

Abstract
The clinical progression of new chemical entities to pharmaceuticals remains hindered by the relatively slow pace of technology development in toxicology and clinical safety evaluation, particularly in vitro approaches, that can be used in the preclinical and early clinical phases of drug development. To alleviate this bottle-neck, we have developed a metabolizing enzyme toxicology assay chip (MetaChip) that combines high-throughput P450 catalysis with cell-based screening on a microscale platform. The MetaChip concept is demonstrated by using sol-gel encapsulated P450s to activate the prodrug cyclophosphamide, which is the major constituent of the anticancer drug Cytoxan, as well as other compounds that are activated by P450 metabolism. The MetaChip provides a high-throughput microscale alternative to currently used in vitro methods for human metabolism and toxicology screening based on liver slices, cultured human hepatocytes, purified microsomal preparations, or isolated and purified P450s. This technology creates opportunities for rapid and inexpensive assessment of ADME/Tox (absorption, distribution, metabolism, excretion/toxicology) at very early phases of drug development, thereby enabling unsuitable candidates to be eliminated from consideration much earlier in the drug discovery process.
Keywords: in situ drug metabolism, in vitro cytotoxicity, P450, sol-gel encapsulation
In the past decade, there has been a dramatic increase in the number of new chemical entities (NCEs) and screenable drug targets as a result of combinatorial chemistry and advances in genomics and proteomics (1–4). Nevertheless, these advances have not translated into an increased number of new drug approvals (5, 6), in part because of the high failure rate due to toxicity of the NCE or its metabolite(s) (7). Furthermore, screening for toxicity at early stages of the drug discovery process is precluded by the large number of compounds available at the lead discovery stage, forcing medicinal chemists to select compounds for lead optimization based on limited information about their toxicological properties. In particular, there remains a lack of in vitro techniques that can adequately mimic human metabolism and therefore assess cell-specific toxicity of NCEs and their metabolites at speeds consistent with high-throughput biological activity screening.
The human body, primarily the liver, contains a variety of enzymes that are involved in the metabolism of the myriad chemicals that comprise today's pharmaceuticals. By far the most important class of metabolic enzymes is the cytochromes P450, which are directly involved in the initial (or “first-pass”) clearance of drugs from the body (8, 9). During this process, drug metabolites are generated, some of which are biologically active in their own right and exert the desired pharmacological effect. For example, conversion of the antihistamine loratadine to descarboethoxyloratadine by CYP2D6 and CYP3A4 is required for biological activity (10). Often, however, drug metabolism can lead to undesirable biological consequences (11). A well known example of a toxic metabolic response is the P450-catalyzed oxidation of the common analgesic acetaminophen to N-acetyl-p-benzoquinone imine (12), which is hepatotoxic and is a major cause of liver failure (13).
A number of cell- and tissue-based in vitro systems have been developed in attempts to mimic human metabolism (14), including isolated liver slices (15, 16), primary hepatocytes (17), and transformed cultured human hepatoma cell lines (e.g., HepG2, Hep 3B, and BC2, among others) (18–21). To address the need for high-throughput, various cell culture techniques have been developed at the microscale, including microfabricated arrays for perfused 3D liver culture (22) and multiwell-plate cell cultures (23). However, cell- and tissue-based systems suffer from several key drawbacks. For example, liver slices are difficult to obtain in consistent quantities and qualities, and they deteriorate rapidly (15, 16); cell lines exhibit variable metabolic activity upon passaging (20) or have a limited lifespan and must be freshly derived (17). Furthermore, both cultured hepatocytes and hepatoma cell lines have low levels of P450 isoforms in the absence of specific inducers (24, 25). Although hepatocytes are useful for approximating liver toxicity, human toxicity can be due to effects of the metabolites on other organs.
In contrast with hepatocytes, a direct mimic of first-pass human metabolism can be achieved by using microsomal or recombinant P450 preparations (26–29). Isolated enzymes, however, must be linked to cell-based screening to assess the toxicity of the generated metabolites. To our knowledge, there is no report of a system or device that enables P450-catalyzed metabolism of drug candidates to be integrated with cell-based screening in high-throughput fashion.
