

Abstract
Flux balance analysis (FBA) with genome-scale metabolic network models (GSMNM) allows systems level predictions of metabolism in a variety of organisms. Different types of predictions with different accuracy levels can be made depending on the applied experimental constraints ranging from measurement of exchange fluxes to the integration of gene expression data. Metabolic network modeling with model organisms has pioneered method development in this field. In addition, model organism GSMNMs are useful for basic understanding of metabolism, and in the case of animal models, for the study of metabolic human diseases. Here, we discuss GSMNMs of most highly used model organisms with the emphasis on recent reconstructions.
Introduction
A metabolic network is a system that converts carbon and energy sources and electron donors and acceptors of an organism into biomass, energy, and byproducts. Deficiencies in this system cause disease when biomass production or energy generation is impaired, or when toxic by-products accumulate. On the other side, engineering of a metabolic system can produce higher yields of biomass or valuable by-products. Thus, a mechanistic understanding of metabolism is crucial for various disciplines, from biomedical to biofuels research [1].
A commonly applied powerful method of metabolic analysis is constraint-based metabolic network modeling at the whole system level [2]. In this approach, all annotated metabolic genes in an organism are first matched to enzymes and then to reactions to obtain gene-protein-reaction associations (GPRs). These GPRs are used to reconstruct a genome-scale metabolic network model (GSMNM), which is then used to calculate the flux distribution over the entire network in any defined condition for the organism (see below). For model organisms, high-quality genomic annotations allow the reconstruction of comprehensive GSMNMs that can be parameterized and validated with publically available experimental datasets. In addition, since many properties of metabolic networks are conserved across taxa, model organism GSMNMs can be used to study human disease with animal models, while plant models can instruct agriculture.
Here, we first summarize the basics of genome-scale metabolic network modeling, and then explore GSMNMs and their applications in common model organisms [3] (Figure 1). GSMNMs mentioned are listed in Table 1, together with the latest human model [4*] for comparison.
Figure 1.
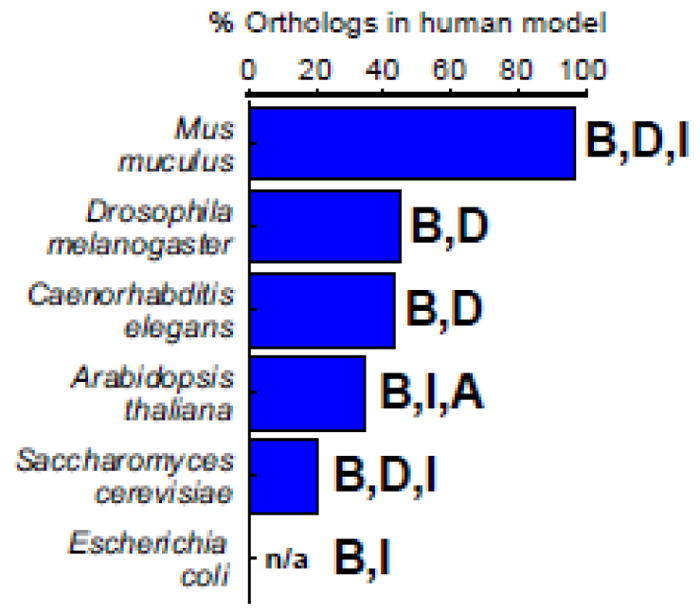
Model organisms reviewed. Percentage of genes in a human genome scale metabolic network model [4*] that have orthologs to each model organism is shown based on [31]. This number is not available for E. coli. Bold letters indicate whether the metabolic network modeling with the indicated organism is relevant to basic understanding (B), human disease (D), industrial applications (I), or agricultural (A) applications.
Table 1.
List of metabolic network models mentioneda.
