

Abstract
Transcriptional co-repressor proteins have emerged as an important facet of cancer etiology. These co-repressor proteins are often altered by loss- or gain-of-function mutations, leading to transcriptional imbalance. Thus, research directed at expanding our present understanding of transcriptional co-repressors could impact the future development of new cancer diagnostics, prognostics, and therapies. In this review, our current understanding of the CtBP co-repressors, and their role in both development and disease, is discussed in detail. Importantly, the role of CtBP1 overexpression in adult tissues in promoting the progression of multiple cancer types through their ability to modulate the transcription of developmental genes ectopically is explored. CtBP1 overexpression is known to be pro-tumorigenic and affects the regulation of gene networks associated with “cancer hallmarks” and malignant behavior including: increased cell survival, proliferation, migration, invasion, and the epithelial-mesenchymal transition (EMT). As a transcriptional regulator of broad developmental processes capable of promoting malignant growth in adult tissues, therapeutically targeting the CtBP1 co-repressor has the potential to be an effective method for the treatment of diverse tumor types. While efforts to develop CtBP1 inhibitors are still in the early stages, the current progress and the future perspectives of therapeutically targeting this transcriptional co-repressor are also discussed.
Keywords: CtBP, transcriptional co-repressors, cancer hallmarks, therapeutic targets, CtBP inhibitors
CtBPs are transcriptional co-repressors that play important roles in development
The C-terminal binding proteins are encoded by two genes, CtBP1 and CtBP2, in mammals (1). CtBP1 has two splicing variants, CtBP1 (also known as CtBP1-L) and CtBP1-S/BARS (also known as CtBP3/BARS) (2). CtBP2 has three splicing variants, CtBP2, CtBP2-S, and RIBEYE (3–5). CtBP1 and 2 are primarily nuclear transcriptional co-repressors evolutionarily conserved from C. elegans to H. sapiens (6) and will be the focus of this review. We will use either CtBP1 or CtBP2 when it is clearly indicated in the research being discussed, and use CtBP otherwise to represent both CtBP1 and CtBP2. BARS and CtBP2-S are largely cytosolic and RIBEYE is mostly localized in synaptic ribbons, and these isoforms likely have functions unrelated to transcription (see reference 1 for more information).
The CtBP proteins were originally identified through their ability to bind to the C-terminus of the adenovirus protein, E1A (7, 8). Subsequently, the CtBP proteins were determined to be a family of transcriptional co-repressors that are recruited by various DNA-binding transcription factors to specific promoter/enhancer regions to carry out diverse functions in both developmental and oncogenic processes (9). CtBP proteins are expressed at high levels during development, participating in axial-patterning, cellular proliferation, and differentiation within many organs including the eyes, heart, brain, placenta vasculature, and muscle (10–13). Loss- and gain-of-function studies have confirmed the CtBP proteins as regulators of sequence-specific DNA-binding transcription factors that control segmentation, epithelial-mesenchymal transition (EMT), and apoptosis (9, 14). Genetically engineered mutations in CtBP have adverse consequences on the development of organs/tissues, confirming their role as critical regulators of organogenesis and tissue morphogenesis (10–12). For example, CtBP1-null mice, albeit viable, have a significantly smaller body size and shorter life span relative to the wild type control (10). CtBP2-null mice are embryonic lethal and often exhibit axial truncations, heart defects, and incomplete neural development (10).
A naturally occurring mutation in the CtBP recognition sequence of the Three Amino acid Loop Extension (TALE) family of homeobox transcription factors is implicated in at least one developmental disorder, consistent with the role of CtBP in organogenesis. Normally, TALE works in concert with CtBP to repress genes activated by TGF-β (15). Mutations within TALE can result in holoprosencephaly (HPE), a defect in craniofacial development due to brain malformations (15). One of the known HPE mutations within the TALE transcription factor is a single amino acid substitution in the CtBP-binding motif, from PLDLS to PLDLC, which abolishes its ability to interact with CtBP. To date, there are no known genetic mutations within CtBP itself. However, a natural mutation within a CtBP-protein partner confirms that the CtBP co-repressors are critical for normal organ development in humans.
Structural and Functional Analyses of CtBP
The crystal structures of the N-terminal two-thirds of both human CtBP1/2 (16, 17) and rat CtBP/BARS (18) have been solved, providing insights into their structure and function, and the possibility of inhibiting this protein family (Figure 1). Considering the substantial sequence homology between CtBP and other NADH-dependent dehydrogenases (8), it is not surprising that the overall structure of CtBP is similar to the D-2-hydroxyacid family of dehydrogenases that utilize nicotinamide adenine dinucleotide (NADH/NAD+) as a co-factor (Figure 1B). CtBP1/2 is composed of two structured domains, the substrate-binding domain (SBD) and the nucleotide-binding domain (NBD), with the C-terminus being predicted to be disordered (Figure 1A) (19). A deep cleft between the two domains encompasses both the active site and the NADH-binding pocket. Previous binding studies have shown that CtBP1 binds NADH with a 100-fold higher affinity than NAD+ (20, 21). Although both NADH and NAD+ are capable of enhancing CtBP1’s dimerization and interaction with target transcription factors, NADH has a more prominent effect (21, 22). Consistent with the in vitro binding data, an increase in nuclear NADH under hypoxic conditions stimulates the recruitment of CtBP to target promoters through transcription factor interactions resulting in enhanced transcriptional repression (23). These findings suggest a role for the CtBP proteins as a metabolic sensor of redox status. Interestingly, Kumar and colleagues demonstrated that mutations disrupting only the enzymatic activity showed no effect on CtBP1 transcriptional regulation using an in vitro reporter assay, suggesting that NADH/NAD+ binding, not the dehydrogenase activity of CtBP1, is important for its repressor activity (17, 24). On the other hand, a study by Zhang and colleagues found different phenotypes for Drosophila CtBP with impaired enzymatic activity (25). Consequently, the true biological importance of the CtBP dehydrogenase activity still remains unclear.
Figure 1. Molecular details of the rat CtBP1-S/E1A peptide interaction.

(A) A domain organization map of CtBP1. Each domain and their corresponding amino acid ranges are listed below the linear map while the binding interfaces and their corresponding amino acid residues or ranges are listed above. (B) The crystal structure of the rat CtBP1-S dimer (blue and purple ribbons) in complex with the PIDLSKK E1A peptide (green) (PDB:1HL3) (18). The NADH cofactor is represented as orange sticks in the center of each CtBP1-S molecule. (C) An electrostatic surface representation of CtBP1-S at the peptide interface. The PIDLSKK peptide is shown as green sticks. (D) A surface representation of CtBP1-S highlighting the location of the two residues, V55 and A41 (orange). Mutation of these residues disrupts the ability of CtBP1-S to interact with E1A. (E) A sequence alignment of different dehydrogenase family members in the CtBP1 region that binds its transcription factor partners. The sequence alignment of CtBP family members and the two other dehydrogenases: lactate (LDH) and malate (MDH) dehydrogenases, was performed using Clustal Omega (122). Residues that make up the hydrophobic binding pocket are italicized, and those that are involved in hydrogen bonds are underlined.
To date, the best known endogenous substrate of CtBP is an intermediate within the methionine-salvage pathway, 4-methylthio-2-oxobutyric acid (MTOB) (26). However, CtBP1 was shown to display only weak catalytic activity, reducing MTOB with a kcat of 0.075/s. Since the enzymatic product, 2-hydroxy-4-methylthiobutyrate (HMTB), has no known biological function other than being reconverted to MTOB, the enzymatic activity of CtBP is currently viewed as secondary to its function as a transcriptional co-repressor.