To address this need, we have developed a metabolizing enzyme toxicology assay chip (MetaChip) that integrates the high-throughput, metabolite-generating capability of P450 catalysis with human cell-based screening on a microarray platform. Unlike previous cell-based microarrays (30), the MetaChip concept is based on the combination of a biocatalytic event (the in situ generation of a drug candidate metabolite or metabolites) with the cell-based screening of the metabolite(s). As a result, P450-generated drug candidate metabolites can be generated and screened against human cell lines on a single microscale platform, which remains useful even if the metabolites are unstable. To demonstrate this concept, we show that several P450 isozymes coupled with an MCF7 breast cancer cell line accurately mimic the prodrug activation of the anticancer therapeutics cyclophosphamide (CP) (Cytoxan) and Tegafur and the generation of cytotoxic metabolites from the simple analgesic acetaminophen. The MetaChip, therefore, provides a high-throughput microscale alternative to currently used in vitro methods of human metabolism and creates opportunities for rapid and inexpensive assessment of the biological activity of P450-generated metabolites at very early phases of drug development.
Methods
Preparation of the MCF7 Cancer Cell Monolayer. MCF7 human breast cancer cells (from the American Type Culture Collection) were grown in DMEM (Sigma) supplemented with 5% heat-inactivated FBS (GIBCO) in T-150 cell-culture flasks in a humidified 5% CO2 incubator (ThermoForma Electron, Marietta, OH) at 37°C. The cell monolayer was prepared by trypsinizing a confluent layer of cells with 4 ml of 0.05% trypsin/0.53 mM EDTA (GIBCO) from the culture flask and resuspending the cells in 20 ml of FBS-supplemented DMEM. After centrifugation at 450 × g for 8 min at 4°C, the supernatant was removed and the cells were resuspended with ≈40 ml of FBS-supplemented DMEM. The cell suspension (3 ml containing ≈4 × 105 cells per ml) was then transferred to a 2.6 × 7.5 cm2 chamber slide (Fisher Scientific), and the slide was incubated for 1 day in the CO2 incubator.
Sol-Gel Encapsulation of Cytochromes P450. Sol solution was prepared by mixing 250 μl of methyltrimethoxysilane (MTMOS) (Aldrich) with 100 μl of HCl (5 mM), followed by sonication for 10 min. The MTMOS/HCl sol solution (40 μl) was mixed with 60 μl of CYP3A4 baculosomes (1.1 nmol of P450 per ml, Invitrogen) and 10 μl of regeneration system (333 mM glucose-6-phosphate/40 units/ml glucose-6-phosphate dehydrogenase in 100 mM potassium phosphate buffer, pH 8). To prevent detachment of sol-gel spots from the glass slide and to ensure hemispherical spots, MTMOS sol solution (2 ml, pH 7) was spin-coated (at 50 × g for 30 s) onto the slide. The MTMOS sol solution containing CYP3A4 was then spotted onto the MTMOS-coated glass slide by using a MicroSys 5100–4SQ microarrayer (Cartesian Technologies, Irvine, CA) and allowed to gel for 24 h at 4°C. A similar method was used to encapsulate other P450 isozymes in the sol-gels. P450 reactions were performed in 525-spot arrays consisting of 15 × 35 spots (30 nl each) by dispensing 60 nl of substrate solution (see below) on top of each sol-gel spot with the microarrayer.
P450 Metabolism with CP in 96-Well Plate Format. Sol-gel reactions were performed in 50 μl of phosphate buffer solution (pH 8) containing 2 mM CP, 1 mM NADP+, and 5 μl of regeneration system added to 10 μl of sol-gel (containing 55 nM P450). Solution-phase reactions were performed as a control in 50 μlof phosphate buffer solution (pH 8) containing 2 mM CP, 110 nM P450, 1 mM NADP+, and 5 μl of regeneration system. The reactions were performed in duplicate in a 96-well plate with orbital shaking at 5 × g at room temperature. Periodically, 50-μl aliquots were taken, mixed sequentially with 20 μl of ZnSO4, 20 μl of Ba(OH)2, and 10 μl of 0.01 M HCl, and centrifuged at 14,000 × g for 20 min to remove the protein. To assay for acrolein, 40 μl of a mixture (6 mg/ml 3-aminophenol/6 mg/ml hydroxylamine hydrochloride freshly dissolved in 1 M HCl) was added to 80 μl of supernatant, and then the samples were heated at 90°C for 30 min to form 7-hydroxyquinoline. The derivatized samples were injected directly onto a C8 column (Waters), and the column was eluted isocratically with 10% acetonitrile and 90% water with 0.5% H3PO4 at a flow rate of 0.5 ml/min by HPLC (Shimadzu LC-10AT). The product eluted at 5.8 min and was monitored at UV 254 nm.
