

Abstract
Resting heart rate is a heritable trait, and an increase in heart rate is associated with increased mortality risk. Genome-wide association study analyses have found loci associated with resting heart rate, at the time of our study these loci explained 0.9% of the variation. This study aims to discover new genetic loci associated with heart rate from Exome Chip meta-analyses.
Heart rate was measured from either elecrtrocardiograms or pulse recordings. We meta-analysed heart rate association results from 104 452 European-ancestry individuals from 30 cohorts, genotyped using the Exome Chip. Twenty-four variants were selected for follow-up in an independent dataset (UK Biobank, N = 134 251). Conditional and gene-based testing was undertaken, and variants were investigated with bioinformatics methods.
We discovered five novel heart rate loci, and one new independent low-frequency non-synonymous variant in an established heart rate locus (KIAA1755). Lead variants in four of the novel loci are non-synonymous variants in the genes C10orf71, DALDR3, TESK2 and SEC31B. The variant at SEC31B is significantly associated with SEC31B expression in heart and tibial nerve tissue. Further candidate genes were detected from long-range regulatory chromatin interactions in heart tissue (SCD, SLF2 and MAPK8). We observed significant enrichment in DNase I hypersensitive sites in fetal heart and lung. Moreover, enrichment was seen for the first time in human neuronal progenitor cells (derived from embryonic stem cells) and fetal muscle samples by including our novel variants.
Our findings advance the knowledge of the genetic architecture of heart rate, and indicate new candidate genes for follow-up functional studies.
Introduction
Increased resting heart rate (HR) is a known risk factor for cardiovascular morbidity and mortality (1–3), including stroke (4) and sudden cardiac death (5,6). Heart rate increased by 20 beats per minute (BPM) is associated with 30-50% higher mortality and appears to be independent of confounder factors (7). High HR increases myocardial oxygen consumption yet lessens oxygen delivery to myocardial tissue. It also increases arterial stiffness and risk of plaque rupture (8). Although HR can be influenced by many non-genetic factors (e.g. exercise, smoking and cardiovascular drugs), the heritability of resting HR is estimated to be 26–32% from family studies (9,10), and 55–63% from twin studies (11).
Several meta-analyses of genome-wide association studies (GWASs) have been undertaken to detect genetic determinants of HR (12–16). There were 21 HR loci previously reported at the time of our study by den Hoed et al. (12) in a GWAS analysis of 180 000 individuals, predominantly of European ancestry. The study implicated 20 candidate genes from follow-up functional studies in Danio rerio and Drosophila melanogaster models. Smaller GWAS analyses have also been performed in Icelandic and Norwegian populations (15), African Americans (13) and genetically isolated European populations (16). The variants discovered by GWAS are common, and are mostly in introns or intergenic regions. Together the previous loci from GWAS at the time of our study only explain a small percentage [0.9% of the variability in HR (12,17)].
To increase our knowledge of genetic determinants influencing HR and discover novel loci, especially rare or low frequency coding variants with larger effects, we meta-analysed data from 104 452 individuals of European-ancestry using the Exome Chip, from cohorts that participated in the Cohorts for Heart & Aging Research in Genomic Epidemiology (CHARGE) EKG consortium. The Exome Chip permits a cost-efficient analysis of coding variants derived from sequencing of >12 000 individuals and includes many rare and low-frequency variants (18). We performed a validation experiment using independent replication samples from UK Biobank data, and bioinformatics investigations to gain an understanding of the new HR loci.
Results
Single-nucleotide variant analysis in individuals of European-ancestry
In the discovery phase, association results of 235 677 single-nucleotide variants (SNVs) from 104 452 individuals were meta-analysed using a fixed-effects model (Supplementary Material, Fig. S1). Two analyses were performed. The first used RR-intervals (RR in milliseconds= 60 000/HR, in beats per minute, according to the inverse relationship between HR and RR). The second used the inverse-normalized residuals of the linear regression RR-interval adjusted for age + sex + body mass index (BMI) as covariates (denoted as RR-INVN). An overview of the study design is provided in Figure 1.
Figure 1.

Schematic flow diagram of the study design. N, sample size; SKAT, SNV-set Kernel Association Test; P, P-value; LD, linkage disequilibrium; SNV, single nucleotide variant; GCTA, Genome-wide Complex Traits Analysis software; 1958BC, 1958 Birth Cohort; UKB, UK Biobank.
We observed a high correlation of effect sizes and P-values between the RR-interval and RR-INVN meta-analyses (r2 = 0.99 and 0.98, respectively; Supplementary Material, Fig. S2). Furthermore, the RR-interval was near-normally distributed, so inverse normalization was deemed unnecessary (Supplementary Material, Fig. S3).
Beta-blockers are clinically known to lower HR, therefore the phenotype measurements of beta-blocker users may be under-estimated, and hence the inclusion of beta-blocker users in our analysis may potentially bias our analysis results. We therefore performed a sensitivity analysis by also meta-analysing a subgroup of cohorts that provided beta-blocker data (N = 48 347; 17 cohorts). Results including or excluding beta-blocker users were highly correlated (r2 of the betas = 0.97; r2 of the P-values = 0.74; Supplementary Material, Fig. S4), suggesting there is little or no bias from including beta-blocker users in the analysis. Therefore we report the meta-analysis results from the full dataset for the RR-interval, to maximize sample size and power.
Replication and meta-analysis with the UK Biobank dataset
To identify novel associated loci, we selected 12 variants with P < 1 × 10−5 that mapped outside the 21 HR loci reported in the previous GWAS (12) for follow-up in an independent dataset. Within each unknown locus, there were no potential secondary SNVs not in linkage disequilibrium (LD) with the lead SNV (r2 < 0.2) and meeting our look-up significance threshold (P < 1 × 10−5). Hence only 12 new lead SNVs were carried forward. We also followed-up 12 potential secondary signals at 9 of the 21 previously reported HR loci (further details on selection criteria are provided in the Materials and Methods) (12). None of the selected variants was in LD (r2 < 0.2) with each other, or with the published SNVs. Thus, a total of 24 variants were taken forward into replication. The UK Biobank dataset provided results for the selected genetic variants (N = 134 251 individuals).
Nine of the 12 previously unknown variants were validated based on exome-wide significance (P ≤ 2.12 × 10−7) in the combined meta-analysis of CHARGE and UK Biobank data, and on Bonferroni-adjusted significance (P ≤ 0.0042 for 12 tests) in the replication dataset alone, with concordant directions of effects taking into account the inverse relationship between the RR-interval from the discovery data and HR from the replication data (Table 1; Fig. 2). Indeed, all nine SNV associations were genome-wide significant in the combined meta-analysis (P < 5.0 × 10−8). Four of our nine validated novel loci were reported in a UK Biobank study (17) that was published after completion of our study (Table 1B). Hence, we present results here for five unreported novel loci (Table 1A;Supplementary Material, Figs S5 and S6).
Table 1.
Heart rate-associated loci identified from Exome Chip analysis
| SNV | Locus | Chr:Pos | EA | EAF | Ndiscovery | BETA-RR (SE) | Pdiscovery | BETA-HR (SE) | Preplication | Pcombined |
|---|---|---|---|---|---|---|---|---|---|---|
| (A) Five unreported novel loci | ||||||||||
| rs17853159a | TESK2 | 1:45810865 | A | 0.07 | 104 452 | −6.03 (1.20) | 5.02 × 10−7 | 0.31 (0.08) | 9.55 × 10−5 | 4.09 × 10−10 |
| rs3087866a | DALRD3 | 3:49054692 | T | 0.25 | 104 452 | 3.29 (0.72) | 4.92 × 10−6 | −0.31 (0.05) | 7.06 × 10−10 | 2.09 × 10−14 |
| rs1635852 | JAZF1 | 7:28189411 | C | 0.50 | 104 452 | 2.96 (0.62) | 2.04 × 10−6 | −0.15 (0.04) | 4.10 × 10−4 | 6.97 × 10−9 |
| rs10857472a | C10orf71 | 10:50534599 | A | 0.45 | 104 452 | −2.97 (0.63) | 2.11 × 10−6 | 0.16 (0.04) | 1.49 × 10−4 | 2.21 × 10−9 |
| rs3793706a,b | SEC31B | 10:102269085 | A | 0.22 | 104 452 | 3.52 (0.75) | 2.54 × 10−6 | −0.19 (0.05) | 2.06 × 10−4 | 3.72 × 10−9 |
| (B) Four loci validated in our study and also recently published in the UK Biobank study | ||||||||||
| rs709209a | RNF207 | 1:6278414 | G | 0.35 | 104 452 | −3.30 (0.66) | 4.94 × 10−7 | 0.27 (0.04) | 2.14 × 10−9 | 5.44 × 10−15 |
| rs6795970a | SCN10A | 3:38766675 | A | 0.40 | 104 452 | 2.97 (0.64) | 3.10 × 10−6 | −0.24 (0.04) | 1.81 × 10−8 | 2.73 × 10−13 |
| rs4282331 | 5p13.3 | 5:30881510 | G | 0.42 | 104 452 | −3.56 (0.63) | 2.03 × 10−8 | 0.26 (0.04) | 2.97 × 10−9 | 3.34 × 10−16 |
| rs12004a | KDELR3 | 22:38877461 | G | 0.30 | 104 452 | 3.30 (0.68) | 1.24 × 10−6 | −0.31 (0.05) | 4.92 × 10−11 | 4.04 × 10−16 |
Due to the inverse relationship between R-R interval and HR the opposite beta directions do relate to concordant directions of effect between discovery and replication. SNV, single-nucleotide variant; Chr:Pos, Chromosome:Position based on HG build 19; EA, effect allele; EAF, effect allele frequency from the discovery data; BETA-RR, beta effect estimate of RR-interval (milliseconds) taken from the ExomeRR discovery data; SE, standard error of the effect estimate; N, sample size analysed per variant (provided for genotyped discovery data only, as replication data was imputed so N = maximum N for all variants); BETA-HR, beta effect for heart rate (in beats per minute) taken from the UK Biobank replication data; P, P-value from either the discovery meta-analysis, the replication data, or the combined meta-analysis of discovery and replication data. Locus name indicates the nearest gene to the HR-associated SNV.