| Organism | Year | Name | Reactionsb | Genes | Metabolitesb,c | Subcellular compartmentsd | Reference |
|---|---|---|---|---|---|---|---|
| Homo sapiens | 2016 | Recon 2.2 | 7785 | 1675 | 2652 (5324) | c,m,n,x,r,g,l,i,e | [4*] |
|
| |||||||
| Escherichia coli | 2011 | iJO1366 | 2583 | 1366 | 1136 (1805) | c,p,e | [14] |
| 2014 | EcoCyc–18.0– GEM | >2286e | 1445 | 1453 (nr) | c,p,ef | [15] | |
|
| |||||||
| Saccharomyces cerevisiae | 2003 | iFF708 | 1379 | 708 | 555 (796) | c,m,e | [20] |
| 2013 | iTO977 | 1562 | 977 | 817 (1353) | c,m,x,e | [21] | |
|
| |||||||
| Caenorhabditis elegans | 2016 | iCEL1273 | 1985 | 1273 | 887 (1401) | c,m,e | [58] |
| 2016 | na | 1922 | 979 | 901 (1646) | c,m,n,i,e | [57] | |
|
| |||||||
| Drosophila melanogaster | 2008 | na | 196 | 211 | 151 (224) | c,m,e | [23] |
|
| |||||||
| Mus muculus | 2005 | na | 1220 | 473 | 786 (872) | c,m,e | [27] |
| 2008 | na | 2037 | 1399 | 1631 (2104) | c,m,e | [35] | |
| 2009 | na | 1344 | nr | nr (1042) | c,m,e | [26] | |
| 2010 | na | 1494 | 724 | 945 (1162) | c,m,e | [25] | |
| 2010 | iMM1415 | 3724 | 1415 | 1503 (2774) | c,m,n,x,r,g,l,e | [32] | |
| 2013 | iSS1393 | 4091 | 1393 | 1536 (2950) | c,m,n,x,r,g,l,e | [33] | |
| 2015 | na | 2916 | 636 | 1097 (2072) | c,m,n,x,r,g,l,e | [34] | |
|
| |||||||
| Arabidoplsis thaliana | 2009 | na | 1406 | na | 1253 (nr) | c,m | [43] |
| 2010 | AraGEM | 1625 | 1419 | 1515 (1748) | c,m,pl,v,x,e | [46] | |
| 2011 | iRS1597 | 1798 | 1597 | 1684 (1820) | c,m,pl,v,x,e | [47] | |
| 2012 | na | 1617 | 1791 | 1188 (1188) | c,m,pl,v,x,r,e | [49] | |
| 2013 | na | 2769 | 2857 | 2371 (2739) | c,m,pl,v,x,e | [44] | |
| 2014 | na | 549 | 627 | 236 (407) | c,m,pl,x,i,l | [50] | |
na, not available; nr, not reported.
Number of reactions and metabolites were obtained from both papers and model files (SBML or XLS), if available. When reported and model-based values did not match, model values were used.
Number of unique metabolites followed by total number of compartmentalized metabolites in parenthesis.
Abbreviations: c, cytosol; m, mitochondria; n, nucleus; x, peroxisome; r, endoplasmic reticulum; g, golgi apparatus; l, lysosome; i, intermembrane space; e, extracellular space; pl, plastid; v, vacuole.
Only the number of unique reactions is reported which is the indicated number.
Since the model is not available, compartmentalization was inferred from the publication based on indirect information.
Mathematical modeling with genome scale metabolic networks
The steps of mathematical network modeling are summarized in Figure 2A. After GPRs are annotated, the reaction list is complemented by necessary transport reactions that carry metabolites between different compartments, exchange reactions that define the input and output of the system, and biomass reactions that represent growth. Next, this network is converted to a mathematical model that describes the mass balance of each metabolite as the difference between the fluxes of reactions that produce and consume it. The combination of mass balance equations for all metabolites yields a linear algebraic equation (Figure 2A) where a stoichiometry matrix (S) is multiplied by the reaction flux vector (v) to obtain the production rates of compounds. Due to the large number of reactions in GSMNMs, kinetic modeling approaches that allow the prediction of metabolite concentrations are not feasible. Instead, the mass balance is solved for an assumed steady state, i.e., a zero sum of fluxes at each compound node, to obtain the flux vector, which describes the metabolic state as a flux distribution across the network. Importantly, the steady state assumption refers to the internal metabolism only, and is therefore not limited to the true steady state established for cells in a continuous flow reactor, but is also valid for any stable metabolic state, as in exponential growth, homeostasis, or growth in a small time interval that can be considered a quasi-steady state.
Figure 2.

Genome-scale metabolic network modeling. (A) Metabolic network modeling is an iterative procedure. Main steps of model development are shown. (B) Cartoon representation of a GSMNM (top) and flux balance analysis (middle and bottom). In the absence of experimental constraints, pure theoretical analyses can be done (middle left). A flux distribution is found but alternate pathways exist. Integration of gene expression data guides the flux distribution to choose from alternate pathways (middle right). Still, alternate solutions may exist. Inclusion of experimental measurements for exchange fluxes can constrain the solution further (bottom left).
There is no single, unique solution to the governing mass balance equation, instead there is a distribution of possible solutions (Figure 2B). To find a biologically meaningful flux distribution, fluxes are constrained by reaction reversibility and any known flux such as uptake rates of nutrients and secretion rates of by-products. In addition, an objective is set for maximizing or minimizing a subset of fluxes and optimized as a linear programming problem (Figure 2A). A typical objective function is maximization of biomass production to represent growth-oriented metabolism. This mathematical method is referred to as constraint-based flux balance analysis (FBA). FBA should not be confused with metabolic flux analysis (MFA), which uses isotope labeling and derives fluxes generally in core pathways by best fitting the flux distribution to the pattern of labeled metabolites in the central carbon metabolism only [5].