As mentioned previously, the CtBP co-repressors were identified through their ability to bind to the C-terminus of the E1A adenoviral protein. This binding is predominately mediated by a conserved peptide motif in E1A, PXDLS (8), which has subsequently been found in many CtBP-binding transcription factor partners (27). The co-crystal of the CtBP1-E1A peptide (PIDLSKK) (18), 1D and 2D 1H-NMR, and biochemical analyses of 20-residue E1A peptides containing the PXDLS residues (28, 29) reveal additional details regarding the CtBP1-PXDLS interaction. In the co-crystal structure, the short peptide binds in an extended conformation docking the isoleucine and leucine side chains into hydrophobic cleft on the SBD end of the molecule (Figure 1C) (18, 30). Individual mutations of two residues lining the recognition cleft on rat CtBP/BARS, A41E and V55R (Figure 1D), abolished the interaction between CtBP/BARS and E1A, thereby confirming the location of the E1A-interface (18). NMR experiments using much longer peptides encompassing the PXDLS motif found that these peptides contain a series of C-terminal β-turns (including the PXDLS motif) and an N-terminal α-helix in solution. Interestingly, upon binding to CtBP1, these peptides undergo a β→α conformational rearrangement that extends the α-helix across the PXDLS motif in place of the β-turn. The significance of this β→α structural rearrangement upon CtBP1 binding within the context of full-length E1A (and possible other transcription factor partners) awaits further investigation.
At the other end of the molecule, CtBP dimerizes through a large predominately hydrophobic dimerization interface within the NBD (Figure 1B). As a result, dimeric CtBP could recruit two PXDLS-containing targets at opposite ends of the dimer. One predominant model of how CtBP fulfills its co-repressor functions suggests that one CtBP molecule within the dimer is recruited to specific promoter regions via their interaction with the PXDLS motif in DNA-binding transcription factors, while the other CtBP protein molecule recruits epigenetic modifiers such as HDAC3/4 and PcG (31–35), also through the PXDLS motif in most cases, thereby facilitating the epigenetic silencing of target promoters. Interestingly, there are several known CtBP partners that do not contain the canonical PXDLS motif (36), possibly interacting through a secondary interface on CtBP. This theory could allow a way for CtBP to distinguish between partners involved in recruiting CtBP to promoters and those involved in epigenetic modifications. For example, HDAC1/2, histone demethylase LSD1, and histone methyltransferase G9, do not contain the PXDLS motif, suggesting they interact with CtBP at an alternative interface or associate with CtBP indirectly. Furthermore, a secondary interface located in a non-conserved region between CtBP1 and CtBP2, such as the N-terminus, could impart differing functions between the two family members, unlike the highly conserved PXDLS binding cleft. Therefore, an additional model has been proposed where the CtBP dimer is targeted to a specific promoter region via two PXDLS-containing transcription factors, where it further recruits additional co-repressor proteins through a different interface. This theory is supported by work from Quinlan and colleagues who found that in addition to its PXDLS motif, ZNF217 is also able to bind CtBP2 through a second Arg-Arg-Thr (RRT) motif, also found in other zinc finger proteins (37). The crystal structure of the RRT containing peptide in complex with CtBP1-S identifies a second site, distinct from the PXDLS binding cleft, for additional protein-protein interactions. However, the RRT binding groove appears redundant to the PXDLS motif, since all known RRT motif-containing proteins also contain PXDLS motifs. In addition to these two binding sites, the unstructured C-terminal region also contains a PDZ-binding domain and a target site for SUMO modification (38–40). Interestingly, SUMO modifications have been shown to interact with HDACs, Co-REST, and LSD1 (41–43). Thus, the C-terminal region of CtBP proteins is also a potential site that could attract additional protein partners to the co-repressor complex.
Due to its numerous protein partners and mechanisms of interaction, it is not surprising that the precise mechanisms by which the CtBP family mediates transcriptional repression is both multifaceted and context-dependent. For example, the CtBP transcriptional complex can mediate transcriptional repression in a manner that is both HDAC-dependent and independent, as CtBP target promoters display both sensitivity and insensitivity to the HDAC inhibitor, trichostatin A (34, 44). In addition to altering the expression levels of proteins, CtBP1 can regulate the expression of numerous miRNAs involved in cell cycle, cell communication, and primary metabolic processes (45). CtBP can also antagonize the activity of the global transcriptional co-activators, p300/CBP, through possible inhibition of its HAT activity (46, 47). An excellent review by Chinnadurai in 2007 details the diverse mechanisms underlying CtBP-mediated transcriptional regulation, mainly examining its role as a transcriptional repressor (1).
Since the earlier studies implicating CtBP as a transcriptional co-repressor, several studies have also revealed context-specific roles of CtBP in transcriptional activation. For instance, CtBP2 has been shown to directly activate the expression levels of Tiam1 in a NADH-dependent manner (48), as well as activate the Transcription factor 4 (TCF-4) signaling pathway (49). In human multidrug resistance (MDR) cancer cell lines, CtBP1 has been shown to directly activate the expression of the MDR1 gene, thereby increasing the levels of the P-glycoprotein and drug resistance (50). In gastrointestinal endocrine cells, CtBP can activate the transcription of NeuroD1 target genes in collaboration with other CtBP-associated proteins, including LSD1 and PCAF (51). In differentiating epidermal keratinocytes, the DNA-binding transcription factor, ZNF750, requires the CtBP complex to regulate both the activation of differentiation genes and the repression of progenitor genes (52). Interestingly, the NADH-unbound form of CtBP has also been implicated in distinct transcriptional activities unique to apo-CtBP, including the interaction with specific transcriptional regulators, p300 and Hdm2 (53, 54), and the transcriptional activation of Wingless pathway targets (55).
The CtBP co-repressors play multiple and context-dependent roles in oncogenic processes
In normal cells, proper temporal and spatial regulation of the myriad of transcription factors ensures a balance between proliferation and differentiation. Perturbation of the regulation of these transcriptional factors, especially those capable of modulating entire gene networks, can promote a tumorigenic phenotype (56). In cancer, transcriptional co-repressors are often altered by either loss- or gain-of-function mutations that lead to transcriptional imbalance (57, 58), thereby co-repressor proteins have emerged as an important facet of cancer etiology (59). Consequently, further research directed to expand our present understanding of transcriptional co-repressors in oncogenesis, could impact the future development of new therapies.
The importance of the CtBP co-repressor complex in multiple developmental programs suggests that the overexpression of CtBP proteins in adult tissues could play a role in both tumorigenesis and tumor progression. Tumorigenic cells frequently exhibit a more embryonic phenotype, having been reprogrammed to activate survival, proliferation, and other cancer hallmark pathways (60), suggesting that inhibiting developmental transcriptional pathways in cancerous tissue may be an effective therapeutic approach. In addition to overexpression, CtBP can also be hyper-activated in cancer cells through other means. Cancer cells typically have higher levels of NADH due to both hypoxia and pseudo-hypoxia (NADH production when oxygen is not limited) (21, 23, 61, 62). NADH binds to the CtBP proteins with a high affinity (Kd = 100 nM) triggering a conformational change that favors its binding to transcriptional repressors (21) and promotes the homo and heterodimerization of CtBP proteins (4, 18, 22, 63). Consistent with these findings, elevated NADH levels under hypoxic conditions associated with solid tumors repress the transcription of the CtBP target gene, E-cadherin, and increases cell migration, both of which are reversed by CtBP knockdown (23). Furthermore, increased NADH levels resulting from high-caloric intake, promotes both prostate and breast carcinogenesis in a CtBP1-dependent manner in vivo (45, 64, 65). Since CtBP1 is the more commonly studied CtBP family member in cancer, the remainder of this review will focus on CtBP1, its known oncogenic roles, and the possibility of therapeutically targeting this protein.
