

Abstract
The genetic code is fixed in aminoacylation reactions catalyzed by aminoacyl-tRNA synthetases. Amino acid discrimination occurs at two sites: one for amino acid activation and aminoacylation and one for editing misactivated amino acids. Although the active site sieves out bulkier amino acids, misactivation occurs with substrates whose side chains are smaller than the cognate one. Paradoxically, although alanyl-tRNA synthetase activates glycine as well as alanine, the sterically larger (than alanine) serine is also misactivated. Here, we report crystal structures of an active fragment of Aquifex aeolicus alanyl-tRNA synthetase complexed, separately, with Mg2+–ATP, alanine, glycine, and serine. Ala and Gly are bound in similar orientations in a side-chain-accommodating pocket, where α-amino and carboxyl groups are stabilized by salt bridges, and the carboxyl by an H-bond from the side chain NH2 of Asn-194. In contrast, whereas the same two salt bridges stabilize bound Ser, H-bonding of the highly conserved (among class II tRNA synthetases) Asn-194 side chain NH2 to the Ser OH, instead of to the carboxyl, forces pocket expansion. Significantly, in the Mg2+–ATP complex, Asn-194 coordinates a Mg2+-α-phosphate bridge. Thus, the sieve for Ser exclusion is broken because of selective pressure to retain Asn-194 for Mg2+–ATP and Ala binding.
Keywords: alanyl-tRNA synthetase, amino acid discrimination, editing, genetic code accuracy, ATP binding
Aminoacyl-tRNA synthetases (AARSs) establish the genetic code by attaching specific amino acids to the 3′ ends of tRNAs bearing the anticodons cognate for those amino acids (1–3). These modular enzymes fall into two classes (I and II), based on the architectures of their catalytic domains (4). For both classes, aminoacylation proceeds in two steps catalyzed by one active site. A bound amino acid such as Ala is activated with ATP to form the enzyme-bound aminoacyl adenylate intermediate (Ala-AMP), releasing inorganic pyrophosphate (step 1). The aminoacyl moiety of the adenylate is then transferred to the 2′- or 3′-OH of the ribose of A76, the universally conserved 3′ nucleotide of tRNA, with the release of AMP (step 2). Misactivation of amino acids occurs occasionally when a wrong amino acid fits into the active site, usually because it is smaller than the cognate one. Here, we describe structural features in a specific AARS (complexed with three different amino acids) that explain a longstanding paradox about activation of a larger amino acid.
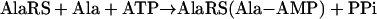 |
[1] |
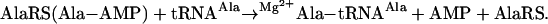 |
[2] |
Those synthetases that fail to reject noncognate amino acids with an accuracy of <1:3,000 (5) possess editing domains that clear the misactivated adenylate (pretransfer editing) or misacylated tRNA (posttransfer editing). Alanyl-tRNA synthetase (AlaRS) misactivates glycine and serine. Whereas misactivation of Gly is unsurprising, how Ser fits into the active site pocket for Ala is not obvious. Still, the enzyme clears misactivated Ser by hydrolytic editing. Prokaryotic AlaRSs are constituted of four modules: the class-defining N-terminal synthetic domain [which binds ATP, amino acid, and tRNA acceptor end with the active site for aminoacyl adenylate synthesis (step 1) and aminoacylation (step 2) (4, 6, 7)], a tRNA recognition module (8, 9), an editing domain (10), and a C-terminal oligomerization domain (6, 9). Although it lacks the editing and oligomerization domains, a 453-aa N-terminal fragment of Aquifex aeolicus AlaRS (AlaRS453) (11) is fully active in adenylate synthesis and aminoacylation. [It is homologous (49% sequence identity) to the functionally well characterized Escherichia coli AlaRS fragment 461N (the N-terminal 461 aa), which is the minimal fragment with aminoacylation activity (6, 10).] The crystal structure of this fragment revealed a catalytic domain with the architecture characteristic of class II synthetases, followed by a helical domain critical for acceptor-stem recognition and a mixed α/β C-domain (Fig. 1).