Indicates that the lead or a proxy SNV (r2>0.8) is a non-synonymous SNV.
Indicates if the lead SNV is predicted to be damaging. Mapping to more than 500 kb from either side of a previously reported HR-associated SNV. A novel locus is a genomic region with no SNVs in LD (r2 < 0.2) with HR-associated SNVs.
Figure 2.

Manhattan plot for the RR-interval discovery meta-analysis in European individuals. The Manhattan plot displays the results from the discovery meta-analysis of RR-intervals from N = 104,452 individuals of European ancestry (from 30 cohorts). On the X axis, P-values are expressed as −log10(P) are plotted according to physical genomic locations by chromosome. The Y-axis is truncated to −log10(P) = 20 with any variants with P < 1 × 10−20 displayed on the −log10(P) = 20 line. The nine novel variants validated from the combined meta-analysis with UK Biobank data are represented by squares. Variants in linkage disequilibrium (LD; r2 > 0.8) with published GWAS variants are highlighted with black circles (12). New secondary variants validated in our analysis are indicated as triangles. Locus names of the novel loci correspond to the nearest annotated gene, with 5p13.3 denoting an intergenic variant. The dashed line indicates a P-value threshold of 1 × 10−5, corresponding to the lookup significance threshold and the continuous line indicates a P-value threshold of 2 × 10−7, corresponding to exome-wide significance.
Twelve of the 21 HR-associated SNVs from the previously reported GWAS (12) were covered on the Exome Chip, either directly or by a proxy SNV in high LD (r2 > 0.8). Our discovery meta-analysis showed strong support for the previous findings, with 11 of the 12 SNVs validated at Bonferroni-adjusted significance (P ≤ 0.0042 for 12 tests), of which nine were validated at exome-wide significance (P < 2 × 10−7; Fig. 2). Only rs4140885 at the TFPI locus was not supported in our data (P = 0.10; Supplementary Material, Table S1).
Independent secondary signals at known loci
All 12 potential secondary signals at loci previously reported by den Hoed et al. (12) were genome-wide significant in the combined meta-analysis (Supplementary Material, Table S2) and are independent to the known SNPs according to LD (r2 < 0.2). We performed a conditional analysis using Genome-wide Complex Traits Analysis (GCTA) to formally identify secondary signals of association. Five of the 12 validated potential secondary SNVs (within CD46, CCDC141, SLC35F1, ACHE and KIAA1755 loci) were selected within the final GCTA model (Supplementary Material, Table S3). At four of the previously reported HR regions the secondary signals that we identified were confirmed to be statistically independent signals of association: CD46 (rs2745967), CCDC141 (rs10497529), SLC35F1 (rs12210810) and KIAA1755 (rs41282820) in addition to the known SNV, as both the published SNV and the new secondary SNV were present in the final GCTA model of jointly independent associated variants. Hence, we identified two distinct signals of association at each of these four known HR loci. However, the published SNV at the ACHE locus (rs13245899) is not covered on the Exome Chip, or by any proxies (Supplementary Material, Table S1), so the GCTA analysis does not include the known variant. As we are not able to condition on the unavailable published SNV and formally test association jointly with the known SNV, we are unable to statistically confirm the total number of independent signals at the ACHE locus.
The secondary SNVs at CCDC141, ACHE and KIAA1755 are non-synonymous variants. Furthermore, the SNVs at CCDC141 and KIAA1755 are low-frequency with minor allele frequencies (MAFs) of 3.6 and 1.7%, respectively. Secondary signals have also recently been observed at four of the five loci (CD46, CCDC141, SLC35F1 and ACHE) in UK Biobank data (17), since completion of our meta-analysis. At CD46, our secondary SNV (rs2745967) is in high LD (r2 = 0.78) with the secondary SNV (rs2745959) reported in UK Biobank, so likely to be the same signal. At CCDC141 our secondary variant is exactly the same SNV as from UK Biobank (rs10497529). Similarly, at SLC35F1, our secondary SNV (rs12210810) is in very high LD (r2 = 0.98), so is likely to be the same signal. Hence at these three known loci (CD46, CCDC141, SLC35F1), all existing data suggest there are two independent signals of association. At the ACHE locus, our secondary SNV (rs542137; ∼38 kb and r2 < 0.2 from the published SNV) is not in LD (r2 < 0.2) with the secondary SNV from UK Biobank (rs140367586; ∼659 kb and r2 < 0.2 from the published SNV). We are unable to clearly determine the number of distinct signals at the ACHE locus from our Exome Chip RR-interval discovery meta-analysis data, without the published SNV being covered on the Exome Chip. The low-frequency non-synonymous variant (rs41282820) at the known KIAA1755 locus is a new, secondary variant, with strong evidence of independent association, it does not overlap with other published findings.
Variance explained
Twelve of the 21 previously reported HR-associated SNVs (12) covered on the Exome Chip explain 1.14% of RR-interval variance (P = 3.96 × 10−10) within the 1958 Birth Cohort study (see Materials and Methods). The added contribution of the lead SNVs at our five unreported novel loci, combined with the 12 previously reported SNVs, increases the variance explained to 1.28% overall (P = 9.17 × 10−11).
Comparison of results between European and non-European populations
To investigate our data from non-European samples [9358 African Americans (AA), 1411 Hispanic (HIS) and 754 Chinese-Americans (CH); Supplementary Material, Table S4], we first extracted results for the 12 of the 21 previously reported HR-associated SNVs covered on the Exome Chip (12). In contrast to previous results for Europeans, only two known HR-SNVs showed evidence of association (P < 0.05), at the GJA1 and MYH6 loci, in the AA population only. This is likely due to a lack of power from the smaller non-European sample sizes, considering the power was calculated to be only 48, 11.7 and 8.5% for AA, HIS and CH, respectively. Concordance in the direction of effects compared with Europeans was only significant for AA, with 92, 64 and 50% concordance, corresponding to P-values of 2.9 × 10−3, 0.16 and 0.23 from binomial tests for AA, HIS and CH, respectively. The lack of support of previous findings from the under-powered non-European data led us to restrict our primary discovery meta-analysis to Europeans only.
We also performed a look-up of the nine validated SNV associations in the non-European samples. Due to the lack of power, and different allele frequencies compared with Europeans, none of the SNVs had results with P < 0.05 within any ancestry (Supplementary Material, Fig. S6), and there was little concordance in effect directions: 56% and P = 0.246 for AA; 33% and P = 0.164 for HIS and CH.
Gene-based tests
Gene-based testing was performed to identify genes which may have multiple rare variant associations. None of the gene-based test results was significant, after excluding the single most significant low-frequency variant from the tests (Supplementary Material, Table S5).
Look-up of UK Biobank HR-SNVs
Since completion of our meta-analysis of Exome Chip genotypes, a genome-wide scan for HR has been completed in UK Biobank (17). This study published 46 new HR loci. Four of these novel loci were simultaneously discovered in our analyses (RNF207, SCN10A, 5p13.3, KDELR3:Table 1B). Among the 42 remaining UK Biobank loci, only five of the lead SNVs were covered on the Exome Chip at r2 ≥ 0.8. Results from our exome RR European-ancestry meta-analyses show support for all five of these loci (P < 0.01; Bonferroni-adjusted significance for five tests; Supplementary Material, Table S6).
HR loci and association with other traits
To provide insights into possible shared aetiologies or mechanisms of disease, we assessed association of our five unreported novel HR-SNVs (and their proxies, r2 ≥ 0.8) with other traits. Genome-wide significant phenotype–genotype associations were observed for three novel loci (Supplementary Material, Table S7). The SNV at the DLRD3 locus was associated with age of menarche. The SNV at the JAZF1 locus was highly pleiotropic, as shown by associations with several autoimmune disorders (systemic lupus erythematosus, Crohn’s disease and selective immunoglobulin A deficiency), height, type 2 diabetes and JAZF1 transcript levels in adipose tissue. The SNV at the SEC31B locus was associated with plasma palmitoleic acid levels and differential exon expression of SEC31B.
Functional annotation of novel HR-SNVs and candidate genes
Four of the five unreported novel HR-SNVs or their proxies (r2 > 0.8) are non-synonymous SNVs in TESK2, DALRD3, C10orf71 and SEC31B (Table 1A). The non-synonymous SNV in SEC31B (rs2295774, c.1096T>G, p.Ser332Ala) is in a conserved region of the protein, and is predicted to be damaging using three different algorithms in ANNOVAR (19). We also investigated whether the novel HR-associated SNVs or their proxies (r2 > 0.8) were associated with changes in expression levels of nearby genes (i.e. as expression quantitative trait loci, or eQTLs) in the Genotype-Tissue Expression database (GTEx) dataset (20). We observed a significant eQTL association at one novel HR locus (Supplementary Material, Table S8). Specifically, the HR increasing allele of the non-synonymous SNV at SEC31B was associated with increased levels of SEC31B in tibial nerves (P = 8.08 × 10−33), lung (P = 1.22 × 10−23), atrial appendage tissue (P = 4.56 × 10−11) and the left ventricle (P = 4.0 × 10−9), tissues which may be regarded as physiologically relevant for HR.
We also observed HR loci to be significantly enriched for DNase I hypersensitive sites (DHSs; Fig. 3). We evaluated regions containing the five unreported novel HR loci and five independent secondary variants at previously reported HR loci (12) together with all 67 published HR-associated SNVs [21 loci reported from the original GWAS (12) plus 46 loci recently published from UK Biobank (17)]. Highest enrichment for DHSs in HR loci occurred within regions that are transcriptionally active in fetal heart tissue and fetal lung, as reported in the UK Biobank study. Moreover, for the first time we found significant enrichment for DHSs in human neuronal progenitor cells (derived from embryonic stem cells) and fetal muscle samples, with the inclusion of our novel loci.
Figure 3.