The oncogenic ramifications of CtBP1 overexpression was first glimpsed in early studies investigating its ability to interact with the adenovirus E1A oncogene (7, 8). Chinnadurai and colleagues reported that mutations within the PLDLS motif of E1A enhanced primary epithelial cell transformation and metastasis in cooperation with the Ras oncogene (66). These studies found that the E1A-CtBP1 interaction restricted tumorigenesis by antagonizing CtBP1 activity. Furthermore, expression of E1A in several cancer cell lines suppresses their oncogenic phenotypes by activating the expression of multiple epithelial genes that are normally suppressed by CtBP1. For example, the E1A-CtBP1 interaction was important for the re-expression of the cell adhesion proteins, E-cadherin, desmoglein-2 and plakoglobin, suggesting that E1A alleviates transcriptional repression of these genes by disrupting CtBP’s ability to interact with its endogenous transcription factor partners (67).
Following these early studies, CtBP1 overexpression has been observed in a number of different cancers, including prostate (64, 68), melanomas (69), colon (70), leukemia (71–73), ovarian (74, 75), and breast cancer (76, 77), among others. In most cases CtBP1 overexpression is pro-tumorigenic. It is of note however, that Takayama and colleagues found that in androgen receptor (AR)-positive prostate cancer cells, CtBP1 exhibited tumor-suppressive effects (78), as opposed to Wang and colleagues who found CtBP1 overexpression promoted cellular proliferation in predominately AR-negative prostate cancer cell lines (68). Additionally, Poser and colleagues found that a large percentage of melanomas express low mRNA levels of CtBP1, increasing their invasive potential (but not cell proliferation) through the down-regulation of the Melanoma Inhibitory Activity (MIA) protein (79, 80). However, on the protein level, Deng and colleagues observed overexpression of CtBP1 in a large percentage of melanomas and found that CtBP1 overexpression contributed to an increase in cellular proliferation and a decrease in genome instability (69). In breast cancer cells, ERα upregulates the expression of multiple growth factors, growth factor receptors, and cell cycle regulators, and downregulates anti-proliferative and pro-apoptotic genes in an estrogen-dependent manner. CtBP1 has been shown to regulate the transcriptional repression of the ERα-repressive genes through p300 (81). However, CtBP1 can also inhibit the ERα-responsive genes through ZNF366 (82), suggesting that CtBP1’s oncogenic role is context-dependent and that the concurrent expression pattern of its transcription factor partners could modulate multiple aspects of its oncogenic properties.
Nevertheless, in the numerous cancers where CtBP1 overexpression is pro-tumorigenic, CtBP1 has been implicated in multiple “hallmarks of cancer” through its transcriptional regulation of gene networks associated with malignant behavior (Figure 2). In these cancers, CtBP1 promotes a broad range of pro-tumorigenic and cancer stem cell (CSC) phenotypes, including increased cell survival, proliferation, migration/invasion, EMT, and other stem cell-like features. This is consistent with its ability to influence tumor evolution and progression through suppressing the expression of multiple epithelial and pro-apoptotic genes (14). In the remainder of this section, we will discuss the oncogenic roles of CtBP1 from the context of the established cancer hallmarks.
Figure 2. The CtBP1 transcriptional co-repressor influences a number of Cancer Hallmarks to carry out its tumor-promoting functions.
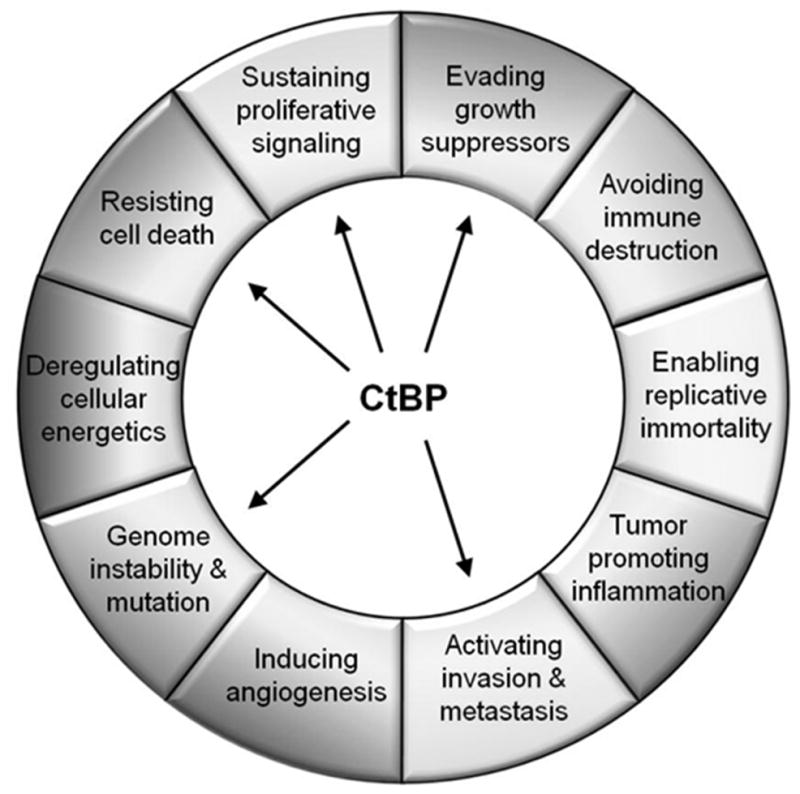
CtBP1 has been shown to play a role in five out of the six original cancer hallmarks and one of the two enabling characteristics, highlighting the potential importance of therapeutically targeting this transcriptional complex.
To date, the most well understood cancer hallmarks associated with CtBP1 overexpression are the evasion of cell death and increased EMT. In addition to these two hallmarks, CtBP1 proteins play critical roles in promoting cellular proliferation, genome instability, as well as evading growth suppressors. In the first hallmark mentioned above (evasion of cell death), CtBP1 can promote cell survival through the transcriptional suppression of multiple pro-apoptotic genes including: Bax, Bik, Bim, Bmf, Noxa, p21, Puma, and PERP (14, 83, 84). CtBP1-null mouse embryonic fibroblasts (MEF) are hypersensitized to apoptosis triggered by diverse stimuli (such as Fas ligand and UV-radiation) as compared to a CtBP1 rescue, and this hypersensitization to apoptosis is mediated through the release of transcriptional suppression of Bax, Noxa, p21 and PERP (14, 83, 84). Additionally, siRNA knockdown of CtBP2 enhanced Bim and Bmf expression in osteosarcoma cells (84). In colon cancer cells, proteasome-dependent CtBP protein degradation, mediated through its interaction with p19ARF, induces p53-independent apoptosis (85). CtBP1 also represses epithelial gene programming associated with insensitivity to anoikis (a specific type of apoptosis triggered by the loss of cell adhesion) by repression of E-cadherin, keratin 8, occluding, and plakoglobin (14, 67). The repression of epithelial genes, such as E-cadherin, is an important process in attaining anoikis insensitivity, allowing cells to survive proceeding detachment from the extracellular matrix (86).