P450 Metabolism with Prodrugs. Metabolites of CP, Tegafur, and acetaminophen were generated on the P450 sol-gels by first preparing a prodrug (or protoxicant) substrate solution that contained 5 μl of NADP+ (10 mM), 5 μl of regeneration system, 25 μl of prodrug (4 mM), and 15 μl of DMEM. Metabolites of prodrugs were generated by spotting 60 nl of prodrug solution onto each 30-nl P450 sol-gel spot. The chamber slide with the MCF7 cell monolayer was immediately manually stamped onto the slide containing the P450 sol-gels and prodrug solutions. For efficient stamping and transfer of the P450 reaction product(s) onto the cell monolayer, the chamber slides contained a 250-μm-high silicone gasket to maintain a suitable distance between the two slides and to minimize drying of the cell monolayer during incubation. During short incubations of the P450 sol-gel spots with the cell monolayer (e.g., up to 6 h), the cell monolayer remains hydrated because of the airtight closure of the P450 slide with the cell monolayer slide. In addition, the liquid on the sol-gel adds hydration to the cell monolayer in regions contacted by the sol-gel during the stamping process. After incubation for 6 h at 37°C, the cell monolayer slide was lifted off the sol-gel slide, washed twice with 1 ml of sterile PBS (GIBCO), transferred to a Petri dish containing 25 ml of DMEM supplemented with 5% FBS, and cultured for 18 h in a CO2 incubator at 37°C. As a control, a solution phase reaction was performed by mixing 5 μl of P450 with 5 μl of NADP+, 5 μl of regeneration system, 10 μl of DMEM, and 25 μl of prodrug (4 mM), spotting 60 nl of this solution onto the MTMOS-coated slide (without sol-gel spots), and finally stamping the MCF7 cell monolayer slide onto the solution-spotted, MTMOS-coated slide. The hydrophobic coating enabled the solution to take on a hemispherical shape.
Cell Staining and Analysis. The cytotoxicity of P450 metabolites was determined by cell staining. A live/dead test kit (Molecular Probes) was used to produce a green fluorescent response from living cells and a red fluorescent signal from dead cells. To that end, 10 μl of ethidium homodimer-1 (2 mM) and 5 μl of calcein AM (4 mM) were added to 2 ml of PBS, and 500 μl of this mixture was applied to each slide containing the cell monolayer. After incubation for 30 min at room temperature, the location of each spot where a P450-catalyzed reaction occurred was detected with an epifluorescent microscope (Micro Video Instruments, Avon, MA) equipped with an FITC-Texas red double filter. In addition, the entire monolayer on the slide was imaged at 532 nm (green) and 635 nm (red) by using a GenePix 4000B microarray scanner (Axon Instruments, Union City, CA). The red fluorescence intensity is linearly proportional to the total number of dead cells and was quantified from the microscopic images with photoshop (Adobe Systems, San Jose, CA) (using the histogram function) and from the array scanner with genepix pro (Axon). Background red fluorescence of fully viable cells (FMin) was subtracted from all fluorescence values in the incubation spots (FReaction), allowing calculation of the percentage of dead cells through use of Eq. 1:
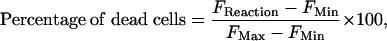 |
[1] |
where FReaction is the red fluorescence intensity of the reaction spot, FMax is the red fluorescence intensity of 100% dead cells (after treatment with 70% methanol for 1 h), and FMin is the red fluorescence intensity of untreated fully viable cells.
Results and Discussion
The MetaChip design involves two key components, as depicted in the schematic of Fig. 1. One component is a sol-gel microarray that contains one or more human P450 isoforms used to generate biologically active metabolites of a lead compound (e.g., a drug, prodrug, or drug candidate). With a standard microarrayer, the volume of the sol-gel spots can be varied from 5 to 100 nl, and the spots can be arrayed in any number of spatially addressable arrangements. The second component is a human cell monolayer housed in a chamber slide, which is used for cytotoxicity screening of the P450-generated metabolite. A solution of lead compound is applied to the sol-gel spots by using the microarrayer, followed by stamping of the cell monolayer onto the sol-gel array. After incubation for sufficient time to allow the synthesis of P450-generated metabolites, the cell monolayer slide is removed, and the cells are stained to determine the percentage of dead cells by using a microarray scanner.