Fig. 1.

Synthetic active site of AlaRS. A ribbon diagram of the crystal structure of A. aeolicus AlaRS453 shows the location of the synthetic active site. Motifs 1 (α1-β1), 2 (β2-β5), and 3 (β11-α7) are colored in pink, orange, and cyan, respectively. Secondary structure nomenclature is shown.
To understand the structural basis of amino acid specificity and the unusual misactivation of serine, we determined the cocrystal structures at high resolution of the A. aeolicus AlaRS453 catalytic fragment complexed with each of Ala, Gly, and Ser. The results support size exclusion as the general basis for discriminating against amino acids larger than Ala. However, the amino acid binding pocket expands in the case of Ser. Results from the other cocrystal structures suggest that Ser misactivation is an evolutionary consequence of the ATP and Ala binding functions of this enzyme.
Materials and Methods
Crystallization of A. aeolicus AlaRS453 with Three Amino Acids and ATP. The His-6-tagged, 53-kDa A. aeolicus AlaRS fragment AlaRS453 was overexpressed in E. coli and purified as described (11). Complexes with each of l-alanine, glycine, and l-serine were crystallized as done for the apo enzyme (11), with the addition of 5 mM amino acid to the crystallization buffer. The complex with ATP was crystallized similarly from a sample containing 2 mM ATP and 5 mM MgCl2.
X-Ray Data Collection, Structure Determination, and Refinement. Cryo x-ray diffraction data (100 K) were collected at the Stanford Synchrotron Research Laboratory (SSRL, Stanford, CA, beamline 11-1) and at the Advanced Light Source (ALS, Berkeley, CA, beamline 8.2.2). All data were processed with denzo and scaled by using scalepack in the hkl2000 package (12).
The structures of the cocrystals with Mg2+–ATP and Ala were determined by molecular replacement by using the standard protocols of cns (13), and with the apo enzyme's structure as a search model. For both complexes, the probability scores for the top two solutions of the rotation function were 175% higher than the next solution. These two solutions were applied in a two-step translation search–one molecule at a time in the asymmetric unit. The translation function solution (packing score 0.65) was rigid-body refined when the Rfree decreased from 36.5 to 33.7 for the Mg2+–ATP complex, and from 33.5 to 30.0 for the Ala complex. Noncrystallographic symmetry (NCS) restraints (13) were then applied to the backbone atoms of the central β strands of the catalytic domain (β2, R60-K66; β5, F90-S97; β9, S172-V177; β10, I192-Q199; β11, P214-L222) and to the tRNA recognition module (residues F242-R423). After one round of simulated annealing (2,000 K), the Rfree decreased further to 28.3 and 25.1 for the Mg2+–ATP and for the Ala complex, respectively. NCS-averaged 2Fo – Fc maps were used to built models for the ligands in o (14). The structures were further refined in cns in two rounds of simulated annealing (2,000 K, with bulk-solvent correction and no NCS restraints), B-factor refinement, and solvent fitting. To ensure correct elucidation of the metal binding sites, we collected data from a crystal of the ATP complex soaked for 4 h in mother liquor, 2 mM MnCl2, and no MgCl2. The crystal belonged to the P41212 space group and diffracted to 2.7 Å (data not shown).
The crystal structures of complexes with Gly and Ser were determined by direct difference Fourier calculation in which the 2.14-Å crystal structure of the apo enzyme [PDB ID code 1RIQ (11)] was used to calculate phases for the observed structure-factor amplitudes. After initial rigid-body refinement, difference Fourier maps showed clear density for the bound amino acids, which were modeled in o, and the structures were refined in cns as mentioned above (13). Simulated annealed Fo – Fc maps, calculated with the active-site region and ligand omitted from the model, were used to check the fitting of the ligand and water molecules in the electron density. All four structures were validated in procheck (15).
