was owing to anomalous splicing, which is expected to create a truncated protein (residues 1–55 and six additional amino acids) because of a frame shift. We also examined induction of the long, monoubiquitinated form of FANCD2 protein (FANCD2-L) (Gregory et al, 2003) by MMC treatment using Western blotting with anti-chicken FANCD2 antiserum. As shown in Figure 1D, the FANCD2-L form existed in untreated wild-type cells, and its proportion was clearly increased after MMC treatment. In contrast, FANCD2-L was not detected in fancc cells before or after MMC treatment (Figure 1D), similar to human fancc cells (Garcia-Higuera et al, 2001).
Figure 1.

Targeted disruption of chicken FANCC loci in DT40 cells. (A) Schematic representation of partial chicken FANCC locus, the gene disruption construct, and the configuration of targeted allele. S, SacI; B, BamHI; H, HindIII. Only a single exon was deleted by the gene disruption. Arrowheads indicate the positions of primers used in RT–PCR. (B) Southern blot analysis of SacI-digested genomic DNA from cells with indicated genotypes using flanking probe as shown in (A). WT, wild type. (C) RT–PCR analysis of total RNA from wild-type and fancc cells. Primers were designed from FANCC sequences of upstream and downstream exons as shown in (A). As a control, the entire coding region of chicken Rad51 was amplified from each RT product. (D) Western blot analysis of whole-cell lysate prepared from wild-type and fancc cells probed with anti-chicken FANCD2 serum. Cells were treated with MMC (500 ng/ml) for 1 h, washed, and lysed 6 h later. L or S, long or short form of FANCD2, respectively.
fancc cells have increased levels of spontaneous SCE
The FANCC-deficient cells grew more slowly than wild-type cells with an increased proportion of dead cells in the culture (data not shown), and the cell cycle distribution examined by BrdU pulse labelling was not altered (data not shown). As expected, the cells were highly sensitive to the DNA crosslinkers MMC and cisplatin, while sensitivity to X-rays or UV irradiation was only mildly increased (Figure 2A). The cisplatin sensitivity could be complemented to wild-type levels by expression of chicken FANCC cDNA, indicating that this defect was indeed caused by FANCC disruption (Figure 2B). Although spontaneous chromosomal breaks were not elevated, MMC-induced aberrations occurred much more frequently in fancc cells (Figure 2C).
Figure 2.
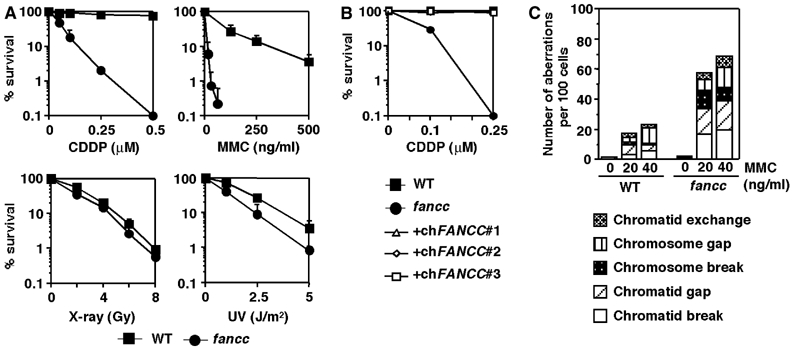
Characterization of fancc cells. (A) Sensitivity curves of cells to various DNA-damaging agents. The fraction of surviving colonies in methylcellulose plates is shown for each agent. Mean and standard deviation (s.d.) of at least three independent experiments are shown. WT, wild-type cells. (B) Complementation of fancc cells with FANCC expression. Three fancc clones expressing chicken FANCC cDNA (+chFANCC#1 to #3) were compared with wild-type and fancc cells in colony survival with cisplatin. (C) Chromosome aberrations after MMC treatment in wild-type and fancc cells. A total of 200 metaphases were scored.
To test whether fancc cells have HR defects, we examined gene targeting at three genomic loci. Wild-type and fancc cells were transfected with linearized targeting vectors and selected in media containing appropriate drugs. After expansion, each colony was examined for targeting events by Southern blot analysis of genomic DNA. Fancc cells had dramatically reduced gene-targeting efficiency compared to wild-type cells (Table I). In contrast, we also found that the frequency of spontaneous SCE in fancc cells was elevated ∼2-fold compared to wild-type cells (Figures 3C and 4B), similar to our fancd2 cells (Yamamoto et al, 2005).
Table 1.
Targeted integration efficiencies in fancc cells
| Targeting vectors |
|||
|---|---|---|---|
| Genotypes | Ku70-His | Xrcc2-puro | Ovalubumin-puro |
| Wild type | 14/34 (41%) | 12/24 (50%) | 10/24 (42%) |
|
fancc |
3/20 (15%) |
1/11 (9%) |
0/35 (0%) |
| Data are numbers of targeted clones per number of clones analyzed by Southern blotting. The percentage of targeted integration events is given in parentheses. | |||
Figure 3.
