

Abstract
Knudson [Knudson, A. G. (1971) Proc. Natl. Acad. Sci. USA 68, 820–823] suggested that progression of retinoblastoma follows from two mutational events. Individuals who inherit one mutated gene copy should follow an age-onset pattern set by only a single rate-limiting step for transformation, whereas normal individuals should follow an age-onset pattern set by two rate-limiting events. Knudson's analysis of inherited and sporadic cases of retinoblastoma supported this prediction. However, retinoblastoma has a peculiar age-onset pattern concentrated in early life, because the retinal tissue completes most of its cell division by 5 years of age. Here, I compare age-specific incidences of inherited and sporadic forms of colon cancer, a much more typical form of human cancer. My simple mathematical analysis based on multistage theory explains the observed differences in age-onset patterns between inherited and sporadic cases. I also analyze recent retinoblastoma data and provide a mathematical analysis and interpretation. My analysis supports Knudson's two-hit theory but is much simpler and easier to understand than the original mathematical theory, which was based on a complicated model of cell division in the retina. My simpler theory for retinoblastoma makes clear the common basis for understanding multistage progression in tissues as different as the retina and colon.
Keywords: cancer epidemiology, inherited predisposition, retinoblastoma
The age of cancer onset follows remarkably simple patterns. The most common cancers arise in epithelial tissue late in life, increasing in incidence with roughly the fifth or sixth power of age (1). By contrast, the incidences of many childhood cancers decline steadily with age as the particular tissues in which they occur slow their rate of cell division with advancing development (2).
The simple regularity in age-specific incidence seems at odds with the great complexity and apparent randomness of environmental and genetic aspects of carcinogenesis. Yet, where one observes simple patterns, one suspects a simple process at some level of explanation.
For the past 50 years, Armitage and Doll's (1) multistage theory of cancer has provided the conceptual foundation for simple explanations of the observed age-specific incidence patterns. The multistage theory suggests that normal tissues are transformed into cancerous ones by means of a series of discrete stages. The stages may be somatic mutations, broad genomic rearrangements, or changes in tissue interactions and environment.
The precise nature of the stages does not affect the predictions of the multistage theory. What matters is that individuals, as they age, move stochastically through the various stages of transformation. Then, at any particular age, there is a regular probability distribution of individuals who have progressed to particular precancerous stages or all of the way to the final, malignant stage. What happens to an individual is highly random and cannot be predicted. By contrast, a distinct and predictable pattern emerges at the population level.
Here, I test the predictions of multistage theory by comparing the age-specific incidences of cancer in individuals who inherit a predisposing genetic mutation versus those individuals who do not inherit such a mutation. It was Knudson's (3) great insight to compare incidences in inherited and noninherited forms of retinoblastoma to test the predictions of multistage theory.
Methods
I extended Knudson's general approach in two ways. First, I compared age-specific incidences in inherited and noninherited forms of colon cancer. I also developed the specific mathematical prediction for this comparison that follows from multistage theory. This analysis and comparison to the data add to the subject because the only similar comparison, which was on retinoblastoma, was for a cancer with unusually simple genetics and a peculiar pattern of childhood onset. By contrast, colon cancer has the age-incidence profile of the common epithelial cancers that account for the great majority of all human cancers.
Second, I analyzed recent retinoblastoma data, again comparing the age-specific incidences for inherited and noninherited forms. I developed a very simple multistage theory that explains the observed differences. Knudson's original papers chose the right comparisons to illuminate the theory and correctly interpreted the data. But the first mathematical analysis (3) did not account for the patterns of cell division in retinal development, and the later papers (4, 5) accounted for cell division but were so mathematically complex that the elegant insight of comparing inherited and noninherited cases was lost in mathematical detail and technical procedures for fitting the model to the data. By contrast, I show how a simple quantitative analysis can bring out Knudson's insight in a more accessible and profound way.
Results and Discussion
Colon Cancer. Individuals who inherit one mutated copy of the APC gene almost invariably develop multiple colon tumors by midlife, causing a disease known as familial adenomatous polyp osis (FAP) (6). In terms of multistage models, individuals with an inherited APC mutation begin life one stage further along than do normal individuals.
Fig. 1A shows the age-specific incidence for individuals with inherited FAP or noninherited (sporadic) colon cancer. On log–log scales, both inherited and sporadic forms show approximately linear increases in incidence with age. The two lines are nearly parallel, with the inherited form occurring at an incidence rate 3–4 orders of magnitude greater than that for sporadic cases. Note that the incidence rate for inherited cases is given relative to the population of individuals carrying the inherited mutation, whereas the sporadic incidence is given relative to the population of noncarriers. Thus, the frequency of carriers does not influence the rate curves.