The loss of cellular adhesion molecules and the acquisition of anoikis resistance are also significant for the second cancer hallmark in which CtBP1 has been implicated, the ability to initiate cancer invasion and metastasis. The acquisition of these traits often leads to a more motile and invasive mesenchymal phenotype. EMT is believed to be a prominent means by which transformed epithelial cells acquire the ability to invade, resist apoptosis, and disseminate (87). In particular, CtBP1 has been shown to promote a more mesenchymal phenotype through the repression of the epithelial marker, E-cadherin, by recruiting the EMT-inducing transcription factor, ZEB1/2, to the E-cadherin promoter (33, 88) and promoting epigenetic modifications through histone deacetylases (34, 89). Interestingly, ZEB1 overexpression and associated E-cadherin downregulation have been found in several cancers, including, breast, uterine, and colon cancers (90–94), thus, the role of CtBP1 in EMT may be particularly critical in these cancers that also overexpress the ZEB family of transcription factors. Interestingly, in hepatocellular carcinomas, both CtBP1 and 2 increase cellular invasion independent of EMT and this action is reversible though their interaction with the INK4 family member, p19ARF (95). However, the exact mechanism through which CtBP1 is able to promote the EMT-independent invasive phenotype is still unclear.
Other hallmarks with which CtBP1 is associated are an increase in cell proliferation and the evasion of growth/tumor suppressor functions. CtBP1 has been shown to negatively regulate numerous tumor suppressor genes including: Phosphatase and Tension Homologue (PTEN) (14, 96), Wnt (97, 98), and several Cyclin-Dependent Kinase Inhibitors (CDKIs) (99–101). PTEN is a known regulator of the cell cycle, and its repression results in increased cell migration and proliferation (102). PTEN expression is repressed by the Snail transcription factor in a CtBP1-dependent manner, which results in a pro-survival effect (103). CtBP1 has also been shown to interact with the Adenomatous Polyposis Coli (APC) tumor suppressor to repress the expression of Wnt target genes (97). In colorectal cancer cells, mutations in APC disrupting the CtBP-APC complex result in aberrant Wnt signaling and cancer progression (98). Finally, CtBP1 modulates the expression and activities of the Ink4 family of tumor suppressors. The Ink4 family of cyclin-dependent kinase inhibitors regulate their onco-suppressive properties by triggering cell cycle arrest (104). Specifically, CtBP2 represses the tumor suppressor p16INK4a and prevents senescence in human esophageal squamous cell carcinomas (100), suggesting that CtBP overexpression promotes the evasion of cell cycle regulation. Deng and colleagues made similar observations in melanoma where the overexpression of CtBP1 repressed the expression of p16INK4a potentially contributing to increased proliferation (69). Moreover, in prostate adenocarcinomas CtBP1 represses the transcription of the p21waf1/cip1 gene, likely through formation of a corepressor complex with PARP, which is disrupted following DNA damage signals. (101). The implications of CtBP1-dependent repression of these proteins highlight a key role for CtBP1 in the evasion of cell cycle checkpoints and enhanced tumor progression. In murine bone marrow progenitor cells, the direct interaction between the AML1/MDS1/EVI1(AME) leukemia-associated fusion gene and CtBP1 is important and required for an increase in cell proliferation and abnormal differentiation (77). Thus, the improper recruitment of CtBP1 to a promoter site is critical for the transformation of these leukemic cells and its disruption may be a way to restore their normal growth and differentiation patterns.
CtBP1 is implicated in the cancer hallmark of genome instability through its effect on the Breast Cancer 1 and 2, early onset (BRCA1 and 2) genes. BRCA1 and 2 play crucial roles in the homologous recombination repair pathway of DNA double-strand breaks and in DNA damage signaling. Deficiencies in BRCA1 lead to accelerated proliferation, abnormal mitosis, increased genomic instability, and tumorigenesis (105). In breast cancer cells, CtBP1 assembles on the BRCA1 promoter as part of a co-repressor complex, containing p130 and histone deacetylase 1 (HDAC1), which alters local histone acetylation at the BRCA1 promoter and subsequent transcription (106). Disruption of this repressor complex (either through estrogen induction or NAD+/NADH ratios) leads to eviction of CtBP1 and HDAC1, increased histone acetylation and subsequent BRCA1 transcription, thereby associating CtBP1 overexpression with genome instability, an enabling characteristic of cancer hallmarks. Furthermore, a decrease in BRCA1 expression levels correlated with an increase in CtBP1 levels in both head and neck cancers and melanomas, where CtBP1 was shown to repress BRCA1, contributing to an increase in DNA damage (69, 107). In the case of BRCA2, its deficiency has also been linked to many cases of breast, pancreatic and ovarian carcinomas (108), and similar to BRCA1, the BRCA2 gene can be epigenetically silenced in breast cancer cells, through the recruitment of the SLUG-CtBP1-HDAC1 repressor complex (109).
The above data illustrate that CtBP1 plays a critical role in the development and progression of multiple cancer types through the regulation of several cancer hallmarks. Understanding and predicting the consequences of altered co-repressor expression patterns in cancer cells has diagnostic and prognostic implications, and disrupting CtBP1-mediated transcriptional repression, in particular, represents a unique therapeutic opportunity to cripple multiple oncogenic pathways.
Targeting the CtBP1 transcriptional co-repressor complex for cancer therapy
As a transcriptional regulator of broad developmental processes capable of promoting malignant growth in adult tissues, the CtBP1 transcriptional co-repressor complex is an attractive therapeutic target for the treatment of diverse tumor types. Although therapeutically targeting transcription factors is typically considered a challenging endeavor, there has been increasing progress made in exploiting various modes of transcriptional regulation including protein-protein interactions, enzymatic activity, DNA binding, and epigenetic alterations. In the remaining section, we will focus on the advantages and feasibility of targeting the CtBP1/transcription factor interaction, the dehydrogenase activity, and the CtBP1-dimerization in CtBP1-mediated tumor progression.
The first approach to decrease CtBP1 activity is to identify small molecules inhibiting the interaction between CtBP1 and its protein partners. Despite the recent advancements in high-throughput fragment-based, in silico, and in vitro screening technologies, identifying inhibitors of protein-protein interactions is still a challenging endeavor. Unlike many ligand-receptor or substrate-enzyme interactions, most protein-protein interactions have not evolved to accommodate small molecules, so the druggability of each protein must be evaluated individually. Nevertheless, mounting efforts from many research groups targeting protein-protein interactions have led to the establishment of a more solid framework regarding the conceptual and technical fundamentals required for the development of a successful protein-protein interaction inhibitor.
Current evidence indicates that small molecules can interfere with “hot spots” on protein interfaces, to disrupt protein-protein interactions (110). The CtBP1-transcription factor interaction has some characteristics that suggest it contains such “hot spots” and is amenable to small molecule intervention, when examining the crystal structure of the CtBP1/PXDLS complex and mutational analyses (18, 28, 29). The PXDLS peptide interacts with the SBD of CtBP1 primarily through two hydrophobic side chains docking into a hydrophobic pocket (Figure 1C) (18). The interface is relatively small and a single amino acid mutation in either the hydrophobic cleft of CtBP1 or the conserved PXDLS motif is able to disrupt the protein interaction and subsequent transcriptional repression (28, 36). In particular, when the residues Pro, Leu, and Ser within the PXDLS motif are mutated to an Ala residue, there is 10–35-fold loss in binding (28). Although the SBD has structural homology to other 2-hydroxyacid dehydrogenases, this interface has little to no similarity compared to two other well-known 2-hydroxyacid dehydrogenases, lactate and malate dehydrogenase (Figure 1E), and currently there are no known dehydrogenases involved in protein-protein interactions through this interface. This suggests that it is possible to develop highly selective small molecules capable of disrupting the CtBP/transcription factor interaction. This hydrophobic groove is also fully conserved between CtBP1 and 2 (Figure 1E). Because these grooves can form pockets more comparable to an enzyme active site, this suggests that small molecule inhibitors could be engineered that bind in this cleft to perturb CtBP-mediated transcriptional repression. This, in combination with the recognized importance of the CtBP1 transcription factor interactions (through single amino acid mutations in the PXDLS sequence or the CtBP1-binding cleft) in gene repression and tumorigenesis (28, 36), suggest a small molecule inhibitor targeting this protein interaction may be an effective therapeutic approach.