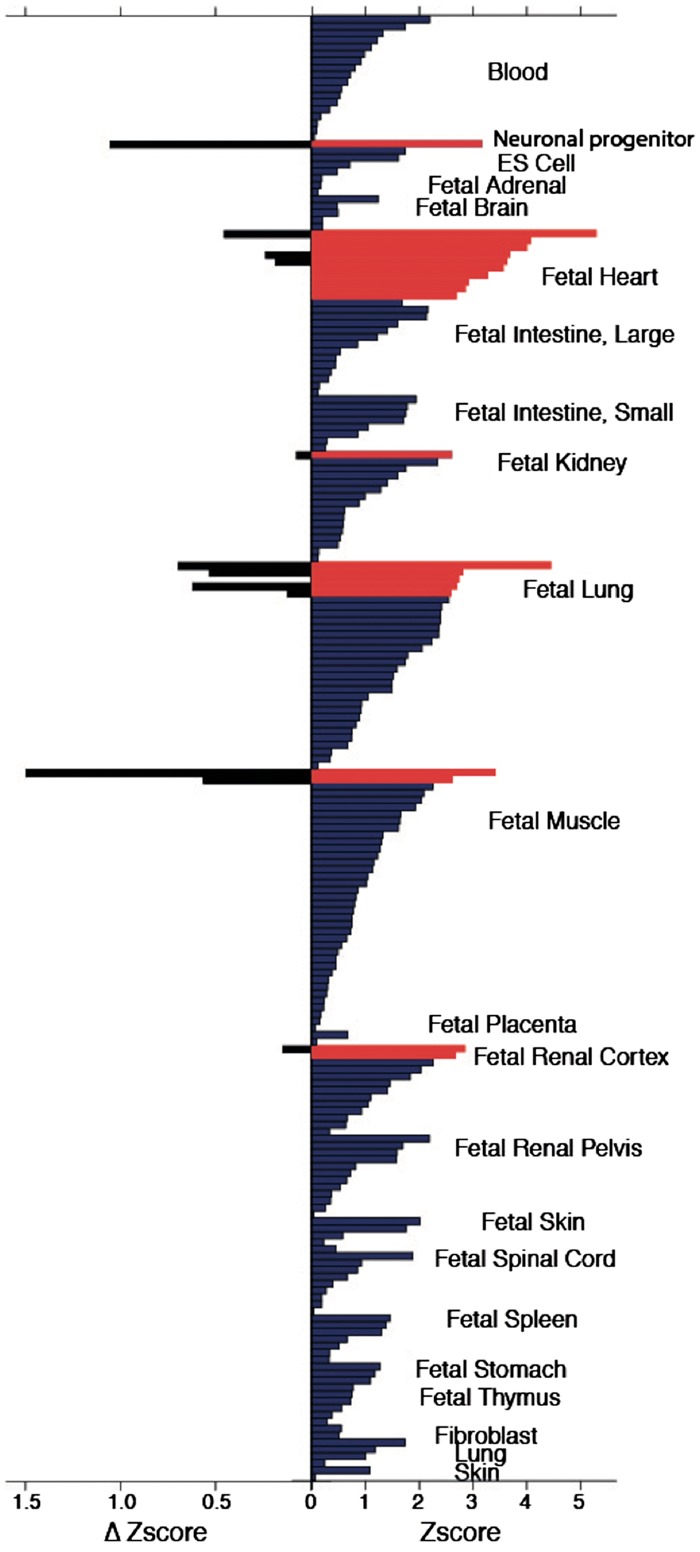
Enrichment of HR-SNVs in DNase I hypersensitive sites of 299 tissue samples. The right panel shows the enrichment of the combined known and novel (all) HR-SNVs in DNase I hypersensitivity sites of 212 Roadmap Epigenome tissue samples (those with positive Z-scores). Enrichment is expressed as a Z-score compared with the distribution of 1000 matched background SNV sets. Significant enrichments are shown in red (Z-score ≥ 2.58, false discovery rate (FDR) <1.5%), enrichments below this threshold are shown in blue. The left panel shows the enrichment difference (ΔZscore= Zscoreall − Zscoreknown) for those tissue samples in which we found significant enrichment using all SNPs and that further show a positive change using all SNVs compared with only known SNVs, with increased enrichment hence due to the novel loci identified.
Pathway analyses
We used Ingenuity pathway analyses to determine whether there was any increased enrichment in HR-associated pathways with the contribution of our five newly identified loci. We identified 16 significantly enriched pathways at P < 1 × 10−4. Most of these pathways are related to the cardiovascular system and involve, for example, supraventricular arrhythmias, dilated cardiomyopathy and HR (Supplementary Material, Table S9).
Coding variants at HR loci
The Exome Chip provides a unique opportunity to search for coding variants within known HR loci. Although GWAS analyses typically identify intron or intergenic variants, Exome Chip analysis may identify HR-associated coding variants, which would point to candidate causal genes. We considered all 67 published HR loci [21 previously reported GWAS loci (12) plus 46 recently published loci from UK Biobank (17)] and extracted all SNVs in high LD with the lead variants (r2 ≥ 0.8), tagging the same association signal, restricted to variants covered on the Exome Chip. We further filtered variants to obtain SNVs that reached exome-wide significance for associations with RR-interval in our primary discovery meta-analysis, to ensure that variants have a highly significant association with the trait. Coding SNVs were identified, using the CHARGE Exome Chip annotation file.
We only observed two such coding variants in two reported loci: CCDC141 and KIAA1755. The published CCDC141 coding variant was previously annotated as being non-synonymous (12), and is predicted to be damaging in our annotation (rs17362588; p.Arg935Trp). The coding SNV at KIAA1755 is the best proxy (r2 ∼ 1) for the published non-synonymous SNV (rs6127471) covered on the Exome Chip (Supplementary Material, Table S1). The original GWAS (12) had reported this signal as non-synonymous. Therefore, our Exome Chip analyses do not reveal any new evidence of likely causal coding variants at well-established HR loci.
Regulatory variants at HR loci
Our analyses of coding variants at all known HR loci indicated that the majority of HR-associated SNVs and the variants in high LD with them are non-coding. We thus investigated which variants could have a causal effect through regulatory chromatin interactions, such as promoter–enhancer contacts. We considered all 67 published HR loci [21 previously reported GWAS loci (12) plus 46 recently published loci from UK Biobank (17)], and the five novel loci reported here. We found variants that potentially affect enhancer function using RegulomeDB (21) and found genes whose promoter regions form significant chromatin interaction with them from right ventricle Hi-C data (22). We found 64 potential target genes in 49 HR loci (4 new loci, 18 loci from the GWAS and 27 loci from the UK Biobank study; Supplementary Material, Table S10). Including these long-range interactors in the candidate causal genes list increased the significance of enrichment for many HR-related terms, such as arrhythmia and cardiac fibrillation in our Ingenuity® Pathway Analysis (IPA®; Supplementary Material, Table S11).
For newly identified loci, the TESK2 promoter had a long-range interaction with the SNVs with highest regulatory potential in the locus, underlining it as a candidate. LOC441204, a gene of unknown function was found to interact with the JAZF1 locus. At the SEC31B locus, there were interactions with two genes, SCD and SLF2. At the C10orf71 locus, MAPK8 showed the most significant interaction.
In the 21 loci from the previously published GWAS (12), we identified significant chromatin contacts for the regulatory SNVs of 18 loci. We found CALCRL, TTN, HTR2B, PLD1 and CHRM2 as strongest interactors at the TFPI, CCDC141, B3GNT7, FNDC3B and CHRM2 loci, respectively, out of these only CALCRL is in LD (r2 > 0.8) with the lead SNV. The previous study (12) functionally tested 31 candidate genes, they found 20 of them to have an HR phenotype in either Drosophila melanogaster or Danio rerio experiments. All five of the strongest interactor genes were amongst the 20 genes with an HR phenotype.
Finally, we found 41 potential causal genes that have not been implicated by previous GWASs. A few of these genes have a cardiac function, including RAPGEF4 (18) and PIM1 (23), whereas some are involved in neuronal development and function, e.g. PBX3, NRNX3. These candidates open up new avenues that may aid our understanding of HR biology.
Discussion
Our meta-analysis of Exome Chip genotypes yielded five unreported novel HR loci, and one unreported independent new secondary signal, which was a low-frequency non-synonymous SNV at the previously reported KIAA1755 locus. Our data strongly supported the association of SNVs at 11 of the 12 previously reported GWAS loci that were covered on the Exome Chip. All lead SNVs at all validated novel loci are common (MAF ≥ 5%) and have similar effect sizes, which are smaller than the effect sizes for the majority of previously reported SNVs (Supplementary Material, Fig. S7). Our study did not yield any rare SNV associations with HR, indicating that much larger sample sizes will be required in future studies to have sufficient power to detect effects of any rare variants and assess their contributions to HR heritability.
The same observation of the need of larger sample sizes applies to the analysis of HR loci identified within Europeans in other ancestries, where the lack of significance and concordance in the results from non-European populations is most likely due to a lack of power, as well as differences in the allele frequencies and LD patterns between Europeans and non-Europeans. As the non-European samples were much smaller, we did not perform a comprehensive comparison across populations or a robust trans-ethnic meta-analysis.