Fig. 1.

Schematic of MetaChip platform and microscopic photographs of sol-gel spots. Shown are 30-nl P450 sol-gel spots (A), 30-nl sol-gel spots with 60 nl of prodrug solution after being stamped by MCF7 cell monolayer (B), and MCF7 cell monolayer after removal from sol-gel array and staining (C).
Sol-Gel Encapsulation of Human Cytochromes P450. The first step toward construction of a viable MetaChip is the optimization of P450 catalysis in sol-gel arrays. Sol-gels offer important advantages for the fabrication of protein-based materials and biocatalytic devices because of their optical transparency, compatibility with various organic moieties, and stability in harsh environments (31–33). Furthermore, the porosity of sol-gels can be precisely controlled, as can the robustness of the sol-gel structure. For the prototype MetaChip, human cytochrome P450 isoform 3A4 (CYP3A4) was encapsulated into 30 nl of MTMOS sol-gels and spotted onto MTMOS-coated microscope slides (0.5-μm-thick coating, as determined by profilometry). The coating ensured that stable sol-gel spots were generated; the spot diameter was 0.6 mm (close to the expected size for hemispherical spots), and the center-to-center distance was 1.2 mm (Fig. 1 A). The catalytic activity (Vmax/Km) of the enzyme was then determined for the oxidation of the fluorogenic substrate dibenzoxymethylfluorescein in aqueous buffer. Under these conditions, (Vmax/Km) for the soluble enzyme was 1.9 ± 0.2 (× 105) M–1·min–1; (Vmax/Km) for the sol-gel-encapsulated enzyme was 6.9 ± 0.3 (× 104) M–1·min–1. Hence, the sol-gel-encapsulated CYP3A4 was ca. one-third as active as its dissolved counterpart, which is a sufficient level of reactivity for MetaChip operation.
P450-Catalyzed Metabolism of CP. To extend these favorable results to a pharmacologically important prodrug, we selected CP as a model nontoxic compound. CP is metabolized to the cytotoxic 4-hydroxycyclophosphamide (4-OH-CP) by both CYP3A4 and CYP2B6 isoforms (34, 35). The metabolism of CP is complex and involves a series of spontaneous and rapid reactions that occur after the initial hydroxylation to 4-OH-CP, including the formation of acrolein and phosphoramide mustard, which is generally believed to be the active cytotoxic metabolite (Eq. 2) (35, 36). Acrolein can be assayed by HPLC after its condensation with 3-aminophenol to give the stable 7-hydroxyquinoline (37). The oxidation of CP to 4-OH-CP was performed in 96-well plates with a total reaction volume of 50 μl per well; the larger reaction size was necessary to follow 7-hydroxyquinoline synthesis by means of HPLC (35).
Figure 3.

CYP3A4 and CYP2B6 catalyzed the oxidation of CP to 4-OH-CP in solution with initial rates of 190 ± 10 and 132 ± 22 nmol/mg of enzyme per min, respectively. The enzymatic reactions ceased after ≈4 h (Fig. 2). Addition of fresh CYP3A4 isoform after 6 h resulted in further generation of 4-OH-CP, indicating that cessation of the oxidation reactions was caused by enzyme deactivation, perhaps due to the deactivating effect of the highly reactive acrolein, which is known to react with free amino groups on proteins (38). Similar reaction profiles were observed for the sol-gel-encapsulated P450 isoforms (Fig. 2), with initial rates of 79.5 ± 5.6 and 60.7 ± 3.4 nmol/mg of enzyme per min for CYP3A4 and CYP2B6, respectively. Hence, the initial rates of sol-gel reactions were ≈45% of those in the solution phase, which is consistent with the relative reactivities observed with dibenzoxymethylfluorescein as a model substrate. Therefore, the sol-gel does not appear to alter the substrate specificity of the P450 isoforms. The conversion of CP to 4-OH-CP by the sol-gel-encapsulated enzyme after 6 h, as reflected by the formation of acrolein, was ≈50% of the solution-phase reaction for CYP3A4 and 70% of the solution-phase reaction for CYP2B6. These results indicate that sol-gel-encapsulated P450 isoforms are effective in catalyzing prodrug activation.