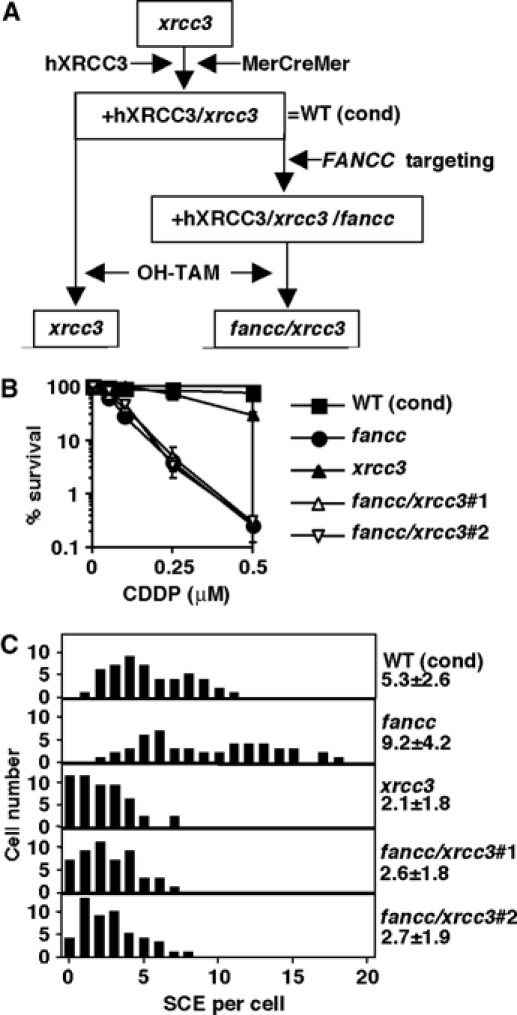
Genetic analysis of fancc mutation combined with xrcc3. (A) Generation of conditional xrcc3 cells and disruption of FANCC in those cells. OH-TAM treatment activates MerCreMer recombinase that removes the human Xrcc3 (hXrcc3)-IRES-EGFP expression cassette. WT (cond), xrcc3 cells that express hXrcc3, GFP, and MerCreMer. (B) CDDP sensitivity curves of cells with indicated genotypes. Fancc cells used were derived from wild-type DT40 cells, not from the conditional xrcc3 mutant. Mean and s.d. from three independent experiments are shown. (C) Spontaneous SCE levels. Numbers represent mean and s.d. of scores from 50 metaphases. In this analysis, we used fancc cells derived from the conditional xrcc3 mutant, which express hXrcc3. Statistical significance was detected between WT (cond) versus fancc, WT (cond) versus xrcc3, and fancc versus xrcc3 (Bonferroni/Dunn test, P<0.0001 in all comparisons). The difference was insignificant between xrcc3 versus fancc/xrcc3 double-mutant clones.
Figure 4.

Genetic analysis of fancc mutation combined with rad18 or blm. (A) CDDP or MMS survival curves of cells with indicated genotypes. Mean and s.d. from three independent experiments are shown. WT, wild type. (B) Spontaneous SCE levels. Numbers represent mean and s.d. of scores from 50 metaphases. Statistical significance was detected between WT versus fancc, WT versus rad18, WT versus fancc/rad18, fancc versus facc/rad18, and rad18 versus fancc/rad18 (Bonferroni/Dunn test, P<0.0001 in all comparisons). (C) Survival curves of cells with indicated genotypes. Mean and s.d. from three independent experiments are shown. (D) Spontaneous SCE levels. Numbers represent mean and s.d. of scores from 50 metaphases. Statistical significance was detected between fancc versus blm, fancc versus fancc/blm#1, and fancc versus fancc/blm#2 (Bonferroni/Dunn test, P<0.0001 in all comparisons). There was no statistically significant difference between blm and fancc/blm double mutants. (E) MMC (20 ng/ml)-induced SCE levels. Numbers represent mean and s.d. of scores from 50 metaphases. Statistical significance was detected between fancc versus blm, fancc versus fancc/blm#1, and fancc versus fancc/blm#2 (Bonferroni/Dunn test, P<0.0001 in all comparisons). There was no statistically significant difference between blm and fancc/blm double mutants.
Elevated SCEs in fancc cells depend on Rad51 paralog Xrcc3
To investigate the mechanism of SCE elevation in fancc cells, and to better define the FA pathway, we performed genetic analysis by disrupting FANCC in cells that are deficient in HR (xrcc3), TLS (rad18), or BLM helicase (blm).
We disrupted FANCC in conditional xrcc3 background (Ishiai et al, 2004), and exposed the cells, as well as the parental cells, to 4-hydroxy tamoxifen (OH-TAM) (Figure 3A) to excise the Xrcc3-EGFP expression cassette by activating the MerCreMer recombinase (Zhang et al, 1998). The removal was ensured by subcloning, and was further verified by the loss of GFP fluorescence and by Southern blotting using human XRCC3 probe (data not shown). Thus, two independent clones of fancc/xrcc3 cells were established.
We compared the fancc/xrcc3 cells with fancc cells in terms of cisplatin sensitivity. Fancc cells were much more cisplatin sensitive to killing compared to xrcc3 cells. However, fancc/xrcc3 cells displayed about the same cisplatin sensitivity as the fancc single mutant, indicating functional overlap between FANCC and XRCC3 (Figure 3B). In addition, spontaneous SCE was clearly decreased in xrcc3, but elevated in fancc cells compared to the parental conditional cells (Figure 3C). Not surprisingly, the SCE frequency in two clones of fancc/xrcc3 cells was similar to that of xrcc3 cells (Figure 3C), indicating that spontaneous SCE in fancc cells is partially Xrcc3-dependent as in wild-type cells (Takata et al, 2001).
Genetic analysis of FANCC and TLS factor Rad18
Since several TLS-deficient vertebrate cells including Rad18-deficient DT40 (Yamashita et al, 2002) have increased SCE (Okada et al, 2002; Sonoda et al, 2003), it is quite possible that elevated SCE in fancc cells might be related to TLS defects. In yeast Saccharomyces cerevisiae, most TLS activity is dependent on Rad6, and its heterodimeric partner Rad18. To examine the relationship between the FA pathway and TLS, the FANCC gene was targeted in rad18 cells (Yamashita et al, 2002). Although both rad18 and fancc cells had high sensitivity to cisplatin or methylmethanesulfonate (MMS), the fancc/rad18 double mutant displayed even higher sensitivity to both agents, indicating their distinct functions (Figure 4A). Moreover, fancc/rad18 cells had even greater increased SCE than either rad18 or fancc cells (Figure 4B). Thus, we conclude that spontaneous SCEs in fancc cells are elevated by a mechanism separate from Rad18 function. It is still possible that FANCC plays a role in Rad18-independent TLS, since not all TLS events require Rad18 in vertebrate cells (Okada et al, 2002).