Results
Two Crystal Forms. For all of the small-ligand complexes of the N-terminal 453-aa fragment of A. aeolicus AlaRS (AlaRS453), two crystal forms (tetragonal and orthorhombic) were grown in the same crystallization drop. [Because the apo protein gave the same two crystal forms (11) (data not shown), the multiplicity of crystal forms does not result from ligand binding per se and is more likely a property of the protein itself.] The tetragonal crystals had one complex per asymmetric unit, whereas the orthorhombic crystals had two (Table 1). The two forms were visually indistinguishable and produced similar quality diffraction data. Therefore, crystals were randomly picked for data collection.
Table 1. Data collection and structure refinement statistics.
| Dataset | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Data collection | ||||
| Ligand | Mg2+-ATP | Alanine | Glycine | Serine |
| Space group | P212121 | P212121 | P41212 | P41212 |
| Unit cell, Å | 173.6, 73.9, 74.0 | 173.0, 73.9, 74.0 | 73.9, 172.9 | 74.1, 173.6 |
| Complexes per a.u. | 2 | 2 | 1 | 1 |
| Wavelength, Å | 0.97976 | 1.0332 | 0.82653 | 0.97976 |
| Resolution, Å | 50.0-2.15 | 50.0-2.08 | 50.0-2.23 | 50.0-2.48 |
| Unique reflections | 50,235 (5,064) | 56,944 (5,680) | 24,242 (2,365) | 17,803 (1,739) |
| Completeness, % | 94.8 (96.9) | 99.8 (99.9) | 99.7 (100.0) | 98.7 (99.8) |
| Redundancy | 4.7 (4.8) | 4.3 (3.4) | 5.4 (5.4) | 13.4 (14.0) |
| Rmerge,* % | 3.5 (25.0) | 8.0 (17.0) | 8.3 (44.4) | 10.9 (38.1) |
| 〈I/σ(I)〉 | 20.4 | 15.3 | 12.0 | 11.5 |
| Structure refinement† | ||||
| No. of reflections: work/free | 44,397/4,965 | 51,940/5,804 | 21,716/2,384 | 15,913/1,761 |
| No. of atoms: protein/water | 7,284/190 | 7,284/361 | 3,642/194 | 3,642/147 |
| Ligands/Mg | 62/6 | 12/0 | 5/0 | 7/0 |
| Rcryst‡/Rfree,§ % | 19.4/22.5 | 18.9/23.6 | 20.7/23.9 | 19.9/23.2 |
| rmsd bond lengths, Å/angles, ° | 0.01/1.056 | 0.01/1.092 | 0.0185/1.431 | 0.0185/1.257 |
| Ramachandran plot, % | ||||
| Favored | 80.0 | 89.2 | 88.7 | 79.1 |
| Allowed | 19.4 | 10.3 | 10.3 | 20.0 |
| Generous | 0.6 | 0.5 | 1.0 | 0.9 |
| Average B factor, Å2 | ||||
| Protein | 30.6/38.8¶ | 22.0/24.1 | 20.2 | 22.2 |
| Ligands | 33.9/38.9 | 19.0/22.8 | 24.4 | 26.9 |
| Ions | 35.4 | — | — | — |
| Water | 32.3 | 25.0 | 21.2 | 46.0 |
Highest-resolution shell information is shown in parentheses.
Rmerge = 100 × (Σh Σi| 〈I(h)〉 - I(h)i|)/Σh Σi I(h)i, where I(h)i is the ith observation of reflection h and 〈I(h)〉 is the mean intensity of all observations of reflection h.
All observed data were used for refinement.
Crystallographic R-factor = 100 × (Σh‖ Fobs(h) | - | Fcalc(h) ‖)/Σh | Fobs(h) |, where Fobs(h) and Fcalc(h) are the observed structure factor amplitude and the structure factor amplitude calculated from the model, respectively.
The free R-factor was calculated by using 90% of the data.
For the orthorhombic crystals, two values are shown for the two molecules in the asymmetric unit (a.u.).