Running FBA on a draft metabolic network model can reveal gaps that prevent biomass production or other reactions from carrying flux. These are to be iteratively corrected before validating the GSMNM with experimental observations (Figure 2A). A validated GSMNM can be used for different types and levels of predictions by varying the constraints used during FBA. In the simplest case, none of the fluxes are known or only uptake or secretions of main metabolites are experimentally obtained. Then network properties can be analyzed using FBA and flux variability analysis (FVA) for hypothesis-driven discovery [2]. In more advanced applications, large experimental datasets are integrated with the GSMNM, such that, flux predictions are constrained by global gene expression [6] or metabolomics [7*] to get an accurate picture of the metabolic state of the organism (Figure 2B).
Reconstructions and applications of model organism metabolic network models
Escherichia coli, the model bacterium
Applications of GSMNMs have been most widespread and successful with microorganisms, which have simple life styles accountable by FBA. This is because a bacterium in a bioreactor can be modeled as an open system that takes provided nutrients as the input and yields biomass and by-products as the output. The pioneering work on constraint-based metabolic network modeling in microorganisms was conducted with early E. coli reconstructions [8,9]. Later, E. coli GSMNMs served for the development of new methods such as dynamic FBA [10], parsimonious enzyme usage FBA (pFBA) [11], and regulatory FBA [12]. A review of most E. coli GSMNMs have recently been provided [13] and will not be repeated here. An example from the previous review [14] and the most recent reconstruction [15] are included in Table 1.
Saccharomyces cerevisiae, the budding yeast as a model eukaryote
There are more than two dozen GSMNMs reconstructed for the budding yeast Saccharomyces cerevisiae to date [16]. This scale of effort reflects the fact that S. cerevisiae is the most studied unicellular eukaryote serving as a general eukaryotic model, and that it is directly used as a workhorse for the production of valuable metabolites such as ethanol [17]. In addition, S. cerevisiae can also be easily grown in controlled bioreactors and modeled as a unicellular open system, although, in contrast to bacteria, the metabolism is compartmentalized to organelles (Table 1). As with E. coli, GSMNMs of the yeast have already been reviewed [16,18*,19] and we show only two representative models in Table 1 [20,21].
Drosophila melanogaster, the fly as a model animal
Unfortunately, a genome-scale metabolic network model is not yet available for the fruit fly Drosophila melanogaster, although a core metabolic model to study the central metabolic properties of the muscle tissues of hypoxia resistant flies was published nearly a decade ago [22]. The model was reconstructed using metabolomics data obtained from dissected thoraxes, and by linking detected metabolites with GPR annotations. Later, the same model was expanded based on highly expressed enzymes in the thorax [23] (Table 1). These modeling efforts showed that FBA is a suitable method to study disease-related metabolic phenotypes in Drosophila. However, a genome-scale reconstruction will be needed to realize the full potential of metabolic network modeling with this organism.
Mus muculus, the model mammal
Several GSMNMs are available for the mouse (Table 1). These models are useful to study human disease, but also have industrial relevance since mouse hybridoma cell lines are employed for the production of biopharmaceuticals such as monoclonal antibodies (MAb) and vaccines [24]. Indeed, initial mouse GSMNMs targeted the modeling of hybridoma cell lines for a rational engineering approach to improve the production yields of MAb [25–27]. These models were tested using batch or continuous cultures of hybridoma cell lines. FBA-based prediction of growth rates and by-products, constrained by metabolomics measurements, were validated and improved over time. These models have further served as knowledge bases for other studies that used the provided biomass composition [28], maintenance energy [29], and other network properties [30].
Another lineage of mouse models was based on a human reconstruction [31]. Reactions in the human network associated with genes that had mouse orthologs were first extracted, which was followed by the arrangement of transport reactions and filling of the created gaps [32]. The final model, called iMM1415, included eight compartments (Table 1). In a later study [33], iMM1415 was not only updated but also extended to include intestine-specific transport and exchange reactions, and was combined with a Bacteroides reconstruction to develop a unified host-microbiota model. This interspecies model was useful in the analysis of synergistic and competitive interactions between the host and bacterial metabolism. The most recent mouse model was developed using a similar approach to iMM1415 reconstruction and was subsequently converted to a germ cell-specific model by integration of transcription levels in germ cells during spermatogenesis [34]. Using FBA constrained by gene expression levels throughout a spermatogenesis period, the authors determined metabolic genes and reactions that are critical for the germ cell differentiation for commitment to meiosis.
In addition to the above GSMNMs, an independent mouse model was semi-automatically reconstructed using the information stored in publicly available databases [35] (Table 1). Although only half of the reactions in this model are able to carry flux because of network gaps, it has been useful as a resource [36*,37].