Annotation of novel HR-SNVs or their close proxies, eQTL analyses and long-range chromatin interactions in heart tissue reveal new potential causal candidate HR genes (Supplementary Material, Tables S10 and S12). At the SEC31B locus there is a predicted damaging non-synonymous variant in SEC31B, and SNVs at this locus are also significantly associated with SEC31B expression levels. Although its precise function is unknown, the SEC31B gene encodes SEC31 homolog B, a COPII coat complex component. SEC31B has been proposed to function in vesicle budding, and cargo export from the endoplasmic reticulum (24). The gene is ubiquitously expressed at low levels, but there are higher levels of expression in the cerebellum. There are 13 transcripts, and thus several predicted SEC31B proteins. The major isoform is 129 kDa, but the HR-associated non-synonymous SNV maps to all SEC31B transcripts. There are no existing mouse models, and the predicted protein does not directly interact with other proteins or pathways currently recognized as being important to HR. Chromatin interactions in heart tissue indicate SCD and SLF2 as two other candidate genes for consideration at this locus. SCD encodes a stearoyl-CoA desaturase, which has a role in myocardial dysfunction (25) and SLF2 encodes the SMC5–SMC6 complex localization factor 2. TESK2, C10orf71 and DALRD3 can be considered as candidates for further analyses, based on the lead SNVs being non-synonymous variants in each gene. TESK2 encodes a serine/threonine protein kinase with an N-terminal protein kinase domain that is structurally similar to the kinase domains of testis-specific protein kinase-1 and the LIM motif-containing protein kinases. TESK2 is ubiquitously expressed, but its function is unknown (26). There is also support for TESK2 from the chromatin interaction analyses. C10orf71 encodes an open reading frame of unknown function that is highly expressed in heart and skeletal muscle. Chromatin interaction analyses indicate MAPK8 as a second candidate gene at the C10orf71 locus, MAPK8 is involved in formation of the heart as well as HR regulation (27,28). DALRD3 encodes a protein with a DALR anticodon-binding domain similar to that of class Ia aminoacyl tRNA synthetases (29).
The conditional analysis results provided one new, unreported association at a previously reported HR locus, KIAA1755 (rs41282820; c.1528C>T or c.1528C>A; p. Arg510Ter, a loss of function variant). KIAA1755 is predicted to encode an uncharacterized protein, and is only characterized at the transcriptional level. The transcript is highly expressed in the brain and nerves, and it is also expressed in the heart.
Our analyses and the recently published UK Biobank analyses (17) discovered a second low-frequency non-synonymous SNV at CCDC141 (rs10497529, c. 442C > T, P. Ala141Val). CCDC141 (also known as CAMDI) encodes the coiled-coil domain containing 141 protein and interacts with DISC1 (disrupted in schizophrenia 1) and MYL2 (phosphorylatable myosin light chain). CCDC141 is highly expressed in heart muscle (30). Knockdown of CCDC141 in neurons leads to abnormal cortical neuronal migration, but there are otherwise limited functional studies of CCDC141 (30). The CCDC141 locus includes TTN (titin), which encodes a major structural protein in striated muscle. TTN mutations are associated with a range of hereditary myopathies (31). Prior work (12) using RNA interference in Drosophila melanogaster has shown that knockdown of TTN leads to significant changes in resting HR and HR post tachypacing, supporting TTN is a causal candidate gene at this locus. The new data described here implicate CCDC141 as a second candidate gene at this locus for functional follow-up.
Enrichment analysis of HR variants in DNase I hypersensitivity sites across nearly 300 tissue samples and cell lines indicated new candidate tissues, such as neuronal progenitors and fetal muscle as being functionally relevant. Our data suggest these tissues should be targeted for future functional studies.
Our long-range regulatory chromatin interaction analyses provided additional support for some of the candidate genes have been experimentally tested previously (12) and shown to have an HR-related phenotype (CALCRL, TTN, HTR2B, PLD1 and CHRM2). By expanding the list of HR loci to include new and published, several new candidate genes are highlighted for functional studies in Supplementary Material, Table S10.
The Exome Chip contains non-synonymous, splicing and stop-coding variants that are thought to alter protein expression and function. Our analyses discovered four novel coding variants, indicating potential candidate causal genes at these loci. Our two-stage study design permitted the robust validation of all our novel loci findings, with a large replication sample size from UK Biobank (N = 134 251) to add together to our European discovery data (N = 104 452) for a large combined meta-analysis. However, due to the Exome Chip covering mainly coding regions, we were not able to compare results with all previous GWAS findings. In conclusion, our results taken together with recent studies (12) indicate HR-associated SNVs are mostly common (MAF > 5%) and have relatively small effect sizes. The maximum effect sizes reported thus far are ∼0.70 BPM per allele and MAF of 1% for SNVs at CCDC141 (rs17362588) and GJA1 (rs1015451). An analysis of much larger sample sizes (1M and above) including rare and common SNVs, and samples across different ancestries may provide further information on the contributions of both coding and non-coding variants, and the importance of rare coding variants in HR.
Materials and Methods
Study populations, phenotypes and exclusions
Thirty cohorts contributed data to the discovery meta-analysis in individuals of European ancestry. Details of all participating cohorts are provided in Supplementary Material, Table S13, including phenotype, cohort ancestry, study design and key references. The UK Biobank study, which was only recently published since the completion of our meta-analysis (17), provided results for replication analyses. Details of this study are also included in Supplementary Material, Table S13.
All participating cohorts either measured RR-intervals from the standard 12-lead electrocardiogram (ECG) or used HR measurements (in beats per minute) from peripheral pulse measurements (Supplementary Material, Table S14), which were converted to the RR-interval scale (in milliseconds) using the inverse relationship formula: RR (ms) = 60 000/HR (BPM). The discovery analysis was undertaken using the RR-interval phenotype. The exclusion criteria included: extreme RR-intervals (< 600 or > 1500 ms), atrial fibrillation on the ECG, a history of myocardial infarction or heart failure, use of non-dihydropyridine calcium-antagonists [Anatomic Therapeutic Chemical (ATC) code C08D], digoxin (ATC code C01AA5), second or third degree atrioventricular block and a pacemaker signal on the ECG. Local ethics committees approved the contributing studies from the CHARGE consortium, and all individuals provided their consent in writing. The UK Biobank study has approval from the North West Multi-centre Research Ethics Committee and has Research Tissue Bank approval.
Study-level genotyping and quality control
All discovery cohorts were genotyped using a human Exome Chip array (exact details of the chip for each study are provided in Supplementary Material, Table S15). Quality control (QC) was done according to CHARGE Exome QC guidelines, including joint variant calling with zCall (32). At the study-level, the sample-level QC consisted of excluding samples of non-European ancestry (for European-ancestry cohorts), samples with call rates <95%, samples with sex discordance or related samples with an unexpected high identical by descent estimate. It was recommended that principal components (PCs) be obtained using variants with MAF ≥ 1%. The variant QC consisted of exclusion of SNVs with call rate < 95%, with Hardy–Weinberg equilibrium values of P < 1 × 10−6, and of variants that were strongly associated with plate assignment.
Study-level statistical analysis
Each cohort performed two SNV association analyses using an additive model implemented with the R package SeqMeta, http://cran.r-project.org/web/packages/seqMeta/index.html. Analyses were stratified by ancestry. One SNV association analysis used an untransformed model with RR-interval as the outcome, adjusted for age, sex, BMI and cohort-specific adjustments. The other SNV association analysis was a model based on the rank-based inverse-normal transformed residuals (RR-INVN), with residuals taken from a linear regression RR-interval adjusted for age, sex and BMI covariates. The RR-INVN analysis was performed to check for potential sensitivity to deviations from normality within the analysis of rare variants. Additional cohort-specific covariate adjustments were also applied, which included for example PCs or family structure.
Central QC and meta-analyses
We performed additional QC checks centrally. For each study, we checked the sample size and the total number of SNVs (monomorphic and polymorphic) and assessed the beta distribution. Within each cohort’s results, all monomorphic SNVs were checked to have non-available results. In order to detect potential strand-flip issues, the cohort-coded effect allele frequencies (EAF) of each SNV were compared with the meta-analysed EAF of a group of CHARGE cohorts (AGES, ARIC, CHS, FHS and WHI). Any discordant SNVs showing cohort-EAF ∼ 0 in at least one study, but meta-EAF ∼ 1, or vice versa, were excluded from the central meta-analysis. A set of approximately 11 000 SNVs that were known to have QC issues from central CHARGE QC were also excluded from the meta-analysis. Quantile–Quantile plots were produced to inspect each cohort. After all QC steps were completed 235 677 SNVs remained. The results from all cohorts were then combined into a discovery meta-analysis using the SeqMeta R package.
Sensitivity analyses
A sensitivity analysis was performed on the use of beta-blockers (ATC code C07) due to the recognized effects of beta-blockers on HR. All cohorts with data on beta-blocker use were re-analysed with exclusion of individuals using beta-blockers at the time of phenotype measurement. Results of this meta-analysis were compared with the results from the same subset of cohorts with beta-blocker users included.
Selection of variants for replication
All SNVs with P < 1 × 10−5 from the discovery meta-analysis in European individuals were considered for follow-up. As a QC step after meta-analysis, we excluded four SNVs with unrealistically high beta values, large standard errors and results that were reported in less than four studies. We defined a novel locus as a genomic region (i) with SNVs not in LD (r2 < 0.2) with any well-established HR-associated SNVs from the previously reported GWAS (12) (Supplementary Material, Table S1), and (ii) mapping to more than 500 kb from either side of a previously reported HR-associated SNV. At the time of our study, there were 21 loci reported from GWAS analyses with HR-associated SNVs (12). A potential secondary signal within a previously reported locus was defined as being within a 1 Mb region centred around the published SNV, but not in LD (r2 < 0.2) with the published SNV in that region. LocusZoom plots were produced for all selected SNVs. Only the lead SNV was carried forward, for each signal being followed up. Specifically, the most significantly associated SNV was selected for any SNVs in pairwise-LD (r2 > 0.2). LD was calculated within UK Biobank genetic data, in order to calculate pairwise-LD for all 21 known SNVs (not only those covered on the Exome Chip).
Replication analyses
We used data from UK Biobank for replication of the selected SNVs (at the time of analysis genetic data were available for 150 000 individuals). The UK Biobank data were analysed with untransformed HR as the phenotype, with no exclusions for medication use. In UK Biobank resting HR was assessed by two methods: first, pulse rate using an automated reading during blood pressure measurement, and second, pulse rate during arterial stiffness measurement using the pulse wave form obtained of the finger with an infra red sensor. When multiple HR measurements were available during the first visit for an individual, these measurements were averaged. In 99.7% of participants at least one single measurement was available. Individuals were excluded with extreme (> 4 SD) values (N = 818). Further details are provided (17). The results of our European exome discovery meta-analysis for RR were combined with the UK Biobank replication results for HR (N = 134 251), and a combined meta-analysis, using sample-size weighted fixed effects meta-analysis in METAL was performed (33). All alleles were aligned between the discovery and replication data, and the inverse relationship between RR-interval and HR was taken into account, i.e. so that a negative beta direction from our discovery data for a decreased effect on RR-interval was made equivalent to a positive beta.