A functional link between FA pathway and BLM in suppressing spontaneous SCE
Next, we deleted FANCC in BLM-deficient cells (Wang et al, 2000a). In contrast to fancc/rad18 cells, we found that the blm and fancc/blm mutants displayed about the same high levels of spontaneous and MMC-induced SCE (Figure 4D and E). Given the functional link suggested by these findings, we measured cell survival of blm, fancc, and fancc/blm cells following treatment with several mutagens. While MMS-treated fancc/blm cells survived more poorly than either single mutant (additive phenotype), survival following cisplatin or MMC treatment was similar between fancc and fancc/blm cells (Figure 4C). These results suggest that FANCC and BLM act in a common pathway in ICL repair as well as in controlling SCE levels. However, in repairing DNA lesions created by MMS, they likely have more distinct functions. In addition, fancc/blm cells grew more slowly than either fancc or blm cells (data not shown), indicating that the requirement of both genes extends to cell viability.
To further verify the potential link between FA proteins and BLM, we created double mutant lacking both FANCD2 and BLM by targeting FANCD2 gene in blm cells. FANCD2 disruption was confirmed by Southern (data not shown) and Western blotting (Supplementary Figure 1A). We found that spontaneous SCE levels and cisplatin sensitivity of the double mutant were essentially the same as those of blm mutant or fancd2 mutant, respectively (Supplementary Figure 1B and C). This result is consistent with the essential role of FANCC in activation and/or monoubiquitination of FANCD2 (Figure 1D).
Defective BLM focus formation in the absence of FANCC and FANCD2
While blm cells have extremely elevated SCEs, the elevation is only ∼2-fold both in fancc and fancd2 cells. As BLM helicase was shown to mediate the ‘dissolution' of double Holliday junctions without crossing over (Wu and Hickson, 2003), it likely suppresses SCE in a direct manner. We hypothesized that increased SCE in fancc and fancd2 cells is due to mildly compromised BLM function by the absence of the FA ‘pathway'. Since BLM resides in a nuclear complex with several FA proteins (Meetei et al, 2003b), we examined the distribution of BLM inside fancc and fancd2 cells. GFP-human BLM (GFP-hBLM) has been extensively utilized in a number of studies to examine its subnuclear localization (Hu et al, 2001; Suzuki et al, 2001; Yankiwski et al, 2001; Stavropoulos et al, 2002). To visualize chicken BLM localization, we transfected a GFP-chicken BLM (GFP-chBLM) expression construct into wild-type, fancc, and fancd2 cells. Stably expressing transfectants were analyzed by FACSCalibur, and clones with equal expression levels were selected for further analysis. GFP-chBLM was fully functional in suppressing SCE in blm DT40 cells (data not shown).
In wild-type cells expressing GFP-chBLM, we observed relatively large foci (5–10 foci per cell) in 2–3% of the untreated cells (Figure 5A and B). These foci might correspond to PML nuclear bodies (Ishov et al, 1999), although we could not confirm this because of the lack of suitable reagents. In addition, we detected a number of less intense, smaller foci in untreated cells (Figure 5A and B). MMC treatment produced brighter and more numerous foci (Figure 5A and B). Some cells had a rather faint patch-like GFP accumulation that seems to be BLM localized in the nucleolus (Figure 5B).
Figure 5.
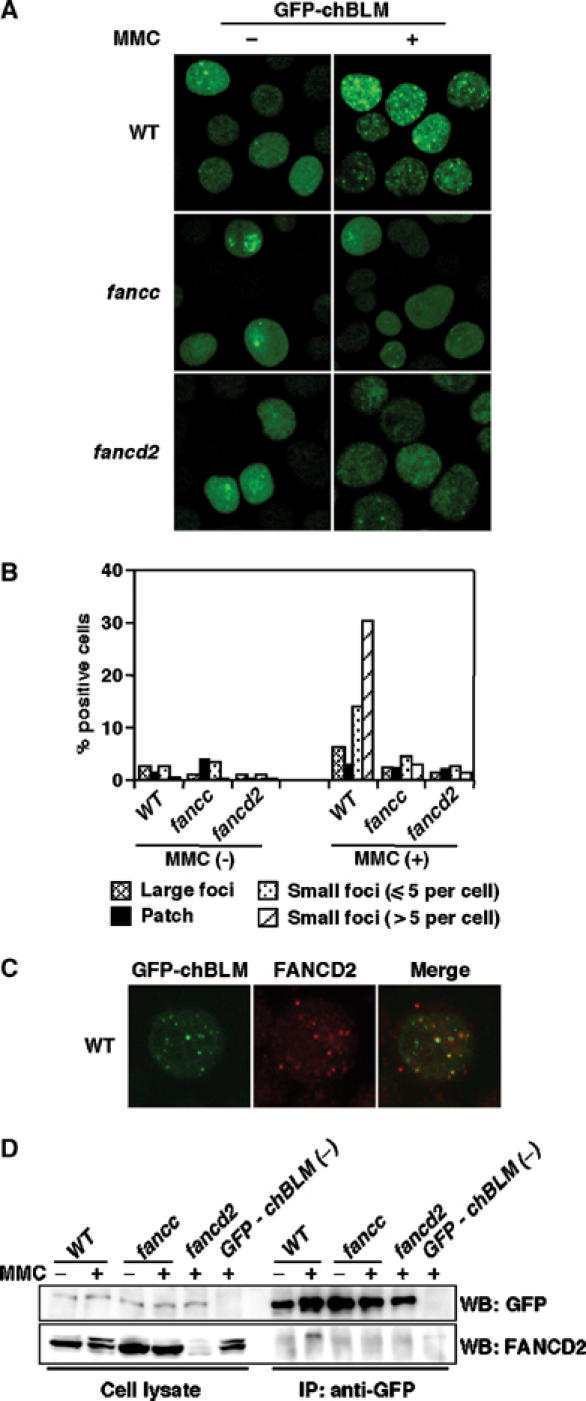
Analysis of GFP-chBLM in wild-type, fancc, and fancd2 DT40 cells. (A) Focus formation of GFP-chBLM. Cells expressing GFP-chBLM were observed before and 7 h after MMC treatment (500 ng/ml, 1 h). (B) Fraction of cells having large foci, indicated number of small foci, and patch-like accumulation. Cells were treated as in (A). At least 200 cells were scored in each preparation. (C) Colocalization of GFP-chBLM and FANCD2 in wild-type cells treated with MMC as in (A). (D) Co-immunoprecipitation of GFP-chBLM with FANCD2. Cells with indicated genotypes were treated with MMC (500 ng/ml for 7 h) or left untreated and lysed. A portion of the lysate (2.5%) was saved as an input control, and the rest was subjected to immunoprecipitation with anti-GFP. The lysates and immunoprecipitates were separated and probed with the indicated antibodies. GFP-chBLM (−), wild-type cells that were not transfected with the GFP-chBLM construct serving as negative control. The experiments were repeated three times with similar results.