Arabidopsis thaliana, the model plant
Flux analysis using FBA and MFA has been particularly popular in plant research, which is not surprising given the broad interest in engineering plant metabolism to improve yields of agriculturally and industrially valuable products [5,38,39]. A relatively large number of GSMNMs are available for the model plant Arabidopsis thaliana (Table 1) [40*,41,42]. The first reconstruction was targeted at and validated by heterotrophic plant cells grown in suspension [43]. This model was later expanded by GPR annotations and compartmentalized into plant organelles [44]. With these modifications the model was able to represent both heterotrophic and photoautotrophic metabolism, which was recently exploited to develop a model capable of simulating metabolism in day-night cycles with two separate compartments [45].
Another lineage of plant GSMNMs was started with the first photosynthetic A. thaliana model, AraGEM [46]. This reconstruction was updated in a study that compared A. thaliana and Zea mays metabolism [47] (Table 1). AraGEM was recently used to create a multi-tissue network that represents a whole plant [48**]. Metabolic networks for multiple plant tissues were derived from the generic model and combined in an organism framework. FBA was done by minimizing photon usage as the objective function for the entire network.
Two additional Arabidopsis GSMNMs have been developed. The most compartmentalized plant model [49] (Table 1) was used to derive the metabolic states of multiple plant organs using organ-specific protein expression datasets. The most recent reconstruction of A. thaliana focused on central metabolism (Table 1) [50]. This small but robust model of leaf cells consisted of manually curated GPRs, and was used to estimate the energetic cost of amino acid and enzyme synthesis during photoautotrophic growth conditions that employed carbon fixation by Rubisco, the most abundant protein in the world.
Caenorhabditis elegans, the nematode as a model animal
The nematode C. elegans (the worm) is a self-reproducing hermaphrodite with a relatively short life cycle. Although the worm has been widely used to study development, neurobiology and aging, it has recently also emerged as a powerful model for studying the effects of diet on metabolism and growth [51]. The laboratory diet of C. elegans typically consists of a pure bacterial culture. Different bacterial species can be fed to the worm, and can be combined with easy genetic screening in both the animal and its diet [52–54]. In addition, C. elegans can be uniquely used to model some human diseases or specific components of the human diet. For instance, the two vitamin B12 dependent enzymes, methionine synthase and methylmalonyl-CoA mutase, are present in both humans and the nematode, but not in flies or yeast (Figure 3). We have recently discovered an alternative pathway to propionic acid breakdown that does not depend on vitamin B12, illustrating the power of this model [55].
Figure 3.

C. elegans is an adequate model to study vitamin B12 metabolism but S. cerevisiae and D. melanogaster are not. Left panel shows two vitamin B12-dependent pathways. C. elegans genes encoding the enzymes are indicated. Recently discovered propionate shunt is also drawn without the details. For the genes in bold font, phylogenetic protein sequence trees are provided on the right panel. Trees were obtained from WormFlux (http://wormflux.umassmed.edu/) and edited for clarity. All reasonable best hits (B) and reciprocal best hits (R) (if available) from human (HSA), S. cerevisiae (SCE), and D. melanogaster (DME) were included in these trees [58]. Thus, S. cerevisiae (SCE) and D. melanogaster (DME) do not have the vitamin B12 enzyme homologs but they have an ortholog for methionine adenosyltransferase. Hits from A. thaliana (ATH) and C. elegans paralogs (P) are also shown. Identities of other organism genes are hidden except for the taxonomy.
The overall function of C. elegans metabolism can be seen as the conversion of bacterial biomass into worm biomass. Unlike any other animal model, C. elegans can be easily grown in liquid cultures as a dense population (hundreds of animals per ml) with both the bacterial diet and a chemically well-defined diet called axenic medium [56]. Thus, the nematode is a unique model animal in its suitability for metabolic network modeling and FBA, and a candidate for repeating the success of GSMNMs with microorganisms in an animal model. Two GSMNMs of C. elegans were recently published [57,58] (Table 1). Both models are able to represent growth with the bacterial and axenic diet and involve peculiar traits of C. elegans metabolism known so far, such as the presence of a glyoxylate shunt in an animal and the absence of a de novo NAD biosynthesis pathway. However, the focus of reconstruction and validation was different for each model.
The first model in Table 1, iCEL1273 [58], was shown to quantitatively account for the growth of C. elegans at pseudo-steady states in two different stages of life, the L4 growing larvae and egg-laying adults. Gene essentiality for growth at any life stage was systematically analyzed based on assumptions on animal physiology, such as the non-redundant functioning of paralogs in different tissues and the minimization of enzyme usage for an optimal metabolic state (implemented by pFBA). In addition, specific phenotypes were shown to be predictable, including the slow-down of growth in the absence of methionine synthase or the lack of vitamin B12 (Figure 3). The utility of this model was shown by analyzing the metabolic state of dormant dauer larvae in comparison to growing larvae. Integration of gene expression data was sufficient for iCEL1273 to predict the observed differences in these states such as low energetic activity, lack of growth, and dependence on stored carbon resources in the dauer state, but reverse in the other. iCEL1273 is available at a dedicated website named WormFlux (http://wormflux.umassmed.edu). This webtool is relatively unique because it shows the systematic annotation of all genes in the organism with a pipeline that uses multiple resources [58], and all metabolic reactions used in the reconstruction. Genes, enzymes, metabolites, and pathways are interlinked, searchable, and linked to other databases.