Fig. 1.

Age-specific incidence of inherited and sporadic colon cancer. (A) Inherited colon cancer (FAP) caused by mutation of the APC gene (•) and sporadic cases (○) per 106 population, shown on a log10 scale. I calculated FAP incidence by analyzing the age distribution of cases combined for males and females as summarized by Ashley (7) from data originally presented by Veale (8). Mutated APC alleles have very high penetrance for FAP, so the incidence at each age can be measured as the number of cases per year divided by the number of individuals who had not developed the disease in earlier years but who do eventually develop the disease. For the sporadic form, I used the incidence of colorectal cancers from the Surveillance, Epidemiology, and End Results (SEER) database combined for white males and females from the period 1973–1977 (http://seer.cancer.gov). (B) Ratio of sporadic colon cancer incidence (IC) to inherited FAP incidence (IF) at each age by using the data in A. This curve is obtained by taking the ratio of the fitted curves in A.
Fig. 1B plots the ratio of sporadic incidence to inherited age-specific incidence, IC/IF. This ratio increases ≈3-fold with age, varying between ≈2 and 6 × 10–4.
What does multistage theory predict for the ratio of sporadic to inherited incidence? It is possible to construct elaborate multistage models based on assumptions about tissue architecture, cell division rate, clonal expansion of mutated cells, and so on. Such mathematical theories can provide significant insight. However, it is difficult to test such models directly, because there are always other factors not included in the model, so any fit between theory and data is as likely to be a matter of fortuitous fitting of parameters as it is to be a matter of capturing the essential processes that explain most of the observed variation.
For these reasons, I prefer to use the simplest theory. Simple theory makes clear predictions about how observations should change when underlying parameters change. For example, how much greater should the incidence of inherited cancers be relative to sporadic cases? How should the ratio of sporadic to inherited cases change with age? If we can consistently succeed with such simple predictions, then perhaps we have captured the major features that control incidence rates. Of course, it is always possible that we could be right for the wrong reasons, that is, other models may also explain the data. But at least we should start with a simple approach and see how well we can succeed with simple explanations.
The simplest multistage model assumes that there are n stages of cancer development (1). The tissues of a normal individual begin life in stage 0 and slowly make stochastic transitions through the stages as age increases. In particular, suppose the rate of transition between stages is u. For individuals in stage n – 1, the rate of incidence will be u, the rate of the final transition. This rate is independent of time, because the transition hits randomly and so is equally likely to occur in any particular year. Thus, the incidence rate at age t is the probability of being in stage n – 1, multiplied by the rate u. If both inherited and noninherited cases must make the same final transition, then the relative incidence rate, R, of noninherited and inherited cases is the relative probability of being in stage n – 1 at age t.
The probability of any single transition over a period of t years is ut, the rate of transition multiplied by the total time elapsed. By using a widely known result from probability theory based on the gamma distribution, the probability at time t of k events happening in a particular order, each event having probability ut, is approximately Pk = (ut)k/k!, as long as ut is not too close to 1.
In individuals born without a mutation, a cell or tissue moving from stage 0 to stage n – 1 requires n – 1 transitions. By contrast, for individuals who inherit a mutation, only n – 2 transitions are required. Thus, the relative probability of being in stage n – 1 for the noninherited versus inherited group is the relative incidence, R, which is
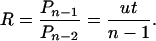 |
If transitions occur as somatic mutations, then the transition rate per year is the mutation rate per cell division, v, multiplied by the number of cell divisions per year, C. Few attempts have been made to measure the somatic mutation rate per gene per cell division. Yeast provide a convenient model of single eukaryotic cells. For yeast, the mutation rate has been estimated at 10–7 – 10–5 (9, 10). In mice, Kohler et al. (11) estimated the frequency of somatic mutations at 1.7 × 10–5. There are ≈101 to 102 cell divisions back to the embryo, so this study suggests a somatic mutation rate per cell division on the order of 10–7 to 10–6. I use the approximate value of 10–6 per gene per cell generation, but this is a very rough estimate at present.