Crystal structures for complexes with Gly and Ser were determined from tetragonal crystals, and structures for complexes with Mg2+–ATP and Ala were from orthorhombic crystals. Structures of all four complexes were similar to each other and to the apo structure of AlaRS453.
Amino Acid Binding Pocket. Amino acid specificity was investigated with crystal structures of AlaRS453 complexed with Ala (Table 1, dataset 2), Gly (dataset 3), and Ser (dataset 4). For each complex, initial difference Fourier electron density maps and simulated-annealing omit maps revealed bound amino acid in the active site, located in a cleft on one face of the central β-sheet and engulfed by the loop of the strand–loop–strand of motif 2 from the cleft's bottom and the loop connecting strands β8 and β9 from its top (Figs. 1 and 2A).
Fig. 2.

Amino acid binding pocket in AlaRS453. (A) Stereoviews of simulated annealed omit Fo – Fc electron density maps for bound Ala (Top, resolution 2.08 Å, contour level 2.4 σ), Gly (Middle, 2.23 Å, 2.1 σ), and Ser (Bottom, 2.48 Å, 1.6 σ) superposed on the refined models. The amino acid and other atoms within a sphere of 3.2 Å were omitted from the model during map calculation. The protein is shown as a ribbon with orientation and colors as in Fig. 1. The bound ligands are shown in stick representation. (B) The active site of AlaRS453 in complex with Ala (Top), Gly (Middle), and Ser (Bottom) from the respective crystal structures. Interactions with protein side chains are shown.
The three amino acids are similarly oriented in their respective binding pockets and make similar interactions with the protein by means of their α-amino and carboxyl groups (Fig. 2B). This orientation is achieved by three interactions with the protein: two salt-bridge interactions and one hydrogen bond. The electropositive α-amino group is stabilized by interaction with the invariant motif 3 Asp-217 side chain carboxyl. This interaction is conserved in class II AARSs, although the spatially conserved acidic side chain may come from a sequentially variable part of the polypeptide chain [e.g., Glu-83 in E. coli HisRS (16), Asp-342 in yeast AspRS (17), Glu-188 and -239 in Thermus thermophilus GlyRS (18), and Glu-240 and 276 in E. coli LysRS (19)]. Mutating the corresponding residue in E. coli AlaRS (Asp-235) to alanine resulted in a 7- and 200-fold lowering in rates of adenylate synthesis and aminoacylation, respectively, thus suggesting a role in the transfer of the aminoacyl group from the adenylate to the tRNA (20). Similarly, one of the carboxylate oxygen atoms of the amino acid substrate makes a salt bridge (water-mediated in the case of Gly) with the side chain of invariant Arg-69 of motif 2. This interaction is also conserved in class II AARSs, although in some cases motif 2 residues other than this universally conserved arginine are involved [e.g., Gln-127 in E. coli HisRS (16), Gln-303 in yeast AspRS (17), and His-204 in T. thermophilus SerRS (21)]. Additionally, this arginine plays a key role in stabilizing the α-phosphate of bound ATP in all class II AARSs, as well as in AlaRS (see below). The third interaction common to all three amino acid substrates is a water-mediated hydrogen bond between the α-amino group of bound amino acids and the NHε of Trp-161. Lastly, one more interaction is seen in the complexes with Ala and Gly, but not in the complex with Ser. That is a hydrogen bond donated by the δ-amine of Asn-194 to the other carboxylate oxygen atom of the bound amino acid (Fig. 2B Top and Middle). In the complex with Ser, this interaction is replaced by a hydrogen bond from an ordered water molecule (Fig. 2B Bottom).