The second C. elegans GSMNM in Table 1 [57] was also quantitatively challenged, this time using measured amino acid concentrations in wild-type versus perturbed worms. The effect of knocking out bcat-1, which codes an enzyme responsible for the initial step of branched chain amino acid breakdown, was simulated to indirectly deduce the changes in amino acid levels using FBA, and verified by experimental observations. The utility of this model was shown by focusing on aging. Integration of gene expression in a time series dataset from younger to older worms successfully predicted the decreasing overall activity of metabolism, and revealed the specific metabolic advantages of long-lived mutants such as the improved activity of the TCA-cycle and ubiquinone biosynthesis at later stages of life. The effective start of metabolic network modeling with the nematode opens up an exciting new field for studying the mechanistic relationships between diet, genotypes, phenotypes and aging at the systems level.
Conclusions and future perspectives
We derived four important conclusions from our review to provide perspective for future GSMNM reconstructions of model organisms and their applications. First, although most of the older GSMNMs in Table 1 are highly cited, the direct use of these models by other groups is rare. The lack of usage may be attributed to the required expertise to run GSMNMs. We believe this can be changed by building devoted web-based tools that allow user-friendly and interactive FBA with GSMNMs. WormFlux [58] has been developed for C. elegans with this purpose and efforts to implement FBA, FVA, and gene expression integration on this webtool are underway.
Second, the repeated use of GSMNMs for a particular organism generally involves a modification, or even a brand new reconstruction, as is clear from the timeline of mouse and Arabidopsis reconstructions (Table 1). We think that modeling modifications are often related to the feedback loops from validation and application stages in Figure 2A. Each time a model is challenged with new research questions or experimental data, potential reconstruction handicaps or false predictions lead to repairs, which improves not only the models but also our understanding of the metabolic network. It is reasonable to expect the same trend for other model organisms for which GSMNMs have recently been reconstructed.
Third, organism-level compartmentalization is needed for multi-cellular organisms and there are advancements towards this direction [59]. The multi-organ framework developed for A. thaliana [48**] is the closest we have to a whole organism model. An additional, more focused multi-compartment model for mouse has become available to simultaneously represent metabolism in liver, muscle and adipose tissue cells [60], although it is important to note that this model was based on a human GSMNM [31]. We advocate the development of compartmentalized models for other organisms, notably C. elegans, because these model organisms are more suitable to high-throughput and large-scale genetic perturbations. Another adequate model system for tissue frameworks would be Drosophila, should GSMNMs become available for this organism.
Finally, other compartmentalized modeling efforts need to consider interspecies metabolic interactions, as exemplified by the host-microbiota model in the mouse [61]. To this end, C. elegans, together with its bacterial diet, provide an ideal model system [51], which has already been used with genetic screening approaches [54]. In the future, our understanding of metabolism will likely continue to be transformed by GSMNMs that characterize multi-cellular model organisms from cellular to whole-organism level and their commensal interactions with other species.
HIGHLIGHTS.
A genome-scale metabolic network model (GSMNM) represents all metabolism.
GSMNMs in model organisms help basic understanding and method development.
Recently, two GSMNMs were developed for Caenorhabditis elegans.
GSMNs of animal models are useful for studying human disease.
A multi-tissue framework of Arabidopsis thaliana inspires whole organism modeling.