Colon epithelial tissue renews itself continuously throughout life. The surface tissue turns over every few days, and stem cells that ultimately renew the tissue probably divide at least once per week or ≈50 times per year. For the number of stages, I use the simplest estimates from epidemiological data, which suggest that the number of stages in colon cancer progression is around n = 6 (1); other specialized models have, for example, put the number at n = 4 (12). All of these numbers are provisional, but they allow us to predict that the ratio of sporadic to inherited incidence rates should be roughly
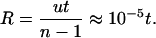 |
The data for inherited FAP and sporadic cases can be compared on the range t = 20–40, so R is predicted to increase over the range 2–4 × 10–4. Fig. 1B shows that the ratio of incidences is of the predicted magnitude and increases with age, although the increase with age is slightly greater than predicted. The nonlinearity between ages 20 and 30 may arise from statistical fluctuations associated with the fewer cases and much lower sample sizes at those ages or from additional aspects of progression not captured in my simple model.
The theory can be refined in many ways, for example, taking account of the number of independent cell lineages at risk for stepping through the various transition stages. But most reasonable assumptions apply both to the inherited and the sporadic rates of transition; therefore, the ratio of incidence rates remains roughly the same under such refinements. Of course, some assumptions will affect the ratio, for example, synergism between different cell lineages progressing through the early stages, which would increase the inherited incidence, because individuals with inherited mutations would have more late-stage cell lineages. But most of such refinements are difficult to test, so we still get the most insight from the simplest theory. The model presented here shows how the most commonly accepted assumptions explain the incidence patterns in a very simple way, suggesting that simple laws govern incidence rates at the population level.
Retinoblastoma. Burch (13) suggested that cancers may arise by multiple mutations (hits) to a cell in which the first mutation is inherited and the later mutations arise somatically. DeMars (14) mentioned a two-hit model for cancer and the possibility that some early onset cancers follow from a combination of inherited and somatic mutations.
Although the two-hit idea and the role of inherited mutations was clearly circulating 1970, this theory had little impact until Knudson's (3) analysis of retinoblastoma. Knudson realized that the two-hit theory predicted different patterns of age-specific incidence between inherited and sporadic cases of retinoblastoma. Individuals who inherit one mutation should follow the age-specific incidence patterns expected when a single somatic mutation causes transformation. Individuals who do not inherit a mutation should follow the age-specific incidence patterns expected when transformation requires two somatic mutations.
Bilateral retinoblastoma, in which tumors develop in both eyes, is an inherited disease. Most unilateral cases occur sporadically. Knudson's two-hit model predicted that bilateral cases follow age-specific patterns consistent with only one somatic hit leading to a tumor, whereas unilateral cases require two somatic hits to form a tumor.
Fig. 2 compares age-specific incidence of bilateral (inherited) and unilateral (sporadic) cases. The typical measure of age-specific incidence is the number of cases in an age group divided by the number of persons at risk in that age group. However, given the small sample sizes and the difficulty of measuring the base population that represents the number of persons at risk, Knudson defined the age-specific incidence as the number of cases not yet diagnosed at a particular age divided by the total number of cases eventually diagnosed, in other words, the fraction of cases not yet diagnosed.
Fig. 2.

Incidence of unilateral and bilateral retinoblastoma. The graph is redrawn from figure 1 of ref. 3.
Knudson (3) fit the bilateral cases to the model log(S) =–k1t, where S is the fraction of cases not diagnosed, k1 is a parameter used to fit the data, and t is age at diagnosis. He fit the unilateral cases to the model log(S) =–k2t2, where k2 is a parameter used to fit the data. The figure shows a reasonable fit for both models, with k1 = 1/30 and k2 = 4 × 10–5.
Knudson (3) gave various theoretical justifications for why inherited and sporadic forms should follow these simple models of incidence, proportional either to t for one hit or t2 for two hits. However, his theoretical arguments in this paper ignored the way the retina actually develops. In a later pair of papers, Knudson and his colleagues (4, 5) produced a theory of incidence that accounts for retinal development. [See also Nowak et al. (15) for a different mathematical approach to understanding how the growth of cell populations influences the dynamics of the two-hit model.]
Consider, for example, an individual who inherits one mutation. All dividing cells in the retina that are at risk for transformation can be transformed by a single additional somatic mutation. As the retina grows, the number of cells at risk for a somatic mutation increases, causing a rise in risk with age. The retina grows to near its final number of cells by ≈60 months. So cell division slows with age, causing a decrease in risk per cell with age. Change in overall risk with age depends on the opposing effects of the rise in cell number and the decline in the rate of cell division.
Hethcote et al. (5) developed a mathematical theory based on cellular processes of retinal development and fit their model to an extended set of data on inherited and sporadic retinoblastoma. The basic pattern in the data remains the same as in Fig. 2, but the later model fits parameters that provide estimates for the somatic mutation rate and for aspects of cell population size and cell division rate.