Surprisingly, in fancc or fancd2 mutant cells, the formation of both large and small GFP-chBLM foci was largely suppressed after MMC treatment. In particular, the number of the small foci per cell and the % positive cells were severely decreased in fancc and fancd2 cells (Figure 5A and B). This reduction was not caused by differences in cell cycle, since both wild-type and mutant cells showed progressive accumulation at late S to G2 phase in an essentially similar way after MMC treatment (data not shown). In untreated cells, the reduction was not as clear as in MMC-treated cells (Figure 5B). These results indicate that nuclear relocalization of BLM is influenced by the FA proteins.
Next, we looked at whether BLM and FANCD2 can colocalize in DNA damage-induced subnuclear foci. FANCD2 foci were detected by staining with anti-chicken FANCD2. As shown in Figure 5C, GFP-chBLM and FANCD2 foci partially colocalized in MMC-treated cells. Among the 200 cells scored 7 h after MMC treatment, ∼40% were positive for FANCD2/GFP-chBLM colocalizing foci, while ∼40 or 10% were positive only for FANCD2 foci or GFP-chBLM foci, respectively. The rest of the cells (∼10%) had no focus formation. Furthermore, we were able to detect FANCD2-L form in anti-GFP immunoprecipitates from wild-type but not fancc cells expressing GFP-chBLM following MMC treatment (Figure 5D). These data suggest that monoubiquitinated FANCD2 physically interacts with BLM either directly or indirectly, resulting in the colocalizing foci. Of note, we could not detect any reproducible mobility change, which may suggest post-translational modifications, in immunoprecipitated GFP-chBLM treated with λ-phosphatase following MMC exposure (data not shown).
To test whether defective focus formation in FA cells is specific to BLM or not, we stably expressed GFP-chRad18 into wild-type and mutant (rad18, fancc, and fancd2) DT40 cells. GFP-chRad18 expression was able to restore cisplatin sensitivity of rad18 mutant to near wild-type levels (data not shown). In all of these transfected cells, GFP-chRad18 protein formed bright and distinct foci in a similar fashion (Supplementary Figure 2), although the percentage of cells with bright foci was quite low even after MMC treatment (2–3% in cells with all genotypes). In addition, colocalization with FANCD2 foci was rather poor (Supplementary Figure 2). These results are consistent with the additive phenotype of fancc/rad18 cells.
In keeping with a previous report (Meetei et al, 2003b), FANCD2 focus formation and FANCD2-L production by MMC treatment were not altered in blm cells (Supplementary Figure 3A and B), whereas, as expected (Garcia-Higuera et al, 2001), we could not detect any FANCD2 focus formation in fancc cells (Supplementary Figure 3A). These data indicate that BLM functions downstream of the FA pathway.
Defective MMC-induced GFP-hBLM relocalization in human FA cells
To extend our observation to human FA cells, we took fibroblast cell lines PD20 (fancd2) and PD331 (fancc) derived from FA patients and transiently transfected them with GFP-hBLM construct (Suzuki et al, 2001). We also included PD20 cells complemented with retrovirus containing human FANCD2 cDNA. As expected, PD20 cells did not express FANCD2, while FANCD2-complemented PD20 or PD331 cells express FANCD2 protein. The FANCD2-L form was not detectable in PD331 cells even after MMC treatment (Figure 6A). Small but bright GFP-hBLM foci were noted in these cells, but the percentage of the focus-positive cells was higher in FANCD2-complemented cells compared to the PD20 or PD331 human mutants. Furthermore, following MMC treatment, the percentage of positive cells with GFP-hBLM foci was increased in the complemented cells but not in PD20 and PD331 cells (Figure 6B and C). We were also able to observe colocalization of FANCD2 and GFP-hBLM foci in the complemented cells following MMC treatment (Figure 6D). Human FA cells displayed accumulation of GFP-hBLM protein in several large nucleolus-like structures, similar to previous reports (Yankiwski et al, 2001; Stavropoulos et al, 2002). Collectively, these results established that the FA pathway determines MMC-induced GFP-BLM relocalization in both human and chicken cells.
Figure 6.

GFP-hBLM focus formation in human FA cells. (A) Expression and MMC-induced monoubiquitination of FANCD2 in human fibroblasts PD20 (fancd2) and PD20 complemented with human FANCD2 cDNA, PD331 (fancc). Cells were treated with MMC (40 ng/ml) for 24 h. Western blot analysis was performed using anti-human FANCD2 monoclonal antibody. An asterisk indicates a nonspecific band. (B) Focus formation of GFP-hBLM. Cells transiently transfected with GFP-hBLM expression construct were treated with MMC as in (A). At least ∼20% of the populations became GFP-positive 24 h after transfection. (C) Percentage of the focus-positive cells among GFP-hBLM-positive populations before and after MMC treatment. Cells with more than four bright foci were scored as positive. At least 300 cells were scored in each preparation. The experiment was repeated three times with similar results. (D) Colocalization of GFP-hBLM and FANCD2 in FANCD2-complemented cells treated with MMC as in (A). Human FANCD2 foci were detected using anti-human FANCD2 polyclonal antibody.