Acknowledgments
This work was funded by the National Institutes of Health grant R21GM108045.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhang C, Hua Q. Applications of Genome-Scale Metabolic Models in Biotechnology and Systems Medicine. Front Physiol. 2015;6:413. doi: 10.3389/fphys.2015.00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Brien EJ, Monk JM, Palsson BO. Using Genome-scale Models to Predict Biological Capabilities. Cell. 2015;161:971–987. doi: 10.1016/j.cell.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edison AS, Hall RD, Junot C, Karp PD, Kurland IJ, Mistrik R, Reed LK, Saito K, Salek RM, Steinbeck C, et al. The Time Is Right to Focus on Model Organism Metabolomes. Metabolites. 2016:6. doi: 10.3390/metabo6010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *4.Swainston N, Smallbone K, Hefzi H, Dobson PD, Brewer J, Hanscho M, Zielinski DC, Ang KS, Gardiner NJ, Gutierrez JM, et al. Recon 2. 2: from reconstruction to model of human metabolism. Metabolomics. 2016;12:109. doi: 10.1007/s11306-016-1051-4. The most recent version of the human GSMNM with many updates that improved the model functionality. For instance, the authors introduced the intramembrane space compartment to by-pass thermodynamically unfeasible loops generating infinite ATP in the older models. This addition allowed the accurate calculation of ATP yields from various Carbon sources. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Junker BH. Flux analysis in plant metabolic networks: increasing throughput and coverage. Curr Opin Biotechnol. 2014;26:183–188. doi: 10.1016/j.copbio.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Machado D, Herrgard M. Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism. PLoS Comput Biol. 2014;10:e1003580. doi: 10.1371/journal.pcbi.1003580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *7.Topfer N, Kleessen S, Nikoloski Z. Integration of metabolomics data into metabolic networks. Front Plant Sci. 2015;6:49. doi: 10.3389/fpls.2015.00049. Regular FBA does not allow prediction of concentrations, and therefore, making sense of metabolomics data with GSMNMs is challenging. This review paper summarizes emerging methods to indirectly integrate metabolomics data with GSMNMs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varma A, Palsson BO. Metabolic capabilities of Escherichia coli: I. synthesis of biosynthetic precursors and cofactors. J Theor Biol. 1993;165:477–502. doi: 10.1006/jtbi.1993.1202. [DOI] [PubMed] [Google Scholar]
- 9.Varma A, Palsson BO. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl Environ Microbiol. 1994;60:3724–3731. doi: 10.1128/aem.60.10.3724-3731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahadevan R, Edwards JS, Doyle FJ., 3rd Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys J. 2002;83:1331–1340. doi: 10.1016/S0006-3495(02)73903-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis NE, Hixson KK, Conrad TM, Lerman JA, Charusanti P, Polpitiya AD, Adkins JN, Schramm G, Purvine SO, Lopez-Ferrer D, et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol Syst Biol. 2010;6:390. doi: 10.1038/msb.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429:92–96. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- 13.McCloskey D, Palsson BO, Feist AM. Basic and applied uses of genome-scale metabolic network reconstructions of Escherichia coli. Mol Syst Biol. 2013;9:661. doi: 10.1038/msb.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orth JD, Conrad TM, Na J, Lerman JA, Nam H, Feist AM, Palsson BO. A comprehensive genome-scale reconstruction of Escherichia coli metabolism--2011. Mol Syst Biol. 2011;7:535. doi: 10.1038/msb.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weaver DS, Keseler IM, Mackie A, Paulsen IT, Karp PD. A genome-scale metabolic flux model of Escherichia coli K-12 derived from the EcoCyc database. BMC Syst Biol. 2014;8:79. doi: 10.1186/1752-0509-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heavner BD, Price ND. Comparative Analysis of Yeast Metabolic Network Models Highlights Progress, Opportunities for Metabolic Reconstruction. PLoS Comput Biol. 2015;11:e1004530. doi: 10.1371/journal.pcbi.1004530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duina AA, Miller ME, Keeney JB. Budding yeast for budding geneticists: a primer on the Saccharomyces cerevisiae model system. Genetics. 2014;197:33–48. doi: 10.1534/genetics.114.163188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *18.Pereira R, Nielsen J, Rocha I. Improving the flux distributions simulated with genome-scale metabolic models of Saccharomyces cerevisiae. Metabolic Engineering Communications. 2016;3:153–163. doi: 10.1016/j.meteno.2016.05.002. The authors compared four yeast models with respect to predictions of fluxes in the central metabolism. Inconsistencies and errors were corrected using proper constraints, mostly on reactions depending on NADPH and NADH. Notably, the early yeast model iFF708 was the best predictor of central metabolic fluxes without these corrections. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez BJ, Nielsen J. Genome scale models of yeast: towards standardized evaluation and consistent omic integration. Integr Biol (Camb) 2015;7:846–858. doi: 10.1039/c5ib00083a. [DOI] [PubMed] [Google Scholar]