At first glance, the more realistic model based on cell populations and cell division may seem more attractive than the original model in ref. 3, which fit the data well but had no biological justification. However, human cancer incidence data are affected by many factors, including environment, cell–cell interactions, tissue structure, and modifying somatic mutations during different phases of tumor development. No model can account for all of these factors, so incidence data can never provide accurate estimates for isolated processes, such as somatic mutation rate or cell division rate.
Specific mathematical models, such as the one by Hethcote et al. (5), provide much insight into the consequences of particular factors, such as mutation and cell division for cancer incidence. However, Knudson's main insight was simply that age-specific incidence of inherited and sporadic retinoblastoma should differ in a characteristic way if cancer arises by two hits to the same cell. He got the data and showed that very simple differences in incidence do occur. The next step was to understand why the observed differences follow the particular patterns that they do. Detailed mathematical theory based on cell division and mutation rates provided insight about the factors involved, but, with regard to data analysis, that theory depended too much on the difficult task of estimating parameters, such as mutation and cell division from highly variable incidence data.
I advocate theory more closely matched to Knudson`s original insight and what one can realistically infer given the nature of the data. According to Knudson's theory, bilateral tumors arise from single hits to somatic cells with an inherited mutation. The rate at which a hit occurs in the developing retina at a particular age depends on many factors, including the number of target cells and the rate of cell division. But good estimates for those factors are not available, so, instead, the observations for bilateral cases at different ages can be used to estimate the rate at which a somatic mutation occurs in the tissue at a particular age, subsuming all of the details that together determine that rate. In particular, the estimates for age-specific bilateral incidence are taken as the estimates for the rate at which second hits occur in the tissue at a particular age. Clearly, this estimation procedure simplifies the real process; for example, bilateral cases require at least one hit in each eye. However, the rate of two second hits leading to bilateral cases is fairly high at ≈10–30% (Fig. 3A); thus, the rate of one second hit is about the same order of magnitude as the rate of two second hits. So I proceeded with the simple approach that IB(t), the incidence of bilateral cases at age t, provides a rough estimate of the rate of second hits to the tissue at age t.
Fig. 3.

Age-specific incidence of retinoblastoma. (A) Bilateral (solid line) and unilateral (dashed line) cases of retinoblastoma per 106 population, shown on a log10 scale. (B) Ratio of unilateral (IU) to bilateral (IB) incidence at each age by using the data in A. For the inherited form, I used 221 reported bilateral cases taken directly from the SEER database (http://seer.cancer.gov) for 1973–2001. To estimate age-specific incidence, I assumed that 65% of carriers eventually developed bilateral tumors based on the estimated penetrance for bilateral retinoblastoma given by Knudson (3). The incidence in each year is approximately the fraction of cases in that year divided by the fraction of individuals who had not developed the disease in earlier years. For the sporadic form, I used the reported incidence of unilateral cases in Young et al. (16), which is also from the SEER database. However, the SEER data do not differentiate between sporadic and hereditary unilateral cases. Based on data from Knudson (3), ≈75% of unilateral cases are sporadic cancers and ≈25% arise from carriers who inherit a mutation.
The incidence of unilateral cases can be written as
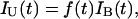 |
where f(t) is the fraction of somatic cells at age t that carry one somatic mutation, and IB(t) is approximately the rate at which the second hit occurs. Knudson's insight was to compare the incidences of unilateral and bilateral cases, so we could study the ratio of unilateral to bilateral incidence at each age
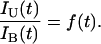 |
In words, the ratio of unilateral to bilateral rates should be roughly f(t), the fraction of cells at time t that carry the first hit in individuals that do not inherit a mutation. For example, if f(t) = 1, then all somatic cells have a first mutation, and the susceptibility is the same as for inherited cases. If f(t) = 0.5, then one-half of the somatic cells carry a first hit, and the susceptibility is one-half that of individuals who inherit the mutation.
The expected number of somatic mutational events suffered by an allele in a particular cell is the mutation rate per cell division, v, multiplied by the number of cell divisions going back to the embryo. Let the number of cell divisions at age t be C(t), so that vC(t) is the expected number of mutational events. For most assumptions, vC(t) < 1, so we can take vC(t) = f(t) as the fraction of cells at time t that carry a somatic mutation, and thus
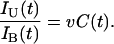 |
As discussed earlier, I used the approximate somatic mutation rate per cell division of v ≈ 10–6. The number of cell divisions, C(t), is roughly in the range of 15–40, because there are probably ≈15–25 cell divisions before the start of retinal development, and it takes ≈15 cellular generations in the retina to make the e15 ≈ 106 to 107 cells in the fully developed retina. Thus, IU(t)/IB(t) ≈ 10–4 to 10–5, and this ratio may increase by a factor of about two during early childhood as C(t) increases from ≈15–25 at the start of retinal development to ≈30–40 in the final cellular generations in the retina.