Discussion
In this study, we carried out genetic analyses between the FA pathway and three proteins involved in DNA damage responses. We revealed an overlap between HR factor Xrcc3 and FANCC in cisplatin sensitivity, which indicates that at least some of FANCC's function in repair of cisplatin damage is linked to HR. This result is consistent with our previous findings that there are HR defects in fancd2 (Yamamoto et al, 2005) and fancg (Yamamoto et al, 2003) DT40 mutant cells. We also found that fancc cells had properties similar to those of fancd2 cells, although the magnitude of the defects was generally more severe in fancd2 than in fancc cells. However, fancc cells appeared to be ∼2-fold more sensitive to MMC than fancd2 cells in colony survival assay (Yamamoto et al, 2005). This difference is consistent with the view that FANCC is required for monoubiquitination of FANCD2 but might have an additional role in MMC tolerance (Kruyt et al, 1998; Cumming et al, 2001).
Several lines of evidence suggest linkage between TLS and the FA pathway (reviewed in Thompson et al, 2005). First, it has been reported that human FA cells display hypomutability at the hprt locus (Papadopoulo et al, 1990). Second, we previously showed lower levels of nontemplated mutations in the immunoglobulin light-chain locus in fancd2 DT40 cells (Yamamoto et al, 2005). These data are consistent with the possibility that the FA pathway promotes TLS and base-substitution mutagenesis at spontaneous or induced DNA lesions. Third, a recent report (Niedzwiedz et al, 2004) indicated that TLS polymerases Rev1 and Rev3 work in a common pathway with FANCC for ICL repair. Collectively, it seems likely that the FA pathway facilitates both TLS and HR processes in response to blocked or broken replication forks, respectively (Thompson et al, 2005).
In contrast to the Rev1/3 study (Niedzwiedz et al, 2004), our present data using cells lacking both FANCC and Rad18 revealed independent functions of these two proteins. Upon replication fork stalling, Rad18 promotes lesion bypass together with its partner Rad6. The Rad6/Rad18 complex carries ubiquitin conjugation and ligation activities, and mediates PCNA monoubiquitination, which is required for switching to the specialized polymerase such as pol eta (Rad30) (Kannouche et al, 2004; Watanabe et al, 2004). Although the complex is thought be a master regulator of TLS in yeast S. cerevisiae, not all of the TLS polymerases are Rad18-dependent in vertebrate cells (Okada et al, 2002). Therefore, our findings do not exclude the possibility that FA proteins and some TLS polymerases operate together.
Interestingly, both FANCC and FANCD2 are dispensable for spontaneous SCEs despite their roles in certain HR-dependent processes, including gene targeting. In fact, both fancc (this paper and Niedzwiedz et al, 2004) and fancd2 (Yamamoto et al, 2005) cells have elevated spontaneous SCE. Since SCE is one form of HR, these results suggest that vertebrate cells possess HR processes that are either dependent on or independent of FA proteins. For example, HR repair of cisplatin damage for cell survival requires both Xrcc3 and FANCC, while Xrcc3 but not FANCC is necessary for spontaneous SCE.
The rate of spontaneous SCE per cell might be influenced by (1) the frequency at which replication forks encounter nicks or blocking lesions, (2) HR capacity, (3) overall activity of TLS, and (4) a factor that determines the frequency of crossing over, for example, BLM helicase (Wu and Hickson, 2003). We tested the contributions of the latter three processes to SCE in fancc cells by making double mutants and found an overlap between BLM and the FA pathway. We further confirmed this relationship by measuring SCE in fancd2/blm double mutants. In addition, both fancc/blm and fancd2/blm cells also displayed the same levels of sensitivity to killing by ICL reagents as those of fancc or fancd2 cells, respectively. These data suggest that the FA pathway and BLM may function in a common pathway to repair ICLs and suppress SCE.
Several lines of evidence support this conclusion. First, ICL damage-induced focus formation of GFP-BLM was defective in both human and chicken fancc and fancd2 cells. Second, we observed colocalization of FANCD2 and GFP-BLM foci in MMC-treated wild-type DT40 and the complemented human fancd2 cells. Third, FANCD2-L form was co-immunoprecipitated with GFP-chBLM in wild-type DT40 but not in fancc cells following MMC treatment. Fourth, BLM is reported to co-immunoprecipitate with the FA core complex in human cells (Meetei et al, 2003b). Fifth, in contrast to GFP-chBLM, Rad18 focus formation was not defective in fancc or fancd2 DT40 cells, consistent with the additive defects found in fancc/rad18 cells. Thus, we conclude that the localization of BLM to stalled/broken replication forks caused by ICLs depends on the FA pathway. In suppressing SCE, BLM requires the FA pathway in a partial manner, since elevation of SCE in fancd2 or fancc cells is not as high as in blm cells. On the other hand, in repairing potentially lethal ICL damage, BLM functions only inside the pathway that depends on FANCD2 and FANCC.
The components of the FA core complex were found in anti-BLM immunoprecipitates (Meetei et al, 2003b). However, FANCD2 appeared not to be tightly bound to the complex (Pace et al, 2002). Since components of the core complex have been shown to associate with chromatin/nuclear matrix upon DNA damage (Qiao et al, 2001), it is possible that a complex containing FANCC helps mobilize BLM to the sites of stalled/broken replication forks induced by DNA damage. Alternatively, FANCC may participate in BLM redistribution only through FANCD2 activation. Monoubiquitinated FANCD2 localized to stalled/broken replication forks may then mediate the formation of BLM foci through physical interaction. This interaction might be indirect, since BLM is found in the BRCA1-containing mega-protein complex (Wang et al, 2000b), and FANCD2-L and BRCA1 co-immunoprecipitate following DNA damage (Garcia-Higuera et al, 2001).