Fig. 2.

Reactivity of CYP3A4 (circles) and CYP2B6 (triangles) in solution (filled symbols) and in sol-gels (open symbols) on CP. Fresh CYP3A4 was added at 6 h, as indicated by the arrow and the dots inside the symbols.
Development and Operation of the MetaChip. To validate the MetaChip concept, a 15 × 35 spot sol-gel array containing CYP3A4 was prepared on a glass microscope slide precoated with MTMOS. A monolayer of MCF-7 breast cancer cells, which are known to respond to the activation of the CP prodrug (36, 39), was used as the screening target. A 60-nl solution of CP (2 mM) in cell culture medium (DMEM) was spotted onto each 30-nl sol-gel spot of the array, followed by stamping of the cell monolayer onto the sol-gel array (Fig. 1B). After incubation for 6 h at 37°C, the cell layer was removed and stained and then scanned by using epifluorescence microscopy and laser array scanning (Figs. 1C and 3, respectively). A control incubation containing all system components except the P450 produced a uniform lawn of green cells (Fig. 3A), indicating fully viable MCF7 cells. By comparison, the complete reaction system in the sol-gels resulted in vivid cytotoxicity (as shown by distinct spots of either red or light yellowish-green color on the stained array) in the region of cell monolayer contact with the sol-gel spots (Fig. 3B). The intensity of the red color was proportional to the fraction of dead cells in the cell monolayer. There was no apparent spot-to-spot contamination; each spot was distinct and separated by viable cells. This observation is consistent with simple diffusion-length calculations, which indicate that CP would diffuse <0.5 mm in 6 h, assuming an effective diffusivity of CP through the cell monolayer of 10–7 cm2·s–1. Moreover, the stamping technique enabled the relatively unstable 4-OH-CP generated by the P450-containing sol-gel spots to diffuse into the cell monolayer. Separate experiments in which the 4-OH-CP was generated from the breakdown of 4-hydroperoxycyclophosphamide [synthesized chemically according to Struck et al. (40)] had verified that 4-OH-CP is indeed toxic to MCF7 cells (data not shown).
Fig. 3.

Microarray scanning pictures of the MCF7 cell monolayer at 635 nm (Left) and 532 nm (Right) for CP activation. Shown are control with sol-gel spots without a P450 isoform (A) and full system with CYP3A4 (B).
Using the microarray scanner, we separately determined the percentages of live and dead cells in the MetaChip array. Sol-gel spots containing CYP3A4 and CYP2B6 with 2 mM CP yielded 99% and 97% cytotoxicity, respectively, in 6-h incubations with MCF7 cells (Fig. 4A). In addition, minimal cytotoxicity (<13% cell death) was observed in control sol-gel spots (without P450) in the presence of CP. Additional control experiments were performed with sol-gel spots containing P450 with DMEM (without CP), which also showed minimal cytotoxicity (<10% cell death; data not shown). This background cytotoxicity may be the result of the sol-gel itself on the MCF7 monolayer.
Fig. 4.

Comparison of cytotoxicity results for the MetaChip and solution-phase reactions. (A) Cytotoxicity of P450-activated CP (as represented by the percentage of dead cells) for solution and sol-gel incubations. Control incubations consisted of all system components, except for a P450 isoform. (B) Effect of CP concentration on the cytotoxicity of MCF7 breast cancer cells for: 3A4 solution (•), 3A4 sol-gel (○), 2B6 solution (▾), and 2B6 sol-gel (▿). (C) Effect of Tegafur concentration on the cytotoxicity of MCF7 breast cancer cells for: 1A2 solution (•), 1A2 sol-gel (○), 3A4 solution (▾), and 3A4 sol-gel (▿). (D) Effect of acetaminophen concentration on the cytotoxicity of MCF7 breast cancer cells for: 3A4 solution (•), 3A4 sol-gel (○), 2B6 solution (▾), and 2B6 sol-gel (▿). In B–D, images from the array scanner are presented. In each 6 × 6 array segment, the columns represent different concentrations of spotted compounds (from left to right: 10, 100, 200, 500, 1,000, and 2,000 μM), and the rows represent replicates.