A novel locus was declared if the lead SNV reached exome-wide significance in the combined meta-analysis of discovery and replication data (P < 2.12 × 10−7) and replicated with Bonferroni-adjusted significance (P < 0.0042 for 12 tests) in the replication data alone. In addition, the directions of effect between the discovery and replication data were required to be concordant, taking into account the inverse relationship between RR from our discovery data and HR from the replication data.
Potential secondary SNVs at known regions were declared as validated if there was an exome-wide significant association in the combined meta-analysis. Variants that validated were subsequently tested for independence from previously reported HR variants in a conditional analysis.
Conditional analysis
In order to determine whether the validated secondary signals at previously reported loci were independent of the published SNV, conditional analysis was performed within GCTA software (34) applying the –cojo method (consisting of conditional and joint analysis with stepwise model selection). The input data were the exome-wide summary statistics from the full discovery meta-analysis of RR-interval in Europeans. The 1958 Birth Cohort Study (1958BC; N = 5815) dataset was used as the reference for genotype data, because it represents one of the largest discovery studies (See Supplementary Material, Table S13). LD was calculated between pairwise SNVs, but any SNVs further than 10 Mb apart were assumed to not be in LD. All autosomal chromosomes were analysed, with MAF restricted to ≥ 0.01%, to allow for low frequency secondary SNVs, whilst taking into account the statistical power achievable. To allow for secondary associations a P-value cut-off of 1 × 10−4 was used as the modelling selection threshold within the GCTA analysis. Results were then extracted for the nine previously reported regions, within which potential secondary signals had been validated from the combined meta-analysis. To be consistent with the look-up threshold for selecting SNVs to carry forward from discovery to replication, results were restricted to SNVs with a significance level of P < 1 × 10−5 from both the discovery meta-analysis and the joint association from GCTA.
Gene-based testing
Gene-based testing was conducted using the primary discovery data in Europeans. Analysis was performed using the SNV-set Kernel Association (SKAT) test within the seqMeta R Package. SKAT tests were performed according to two different MAF filters of 1% and of 5%, and three different levels of variant filtering, based on annotations within the CHARGE Exome SNP Info annotation file: (i) all variants, (ii) variants deemed predicted to be damaging (24) and (iii) variants that were non-synonymous or leading to abnormal splicing. For gene-based tests we adjusted for multiple testing using the Bonferroni correction, according to the number of genes tested. The gene-wide significance level was calculated as 1.98 × 10−6 for 25 241 tests (i.e. the number of genes on the Exome Chip). For any genes attaining significance, the gene-based tests were repeated with exclusion of the most significantly associated lead variant, in order to confirm that the association was due to multiple rare variants.
Non-European ancestry analyses
Association results were also received for non-European samples. Analysis and QC were performed as described for the European data. A meta-analysis was performed centrally in seqMeta for AA ancestry, combining data from the five AA cohorts. Study-level results remained for HIS and CH ancestries (from only the MESA cohort), in order to consider the three non-European ancestries (AA, CH and HIS) separately from stratified analyses. Due to the smaller sample sizes, power calculations were performed using the Genetic Power Calculator (35), based on the average percent trait variance explained per locus being 0.04%, according to the recently published results from 64 validated HR loci explaining ∼2.5% of HR variance (17). To assess the level of heterogeneity by ancestry in non-European data, we performed a look-up of SNVs at the 12 published HR loci covered on the Exome Chip, extracting results for these variants from each of the AA, CH and HIS results. We restricted our primary discovery analysis to Europeans only after finding a lack of significant validation and concordance between EUR and non-EUR data for previously reported HR variants. As a secondary analysis, we performed look-ups of all validated novel loci within the non-European data. The forest plots for all validated novel loci display non-European results, to serve as a comparison to results within Europeans. In addition to calculating the percentage of concordance of effect directions for each ancestry compared with Europeans, a Binomial sign test was also performed in R. This test was based on the number of SNVs with consistent effect directions, and it was done to determine whether the concordance was higher than expected by chance alone, using P < 0.05 to declare significant concordance.
Variance explained
The percentage variance explained for RR-interval was calculated using data from all subjects in the 1958BC study. The SNV genotypes were extracted from the 1958BC Exome Chip data and considered in two different sets: the 12 previously reported SNVs covered on the chip including proxies (r2 ≥ 0.8; see Supplementary Material, Table S1); and the lead SNVs from the five unreported novel loci (see Table 1A). First, RR-interval was regressed in a linear model against the sex and BMI covariates (not age, as all 1958BC subjects are of same age). Then the trait residuals from this first model were used as the phenotype in a second linear regression model, with all SNVs in the given set analysed jointly as multiple predictors, and adjusted for the top 10 PCs. The percentage trait variance explained by the set of SNPs was estimated from this second model, according to the adjusted R2 value.
HR loci annotation
For the purposes of annotation, all signals were expanded to include SNVs in LD. LD was calculated within the UK Biobank full genetic dataset using PLINK (v1.9). All variants with an r2 ≥ 0.8 within 500 kb downstream or upstream of the SNVs of interest were identified. These variants were annotated using ANNOVAR [vJun2015 (19)]. ANNOVAR functionally annotates variants, provides their conservation score, identifies SNVs that may cause protein-coding changes and reports their damaging prediction scores. Various prediction scores are available in ANNOVAR, including SIFT, PolyPhen and MutationTaster, among others.
We investigated the unreported novel SNVs and their proxies (r2 ≥ 0.8) across 44 tissues available in the GTEx dataset (20) for eQTLs. We reviewed the results for SNV-eQTL associations across all tissues, focusing on the heart, nerve, lung, muscle, adrenal and brain tissues which may be relevant tissues for HR based on known physiology of HR and our results from the enrichment analysis. Genes reported as eQTLs are based on study-specific significance thresholds (P-values < 10−8) and r2 ≥ 0.8 between HR-SNV and top-eQTL SNV (the SNV most significantly associated with transcript).
PhenoScanner
PhenoScanner (36) was used to identify variants that are associated with other traits. All proxy SNVs in high LD (r2 ≥ 0.8) with the lead SNVs at our five unreported novel loci were investigated in the PhenoScanner 1000 Genomes reference dataset. Results were filtered to those reaching a genome-wide significance P-value ≤ 5 × 10−8.
Potential candidate genes at new HR loci
Candidate genes at each locus were compiled using LD information, ANNOVAR-derived annotation and eQTL lookup results. A literature review was conducted for potential candidate genes at each new HR locus. Sources of information included: published articles, GeneCards, Online Mendelian Inheritance in Man®, the Human Protein Atlas, STRING and UniProt. We searched for information on the corresponding mouse models via the International Mouse Phenotyping Consortium and the Jackson Laboratory online catalogue. URLs for each of the sources is provided in the URL section below.
Pathway analyses
Pathway analyses were performed using QIAGEN’s IPA® (QIAGEN Redwood City) software. In order to distinguish the pathway enrichment contribution of novel loci from known HR loci, two sets of analyses were conducted. The first analysis captured the total known signal to date, investigating all 67 loci currently published, which include the 21 loci from the previously reported GWAS (12) and the 46 loci recently published from UK Biobank (17) since the completion of our meta-analysis. The second analysis included our five unreported novel loci in addition to all the previously reported loci. In each case, the analysis included all genes annotated from the lead SNVs and their proxies (r2 ≥ 0.8). Results were filtered for pathway enrichment of P-values ≤ 10−4. We specifically report the pathways for which enrichment is increased with the inclusion of genes from our novel loci.
Enrichment in DHSs
To identify the tissues in which HR-associated SNVs are active, we used FORGE to look for enrichment of DHSs in 299 tissue samples from the Roadmap Epigenome Project (37). FORGE calculates enrichment for overlap of HR variants with DHS by comparison with overlap of DHSs with 1000 matched background variant sets (matching distance to transcriptional start sites, GC content and MAF).
We performed two different enrichment analyses. First, we did a ‘known’ analysis using all 67 currently published lead SNVs to date [21 previously reported from the original GWAS (12) and 46 new loci from the recently published UK Biobank study (17)]. Second, we did an ‘all’ analysis using the lead SNVs at our five unreported novel loci and the five independent secondary SNVs that we found at previously reported loci; together with the 67 known signals, denoted as the ‘all’ analysis. We compared the enrichment results of the two analyses, in order to identify any new enrichment due to the inclusion of our novel loci. The enrichment is expressed as Z-score statistics. A Z-score of 2.58 was used as a threshold for statistical significance, which corresponds to false discovery rate (FDR) < 1.5%. We calculated the Z-scoreall − Z-scoreknown (ΔZ-score) for those tissue samples that were found statistically significant in the ‘all’ analysis in order to assess the effect of the 10 new, additional SNVs from our study.
Regulatory potential of SNVs
We selected the HR-associated SNVs and proxies in LD (r2 ≥ 0.8; calculated using the UK Biobank full genetic dataset) that were identified in this study, and from the previous GWAS (16) and UK Biobank studies (17) for annotation. To identify the potential regulatory variants, we retrieve the functional confidence score for SNVs from the RegulomeDb database (21). RegulomeDb assigns a functional confidence score to each SNV by overlapping them with functional genomic data mainly from ENCODE (e.g. DNase I hypersensitivity, DNase I footprinting, ChIP-seq), with eQTL data and with computational prediction (e.g. TF-binding sites and their disruption). We considered any SNP with at least one functional annotation to have regulatory potential (this corresponds to functional confidence scores: 1a-6).