Screening of pharmacologically active compounds using an AlphaScreen assay led to the identification of a selective inhibitor of the CtBP1-E1A complex, NSC95397 (Figure 3A) (111). NSC95397 clearly inhibited the CtBP1-transcription factor interaction in both in vitro assays and in a luciferase reporter assay using a CtBP1-specific promoter. Although NSC95397 displayed specificity towards the CtBP1-E1A interaction in secondary protein-protein interaction, enzymatic, and in vitro assays, NSC95397 does have other known activities. Considering that NSC95397 also targets cdc25 (112) and the spliceosome (113), the effect of NSC95397 in cell culture could come from the inhibition of multiple pathways by this compound. For NSC95397 to be a useful CtBP1 inhibitor, as a chemical probe or therapeutic agent, future medicinal chemistry efforts will be required to improve the potency and specificity of this compound. Importantly, these results suggest that a larger high throughput screening (HTS) campaign maybe be a useful strategy for generating additional and more promising inhibitors of CtBP1.
Figure 3. Chemical structures of the current compounds identified as inhibitors of CtBP1 transcriptional activity.

(A) NSC95397 was discovered through high-throughput screening. (B) 2-keto-4-methylthiobutyrate (MTOB) was identified through screening known substrates of other dehydrogenases. (C) Phenylpyruvate and 2-hydroxyimino-3-phenylproanoic acid are MTOB derivatives generated through structure-based design. (D) The cyclic peptide-61 (CP61) amino acid sequence identified from screening a cyclic peptide library.
A second opportunity for targeting the CtBP1 co-repressor activity is through its dehydrogenase activity. Structurally, the CtBP proteins have a defined active site capable of accommodating either an endogenous substrate or a small molecule inhibitor (16). However, the structural similarity and identical catalytic mechanism between CtBP1 and other NADH-dependent dehydrogenases could make the identification of specific enzymatic inhibitors challenging. Nonetheless, efforts have been made targeting the enzymatic activity of CtBP1 as a means to inhibit CtBP1-mediated tumorigenesis. The compound, 2-keto-4-methylthiobutyrate (MTOB) (Figure 3B), and a few of its derivatives are the most thoroughly investigated substrate-based inhibitors of the CtBP family (114, 115). These substrates were originally identified through the enzymatic screening of other known dehydrogenase substrates and a few derivatives. MTOB displays a biphasic saturation curve with substrate inhibition at higher concentrations. Treatment with high concentrations (4 and 10 mM) of MTOB can disrupt CtBP1 recruitment to target promoters thereby antagonizing CtBP1-mediated transcriptional regulation (65, 116). In HCT116 colon cancer cells, MTOB can induce apoptosis through eviction of CtBP1 from the Bik promoter (116). Additionally, MTOB can shift breast cancer cells from a more mesenchymal to an epithelial phenotype by preventing CtBP localization to target promoters (65). The crystal structure of MTOB bound within the active site of CtBP1/2 does not reveal any large conformational changes from the apo structure (16). Furthermore, MTOB is unable to disrupt the CtBP1-E1A interaction in an Alphascreen or fluorescence polarization assay at 100 μM (111). A possible explanation for the mechanism of inhibition is that MTOB alters the NADH/NAD+ ratio by consuming NADH in the dehydrogenase reaction, consequently shifting the monomeric and dimeric state of CtBP1, thereby regulating its transcriptional activities.
Although high MTOB concentrations are required for inhibition of CtBP transcriptional repression, MTOB’s clear anti-tumorigenic effect on cancer cells provides evidence that small molecules could be developed to treat tumors dependent upon CtBP activity. Very recently, using a structure-guided approach, Hilbert and colleagues designed several high affinity MTOB derivatives by taking advantage of an active site tryptophan unique to CtBP1 and stabilizing the carboxylic acid moiety of MTOB (Figure 3C) (16). Isothermal calorimetry and enzymatic experiments revealed that these chemical modifications improved the binding affinity 1000-fold over MTOB and were also able to inhibit CtBP1 enzymatic activity. However, the in vivo impact of these enzymatic inhibitors on CtBP1-mediated transcription and tumorigenesis has yet to be determined.
A third approach to inhibit CtBP1 activity is to target the CtBP dimerization interface. NADH-dependent dimerization of CtBP1 is essential for many of its co-repressive functions (117). CtBP1 homodimerizes and heterodimerizes with CtBP2 through a predominately large hydrophobic interface in its nucleotide-binding domain, to unite DNA-associated transcription factors with epigenetic regulators. Birts and colleagues exploited this feature and identified another class of CtBP1 inhibitors through screening a cyclic peptide library (Figure 3D) (118). The cyclic peptide, CP61, inhibits CtBP dimerization and NADH binding in vitro and in vivo with an IC50 value of 19 μM. After fusion to the TAT cell-penetrating peptide (CPP), CP61 can internalize into cells and antagonize CtBP-associated proliferation and maintenance of mitotic fidelity in the highly glycolytic MCF-7 breast cancer cells. However, the direct effect of the peptide on either the enzymatic or transcriptional activity of CtBP1 was not explored in this study. Regardless, this work further demonstrates the validity of the CtBP1 co-repressor as a target for anti-cancer therapeutics, and while the full implication of this cyclic peptide on the repressive activity of CtBP1 in cancer cells remains to be explored, this peptide could serve as a useful tool to further our understanding of the biological importance of the monomeric and dimeric forms of CtBP1.
A major advantage of targeting CtBP1 is that CtBP1 expression levels are generally low or absent in many adult tissues, with the exception of the brain, adipose tissue, and skeletal muscle (3, 119, 120), therefore negative side effects in response to a CtBP1-targeting therapy would likely be limited. CtBP is expressed in both the nucleus and the pre-synaptic terminals of cultured hippocampal neurons, suggesting a potential function for CtBP in learning and memory that is distinct from its role as a transcriptional co-repressor, however a mechanistic understanding of the non-transcriptional role of CtBP is lacking (119). Furthermore, treatment of primary cerebellar granule neurons with 5 mM MTOB, induces significant neuronal apoptosis, suggesting that the enzymatic activity of CtBP may play a role in the survival of primary neurons (84). In adipose tissue, CtBP has been shown to induce the phenotypic conversion of white adipocytes to a brown phenotype through the transcriptional repression of a set of white fat genes (120). White adipose tissue secretes stress-related cytokines that can contribute to insulin resistance in obese subjects unlike brown adipose tissue (121). Although the full physiological effect of CtBP inhibition in either of these systems is not yet fully understood, one would want to carefully monitor these tissues in response to any inhibitors developed targeting the CtBP transcriptional complex. In general, because we do not anticipate that the inhibition of CtBP1 transcriptional repression will have an adverse effect in most tissues (due to low CtBP1 expression levels in adult tissue), inhibitors targeting the CtBP1 complex are overall expected to be well tolerated.