The sensitivity of the MetaChip was evaluated by varying the CP concentration. To that end, 60 nl of CP solutions (0–2 mM) were dispensed onto the 30-nl sol-gel spots of the 15 × 35 array, and the slide was immediately stamped with the cell monolayer slide and incubated for 6 h at 37°C. Both CYP3A4 and CYP2B6 were used, and a comparison was made to solution-phase CP oxidation by spotting 60-nl solutions of both P450 isozymes directly onto an MTMOS-coated slide (without sol-gel spots) and stamping the slide with the cell monolayer slide. As shown in Fig. 4B, the sol-gel and solution-phase systems yielded similar cytotoxicity profiles as a function of CP concentration for both CYP3A4 and CYP2B6. Based on cytotoxicity staining, the LD50 of CP, calculated as an average of the CYP3A4 and CYP2B6 isoforms, was 174 ± 21 μM for the sol-gel-encapsulated P450s, compared with 240 ± 36 μM for solution-phase spots. In addition to solution-phase spots, 96-well plate solution-phase assays were used to measure the LD50 of CP. The values for CYP3A4 and CYP2B6 were 214 ± 19 and 257 ± 13 μM, respectively, which are intermediate to those obtained with the sol-gel and solution arrays. Hence, the MetaChip produced a cytotoxic response profile close to that of more conventional solution-phase P450 incubations.
Demonstration of the MetaChip concept was extended beyond CP to include 5-fluoro-1-(tetrahydro-2-furfuryl)-uracil (Tegafur), which yields the potent anticancer compound 5-fluorouracil upon P450-catalyzed oxidation (41, 42), and the common analgesic acetaminophen, which is oxidized to the cytotoxic N-acetyl-p-benzoquinone-imine by P450 catalysis (43, 44). In these cases, CYP1A2 and CYP3A4 were used in 30-nl sol-gel spots with 60 nl of substrate solution (0–2 mM). As with CP, the MCF7 cell monolayer slide was stamped onto the sol-gel spots and kept in place for 6 h. The sol-gel spots containing either of the two P450 isozymes yielded measurable cytotoxicity with 2 mM Tegafur or acetaminophen (Fig. 4 C and D). As with CP, the P450-containing sol-gel spots showed comparable cytotoxicity to those with solution-phase P450 reactions.
In conclusion, we have developed an in vitro, high-throughput tool that is ideal for the rapid detection of P450-generated cytotoxic metabolites. The MetaChip offers several clear advantages over more conventional toxicology screens that use multiwell plates, including highly active and stable human P450s scaled down to 30-nl reaction volumes and the in situ coupling of metabolite synthesis and cytotoxicity screening, which allows rapid cytotoxicity testing of individual- or multiple P450-generated metabolites, similar to what occurs in the human liver. These advantages are important for the acceleration of human toxicology assays, whereby an increasingly large number of drug candidates generated through combinatorial chemistry must be rapidly screened for both bioactivity and toxicity. Furthermore, we envision that the MetaChip concept can ultimately comprise the entire human P450 inventory and be used in conjunction with variable multiisoform mixtures to provide predictive metabolic profiles and cell-specific toxicity data for different populations of individuals. Such a capability is critical for the design of patient-specific treatment regimens in the emerging practice of personalized medicine, as well as for the identification of pharmacologically safe and effective lead compounds for advancement to clinical trials. Finally, the MetaChip concept can be extended to include P450-catalyzed drug activation for enhanced biological activity, rather than toxicity, as well as cell-based screening in which pharmacological activity is linked to growth inhibition rather than cell death.
Acknowledgments
We are grateful to Isabelle Chevralot, Adam Meadows, and Philippa Reader for helpful suggestions and assistance with the cell culture experiments. This work was supported by National Institutes of Health Grant ES-012619 and the National Science Foundation.
Author contributions: J.S.D. and D.S.C. designed research; M.-Y.L. and C.B.P. performed research; and M.-Y.L., J.S.D., and D.S.C. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CP, cyclophosphamide; MTMOS, methyltrimethoxysilane; 4-OH-CP, 4-hydroxycyclophosphamide.