In summary, this study provides genetic evidence that defines relationships between the FA pathway and processes required to resolve stalled or broken replication forks. We found that BLM and FANCC act in a common pathway in suppressing spontaneous SCEs and in repairing ICL. The overlap between FA and BS is perhaps unexpected and surprising, given the highly distinct clinical and cellular features of these disorders. We suggest that FANCD2 utilizes BLM as its effector molecule by regulating BLM relocalization in the vertebrate nucleus.
Materials and methods
Cell culture and transfection
Wild-type and mutant DT40 cells were cultured at 39.5°C with 5% CO2 using RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 1% chicken serum, 2-mercaptoethanol (50 μM), and L-glutamine (2 mM) as described (Yamamoto et al, 2003). Transfection by electroporation and subsequent selection were performed as described (Yamamoto et al, 2003). BLM-deficient (Wang et al, 2000a), RAD18-deficient (Yamashita et al, 2002), and conditional XRCC3-deficient (Ishiai et al, 2004) DT40 mutant cells were previously described.
SV-40 transformed FA-D2 fibroblasts PD20, PD20 complemented with human FANCD2, and FA-C fibroblasts PD331 were kind gifts from Dr Barbara A Cox (Fanconi Anemia Cell Repository, The Oregon Health and Science University). These cells were cultured at 37°C with 5% CO2 using α-minimal essential medium supplemented with 10% FCS. Transient transfection was performed using Lipofectamine Plus (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol.
Chicken FANCC cloning and gene targeting vectors
An EST clone containing full-length chicken FANCC cDNA was obtained by searching chicken EST database. A portion of genomic chicken FANCC was PCR-amplified using primers designed on the basis of the cDNA sequence, and subcloned into TOPO-pCR-XL vector (Invitrogen). FANCC targeting vector was constructed by replacing an ∼1.5 kb genomic fragment containing a single exon with a resistance gene cassette. The targeting vector with an appropriate marker cassette was introduced into wild-type, conditional XRCC3-deficient, RAD18-deficeint, and BLM-deficient DT40 cells. The targeting event is expected to result in deletion of FANCC amino-acid residues 56–84. By splicing between residual exons, it should create a frame shift with immediate protein truncation. To obtain isogenic sets of Xrcc3-deficient and FANCC/Xrcc3 double-deficient clones, conditional xrcc3 cells and FANCC-disrupted conditional xrcc3 cells were cultured for ∼48 h in the presence of OH-TAM and subcloned by limiting dilution.
The targeting vectors for FANCD2 deletion were previously described (Yamamoto et al, 2005). FANCD2 alleles were disrupted by sequential tranfections into BLM-deficient cells. The targeting vectors to measure efficiency in gene targeting were previously described (Yamamoto et al, 2003).
Expression vectors
Chicken BLM cDNA was kindly provided by Dr Takehisa Matsumoto (RIKEN). Chicken Rad18 (Yamashita et al, 2002) was PCR-amplified from DT40 cDNA. To generate expression vectors, chicken FANCC was subcloned into pcDNA4/HisMax (Invitrogen) in-frame. Chicken BLM or Rad18 was inserted into pEGFP-C1 (Clontech, Palo Alto, CA, USA) to create the GFP-chBLM or GFP-chRad18 expression vector, respectively. GFP-hBLM expression vector was previously described (Suzuki et al, 2001).
RT–PCR analysis
Total RNA was isolated using TRIzol (Invitrogen), and converted to cDNA by reverse transcriptase (RT) (Invitrogen). A part of chicken FANCC or Rad51 was amplified with Gold Taq polymerase (Applied Biosystems Inc., Foster City, CA, USA).
Sensitivity of cells to irradiations and drugs
Colony formation was assayed in medium containing 1.4% methylcellulose. Cells were irradiated with X-rays (Linear Accelerator, Mitsubishi Electric Inc., Tokyo, Japan) or UV, or exposed to MMC (Kyowa Hakko, Tokyo, Japan) for 1 h and then plated (Yamamoto et al, 2003). Sensitivity to cisplatin (Nippon Kayaku, Tokyo, Japan) (Yamamoto et al, 2003) or MMS (Sigma-Aldrich, St Louis, MO, USA) was assayed by continuous exposure.
Immunoprecipitation, Western blot analysis, and subnuclear focus assay
For immunoprecipitation, cells were lysed in lysis buffer (0.5% Triton X-100, 50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 1 mM DTT, 10 mM NaF, 1 mM Na orthovanadate, 1 mM PMSF) supplemented with proteinase inhibitor cocktail (complete EDTA-free tablet) (Roche Diagnostics, Basel, Switzerland). GFP-chBLM was immunoprecipitated using polyclonal rabbit anti-GFP antibody (BD Biosciences, San Jose, CA, USA) and Protein G Plus/Protein A-agarose (EMD Biosciences, Inc., San Diego, CA, USA). Anti-chicken FANCD2 serum was described previously (Yamamoto et al, 2005). Anti-human FANCD2 antibodies were purchased from Abcam (Cambridge, UK). Immunoprecipitates or whole-cell lysates were separated with 6% SDS–PAGE, and then transferred to a membrane, and detected by antibodies using ECL plus Western blotting detection reagents (Amersham Biosciences, Piscataway, NJ, USA). To visualize subnuclear focus formation of chicken or human FANCD2, cytospin slides or coverslips were fixed and stained with the antiserum. BLM focus was detected as GFP-BLM. Images were captured using TCS SP2 laser scanning confocal microscopy (Leica microsystems, Bannockburn, IL, USA).