The binding pocket for the Ala side chain is hydrophobic. Invariant Met-92 (from motif 2), highly conserved Val-196 (from strand β10) and Gly-219 (from motif 3) line the pocket from the bottom, top, and side, respectively, and make van der Waals contacts with the methyl group of Ala (Fig. 2B Top). Gly fits in this pocket and sits deeper in the pocket to make a contact with Met-92 by means of its Cα atom (Fig. 2B Middle). In the case of Ser, the pocket is expanded. Whereas the Cβ occupies the same position as that of Ala and makes similar contacts, the hydroxyl group H-bonds with the repositioned Asn-194 side chain carbonyl (Fig. 2B Bottom). This hydrogen bond forces pocket expansion and allows Ser to be accommodated. In all class II AARSs, a similarly positioned side chain (usually positively charged) makes a similar interaction with the backbone of the bound amino acid substrate [e.g., Asn-424 in E. coli LysRS (19)], or in close proximity if the amino alcohol analog of the substrate was bound instead [e.g., Arg-259 in E. coli HisRS (16)]. However, in those cases, no interaction was seen with the amino acid side chain.
Conformational Change Upon Amino Acid Binding. Ala-induced conformational changes are associated with interactions with the Ala backbone atoms. The side chain of Arg-69 moves a distance of 2.5 Å (distance averaged over all side-chain atoms) from its position in the apo structure to form the salt-bridge to the carboxyl group (Fig. 3A). Further, the lodging of the electropositive amino group of Ala causes conserved Met-43 to shift away by 1 Å to avoid an unfavorable interaction, while switching a van der Waals contact from Met-163 to Val-44 and Ala-41. None of these changes is caused by the binding of the Ala side chain methyl group, which fits in the pocket in a lock-and-key fashion.
Fig. 3.

Conformational changes in the synthetic active site upon amino acid substrate binding. (A) Alanine-induced conformational changes. (B) Serine-induced conformational changes. Cyan ribbon and side chains, apo AlaRS453 structure. Yellow ribbon and colored side chains, AlaRS453 structure with substrate.
Gly and Ser binding induces similar conformational changes as seen with Ala, but with an additional change seen only in the case of Ser. This change is in the position of Asn-194. In the apo structure, Asn-194 participates in a network of water-mediated hydrogen bonds by means of its Oδ atom with Ser-172 in the adjacent strand β9. In the Ala or Gly complexes, Asn-194 maintains the same orientation as in the apo structure, with its δ-NH2 additionally interacting with the bound substrate's carboxylate. These interactions are broken upon Ser binding, and the Asn-194 side chain δ-NH2 moves by about 1.0 Å toward the amino acid binding site to H-bond with the OH of bound Ser (Fig. 3B). Thus, binding of the noncognate amino acid Ser occurs through a partial induced-fit mechanism that pushes open the pocket for side chain accommodation.
Complex with ATP. The crystal structure of the Mg2+–ATP complex with AlaRS453 was determined by molecular replacement (Table 1, dataset 1). A difference Fourier Fo – Fc map, calculated with the model of the apo structure as a source of phase information, showed clear electron density for bound ATP and three magnesium ions in each of the two active sites in the asymmetric unit (Fig. 4A).
Fig. 4.

ATP binding by AlaRS. (A) Simulated annealed omit Fo – Fc electron density map (resolution, 2.15 Å; contour, 2.8 σ) for the active site region of AlaRS453/Mg2+–ATP complex, superimposed on the refined model. The model for ATP, magnesium ions, and surrounding atoms within a sphere of 3.2 Å was omitted from map calculation. (B) Similar view showing active site residues involved in ATP or magnesium binding. Model colors are as in Fig. 1. Bound magnesium ions and water molecules are shown as gray and red spheres, respectively. For clarity, some water molecules and interactions with the ribose are not shown. (C) Schematic of the interactions between enzyme, ATP, and magnesium. Residues from motifs 2 and 3 are shown in orange and cyan, respectively. Residues in black belong to strands in the central β-sheet of the active-site domain. Side chain conservation patterns among AlaRS sequences from 80 organisms are shown in brackets (percentage occurrence shown only for side chains present in >4% of the sequences). Side chains without adjacent bracketed numbers are invariant.