These rough calculations lead to two qualitative predictions. First, the ratio of unilateral to bilateral age-specific incidence should be ≈10–4 to 10–5. Second, the ratio of unilateral to bilateral incidence should approximately double with age over the period of retinal growth as the number of cellular generations, C(t), increases with time.
Fig. 3B shows that the ratio of unilateral and bilateral incidence is in the predicted range of 10–4 to 10–5, roughly the somatic mutation rate multiplied by the number of cellular generations. This ratio approximately doubles from the earliest age of 0–1 to the latest age of 2–3 at which sufficient numbers of bilateral cases occur to estimate incidence rates. The increase of this ratio supports the prediction that unilateral incidence increases relative to bilateral incidence as the number of cellular generations increases.
Conclusions
Knudson's insight was to test multistage theory by comparing the age-specific incidences of inherited and sporadic forms. The original mathematical theory for retinoblastoma was precise (5) but so complicated that it did not provide a clear and accessible test of the simple predicted comparison. In this paper, I developed simple mathematical analyses to emphasize the comparison between inherited and sporadic age-specific incidences in colon cancer and retinoblastoma. My mathematical models explain the observations and show the common aspects between these very different cancers.
For complex biological problems, such as cancer, comparative predictions and simple mathematical analyses provide more insight than complex and mechanistically more detailed mathematical models. The complex models require fitting several parameters from the data, and such parameter estimates based on highly variable data rarely provide accurate measures of the true processes. By contrast, comparative predictions, such as Knudson's analysis of sporadic versus inherited retinoblastoma, show how a simple difference in biological process can explain major differences in observed outcomes.
Acknowledgments
This research was supported by National Science Foundation Grant DEB-0089741 and National Institutes of Health Grant AI24424.
Author contributions: S.F. designed research, performed research, analyzed data, and wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: FAP, familial adenomatous polyposis; SEER, Surveillance, Epidemiology, and End Results.
References
- 1.Armitage, P. & Doll, R. (1954) Br. J. Cancer 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knudson, A. G. (2001) Nat. Rev. Cancer 1, 157–162. [DOI] [PubMed] [Google Scholar]
- 3.Knudson, A. G. (1971) Proc. Natl. Acad. Sci. USA 68, 820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knudson, A. G., Hethcote, H. W. & Brown, B. W. (1975) Proc. Natl. Acad. Sci. USA 72, 5116–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hethcote, H. W. & Knudson, A. G. (1978) Proc. Natl. Acad. Sci. USA 75, 2453–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kinzler, K. W. & Vogelstein, B. (2002) in The Genetic Basis of Human Cancer, eds. Vogelstein B. & Kinzler K. W. (McGraw–Hill, New York), 2nd Ed., pp. 583–612.
- 7.Ashley, D. J. (1969) J. Med. Genet. 6, 376–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veale, A. M. O. (1965) Intestinal Polyposis (Cambridge Univ. Press, Cambridge, U.K.).
- 9.Lichten, M. & Haber, J. E. (1989) Genetics 123, 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan, L. W. & Keil, R. L. (1990) Genetics 124, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohler, S. W., Provost, G. S., Fieck, A., Kretz, P. L., Bullock, W. O., Sorge, J. A., Putman, D. L. & Short, J. M. (1991) Proc. Natl. Acad. Sci. USA 88, 7958–7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luebeck, E. G. & Moolgavkar, S. H. (2002) Proc. Natl. Acad. Sci. USA 99, 15095–15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burch, P. R. (1963) Nature 197, 1042–1045. [DOI] [PubMed] [Google Scholar]
- 14.DeMars, R. (1970) in Genetic Concepts and Neoplasia: Proceedings of the 23rd Symposium on Fundamental Cancer Research (Williams & Wilkins, Baltimore), pp. 105–106.
- 15.Nowak, M. A., Michor, F., Komarova, N. L. & Iwasa, Y. (2004) Proc. Natl. Acad. Sci. USA 101, 10635–10638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young, J. L., Smith, M. A., Roffers, S. D., Liff, J. M. & Bunin, G. R. (1999) in Cancer Incidence and Survival Among Children and Adolescents: United States SEER Program 1975–1995, eds. Ries, L. A. G., Smith, M. A., Gurney, J. G., Linet, M., Tamra, T. Young, J. L. & Bunin, G. R. (Natl. Cancer Inst., Bethesda).