- 20.Forster J, Famili I, Fu P, Palsson BO, Nielsen J. Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 2003;13:244–253. doi: 10.1101/gr.234503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osterlund T, Nookaew I, Bordel S, Nielsen J. Mapping condition-dependent regulation of metabolism in yeast through genome-scale modeling. BMC Syst Biol. 2013;7:36. doi: 10.1186/1752-0509-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feala JD, Coquin L, McCulloch AD, Paternostro G. Flexibility in energy metabolism supports hypoxia tolerance in Drosophila flight muscle: metabolomic and computational systems analysis. Mol Syst Biol. 2007;3:99. doi: 10.1038/msb4100139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coquin L, Feala JD, McCulloch AD, Paternostro G. Metabolomic and flux-balance analysis of age-related decline of hypoxia tolerance in Drosophila muscle tissue. Mol Syst Biol. 2008;4:233. doi: 10.1038/msb.2008.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boghigian BA, Seth G, Kiss R, Pfeifer BA. Metabolic flux analysis and pharmaceutical production. Metab Eng. 2010;12:81–95. doi: 10.1016/j.ymben.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Selvarasu S, Karimi IA, Ghim GH, Lee DY. Genome-scale modeling and in silico analysis of mouse cell metabolic network. Mol Biosyst. 2010;6:152–161. doi: 10.1039/b912865d. [DOI] [PubMed] [Google Scholar]
- 26.Selvarasu S, Wong VV, Karimi IA, Lee DY. Elucidation of metabolism in hybridoma cells grown in fed-batch culture by genome-scale modeling. Biotechnol Bioeng. 2009;102:1494–1504. doi: 10.1002/bit.22186. [DOI] [PubMed] [Google Scholar]
- 27.Sheikh K, Forster J, Nielsen LK. Modeling hybridoma cell metabolism using a generic genome-scale metabolic model of Mus musculus. Biotechnol Prog. 2005;21:112–121. doi: 10.1021/bp0498138. [DOI] [PubMed] [Google Scholar]
- 28.Nargund S, Qiu J, Goudar CT. Elucidating the role of copper in CHO cell energy metabolism using (13)C metabolic flux analysis. Biotechnol Prog. 2015;31:1179–1186. doi: 10.1002/btpr.2131. [DOI] [PubMed] [Google Scholar]
- 29.Vazquez A. Metabolic states following accumulation of intracellular aggregates: implications for neurodegenerative diseases. PLoS One. 2013;8:e63822. doi: 10.1371/journal.pone.0063822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bekaert M, Conant GC. Gene duplication and phenotypic changes in the evolution of mammalian metabolic networks. PLoS One. 2014;9:e87115. doi: 10.1371/journal.pone.0087115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson BO. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1777–1782. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sigurdsson MI, Jamshidi N, Steingrimsson E, Thiele I, Palsson BO. A detailed genome-wide reconstruction of mouse metabolism based on human Recon 1. BMC Syst Biol. 2010;4:140. doi: 10.1186/1752-0509-4-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinken A, Sahoo S, Fleming RM, Thiele I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes. 2013;4:28–40. doi: 10.4161/gmic.22370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitmore LS, Ye P. Dissecting Germ Cell Metabolism through Network Modeling. PLoS One. 2015;10:e0137607. doi: 10.1371/journal.pone.0137607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quek LE, Nielsen LK. On the reconstruction of the Mus musculus genome-scale metabolic network model. Genome Inform. 2008;21:89–100. [PubMed] [Google Scholar]
- *36.Martinez VS, Buchsteiner M, Nielsen LK, Quek LE. Dynamic metabolic flux analysis using B-splines to study the effects of temperature shift on CHO cell metabolism. Metabolic Engineering Communications. 2015;2:46–57. doi: 10.1016/j.meteno.2015.06.001. A good example of how GSMNMs of model organisms serve as resources. The central metabolic network of CHO cells were derived from a mouse GSMNM and used for performing MFA. In addition, the authors introduced a method to apply MFA to a time series in a batch culture rather than being limited to steady state or mid-exponential growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sridharan GV, Choi K, Klemashevich C, Wu C, Prabakaran D, Pan LB, Steinmeyer S, Mueller C, Yousofshahi M, Alaniz RC, et al. Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nat Commun. 2014;5:5492. doi: 10.1038/ncomms6492. [DOI] [PubMed] [Google Scholar]
- 38.Baghalian K, Hajirezaei MR, Schreiber F. Plant metabolic modeling: achieving new insight into metabolism and metabolic engineering. Plant Cell. 2014;26:3847–3866. doi: 10.1105/tpc.114.130328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kruger NJ, Ratcliffe RG. Fluxes through plant metabolic networks: measurements, predictions, insights and challenges. Biochem J. 2015;465:27–38. doi: 10.1042/BJ20140984. [DOI] [PubMed] [Google Scholar]