Because similarly located magnesium ions were seen in crystal structures of all other class II AARSs [except HisRS (16)], we assigned the density in the difference Fourier map to magnesium. An electron density map from a MnCl2-soaked crystal showed increased density for the electron-rich Mn2+ ions at the same locations seen in crystals containing Mg2+ (data not shown).
As observed with all class II AARSs, bound ATP adopts a bent conformation with the β- and γ-phosphates folded toward the adenine ring into a U-shaped structure. The hydrogen-bond network and electrostatic interactions between the enzyme, ATP, and three bound magnesium ions are also similar to those previously observed in class II enzymes (16–19, 22, 23). Interactions are with residues from motifs 2 and 3, as well as with conserved side chains from strands β9 and β10 of the central β-sheet (Fig. 4 B and C). The adenine ring is held in position by two H-bonds with Asp-76 and His-87 from the motif 2 loop, as well as by an iminoaromatic stacking between the ring of Phe-90 and the guanidino group of Arg-224, all invariant or highly conserved in AlaRS (Fig. 4C). The ribose moiety is in 3′-endo conformation and donates (by means of its 3′-hydroxyl) a hydrogen bond to the conserved Glu-191 side chain located in strand β10. The 2′- and 4′-hydroxyl groups hydrogen bond with protein backbone atoms.
The three phosphates of ATP are held in place through Mg2+ coordination and direct interactions with the protein. The α- and β-phosphates are bridged to the protein by the principal magnesium ion (Mg1), which in turn is fully coordinated in a pentagonal bipyramidal geometry with the phosphate oxygen atoms, the highly conserved side chains Glu-174, Glu-191, and Asn-194, and a water molecule. The β- and γ-phosphates are bridged on opposite sides by Mg2 and Mg3, which are coordinated by direct or water-mediated interactions with Glu-174, Glu-191, Asp-186 in the central β-sheet, and Asp-76 in the motif 2 loop. The α- and γ-phosphates are further stabilized by the universally conserved motif 2 Arg-69 and motif 3 Arg-224, respectively, as also seen in other class II AARSs. His-86 from the motif 2 loop provides an additional interaction with the γ-phosphate that is not observed in other class II enzymes.
Movement of the phosphate recognition groups of Arg-69 and His-86 toward their ligands by 1 and 2 Å, respectively, are the main ATP-induced changes (Fig. 5). Further, Glu-191 and Glu-174 move by 0.5 and 1.5 Å, respectively, from their positions in the magnesium-free apo structure (where they hydrogen bond to ordered water molecules) to form a cage around the magnesium ions. Finally, the side chain of Asp-76 moves 0.9 Å to hydrogen bond with the adenine base 6-amino group, replacing a bound water molecule in the apo-enzyme structure. These changes are consistent with the induced-fit mechanism of ATP binding, known for class II AARSs (18, 19, 22–25).
Fig. 5.

ATP-induced conformational changes in AlaRS453 active site. Cyan ribbon and side chains, apo AlaRS453; yellow ribbon and colored side chains, complex with Mg2+–ATP.
Discussion
Results from previous mutagenesis studies of the E. coli enzyme support the role of the motif 2 loop of AlaRS in amino acid activation. D76A and F90A (corresponding to Asp-76 and Phe-90 in A. aeolicus) mutants of E. coli AlaRS failed to complement an alaS-null strain due to a dramatic reduction in kcat for amino acid activation. The Km for alanine was minimally affected by the D76A or F90A substitutions (26). These results suggest that, although ATP interactions with Asp-76 and Phe-90 may be individually dispensable for ATP binding, they are required for stabilizing a transition state for adenylate formation and/or stabilization of the leaving Mg2+-pyrophosphate by Asp-76.