References
- 1.Geysen, H. M., Schoenen, F., Wagner, D. & Wagner, R. (2003) Nat. Rev. Drug Discov. 2, 222–230. [DOI] [PubMed] [Google Scholar]
- 2.Myers, P. L. (1997) Curr. Opin. Biotechnol. 2, 701–707. [DOI] [PubMed] [Google Scholar]
- 3.Bleicher, K. H., Bohm, H. J., Muller, K. & Alanine, A.I. (2003) Nat. Rev. Drug Discov. 2, 369–378. [DOI] [PubMed] [Google Scholar]
- 4.Petricoin, E. F., Zoon, K. C., Kohn, E. C., Barrett, J. C. & Liotta, L. A. (2002) Nat. Rev. Drug Discov. 1, 683–695. [DOI] [PubMed] [Google Scholar]
- 5.Gombar, V. K., Silver, L. S. & Zhao, Z. (2003) Top. Med. Chem. 3, 1205–1225. [DOI] [PubMed] [Google Scholar]
- 6.Li, A. P. (2001) Drug Discov. Today 6, 357–366. [DOI] [PubMed] [Google Scholar]
- 7.MacCoss, M. & Ballie, T. A. (2004) Science 303, 1810–1813. [DOI] [PubMed] [Google Scholar]
- 8.Guengerich, F. P. (2001) Chem. Res. Toxicol. 14, 611–650. [DOI] [PubMed] [Google Scholar]
- 9.Lewis, D. F. (2004) Pharmacogenomics 5, 305–318. [DOI] [PubMed] [Google Scholar]
- 10.Yumibe, N., Huie, K., Chen, K. J., Snow, M., Clement, R. P. & Cayen, M. N. (1996) Biochem. Pharmacol. 51, 165–172. [DOI] [PubMed] [Google Scholar]
- 11.Villeneuve, J. P. & Pichette, V. (2004) Curr. Drug Metab. 5, 273–282. [DOI] [PubMed] [Google Scholar]
- 12.Nelson, S. D. (1995) Drug Metab. Rev. 27, 147–177. [DOI] [PubMed] [Google Scholar]
- 13.James, L. P., Mayeux, P. R. & Hinson, J. A. (2003) Drug Metab. Dispos. 31, 1499–1506. [DOI] [PubMed] [Google Scholar]
- 14.Brandon, E. F. A., Raap, C. D., Meijerman, I., Beijnen, J. H. & Schellens, J. H. M. (2003) Toxicol. Appl. Pharmacol. 189, 233–246. [DOI] [PubMed] [Google Scholar]
- 15.Onderwater, R. C., Commandeur, J. N. & Vermeulen, N. P. (2004) Toxicology 197, 81–91. [DOI] [PubMed] [Google Scholar]
- 16.Martignoni, M., Monshouwer, M., de Kanter, R., Pezzetta, D., Moscone, A. & Grossi, P. (2004) Toxicol. In Vitro 18, 121–128. [DOI] [PubMed] [Google Scholar]
- 17.McGinnity, D. F., Soars, M. G., Urbanowicz, R. & Riley, R. J. (2004) Drug Metab. Dispos. 32, 1247–1253. [DOI] [PubMed] [Google Scholar]
- 18.Hewitt, N. J. & Hewitt, P. (2004) Xenobiotica 34, 243–256. [DOI] [PubMed] [Google Scholar]
- 19.Urani, C., Doldi, M., Crippa, S. & Camatini, M. (1998) Chemosphere 37, 2785–2795. [DOI] [PubMed] [Google Scholar]
- 20.Peng, F. C., Chaing, H. H., Tang, S. H., Chen, P. C. & Lu, S. C. (2004) J. Toxicol. Environ. Health A 67, 109–124. [DOI] [PubMed] [Google Scholar]
- 21.Fabre, N., Arrivet, E., Trancard, J., Bichet, N., Roome, N. O., Prenez, A. & Vericat, J. A. (2003) Cell Biol. Toxicol. 19, 71–82. [DOI] [PubMed] [Google Scholar]
- 22.Powers, M. J., Domansky, K., Kaazemour-Mofrad, M. R., Kalezi, A., Capitano, A., Upadhyaya, A., Kurzawski, P., Wack, K. E., Stolz, D. B., Kamm, R. & Griffith, L. G. (2002) Biotechnol. Bioeng. 78, 257–269. [DOI] [PubMed] [Google Scholar]
- 23.Boisclair, Y. R., Wang, J., Shi, J., Hurst, K. R. & Ooi, G. T. (2000) J. Biol. Chem. 275, 3841–3847. [DOI] [PubMed] [Google Scholar]