Long-range regulatory contacts
Using significant long-range chromatin interactions as identified by Fit-Hi-C in right ventricle Hi-C data [40 kb resolution (22)], we annotated the potential regulatory SNVs with potential target genes, whose promoter is in contact with the given SNV. Where the 40-kb genomic region containing the SNV had more significant promoter interactions, we show the genes in order of most significant interaction to least significant. For every locus, we took the gene that had the most significant promoter interaction with a regulatory SNV, and using IPA®, we assessed which pathways were affected, and specifically those that were enriched compared to using only genes in LD with HR-SNVs.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
Individual studies
1958BC: We are grateful for using the British 1958 Birth Cohort DNA collection.
ARIC: The authors thank the staff and participants of the ARIC study for their important contributions.
ASCOT: We thank all ASCOT trial participants, physicians, nurses, and practices in the participating countries for their important contribution to the study. In particular we thank Clare Muckian and David Toomey for their help in DNA extraction, storage and handling. We would also like to acknowledge the Barts and The London Genome Centre staff for genotyping the Exome Chip array.
BRIGHT: The BRIGHT study is extremely grateful to all the patients who participated in the study and the BRIGHT nursing team.
CROATIA-Korcula: We would like to acknowledge the contributions of the recruitment team in Korcula, the administrative teams in Croatia and Edinburgh and the people of Korcula. Exome array genotyping was performed at the Wellcome Trust Clinical Research Facility Genetics Core at Western General Hospital, Edinburgh, UK.
ERF: We are grateful to all study participants and their relatives, general practitioners and neurologists for their contributions to the ERF study and to P. Veraart for her help in genealogy, J. Vergeer for the supervision of the laboratory work and P. Snijders for his help in data collection.
GoDARTs: We are grateful to all the participants in this study, the general practitioners, the Scottish School of Primary Care for their help in recruiting the participants, and to the whole team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. The study complies with the Declaration of Helsinki. We acknowledge the support of the Health Informatics Centre, University of Dundee for managing and supplying the anonymized data and NHS Tayside, the original data owner.
GRAPHIC: This work falls under the portfolio of research supported by the NIHR Leicester Cardiovascular Biomedical Research Unit.
GS:SFHS: We are grateful to all the families who took part, the general practitioners and the Scottish School of Primary Care for their help in recruiting them, and the whole Generation Scotland team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, healthcare assistants and nurses. Ethics approval for the study was given by the NHS Tayside committee on research ethics (reference 05/S1401/89).
HELIC: The MANOLIS cohort is named in honour of Manolis Giannakakis, 1978–2010. We thank the residents of the Mylopotamos villages, and of the Pomak villages, for taking part. The HELIC study has been supported by many individuals who have contributed to sample collection (including Antonis Athanasiadis, Olina Balafouti, Christina Batzaki, Georgios Daskalakis, Eleni Emmanouil, Chrisoula Giannakaki, Margarita Giannakopoulou, Anastasia Kaparou, Vasiliki Kariakli, Stella Koinaki, Dimitra Kokori, Maria Konidari, Hara Koundouraki, Dimitris Koutoukidis, Vasiliki Mamakou, Eirini Mamalaki, Eirini Mpamiaki, Maria Tsoukana, Dimitra Tzakou, Katerina Vosdogianni, Niovi Xenaki, Eleni Zengini), data entry (Thanos Antonos, Dimitra Papagrigoriou, Betty Spiliopoulou), sample logistics (Sarah Edkins, Emma Gray), genotyping (Robert Andrews, Hannah Blackburn, Doug Simpkin, Siobhan Whitehead), research administration (Anja Kolb-Kokocinski, Carol Smee, Danielle Walker) and informatics (Martin Pollard, Josh Randall).
JHS: We thank the Jackson Heart Study (JHS) participants and staff for their contributions to this work. The JHS is supported by contracts HHSN268201300046C, HHSN268201300047C, HHSN 268201300048C, HHSN268201300049C, HHSN268201300050C from the National Heart, Lung, and Blood Institute and the National Institute on Minority Health and Health Disparities.
KORA: We thank all KORA participants and staff members as well as Nadine Lindemann and Franziska Scharl involved in the generation of the SNP data.
Lifelines: We thank U. Bultmann (1), J.M. Geleijnse (2), P. van der Harst (3), S. Mulder (4), J.G.M. Rosmalen (5), E.F.C. van Rossum (6), H.A. Smit (7), M. Swertz (8), E.A.L.M. Verhagen (9)
(1) Department of Social Medicine, University Medical Center Groningen, University of Groningen, The Netherlands, (2) Department of Human Nutrition, Wageningen University, The Netherlands, (3) Department of Cardiology, University Medical Center Groningen, University of Groningen, The Netherlands, (4) Lifelines Cohort Study, The Netherlands, (5) Interdisciplinary Center of Psychopathology of Emotion Regulation (ICPE), Department of Psychiatry, University Medical Center Groningen, University of Groningen, The Netherlands, (6) Department of Endocrinology, Erasmus Medical Center, Rotterdam, The Netherlands, (7) Department of Public Health, University Medical Center Utrecht, The Netherlands, (8) Department of Genetics, University Medical Center Groningen, University of Groningen, The Netherlands, (9) Department of Public and Occupational Health, VU Medical Center, Amsterdam, The Netherlands.
NEO: The authors of the NEO study thank all individuals who participated in the Netherlands Epidemiology in Obesity study, all participating general practitioners for inviting eligible participants and all research nurses for collection of the data. We thank the NEO study group, Pat van Beelen, Petra Noordijk and Ingeborg de Jonge for the coordination, lab and data management of the NEO study. The genotyping in the NEO study was supported by the Centre National de Génotypage (Paris, France), headed by Jean-Francois Deleuze.
RS: The generation and management of the Illumina Exome Chip v1.0 array data for the Rotterdam Study (RS-I) was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, Rotterdam, The Netherlands. The authors are grateful to the study participants, the staff from the Rotterdam Study and the participating general practitioners and pharmacists. We thank Ms Mila Jhamai, Ms Sarah Higgins and Mr Marijn Verkerk for their help in creating the Exome Chip database, and Carolina Medina-Gomez, MSc, Lennard Karsten, MSc and Linda Broer PhD for QC and variant calling. Variants were called using the best practice protocol developed by Grove et al. as part of the CHARGE consortium Exome Chip central calling effort
SardiNIA: We thank all the volunteers who generously participated in this study and made this research possible.
SHIP: We thank all SHIP and SHIP-TREND participants and staff members as well as the genotyping staff involved in the generation of the SNP data.
WHI: The authors thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A full listing of WHI investigators can be found at: http://www.whi.org/researchers/Documents%20%20Write% 20a%20Paper/WHI%20Investigator%20Long%20List.pdf
YFS: The expert technical assistance in the statistical analyses by Irina Lisinen is gratefully acknowledged.
UK Biobank: This research has been conducted using the UK Biobank Resource Application Number 9628.
Conflicts of Interest statement. Dr B.M.P. serves on the DSMB of a clinical trial funded by the manufacturer (Zoll LifeCor) and on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. Dr P.T.E. is the PI on a grant from Bayer HealthCare to the Broad Institute focused on the genetics and therapeutics of atrial fibrillation. Dr N.P. has received financial support from several pharmaceutical companies that manufacture either blood pressure lowering or lipid lowering agents or both and consultancy fees. Dr P.S. has received research awards from Pfizer. Dr M.J.C. is Chief Scientist for Genomics England, a UK government company.
Funding
Individual investigators
B.M holds a Medical Research Council eMedLab Medical Bioinformatics Career Development Fellowship, funded from Medical Research Council award MR/L016311/1. Part of this project was enabled through access to the Medical Research Council eMedLab Medical Bioinformatics infrastructure, Medical Research Council award MR/L016311/1. R.N.E. is supported by the Netherlands organization for health research and development (ZonMw grant 90.700.441). Niek Verweij is supported by ICIN-Netherlands Heart Institute and Marie Sklodowska-Curie Global Fellowship, call: H2020-MSCA-IF-2014, Project ID: 661395. N.J.S is supported by the British Heart Foundation and N.J.S is a NIHR Senior Investigator. C.P.N is supported by the British Heart Foundation. The work of Marten van den Berg, Bruno H. Stricker and Mark Eijgelsheim is supported by grants from the Netherlands Organisation for Health Research and Development [ZonMw grant Priority Medicines Elderly 113102005 to ME; and ZonMw grant DoelmatigheidsOnderzoek 80- 82500-98-10208 to BHS]. Ilonca Vaartjes: Dutch Heart Foundation grant DHF project 'Facts and Figures'. Folkert W. Asselbergs is supported by a Dekker scholarship-Junior Staff Member 2014T001 Netherlands Heart Foundation and UCL Hospitals NIHR Biomedical Research Centre. Niels Grarup, Torben Hansen and Oluf Pedersen: The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation (www.metabol.ku.dk). Steven A. Lubitz: National Institutes Health grant K23HL114724 and a Doris Duke Charitable Foundation Clinical Scientist Development Award 2014105. P.B.M, M.J.C, H.R.W, C.P.C, P.D and A.T, wish to acknowledge the NIHR Cardiovascular Biomedical Research Unit at Barts and The London, Queen Mary University of London, UK for support. M.J.C is a Senior National Institute for Health Research Investigator. Nona Sotoodehnia is supported by National Institutes Health grants R01 HL116747 and R01 HL111089. P.B.M, and A.T, are supported by the Medical Research Council grant: MR/N025083/1.
Contributing studies
1958BC: Sample collection funded by the Medical Research Council grant G0000934 and the Wellcome Trust grant 068545/Z/02. Genotyping was funded by the Wellcome Trust.
AGES: This study has been funded by National Institutes Health contracts N01-AG-1-2100 and National Institutes Health: 271201200022C, the NIA Intramural Research Program, Hjartavernd (the Icelandic Heart Association), and the Althingi (the Icelandic Parliament). The study is approved by the Icelandic National Bioethics Committee, VSN: 00-063. The researchers are indebted to the participants for their willingness to participate in the study.
ARIC: The Atherosclerosis Risk in Communities (ARIC) study is carried out as a collaborative study supported by the National Heart, Lung, and Blood Institute (NHLBI) contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). Funding support for Building on GWAS for NHLBI-diseases: the U.S. CHARGE consortium was provided by the National Institutes Health through the American Recovery and Reinvestment Act of 2009 (ARRA) (5RC2HL102419).