The patch of acidic residues involved in Mg2+-coordinated ATP binding is also highly conserved in sequences of AlaRS (Fig. 4C). All substitutions maintain side chains with a negative charge, or the ability to form a hydrogen bond. These features are needed for direct or water-mediated interaction with divalent cations. Alanine replacements of the residues homologous to Glu-174 or Glu-191 in E. coli AlaRS (Glu-183 and Glu-209) showed that an E183A mutant of E. coli AlaRS failed to complement the alaS-null strain whereas an E183D mutant had weak complementation activity (20). The E209A mutant, however, was functional in complementation assays, suggesting that Mg2 plays a secondary role (as compared with Mg1) in properly orienting the α-phosphate for nucleophilic attack.
Most significant is Asn-194, which also plays a key role in accommodation of the Ser side chain. Asn-194 plays a dual role, acting as a hydrogen-bond acceptor from the side chain of bound Ser in the noncognate amino acid complex, and stabilizing the α-phosphate of bound ATP by coordinating the principal Mg2+ ion in the Mg2+–ATP complex. Asn-194 is highly conserved in AlaRS, and is substituted only by T/Y/S/E, all of which can coordinate Mg2+ (either directly or indirectly by means of a water molecule) as well as hydrogen bond to the hydroxyl group of a bound Ser. An alanine substitution of the corresponding residue in E. coli AlaRS (Asn-212) yielded enzyme that complemented the null strain (20). No kinetic data were obtained for the mutant protein.
Further insight into the dual role of Asn-194 could be obtained from the structure of the complex with seryl-adenylate. Although diffracting crystals of AlaRS453 grew in the presence of magnesium and equimolar amounts of Ser and ATP, only the U-shaped ATP was found bound in the crystal structure.
Overall, the observations are consistent with the Ser side-chain binding pocket being a byproduct of selective pressure to bind ATP. Amino acid specificity of AlaRS is achieved by the hydrophobic nature of the Ala side-chain binding pocket and steric exclusion by the small binding pocket that cannot allow a larger hydrophobic side chain, such as those for Phe, Val, Ile, or Leu. Ser bypasses this exclusion, by H-bond donation from the Ser hydroxyl group to nearby Asn-194, which normally has a role in ATP binding and in H-bonding through its δ-NH2 to the carboxyl of Ala. Thus, the sieve of editing is broken by selective pressure to retain Asn-194 for ATP and Ala binding.
Acknowledgments
We thank Drs. Xiaoping Dai and Marc Elsliger for technical help, Drs. William Waas and Kirk Beebe for valuable discussions, and Professor Ya-Ming Hou (Thomas Jefferson University) for helpful comments on the paper. We thank Dr. Corie Ralston at the Advanced Light Source (ALS) for help in data collection. This work was supported by National Institutes of Health (NIH) Grant GM 15539 and by a fellowship from the National Foundation for Cancer Research. ALS is supported by the U.S. Department of Energy (DOE) under Contract DE-AC03-76SF00098. The Stanford Synchrotron Research Laboratory Structural Molecular Biology Program is supported by DOE, NIH, and the National Institute of General Medical Sciences.
Author contributions: M.A.S. and P.R.S. designed research; M.A.S. performed research; M.A.S. analyzed data; and M.A.S. and P.R.S. wrote the paper.
Abbreviations: AlaRS, alanyl-tRNA synthetase; ARS453, 453-aa N-terminal fragment of AlaRS; AARS, aminoacyl-tRNA synthetase.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 1YFR, 1YFS, 1YFT, and 1YGB).