- *40.Basler G, Kuken A, Fernie AR, Nikoloski Z. Photorespiratory Bypasses Lead to Increased Growth in Arabidopsis thaliana: Are Predictions Consistent with Experimental Evidence? Front Bioeng Biotechnol. 2016;4:31. doi: 10.3389/fbioe.2016.00031. Three Arabidopsis models are compared with respect to their predictive power and it is shown that experimental observations of efficient growth in A. thaliana strains with engineered photorespirotory systems were qualitatively captured by all models when proper constraints were used during FBA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukushima A, Kanaya S, Nishida K. Integrated network analysis and effective tools in plant systems biology. Front Plant Sci. 2014;5:598. doi: 10.3389/fpls.2014.00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi H, Schwender J. Mathematical models of plant metabolism. Curr Opin Biotechnol. 2016;37:143–152. doi: 10.1016/j.copbio.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 43.Poolman MG, Miguet L, Sweetlove LJ, Fell DA. A genome-scale metabolic model of Arabidopsis and some of its properties. Plant Physiol. 2009;151:1570–1581. doi: 10.1104/pp.109.141267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheung CY, Williams TC, Poolman MG, Fell DA, Ratcliffe RG, Sweetlove LJ. A method for accounting for maintenance costs in flux balance analysis improves the prediction of plant cell metabolic phenotypes under stress conditions. Plant J. 2013;75:1050–1061. doi: 10.1111/tpj.12252. [DOI] [PubMed] [Google Scholar]
- 45.Cheung CY, Poolman MG, Fell DA, Ratcliffe RG, Sweetlove LJ. A Diel Flux Balance Model Captures Interactions between Light and Dark Metabolism during Day-Night Cycles in C3 and Crassulacean Acid Metabolism Leaves. Plant Physiol. 2014;165:917–929. doi: 10.1104/pp.113.234468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Oliveira Dal’Molin CG, Quek LE, Palfreyman RW, Brumbley SM, Nielsen LK. AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol. 2010;152:579–589. doi: 10.1104/pp.109.148817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saha R, Suthers PF, Maranas CD. Zea mays iRS1563: a comprehensive genome-scale metabolic reconstruction of maize metabolism. PLoS One. 2011;6:e21784. doi: 10.1371/journal.pone.0021784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **48.Gomes de Oliveira Dal’Molin C, Quek LE, Saa PA, Nielsen LK. A multi-tissue genome-scale metabolic modeling framework for the analysis of whole plant systems. Front Plant Sci. 2015;6:4. doi: 10.3389/fpls.2015.00004. The authors present a multi-tissue plant model that not only represents leaf, stem, and root systems in a framework of GSMNMs, but is also compartmentalized further into two parts to simulate differential metabolism during day and night cycles. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mintz-Oron S, Meir S, Malitsky S, Ruppin E, Aharoni A, Shlomi T. Reconstruction of Arabidopsis metabolic network models accounting for subcellular compartmentalization and tissue-specificity. Proc Natl Acad Sci U S A. 2012;109:339–344. doi: 10.1073/pnas.1100358109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arnold A, Nikoloski Z. Bottom-up Metabolic Reconstruction of Arabidopsis and Its Application to Determining the Metabolic Costs of Enzyme Production. Plant Physiol. 2014;165:1380–1391. doi: 10.1104/pp.114.235358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yilmaz LS, Walhout AJ. Worms, bacteria, and micronutrients: an elegant model of our diet. Trends Genet. 2014;30:496–503. doi: 10.1016/j.tig.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacNeil LT, Watson E, Arda HE, Zhu LJ, Walhout AJ. Diet-induced developmental acceleration independent of TOR and insulin in C. elegans. Cell. 2013;153:240–252. doi: 10.1016/j.cell.2013.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watson E, MacNeil LT, Arda HE, Zhu LJ, Walhout AJ. Integration of metabolic and gene regulatory networks modulates the C. elegans dietary response. Cell. 2013;153:253–266. doi: 10.1016/j.cell.2013.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watson E, MacNeil LT, Ritter AD, Yilmaz LS, Rosebrock AP, Caudy AA, Walhout AJ. Interspecies systems biology uncovers metabolites affecting C. elegans gene expression and life history traits. Cell. 2014;156:759–770. doi: 10.1016/j.cell.2014.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watson E, Olin-Sandoval V, Hoy MJ, Li CH, Louisse T, Yao V, Mori A, Holdorf AD, Troyanskaya OG, Ralser M, et al. Metabolic network rewiring of propionate flux compensates vitamin B12 deficiency in C. elegans. Elife. 2016:5. doi: 10.7554/eLife.17670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Samuel TK, Sinclair JW, Pinter KL, Hamza I. Culturing Caenorhabditis elegans in axenic liquid media and creation of transgenic worms by microparticle bombardment. J Vis Exp. 2014:e51796. doi: 10.3791/51796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gebauer J, Gentsch C, Mansfeld J, Schmeisser K, Waschina S, Brandes S, Klimmasch L, Zamboni N, Zarse K, Schuster S, et al. A Genome-Scale Database and Reconstruction of Caenorhabditis elegans Metabolism. Cell Syst. 2016;2:312–322. doi: 10.1016/j.cels.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 58.Yilmaz LS, Walhout AJ. A Caenorhabditis elegans Genome-Scale Metabolic Network Model. Cell Syst. 2016;2:297–311. doi: 10.1016/j.cels.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.do Martins Conde PR, Sauter T, Pfau T. Constraint Based Modeling Going Multicellular. Front Mol Biosci. 2016;3:3. doi: 10.3389/fmolb.2016.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kumar A, Harrelson T, Lewis NE, Gallagher EJ, LeRoith D, Shiloach J, Betenbaugh MJ. Multi-tissue computational modeling analyzes pathophysiology of type 2 diabetes in MKR mice. PLoS One. 2014;9:e102319. doi: 10.1371/journal.pone.0102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heinken A, Thiele I. Systems biology of host-microbe metabolomics. Wiley Interdiscip Rev Syst Biol Med. 2015;7:195–219. doi: 10.1002/wsbm.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]