ASCOT: The ASCOT study and the collection of the ASCOT DNA repository was supported by Pfizer, New York, NY, USA, Servier Research Group, Paris, France; and by Leo Laboratories, Copenhagen, Denmark. Genotyping of the Exome Chip in ASCOT-SC and ASCOT-UK was funded by the National Institutes of Health Research (NIHR).
BRIGHT: This work was supported by the Medical Research Council grant number: G9521010D; and by the British Heart Foundation, grant number: PG/02/128. A.F.D. was supported by the British Heart Foundation (Grant Numbers RG/07/005/23633, SP/08/005/25115); and by the European Union Ingenious HyperCare Consortium: Integrated Genomics, Clinical Research, and Care in Hypertension (grant number LSHM-C7-2006- 037093). The BRIGHT study is extremely grateful to all the patients who participated in the study and the BRIGHT nursing team. The Exome Chip genotyping was funded by Wellcome Trust Strategic Awards, 083948 and 085475. We would also like to thank the Barts Genome Centre staff for their assistance with this project.
CHS: The Cardiovascular Health Study research was supported by NHLBI contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086; and NHLBI grants U01HL080295, R01HL087652, R01HL105756, R01HL103612, R01HL120393, and R01HL130114 with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided through National Institutes Health R01AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR000124, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CROATIA-Korcula: This study was funded by the Medical Research Council UK and the Ministry of Science, Education and Sport in the Republic of Croatia (Medical Research Council grant number 108-1080315-0302).
ERF: The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP grant number 018947 (LSHGCT- 2006-01947) and also received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013)/grant agreement HEALTH-F4-2007-201413 by the European Commission under the programme Quality of Life and Management of the Living Resources of 5th Framework Programme (no. QLG2-CT-2002-01254). The ERF study was further supported by ENGAGE consortium and CMSB. High-throughput analysis of the ERF data was supported by joint grant from Netherlands Organisation for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043).
FHS: The National Heart, Lung and Blood Institute’s Framingham Heart Study is supported by contract N01-HC-25195 and HHSN268201500001I
GOCHA: The Genetics of Cerebral Hemorrhage with Anticoagulation was carried out as a collaborative study supported by grants National Institutes Health grant R01NS073344, R01NS059727, and 5K23NS059774 from the NIHNational Institute of Neurological Disorders and Stroke (NIH-NINDS)
GoDARTs: The Wellcome Trust United Kingdom Type 2 Diabetes Case Control Collection (GoDARTS) was funded by The Wellcome Trust (grant numbers: 072960/Z/03/Z, 084726/Z/08/Z, 084727/Z/08/Z, 085475/Z/08/Z, 085475/B/08/Z) and as part of the EU IMISUMMIT program.
GRAPHIC: The GRAPHIC Study was funded by the British Heart Foundation (BHF/RG/2000004). CPN and NJS are supported by the British Heart Foundation.
GS:SFHS: Generation Scotland received core funding from the Chief Scientist Office of the Scottish Government Health Directorate CZD/16/6 and the Scottish Funding Council HR03006. Genotyping of the GS:SFHS samples was carried out by the Genetics Core Laboratory at the Wellcome Trust Clinical Research Facility, Edinburgh, Scotland and was funded by the UK s Medical Research Council.
HELIC: This work was funded by the Wellcome Trust, grant number: 098051 and the European Research Council (ERC-2011-StG 280559-SEPI)
Inter99: The Inter99 was initiated by Torben J�sen (PI), Knut Borch-Johnsen (co-PI), Hans Ibsen and Troels F. Thomsen. The steering committee comprises the former two and Charlotta Pisinger. The study was financially supported by research grants from the Danish Research Council, the Danish Centre for Health Technology Assessment, Novo Nordisk Inc., Research Foundation of Copenhagen County, Ministry of Internal Affairs and Health, the Danish Heart Foundation, the Danish Pharmaceutical Association, the Augustinus Foundation, the Ib Henriksen Foundation, the Becket Foundation, and the Danish Diabetes Association.
KORA: The KORA study was initiated and financed by the Helmholtz Zentrum München German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Center of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ.
LifeLines: The LifeLines Cohort Study, and generation and management of GWAS genotype data for the LifeLines Cohort Study is supported by the Netherlands Organization of Scientific Research NWO (grant 175.010.2007.006), the Economic Structure Enhancing Fund (FES) of the Dutch government, the Ministry of Economic Affairs, the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the Northern Netherlands Collaboration of Provinces (SNN), the Province of Groningen, University Medical Center Groningen, the University of Groningen, Dutch Kidney Foundation and Dutch Diabetes Research Foundation.
MESA: This research was supported by the Multi-Ethnic Study of Atherosclerosis (MESA) contracts HHSN2682015000031, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01- HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC- 95167, N01-HC-95168, N01-HC-95169 and by grants UL1-TR-000040, UL1-TR-001079, and UL1-RR-025005 from National Center Research Resources (NCRR). Funding for MESA Family was provided by National Institutes of Health grants R01-HL-071205, R01-HL- 071051, R01-HL-071250, R01-HL-071251, R01-HL-071252, R01-HL-071258, and R01-HL071259, and by UL1-RR-025005 and UL1RR033176 from NCRR. Funding for MESA SHARe genotyping was provided by NHLBI Contract N02-HL-6-4278. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR000124, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
NEO: The NEO study is supported by the participating Departments, the Division and the Board of Directors of the Leiden University Medical Center, and by the Leiden University, Research Profile Area Vascular and Regenerative Medicine. Dennis Mook-Kanamori is supported by Dutch Science Organization (ZonMW-VENI Grant 916.14.023).
PROSPER: The PROSPER study was supported by an investigator initiated grant obtained from Bristol-Myers Squibb. Prof. Dr. J. W. Jukema is an Established Clinical Investigator of the Netherlands Heart Foundation (grant 2001 D 032). Support for genotyping was provided by the seventh framework program of the European commission (grant 223004) and by the Netherlands Genomics Initiative (Netherlands Consortium for Healthy Aging grant 050-060-810).
RS: The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. The Exome Chip array data set was funded by the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, from the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO)-sponsored Netherlands Consortium for Healthy Aging (NCHA; project nr. 050-060- 810); the Netherlands Organization for Scientific Research (NWO; project number 184021007) and by the Rainbow Project (RP10; Netherlands Exome Chip Project) of the Biobanking and Biomolecular Research Infrastructure Netherlands (BBMRINL; www.bbmri.nl).
SardiNIA: This research was supported by National Human Genome Research Institute grants HG005581, HG005552, HG006513, HG007022 and HG007089; by National Heart, Lung, and Blood Institute grant HL117626; by the Intramural Research Program of the US National Institutes of Health, National Institute on Aging, contracts N01-AG-1-2109 and HHSN271201100005C; by Sardinian Autonomous Region (L.R. 7/2009) grant cRP3-154
SHIP: SHIP (Study of Health in Pomerania) and SHIP-TREND both represent populationbased studies. SHIP is supported by the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung (BMBF); grants 01ZZ9603, 01ZZ0103, and 01ZZ0403) and the German Research Foundation (Deutsche Forschungsgemeinschaft (DFG); grant GR 1912/5-1). SHIP and SHIP-TREND are part of the Community Medicine Research net (CMR) of the Ernst-Moritz-Arndt University Greifswald (EMAU) which is funded by the BMBF as well as the Ministry for Education, Science and Culture and the Ministry of Labor, Equal Opportunities, and Social Affairs of the Federal State of Mecklenburg-West Pomerania. The CMR encompasses several research projects that share data from SHIP. The EMAU is a member of the Center of Knowledge Interchange (CKI) program of the Siemens AG. SNP typing of SHIP and SHIP-TREND using the Illumina Infinium HumanExome BeadChip (version v1.0) was supported by the Federal Ministry of Education and Research (BMBF) grant 03Z1CN22.
TwinsUK: This work was funded by a grant from the British Heart Foundation (PG/12/38/29615). The TwinsUK study was funded by the Wellcome Trust; European Community s Seventh Framework Programme (FP7/2007-2013). The study also receives support from the National Institute for Health Research (NIHR) BioResource Clinical Research Facility and Biomedical Research Centre based at Guy's and St Thomas’ NHS Foundation Trust and King's College London
UHP: The Utrecht Health Project received grants from the Ministry of Health, Welfare and Sports (VWS), the University of Utrecht, the Province of Utrecht, the Dutch Organisation of Care. Research, the University Medical Centre of Utrecht, and the Dutch College of Healthcare Insurance Companies. The Exome Chip data were generated in a research project that was financially supported by Biobanking and Biomolecular resources Research Infrastructure (BBMRI-NL, a Research Infrastructure financed by the Dutch government (NWO 184.021.007).
WHI: The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C.
YFS: The Young Finns Study has been financially supported by the Academy of Finland: grants 286284, 134309 (Eye), 126925, 121584, 124282, 129378 (Salve), 117787 (Gendi), and 41071 (Skidi); the Social Insurance Institution of Finland; Kuopio, Tampere and Turku University Hospital Medical Funds (grant X51001); Juho Vainio Foundation; Paavo Nurmi Foundation; Finnish Foundation for Cardiovascular Research; Finnish Cultural Foundation; Tampere Tuberculosis Foundation; Emil Aaltonen Foundation; Yrjö Jahnsson Foundation; Signe and Ane Gyllenberg Foundation; and Diabetes Research Foundation of Finnish Diabetes Association. Funding to pay the Open Access publication charges for this article was provided by the Medical Research Council Grant Number: MR/N025083/1.
URLs
http://www.gtexportal.org; date last accessed January 10, 2017.
http://www.phenoscanner.medschl.cam.ac.uk; date last accessed January 11, 2017.
http://www.genecards.org; date last accessed March 10, 2017.
http://omim.org; date last accessed August 26, 2016.
http://www.proteinatlas.org/; date last accessed March 10, 2017.
http://string-db.org/; date last accessed November 5, 2016.
http://www.uniprot.org; date last accessed March 10, 2017.
http://www.internationalgenome.org/forge-analysis/; date last accessed November 29, 2016.