References
- 1.Carter, C. W., Jr. (1993) Annu. Rev. Biochem. 62, 715–748. [DOI] [PubMed] [Google Scholar]
- 2.Ibba, M. & Söll, D. (2000) Annu. Rev. Biochem. 69, 617–650. [DOI] [PubMed] [Google Scholar]
- 3.Martinis, S. A., Plateau, P., Cavarelli, J. & Florentz, C. (1999) Biochimie 81, 683–700. [DOI] [PubMed] [Google Scholar]
- 4.Eriani, G., Delarue, M., Poch, O., Gangloff, J. & Moras, D. (1990) Nature 347, 203–206. [DOI] [PubMed] [Google Scholar]
- 5.Jakubowski, H. & Goldman, E. (1992) Microbiol. Rev. 56, 412–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jasin, M., Regan, L. & Schimmel, P. (1983) Nature 306, 441–447. [DOI] [PubMed] [Google Scholar]
- 7.Delarue, M. & Moras, D. (1993) BioEssays 15, 675–687. [DOI] [PubMed] [Google Scholar]
- 8.Regan, L., Bowie, J. & Schimmel, P. (1987) Science 235, 1651–1653. [DOI] [PubMed] [Google Scholar]
- 9.Ribas de Pouplana, L. & Schimmel, P. (1997) Biochemistry 36, 15041–15048. [DOI] [PubMed] [Google Scholar]
- 10.Beebe, K., Ribas de Pouplana, L. & Schimmel, P. (2003) EMBO J. 22, 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swairjo, M. A., Otero, F. J., Yang, X. L., Lovato, M. A., Skene, R. J., McRee, D. E., Ribas de Pouplana, L. & Schimmel, P. (2004) Mol. Cell 13, 829–841. [DOI] [PubMed] [Google Scholar]
- 12.Otwinowski, Z. & Minor, W. (1997) in Methods Enzymol., eds. Carter, C. W., Jr., & Sweet, R. W. (Academic, San Diego), Vol. 276, pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 13.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 14.Jones, T. A. & Kjelgaard, M. (1992) O: The Manual (Uppsala University, Uppsala).
- 15.Vaguine, A. A., Richelle, J. & Wodak, S. J. (1999) Acta Crystallogr. D 55, 191–205. [DOI] [PubMed] [Google Scholar]
- 16.Arnez, J. G., Augustine, J. G., Moras, D. & Francklyn, C. S. (1997) Proc. Natl. Acad. Sci. USA 94, 7144–7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavarelli, J., Eriani, G., Rees, B., Ruff, M., Boeglin, M., Mitschler, A., Martin, F., Gangloff, J., Thierry, J.-C. & Moras, D. (1994) EMBO J. 13, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnez, J. G., Dock-Bregeon, A. C. & Moras, D. (1999) J. Mol. Biol. 286, 1449–1459. [DOI] [PubMed] [Google Scholar]
- 19.Desogus, G., Todone, F., Brick, P. & Onesti, S. (2000) Biochemistry 39, 8418–8425. [DOI] [PubMed] [Google Scholar]
- 20.Shi, J. P., Musier-Forsyth, K. & Schimmel, P. (1994) Biochemistry 33, 5312–5318. [DOI] [PubMed] [Google Scholar]
- 21.Belrhali, H., Yaremchuk, A., Tukalo, M., Larsen, K., Berthet-Colominas, C., Leberman, R., Beijer, B., Sproat, B., Als-Nielsen, J., Grubel, G., et al. (1994) Science 263, 1432–1436. [DOI] [PubMed] [Google Scholar]
- 22.Belrhali, H., Yaremchuk, A., Tukalo, M., Berthet-Colominas, C., Rasmussen, B., Bosecke, P., Diat, O. & Cusack, S. (1995) Structure 3, 341–352. [DOI] [PubMed] [Google Scholar]
- 23.Reshetnikova, L., Moor, N., Lavrik, O. & Vassylyev, D. G. (1999) J. Mol. Biol. 287, 555–568. [DOI] [PubMed] [Google Scholar]
- 24.Cavarelli, J., Rees, B., Ruff, M., Thierry, J.-C. & Moras, D. (1993) Nature 362, 181–184. [DOI] [PubMed] [Google Scholar]
- 25.Berthet-Colominas, C., Seignovert, L., Hartlein, M., Grotli, M., Cusack, S. & Leberman, R. (1998) EMBO J. 17, 2947–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis, M. W., Buechter, D. D. & Schimmel, P. (1994) Biochemistry 33, 9904–9911. [DOI] [PubMed] [Google Scholar]