In conclusion, targeting the CtBP1 co-repressor complex still remains a relatively unexplored opportunity. To date, much progress has been made to understand the role of the CtBP-transcriptional complex in tumorigenesis and it remains an appealing target due to its limited expression in most adult tissues and its ability to reactivate developmental programs critical for tumorigenesis/metastasis when aberrantly re-expressed. Current CtBP1 inhibitors are promising leads that target CtBP1 activity from unique angles, each of which could potentially be developed into highly specific and effective therapeutic agents targeting tumors overexpressing CtBP1.
Acknowledgments
Grant Support:
The work of the authors of this review article is supported by a Senior Research Training Fellowship from the American Lung Association (RT-415543 to M.A. Blevins), a Training Fellowship from the National Institutes of Health (T32CA174648-01 to M.A. Blevins), a Research Grant from the VA Eastern Colorado Health Care System (5I01BX002370-03 to Q. Zhang) and a Research Grant from the National Institute of Health (R21CA185752 to R. Zhao).
The authors would like to acknowledge and thank Dr Qinghong Zhang for her contribution to the overall concept, design, and review of the manuscript.
Footnotes
Disclosure of Potential Conflicts of Interest:
No potential conflicts of interest.
References
- 1.Chinnadurai G. Transcriptional regulation by C-terminal binding proteins. Int J Biochem Cell Biol. 2007;39(9):1593–607. doi: 10.1016/j.biocel.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 2.Spano S, Silletta MG, Colanzi A, Alberti S, Fiucci G, Valente C, et al. Molecular cloning and functional characterization of brefeldin A-ADP-ribosylated substrate. A novel protein involved in the maintenance of the Golgi structure. J Biol Chem. 1999 Jun 18;274(25):17705–10. doi: 10.1074/jbc.274.25.17705. [DOI] [PubMed] [Google Scholar]
- 3.Katsanis N, Fisher EM. A novel C-terminal binding protein (CTBP2) is closely related to CTBP1, an adenovirus E1A-binding protein, and maps to human chromosome 21q21.3. Genomics. 1998 Jan 15;47(2):294–9. doi: 10.1006/geno.1997.5115. [DOI] [PubMed] [Google Scholar]
- 4.Verger A, Quinlan KG, Crofts LA, Spano S, Corda D, Kable EP, et al. Mechanisms directing the nuclear localization of the CtBP family proteins. Mol Cell Biol. 2006 Jul;26(13):4882–94. doi: 10.1128/MCB.02402-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmitz F, Konigstorfer A, Sudhof TC. RIBEYE, a component of synaptic ribbons: a protein’s journey through evolution provides insight into synaptic ribbon function. Neuron. 2000 Dec;28(3):857–72. doi: 10.1016/s0896-6273(00)00159-8. [DOI] [PubMed] [Google Scholar]
- 6.Chen S, Whetstine JR, Ghosh S, Hanover JA, Gali RR, Grosu P, et al. The conserved NAD(H)-dependent corepressor CTBP-1 regulates Caenorhabditis elegans life span. Proc Natl Acad Sci U S A. 2009 Feb 3;106(5):1496–501. doi: 10.1073/pnas.0802674106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyd JM, Subramanian T, Schaeper U, La Regina M, Bayley S, Chinnadurai G. A region in the C-terminus of adenovirus 2/5 E1a protein is required for association with a cellular phosphoprotein and important for the negative modulation of T24-ras mediated transformation, tumorigenesis and metastasis. Embo J. 1993 Feb;12(2):469–78. doi: 10.1002/j.1460-2075.1993.tb05679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaeper U, Boyd JM, Verma S, Uhlmann E, Subramanian T, Chinnadurai G. Molecular cloning and characterization of a cellular phosphoprotein that interacts with a conserved C-terminal domain of adenovirus E1A involved in negative modulation of oncogenic transformation. Proc Natl Acad Sci U S A. 1995 Nov 7;92(23):10467–71. doi: 10.1073/pnas.92.23.10467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chinnadurai G. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol Cell. 2002 Feb;9(2):213–24. doi: 10.1016/s1097-2765(02)00443-4. [DOI] [PubMed] [Google Scholar]
- 10.Hildebrand JD, Soriano P. Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol Cell Biol. 2002 Aug;22(15):5296–307. doi: 10.1128/MCB.22.15.5296-5307.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoang CQ, Burnett ME, Curtiss J. Drosophila CtBP regulates proliferation and differentiation of eye precursors and complexes with Eyeless, Dachshund, Dan, and Danr during eye and antennal development. Dev Dyn. 2010 Sep;239(9):2367–85. doi: 10.1002/dvdy.22380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Hateren N, Shenton T, Borycki AG. Expression of avian C-terminal binding proteins (Ctbp1 and Ctbp2) during embryonic development. Dev Dyn. 2006 Feb;235(2):490–5. doi: 10.1002/dvdy.20612. [DOI] [PubMed] [Google Scholar]
- 13.Furusawa T, Moribe H, Kondoh H, Higashi Y. Identification of CtBP1 and CtBP2 as corepressors of zinc finger-homeodomain factor deltaEF1. Mol Cell Biol. 1999 Dec;19(12):8581–90. doi: 10.1128/mcb.19.12.8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grooteclaes M, Deveraux Q, Hildebrand J, Zhang Q, Goodman RH, Frisch SM. C-terminal-binding protein corepresses epithelial and proapoptotic gene expression programs. Proc Natl Acad Sci U S A. 2003 Apr 15;100(8):4568–73. doi: 10.1073/pnas.0830998100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melhuish TA, Wotton D. The interaction of the carboxyl terminus-binding protein with the Smad corepressor TGIF is disrupted by a holoprosencephaly mutation in TGIF. J Biol Chem. 2000 Dec 15;275(50):39762–6. doi: 10.1074/jbc.C000416200. [DOI] [PubMed] [Google Scholar]
- 16.Hilbert BJ, Grossman SR, Schiffer CA, Royer WE., Jr Crystal structures of human CtBP in complex with substrate MTOB reveal active site features useful for inhibitor design. FEBS Lett. 2014 May 2;588(9):1743–8. doi: 10.1016/j.febslet.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar V, Carlson JE, Ohgi KA, Edwards TA, Rose DW, Escalante CR, et al. Transcription corepressor CtBP is an NAD(+)-regulated dehydrogenase. Mol Cell. 2002 Oct;10(4):857–69. doi: 10.1016/s1097-2765(02)00650-0. [DOI] [PubMed] [Google Scholar]
- 18.Nardini M, Spano S, Cericola C, Pesce A, Massaro A, Millo E, et al. CtBP/BARS: a dual-function protein involved in transcription co-repression and Golgi membrane fission. Embo J. 2003 Jun 16;22(12):3122–30. doi: 10.1093/emboj/cdg283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nardini M, Svergun D, Konarev PV, Spano S, Fasano M, Bracco C, et al. The C-terminal domain of the transcriptional corepressor CtBP is intrinsically unstructured. Protein Sci. 2006 May;15(5):1042–50. doi: 10.1110/ps.062115406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fjeld CC, Birdsong WT, Goodman RH. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc Natl Acad Sci U S A. 2003 Aug 5;100(16):9202–7. doi: 10.1073/pnas.1633591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science. 2002 Mar 8;295(5561):1895–7. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