Analysis of chromosomal aberrations and SCE
Chromosome analysis was performed as previously described (Takata et al, 1998, 2000; Yamamoto et al, 2003). For SCE analysis with or without MMC (20 ng/ml), colcemid (0.1 μg/ml) was added for the last 2 h of 16 h incubation in the presence of BrdU, and scoring was performed on coded slides as previously described (Takata et al, 2000). Statistical analysis was carried out by an analysis of variance (ANOVA) with the Bonferroni/Dunn multiple comparison test for intergroup comparison using Statview software (SAS Institute Inc., Cary, NC, USA).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We thank Ms Masayo Kimura and Keiko Namikoshi for expert technical assistance, Ms Kazuko Hikasa for secretarial assistance, Dr Takehisa Matsumoto (RIKEN) for providing chicken BLM cDNA, and Dr Barbara A Cox (Fanconi Anemia Cell Repository, The Oregon Health and Science University) for providing human FA cells. This work was supported in part by Grants-in-aid from the Ministry of Education, Science, Sports, and Culture of Japan (MT). Financial support was also provided by the Kawasaki Medical School (Project Research Grants No. 13-211, 14-203, and 15-201A), The Naito Foundation, The Takeda Foundation, and The Sagawa Foundation for Promotion of Cancer Research. A portion of this work was performed under the auspices of the US Department of Energy by the University of California, Lawrence Livermore National Laboratory under contract no. W-7405-Eng-48, with funding provided by the DOE Low-Dose Program and NCI/NIH Grant CA89405 (LHT).
References
- Bachrati CZ, Hickson ID (2003) RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J 374: 577–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaganti RS, Schonberg S, German J (1974) A manifold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc Natl Acad Sci USA 71: 4508–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MM (2001) Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annu Rev Genet 35: 53–82 [DOI] [PubMed] [Google Scholar]
- Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ (2000) The importance of repairing stalled replication forks. Nature 404: 37–41 [DOI] [PubMed] [Google Scholar]
- Cromie GA, Connelly JC, Leach DR (2001) Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Mol Cell 8: 1163–1174 [DOI] [PubMed] [Google Scholar]
- Cumming RC, Lightfoot J, Beard K, Youssoufian H, O'Brien PJ, Buchwald M (2001) Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nat Med 7: 814–820 [DOI] [PubMed] [Google Scholar]
- D'Andrea AD, Grompe M (2003) The Fanconi anaemia/BRCA pathway. Nat Rev Cancer 3: 23–34 [DOI] [PubMed] [Google Scholar]
- Dronkert ML, Beverloo HB, Johnson RD, Hoeijmakers JH, Jasin M, Kanaar R (2000) Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol Cell Biol 20: 3147–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J (1995) The Bloom's syndrome gene product is homologous to RecQ helicases. Cell 83: 655–666 [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7: 249–262 [DOI] [PubMed] [Google Scholar]
- German J (1993) Bloom syndrome: a mendelian prototype of somatic mutational disease. Medicine (Baltimore) 72: 393–406 [PubMed] [Google Scholar]
- Gregory RC, Taniguchi T, D'Andrea AD (2003) Regulation of the Fanconi anemia pathway by monoubiquitination. Semin Cancer Biol 13: 77–82 [DOI] [PubMed] [Google Scholar]
- Grompe M, D'Andrea A (2001) Fanconi anemia and DNA repair. Hum Mol Genet 10: 2253–2259 [DOI] [PubMed] [Google Scholar]
- Hickson ID (2003) RecQ helicases: caretakers of the genome. Nat Rev Cancer 3: 169–178 [DOI] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D'Andrea AD (2002) Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297: 606–609 [DOI] [PubMed] [Google Scholar]
- Hu P, Beresten SF, van Brabant AJ, Ye TZ, Pandolfi PP, Johnson FB, Guarente L, Ellis NA (2001) Evidence for BLM and topoisomerase IIIalpha interaction in genomic stability. Hum Mol Genet 10: 1287–1298 [DOI] [PubMed] [Google Scholar]
- Ishiai M, Kimura M, Namikoshi K, Yamazoe M, Yamamoto K, Arakawa H, Agematsu K, Matsushita N, Takeda S, Buerstedde JM, Takata M (2004) DNA crosslink repair protein SNM1A interacts with PIAS1 in nuclear focus formation. Mol Cell Biol 24: 10733–10741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF III, Maul GG (1999) PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol 147: 221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joenje H, Patel KJ (2001) The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet 2: 446–457 [DOI] [PubMed] [Google Scholar]
- Johnson RD, Jasin M (2000) Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 19: 3398–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannouche PL, Wing J, Lehmann AR (2004) Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 14: 491–500 [DOI] [PubMed] [Google Scholar]
- Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y (1999) Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet 22: 82–84 [DOI] [PubMed] [Google Scholar]
- Kruyt FA, Hoshino T, Liu JM, Joseph P, Jaiswal AK, Youssoufian H (1998) Abnormal microsomal detoxification implicated in Fanconi anemia group C by interaction of the FAC protein with NADPH cytochrome P450 reductase. Blood 92: 3050–3056 [PubMed] [Google Scholar]
- Levitus M, Rooimans MA, Steltenpool J, Cool NF, Oostra AB, Mathew CG, Hoatlin ME, Waisfisz Q, Arwert F, De Winter JP, Joenje H (2003) Heterogeneity in Fanconi anemia: evidence for two new genetic subtypes. Blood 103: 2498–2503 [DOI] [PubMed] [Google Scholar]
- Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, Hoatlin ME, Joenje H, Wang W (2003a) A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 35: 165–170 [DOI] [PubMed] [Google Scholar]
- Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, Rooimans MA, Bier P, Hoatlin M, Pals G, de Winter JP, Wang W, Joenje H (2004) X-linked inheritance of Fanconi anemia complementation group B. Nat Genet 36: 1219–1224 [DOI] [PubMed] [Google Scholar]
- Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W (2003b) A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 23: 3417–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda I, Shan Z, Schartl M, Burt DW, Koehler M, Nothwang H, Grutzner F, Paton IR, Windsor D, Dunn I, Engel W, Staeheli P, Mizuno S, Haaf T, Schmid M (1999) 300 million years of conserved synteny between chicken Z and human chromosome 9. Nat Genet 21: 258–259 [DOI] [PubMed] [Google Scholar]
- Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ (2004) The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell 15: 607–620 [DOI] [PubMed] [Google Scholar]
- Okada T, Sonoda E, Yamashita YM, Koyoshi S, Tateishi S, Yamaizumi M, Takata M, Ogawa O, Takeda S (2002) Involvement of vertebrate polkappa in Rad18-independent postreplication repair of UV damage. J Biol Chem 277: 48690–48695 [DOI] [PubMed] [Google Scholar]
- Pace P, Johnson M, Tan WM, Mosedale G, Sng C, Hoatlin M, de Winter J, Joenje H, Gergely F, Patel KJ (2002) FANCE: the link between Fanconi anaemia complex assembly and activity. EMBO J 21: 3414–3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulo D, Guillouf C, Mohrenweiser H, Moustacchi E (1990) Hypomutability in Fanconi anemia cells is associated with increased deletion frequency at the HPRT locus. Proc Natl Acad Sci USA 87: 8383–8387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, Haber JE (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63: 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao F, Moss A, Kupfer GM (2001) Fanconi anemia proteins localize to chromatin and the nuclear matrix in a DNA damage- and cell cycle-regulated manner. J Biol Chem 276: 23391–23396 [DOI] [PubMed] [Google Scholar]
- Rouse J, Jackson SP (2002) Interfaces between the detection, signaling, and repair of DNA damage. Science 297: 547–551 [DOI] [PubMed] [Google Scholar]
- Sasaki MS (1975) Is Fanconi's anaemia defective in a process essential to the repair of DNA cross links? Nature 257: 501–503 [DOI] [PubMed] [Google Scholar]
- Sasaki MS, Tonomura A (1973) A high susceptibility of Fanconi's anemia to chromosome breakage by DNA cross-linking agents. Cancer Res 33: 1829–1836 [PubMed] [Google Scholar]
- Sonoda E, Okada T, Zhao GY, Tateishi S, Araki K, Yamaizumi M, Yagi T, Verkaik NS, van Gent DC, Takata M, Takeda S (2003) Multiple roles of Rev3, the catalytic subunit of polzeta in maintaining genome stability in vertebrates. EMBO J 22: 3188–3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Morrison C, Yamaguchi-Iwai Y, Takata M, Takeda S (1999) Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol Cell Biol 19: 5166–5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulos DJ, Bradshaw PS, Li X, Pasic I, Truong K, Ikura M, Ungrin M, Meyn MS (2002) The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum Mol Genet 11: 3135–3144 [DOI] [PubMed] [Google Scholar]
- Suzuki H, Seki M, Kobayashi T, Kawabe Y, Kaneko H, Kondo N, Harata M, Mizuno S, Masuko T, Enomoto T (2001) The N-terminal internal region of BLM is required for the formation of dots/rod-like structures which are associated with SUMO-1. Biochem Biophys Res Commun 286: 322–327 [DOI] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Sonoda E, Fukushima T, Morrison C, Albala JS, Swagemakers SM, Kanaar R, Thompson LH, Takeda S (2000) The Rad51 paralog Rad51B promotes homologous recombinational repair. Mol Cell Biol 20: 6476–6482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17: 5497–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S (2001) Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol 21: 2858–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauchi H, Kobayashi J, Morishima K, van Gent DC, Shiraishi T, Verkaik NS, vanHeems D, Ito E, Nakamura A, Sonoda E, Takata M, Takeda S, Matsuura S, Komatsu K (2002) Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 420: 93–98 [DOI] [PubMed] [Google Scholar]
- Thompson LH, Hinz JM, Yamada NA, Jones NJ (2005) How Fanconi anemia proteins promote the four Rs: replication, recombination, repair, and recovery. Environ Mol Mutagen, in press [DOI] [PubMed] [Google Scholar]
- Venkitaraman AR (2002) Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 108: 171–182 [DOI] [PubMed] [Google Scholar]
- Venkitaraman AR (2004) Tracing the network connecting brca and Fanconi anaemia proteins. Nat Rev Cancer 4: 266–276 [DOI] [PubMed] [Google Scholar]
- Wang W, Seki M, Narita Y, Sonoda E, Takeda S, Yamada K, Masuko T, Katada T, Enomoto T (2000a) Possible association of BLM in decreasing DNA double strand breaks during DNA replication. EMBO J 19: 3428–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J (2000b) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 14: 927–939 [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M (2004) Rad18 guides pol η to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J 23: 3886–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SC (2003) Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 4: 435–445 [DOI] [PubMed] [Google Scholar]
- Wu L, Davies SL, Levitt NC, Hickson ID (2001) Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J Biol Chem 276: 19375–19381 [DOI] [PubMed] [Google Scholar]
- Wu L, Hickson ID (2003) The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426: 870–874 [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Hirano S, Ishiai M, Morishima K, Kitao S, Namikoshi K, Kimura M, Matsushita N, Arakawa H, Buerstedde JM, Komatsu K, Thompson LH, Takata M (2005) Fanconi anemia protein FANCD2 promotes immunoglobulin gene conversion and DNA repair through a mechanism related to homologous recombination. Mol Cell Biol, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Ishiai M, Matsushita N, Arakawa H, Lamerdin JE, Buerstedde JM, Tanimoto M, Harada M, Thompson LH, Takata M (2003) Fanconi anemia FANCG protein in mitigating radiation- and enzyme-induced DNA double-strand breaks by homologous recombination in vertebrate cells. Mol Cell Biol 23: 5421–5430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita YM, Okada T, Matsusaka T, Sonoda E, Zhao GY, Araki K, Tateishi S, Yamaizumi M, Takeda S (2002) RAD18 and RAD54 cooperatively contribute to maintenance of genomic stability in vertebrate cells. EMBO J 21: 5558–5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankiwski V, Noonan JP, Neff NF (2001) The C-terminal domain of the Bloom syndrome DNA helicase is essential for genomic stability. BMC Cell Biol 2: 11; doi:10.1186/1471-2121-2-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD (1996) Positional cloning of the Werner's syndrome gene. Science 272: 258–262 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wienands J, Zurn C, Reth M (1998) Induction of the antigen receptor expression on B lymphocytes results in rapid competence for signaling of SLP-65 and Syk. EMBO J 17: 7304–7310 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3