- 24.Madan, A., Dehaan, R., Mudra, D., Carroll, K., Lecluyse, E. & Parkinson, A. (1999) Drug Metab. Dispos. 27, 327–335. [PubMed] [Google Scholar]
- 25.Fardel, O., Morel, F., Ratanasavanh, D., Fautrel, A., Beaune, P. & Guillouzo, A. (1992) Cell. Mol. Aspects Cirrhosis 216, 327–330. [Google Scholar]
- 26.Samuel, K., Yin, W., Stearns, R. A., Tang, Y. S., Chaudhary, A. G., Jewell, J. P., Lanza, T., Lin, L. S., Hagmann, W. K., Evans, D. C. & Kumar, S. (2003) J. Mass Spectrom. 38, 211–221. [DOI] [PubMed] [Google Scholar]
- 27.Smith, K. S., Smith, P. L., Heady, T. N., Trugman, J. M., Harman, W. D. & Macdonald, T. L. (2003) Chem. Res. Toxicol. 16, 123–128. [DOI] [PubMed] [Google Scholar]
- 28.Ong, C. E., Coulter, S., Birkett, D. J., Bhasker, C. R. & Miners, J. O. (2000) J. Clin. Pharmacol. 50, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gut, I., Danielova, V., Holubova, J., Soucek, P. & Kluckova, H. (2000) Arch. Toxicol. 74, 437–446. [DOI] [PubMed] [Google Scholar]
- 30.Ziauddin, J. & Sabatini, D. M. (2001) Nature 411, 107–110. [DOI] [PubMed] [Google Scholar]
- 31.Gill, I. & Ballesteros, A. (2000) Trends Biotechnol. 18, 282–296. [DOI] [PubMed] [Google Scholar]
- 32.Gill, I. (2001) Chem. Mater. 13, 3404–3421. [Google Scholar]
- 33.Rupcich, N., Goldstein, A. & Brennan, J. D. (2003) Chem. Mater. 15, 1803–1811. [Google Scholar]
- 34.Anderson, D., Bishop, J. B., Garner, R. C., Ostrosky-Wegman, P. & Selby, P. B. (1995) Mutation Res. 330, 115–181. [DOI] [PubMed] [Google Scholar]
- 35.Huang, Z., Roy, P. & Waxman, D. J. (2000) Biochem. Pharmacol. 59, 961–972. [DOI] [PubMed] [Google Scholar]
- 36.Simoes-Wuest, A. P., Schuerpf, T., Hall, J., Stahel, R. A. & Zangemeister-Wittke, U. (2002) Breast Cancer Res. Treat. 76, 157–166. [DOI] [PubMed] [Google Scholar]
- 37.Bohnenstengel, F., Eichelbaum, M., Golbs, E. & Kroemer, H. K. (1997) J. Chromatogr. B Biomed. Sci. Appl. 692, 163–168. [DOI] [PubMed] [Google Scholar]
- 38.Uchida, K., Kanematsu, M., Sakai, K., Matsuda, T., Hattori, N., Mizuno, Y., Suzuki, D., Miyata, T., Noguchi, N., Niki, E. & Osawa, T. (1998) Proc. Natl. Acad. Sci. USA 95, 4882–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morrow, C. S., Smitherman, P. K. & Townsend, A. J. (1998) Biochem. Pharmacol. 56, 1013–1022. [DOI] [PubMed] [Google Scholar]
- 40.Struck, R. F., Thorpe, M. C., Coburn, W. C. & Laster, W. R. (1974) J. Am. Chem. Soc. 96, 313–315. [DOI] [PubMed] [Google Scholar]
- 41.Komatsu, T., Yamazaki, H., Shimada, N., Nakajima, M. & Yokoi, T. (2000) Drug Metab. Dispos. 28, 1457–1463. [PubMed] [Google Scholar]
- 42.Li, D., Jang, S. H., Kim, J. H., Wientjes, M. G. & Au, J. L. S. (2003) Pharm. Res. 20, 45–50. [DOI] [PubMed] [Google Scholar]
- 43.Patten, C. J., Thomas, P. E., Guy, R. L., Lee, M. J., Gonzalez, F. J., Guengerich, F. P. & Yang, C. S. (1993) Chem. Res. Toxicol. 6, 511–518. [DOI] [PubMed] [Google Scholar]
- 44.Hazai, E., Vereczkey, L. & Monostory, K. (2002) Biochem. Biophys. Res. Commun. 291, 1089–1094. [DOI] [PubMed] [Google Scholar]