- 22.Balasubramanian P, Zhao LJ, Chinnadurai G. Nicotinamide adenine dinucleotide stimulates oligomerization, interaction with adenovirus E1A and an intrinsic dehydrogenase activity of CtBP. FEBS Lett. 2003 Feb 27;537(1–3):157–60. doi: 10.1016/s0014-5793(03)00119-4. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Q, Wang SY, Nottke AC, Rocheleau JV, Piston DW, Goodman RH. Redox sensor CtBP mediates hypoxia-induced tumor cell migration. Proc Natl Acad Sci U S A. 2006 Jun 13;103(24):9029–33. doi: 10.1073/pnas.0603269103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sutrias-Grau M, Arnosti DN. CtBP contributes quantitatively to Knirps repression activity in an NAD binding-dependent manner. Mol Cell Biol. 2004 Jul;24(13):5953–66. doi: 10.1128/MCB.24.13.5953-5966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang YW, Arnosti DN. Conserved catalytic and C-terminal regulatory domains of the C-terminal binding protein corepressor fine-tune the transcriptional response in development. Mol Cell Biol. Jan;31(2):375–84. doi: 10.1128/MCB.00772-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Achouri Y, Noel G, Van Schaftingen E. 2-Keto-4-methylthiobutyrate, an intermediate in the methionine salvage pathway, is a good substrate for CtBP1. Biochem Biophys Res Commun. 2007 Jan 26;352(4):903–6. doi: 10.1016/j.bbrc.2006.11.111. [DOI] [PubMed] [Google Scholar]
- 27.Byun JS, Gardner K. C-Terminal Binding Protein: A Molecular Link between Metabolic Imbalance and Epigenetic Regulation in Breast Cancer. Int J Cell Biol. 2013;2013:647975. doi: 10.1155/2013/647975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molloy DP, Milner AE, Yakub IK, Chinnadurai G, Gallimore PH, Grand RJ. Structural determinants present in the C-terminal binding protein binding site of adenovirus early region 1A proteins. J Biol Chem. 1998 Aug 14;273(33):20867–76. doi: 10.1074/jbc.273.33.20867. [DOI] [PubMed] [Google Scholar]
- 29.Molloy DP, Barral PM, Gallimore PH, Grand RJ. The effect of CtBP1 binding on the structure of the C-terminal region of adenovirus 12 early region 1A. Virology. 2007 Jul 5;363(2):342–56. doi: 10.1016/j.virol.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 30.Lundblad J. Madame Curie Bioscience Database [Internet] Landes Biosciences; 2000. Structural Determinants of CtBP Function. [Google Scholar]
- 31.Dahiya A, Wong S, Gonzalo S, Gavin M, Dean DC. Linking the Rb and polycomb pathways. Mol Cell. 2001 Sep;8(3):557–69. doi: 10.1016/s1097-2765(01)00346-x. [DOI] [PubMed] [Google Scholar]
- 32.Kuppuswamy M, Vijayalingam S, Zhao LJ, Zhou Y, Subramanian T, Ryerse J, et al. Role of the PLDLS-binding cleft region of CtBP1 in recruitment of core and auxiliary components of the corepressor complex. Mol Cell Biol. 2008 Jan;28(1):269–81. doi: 10.1128/MCB.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Postigo AA, Dean DC. ZEB represses transcription through interaction with the corepressor CtBP. Proc Natl Acad Sci U S A. 1999 Jun 8;96(12):6683–8. doi: 10.1073/pnas.96.12.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, et al. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003 Apr 17;422(6933):735–8. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 35.Sewalt RG, Gunster MJ, van der Vlag J, Satijn DP, Otte AP. C-Terminal binding protein is a transcriptional repressor that interacts with a specific class of vertebrate Polycomb proteins. Mol Cell Biol. 1999 Jan;19(1):777–87. doi: 10.1128/mcb.19.1.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quinlan KG, Verger A, Kwok A, Lee SH, Perdomo J, Nardini M, et al. Role of the C-terminal binding protein PXDLS motif binding cleft in protein interactions and transcriptional repression. Mol Cell Biol. 2006 Nov;26(21):8202–13. doi: 10.1128/MCB.00445-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quinlan KG, Nardini M, Verger A, Francescato P, Yaswen P, Corda D, et al. Specific recognition of ZNF217 and other zinc finger proteins at a surface groove of C-terminal binding proteins. Mol Cell Biol. 2006 Nov;26(21):8159–72. doi: 10.1128/MCB.00680-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riefler GM, Firestein BL. Binding of neuronal nitric-oxide synthase (nNOS) to carboxyl-terminal-binding protein (CtBP) changes the localization of CtBP from the nucleus to the cytosol: a novel function for targeting by the PDZ domain of nNOS. J Biol Chem. 2001 Dec 21;276(51):48262–8. doi: 10.1074/jbc.M106503200. [DOI] [PubMed] [Google Scholar]
- 39.Kagey MH, Melhuish TA, Wotton D. The polycomb protein Pc2 is a SUMO E3. Cell. 2003 Apr 4;113(1):127–37. doi: 10.1016/s0092-8674(03)00159-4. [DOI] [PubMed] [Google Scholar]
- 40.Lin X, Sun B, Liang M, Liang YY, Gast A, Hildebrand J, et al. Opposed regulation of corepressor CtBP by SUMOylation and PDZ binding. Mol Cell. 2003 May;11(5):1389–96. doi: 10.1016/s1097-2765(03)00175-8. [DOI] [PubMed] [Google Scholar]
- 41.Gill G. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 2004 Sep 1;18(17):2046–59. doi: 10.1101/gad.1214604. [DOI] [PubMed] [Google Scholar]
- 42.Rosendorff A, Sakakibara S, Lu S, Kieff E, Xuan Y, DiBacco A, et al. NXP-2 association with SUMO-2 depends on lysines required for transcriptional repression. Proc Natl Acad Sci U S A. 2006 Apr 4;103(14):5308–13. doi: 10.1073/pnas.0601066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ouyang J, Shi Y, Valin A, Xuan Y, Gill G. Direct binding of CoREST1 to SUMO-2/3 contributes to gene-specific repression by the LSD1/CoREST1/HDAC complex. Mol Cell. 2009 Apr 24;34(2):145–54. doi: 10.1016/j.molcel.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryu JR, Arnosti DN. Functional similarity of Knirps CtBP-dependent and CtBP-independent transcriptional repressor activities. Nucleic Acids Res. 2003 Aug 1;31(15):4654–62. doi: 10.1093/nar/gkg491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Luca P, Dalton GN, Scalise GD, Moiola CP, Porretti J, Massillo C, et al. CtBP1 associates metabolic syndrome and breast carcinogenesis targeting multiple miRNAs. Oncotarget. 2016 Apr 5;7(14):18798–811. doi: 10.18632/oncotarget.7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meloni AR, Lai CH, Yao TP, Nevins JR. A mechanism of COOH-terminal binding protein-mediated repression. Mol Cancer Res. 2005 Oct;3(10):575–83. doi: 10.1158/1541-7786.MCR-05-0088. [DOI] [PubMed] [Google Scholar]