References
- 1. Aladin A.I., Whelton S.P., Al-Mallah M.H., Blaha M.J., Keteyian S.J., Juraschek S.P., Rubin J., Brawner C.A., Michos E.D. (2014) Relation of resting heart rate to risk for all-cause mortality by gender after considering exercise capacity (the Henry Ford exercise testing project). Am. J. Cardiol., 114, 1701–1706. [DOI] [PubMed] [Google Scholar]
- 2. Carlson N., Dixen U., Marott J.L., Jensen M.T., Jensen G.B. (2014) Predictive value of casual ECG-based resting heart rate compared with resting heart rate obtained from Holter recording. Scand. J. Clin. Lab. Invest., 74, 163–169. [DOI] [PubMed] [Google Scholar]
- 3. Fox K., Bousser M.G., Amarenco P., Chamorro A., Fisher M., Ford I., Hennerici M.G., Mattle H.P., Rothwell P.M. (2013) Heart rate is a prognostic risk factor for myocardial infarction: a post hoc analysis in the PERFORM (Prevention of cerebrovascular and cardiovascular Events of ischemic origin with teRutroban in patients with a history oF ischemic strOke or tRansient ischeMic attack) study population. Int. J. Cardiol., 168, 3500–3505. [DOI] [PubMed] [Google Scholar]
- 4. Woodward M., Webster R., Murakami Y., Barzi F., Lam T.H., Fang X., Suh I., Batty G.D., Huxley R., Rodgers A. (2014) The association between resting heart rate, cardiovascular disease and mortality: evidence from 112,680 men and women in 12 cohorts. Eur. J. Prev. Cardiol., 21, 719–726. [DOI] [PubMed] [Google Scholar]
- 5. Jouven X., Zureik M., Desnos M., Guerot C., Ducimetiere P. (2001) Resting heart rate as a predictive risk factor for sudden death in middle-aged men. Cardiovasc. Res., 50, 373–378. [DOI] [PubMed] [Google Scholar]
- 6. Teodorescu C., Reinier K., Uy-Evanado A., Gunson K., Jui J., Chugh S.S. (2013) Resting heart rate and risk of sudden cardiac death in the general population: influence of left ventricular systolic dysfunction and heart rate-modulating drugs. Heart Rhythm, 10, 1153–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seccareccia F., Pannozzo F., Dima F., Minoprio A., Menditto A., Lo Noce C., Giampaoli S. and Malattie Cardiovascolari Aterosclerotiche Istituto Superiore di Sanita Project (2001) Heart rate as a predictor of mortality: the MATISS project. Am. J. Public Health, 91, 1258–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fox K.M., Ferrari R. (2011) Heart rate: a forgotten link in coronary artery disease? Nat. Rev. Cardiol., 8, 369–379. [DOI] [PubMed] [Google Scholar]
- 9. Singh J.P., Larson M.G., O’Donnell C.J., Tsuji H., Evans J.C., Levy D. (1999) Heritability of heart rate variability: the Framingham Heart Study. Circulation, 99, 2251–2254. [DOI] [PubMed] [Google Scholar]
- 10. Martin L.J., Comuzzie A.G., Sonnenberg G.E., Myklebust J., James R., Marks J., Blangero J., Kissebah A.H. (2004) Major quantitative trait locus for resting heart rate maps to a region on chromosome 4. Hypertension, 43, 1146–1151. [DOI] [PubMed] [Google Scholar]
- 11. Wang B., Liao C., Zhou B., Cao W., Lv J., Yu C., Gao W., Li L. (2015) Genetic contribution to the variance of blood pressure and heart rate: a systematic review and meta-regression of twin studies. Twin Res. Hum. Genet., 18, 158–170. [DOI] [PubMed] [Google Scholar]
- 12. den Hoed M., Eijgelsheim M., Esko T., Brundel B.J., Peal D.S., Evans D.M., Nolte I.M., Segre A.V., Holm H., Handsaker R.E.. et al. (2013) Identification of heart rate-associated loci and their effects on cardiac conduction and rhythm disorders. Nat. Genet., 45, 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deo R., Nalls M.A., Avery C.L., Smith J.G., Evans D.S., Keller M.F., Butler A.M., Buxbaum S.G., Li G., Miguel Quibrera P.. et al. (2013) Common genetic variation near the connexin-43 gene is associated with resting heart rate in African Americans: a genome-wide association study of 13,372 participants. Heart Rhythm, 10, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eijgelsheim M., Newton-Cheh C., Sotoodehnia N., de Bakker P.I., Muller M., Morrison A.C., Smith A.V., Isaacs A., Sanna S., Dorr M.. et al. (2010) Genome-wide association analysis identifies multiple loci related to resting heart rate. Hum. Mol. Genet., 19, 3885–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holm H., Gudbjartsson D.F., Arnar D.O., Thorleifsson G., Thorgeirsson G., Stefansdottir H., Gudjonsson S.A., Jonasdottir A., Mathiesen E.B., Njolstad I.. et al. (2010) Several common variants modulate heart rate, PR interval and QRS duration. Nat. Genet., 42, 117–122. [DOI] [PubMed] [Google Scholar]
- 16. Marroni F., Pfeufer A., Aulchenko Y.S., Franklin C.S., Isaacs A., Pichler I., Wild S.H., Oostra B.A., Wright A.F., Campbell H.. et al. (2009) A genome-wide association scan of RR and QT interval duration in 3 European genetically isolated populations: the EUROSPAN project. Circ. Cardiovasc. Genet., 2, 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eppinga R.N., Hagemeijer Y., Burgess S., Hinds D.A., Stefansson K., Gudbjartsson D.F., van Veldhuisen D.J., Munroe P.B., Verweij N., van der Harst P. (2016) Identification of genomic loci associated with resting heart rate and shared genetic predictors with all-cause mortality. Nat. Genet., 48, 1557–1563. [DOI] [PubMed] [Google Scholar]
- 18. Sugawara K., Shibasaki T., Takahashi H., Seino S. (2016) Structure and functional roles of Epac2 (Rapgef4). Gene, 575, 577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang K., Li M., Hakonarson H. (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res., 38, e164.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. GTEx Consortium (2013) The Genotype-Tissue Expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S.. et al. (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res., 22, 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmitt A.D., Hu M., Jung I., Xu Z., Qiu Y., Tan C.L., Li Y., Lin S., Lin Y., Barr C.L.. et al. (2016) A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep., 17, 2042–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wallner M., Kolesnik E., Ablasser K., Khafaga M., Wakula P., Ljubojevic S., Thon-Gutschi E.M., Sourij H., Kapl M., Edmunds N.J.. et al. (2015) Exenatide exerts a PKA-dependent positive inotropic effect in human atrial myocardium: GLP-1R mediated effects in human myocardium. J. Mol. Cell Cardiol., 89, 365–375. [DOI] [PubMed] [Google Scholar]
- 24. Stankewich M.C., Stabach P.R., Morrow J.S. (2006) Human Sec31B: a family of new mammalian orthologues of yeast Sec31p that associate with the COPII coat. J. Cell Sci., 119, 958–969. [DOI] [PubMed] [Google Scholar]
- 25. Dobrzyn P., Dobrzyn A., Miyazaki M., Ntambi J.M. (2010) Loss of stearoyl-CoA desaturase 1 rescues cardiac function in obese leptin-deficient mice. J. Lipid Res., 51, 2202–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rosok O., Pedeutour F., Ree A.H., Aasheim H.C. (1999) Identification and characterization of TESK2, a novel member of the LIMK/TESK family of protein kinases, predominantly expressed in testis. Genomics, 61, 44–54. [DOI] [PubMed] [Google Scholar]
- 27. Rose B.A., Force T., Wang Y. (2010) Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol. Rev., 90, 1507–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen Q., Xu T., Li D., Pan D., Wu P., Luo Y., Ma Y., Liu Y. (2016) JNK/PI3K/Akt signaling pathway is involved in myocardial ischemia/reperfusion injury in diabetic rats: effects of salvianolic acid A intervention. Am. J. Transl. Res., 8, 2534–2548. [PMC free article] [PubMed] [Google Scholar]
- 29. Grinchuk O.V., Jenjaroenpun P., Orlov Y.L., Zhou J., Kuznetsov V.A. (2010) Integrative analysis of the human cis-antisense gene pairs, miRNAs and their transcription regulation patterns. Nucleic Acids Res., 38, 534–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fukuda T., Sugita S., Inatome R., Yanagi S. (2010) CAMDI, a novel disrupted in schizophrenia 1 (DISC1)-binding protein, is required for radial migration. J. Biol. Chem., 285, 40554–40561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. LeWinter M.M., Granzier H.L. (2013) Titin is a major human disease gene. Circulation, 127, 938–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grove M.L., Yu B., Cochran B.J., Haritunians T., Bis J.C., Taylor K.D., Hansen M., Borecki I.B., Cupples L.A., Fornage M.. et al. (2013) Best practices and joint calling of the HumanExome BeadChip: the CHARGE Consortium. PLoS One, 8, e68095.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Willer C.J., Li Y., Abecasis G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang J., Ferreira T., Morris A.P., Medland S.E., Madden P.A., Heath A.C., Martin N.G., Montgomery G.W., Weedon M.N., Loos R.J.. et al. (2012) Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet., 44, 369–375, S361–S363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Purcell S., Cherny S.S., Sham P.C. (2003) Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics, 19, 149–150. [DOI] [PubMed] [Google Scholar]
- 36. Staley J.R., Blackshaw J., Kamat M.A., Ellis S., Surendran P., Sun B.B., Paul D.S., Freitag D., Burgess S., Danesh J.. et al. (2016) PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics, 32, 3207–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dunham I., Kulesha E., Iotchkova V., Morganella S., Birney E. (2014) FORGE: a tool to discover cell specific enrichments of GWAS associated SNPs in regulatory regions. bioRxiv doi:http://dx.doi.org/10.1101/013045.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.