- 47.Senyuk V, Sinha KK, Nucifora G. Corepressor CtBP1 interacts with and specifically inhibits CBP activity. Arch Biochem Biophys. 2005 Sep 15;441(2):168–73. doi: 10.1016/j.abb.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 48.Paliwal S, Ho N, Parker D, Grossman SR. CtBP2 Promotes Human Cancer Cell Migration by Transcriptional Activation of Tiam1. Genes Cancer. 2012 Jul;3(7–8):481–90. doi: 10.1177/1947601912463695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel J, Baranwal S, Love IM, Patel NJ, Grossman SR, Patel BB. Inhibition of C-terminal binding protein attenuates transcription factor 4 signaling to selectively target colon cancer stem cells. Cell Cycle. 2014;13(22):3506–18. doi: 10.4161/15384101.2014.958407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin W, Scotto KW, Hait WN, Yang JM. Involvement of CtBP1 in the transcriptional activation of the MDR1 gene in human multidrug resistant cancer cells. Biochem Pharmacol. 2007 Sep 15;74(6):851–9. doi: 10.1016/j.bcp.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ray SK, Li HJ, Metzger E, Schule R, Leiter AB. CtBP and associated LSD1 are required for transcriptional activation by NeuroD1 in gastrointestinal endocrine cells. Mol Cell Biol. 2014 Jun;34(12):2308–17. doi: 10.1128/MCB.01600-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boxer LD, Barajas B, Tao S, Zhang J, Khavari PA. ZNF750 interacts with KLF4 and RCOR1, KDM1A, and CTBP1/2 chromatin regulators to repress epidermal progenitor genes and induce differentiation genes. Genes Dev. 2014 Sep 15;28(18):2013–26. doi: 10.1101/gad.246579.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mirnezami AH, Campbell SJ, Darley M, Primrose JN, Johnson PW, Blaydes JP. Hdm2 recruits a hypoxia-sensitive corepressor to negatively regulate p53-dependent transcription. Curr Biol. 2003 Jul 15;13(14):1234–9. doi: 10.1016/s0960-9822(03)00454-8. [DOI] [PubMed] [Google Scholar]
- 54.Kim JH, Cho EJ, Kim ST, Youn HD. CtBP represses p300-mediated transcriptional activation by direct association with its bromodomain. Nat Struct Mol Biol. 2005 May;12(5):423–8. doi: 10.1038/nsmb924. [DOI] [PubMed] [Google Scholar]
- 55.Bhambhani C, Chang JL, Akey DL, Cadigan KM. The oligomeric state of CtBP determines its role as a transcriptional co-activator and co-repressor of Wingless targets. EMBO J. 2011 May 18;30(10):2031–43. doi: 10.1038/emboj.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karamouzis MV, Gorgoulis VG, Papavassiliou AG. Transcription factors and neoplasia: vistas in novel drug design. Clin Cancer Res. 2002 May;8(5):949–61. [PubMed] [Google Scholar]
- 57.Vaiopoulos AG, Kostakis ID, Athanasoula K, Papavassiliou AG. Targeting transcription factor corepressors in tumor cells. Cell Mol Life Sci. 2012 Jun;69(11):1745–53. doi: 10.1007/s00018-012-0986-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Battaglia S, Maguire O, Campbell MJ. Transcription factor co-repressors in cancer biology: roles and targeting. Int J Cancer. 2010 Jun 1;126(11):2511–9. doi: 10.1002/ijc.25181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grivas PD, Papavassiliou AG. Transcriptional corepressors in cancer: emerging targets for therapeutic intervention. Cancer. 2012 Mar 15;119(6):1120–8. doi: 10.1002/cncr.27908. [DOI] [PubMed] [Google Scholar]
- 60.Hadjimichael C, Chanoumidou K, Papadopoulou N, Arampatzi P, Papamatheakis J, Kretsovali A. Common stemness regulators of embryonic and cancer stem cells. World J Stem Cells. 2015 Oct 26;7(9):1150–84. doi: 10.4252/wjsc.v7.i9.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sattler UG, Walenta S, Mueller-Klieser W. A bioluminescence technique for quantitative and structure-associated imaging of pyruvate. Lab Invest. 2007 Jan;87(1):84–92. doi: 10.1038/labinvest.3700493. [DOI] [PubMed] [Google Scholar]
- 62.Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cell Mol Life Sci. 2008 Dec;65(24):3981–99. doi: 10.1007/s00018-008-8224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thio SS, Bonventre JV, Hsu SI. The CtBP2 co-repressor is regulated by NADH-dependent dimerization and possesses a novel N-terminal repression domain. Nucleic Acids Res. 2004;32(5):1836–47. doi: 10.1093/nar/gkh344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moiola CP, De Luca P, Zalazar F, Cotignola J, Rodriguez-Segui SA, Gardner K, et al. Prostate tumor growth is impaired by CtBP1 depletion in high-fat diet-fed mice. Clin Cancer Res. 2014 Aug 1;20(15):4086–95. doi: 10.1158/1078-0432.CCR-14-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Di LJ, Byun JS, Wong MM, Wakano C, Taylor T, Bilke S, et al. Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat Commun. 2013;4:1449. doi: 10.1038/ncomms2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Subramanian T, Zhao LJ, Chinnadurai G. Interaction of CtBP with adenovirus E1A suppresses immortalization of primary epithelial cells and enhances virus replication during productive infection. Virology. 2013 Sep 1;443(2):313–20. doi: 10.1016/j.virol.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grooteclaes ML, Frisch SM. Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene. 2000 Aug 3;19(33):3823–8. doi: 10.1038/sj.onc.1203721. [DOI] [PubMed] [Google Scholar]
- 68.Wang R, Asangani IA, Chakravarthi BV, Ateeq B, Lonigro RJ, Cao Q, et al. Role of transcriptional corepressor CtBP1 in prostate cancer progression. Neoplasia. 2012 Oct;14(10):905–14. doi: 10.1593/neo.121192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deng H, Liu J, Deng Y, Han G, Shellman YG, Robinson SE, et al. CtBP1 is expressed in melanoma and represses the transcription of p16INK4a and Brca1. J Invest Dermatol. 2013 May;133(5):1294–301. doi: 10.1038/jid.2012.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pena C, Garcia JM, Garcia V, Silva J, Dominguez G, Rodriguez R, et al. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int J Cancer. 2006 Nov 1;119(9):2098–104. doi: 10.1002/ijc.22083. [DOI] [PubMed] [Google Scholar]
- 71.Dukers DF, van Galen JC, Giroth C, Jansen P, Sewalt RG, Otte AP, et al. Unique polycomb gene expression pattern in Hodgkin’s lymphoma and Hodgkin’s lymphoma-derived cell lines. Am J Pathol. 2004 Mar;164(3):873–81. doi: 10.1016/S0002-9440(10)63175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc Natl Acad Sci U S A. 2003 Jul 8;100(14):8342–7. doi: 10.1073/pnas.1436338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Palmer S, Brouillet JP, Kilbey A, Fulton R, Walker M, Crossley M, et al. Evi-1 transforming and repressor activities are mediated by CtBP co-repressor proteins. J Biol Chem. 2001 Jul 13;276(28):25834–40. doi: 10.1074/jbc.M102343200. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Y, Kwok JS, Choi PW, Liu M, Yang J, Singh M, et al. Pinin interacts with C-terminal binding proteins for RNA alternative splicing and epithelial cell identity of human ovarian cancer cells. Oncotarget. 2016 Feb 8; doi: 10.18632/oncotarget.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.May T, Yang J, Shoni M, Liu S, He H, Gali R, et al. BRCA1 expression is epigenetically repressed in sporadic ovarian cancer cells by overexpression of C-terminal binding protein 2. Neoplasia. 2013 Jun;15(6):600–8. doi: 10.1593/neo.121674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Birts CN, Harding R, Soosaipillai G, Halder T, Azim-Araghi A, Darley M, et al. Expression of CtBP family protein isoforms in breast cancer and their role in chemoresistance. Biol Cell. 2010 Jan;103(1):1–19. doi: 10.1042/BC20100067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Senyuk V, Chakraborty S, Mikhail FM, Zhao R, Chi Y, Nucifora G. The leukemia-associated transcription repressor AML1/MDS1/EVI1 requires CtBP to induce abnormal growth and differentiation of murine hematopoietic cells. Oncogene. 2002 May 9;21(20):3232–40. doi: 10.1038/sj.onc.1205436. [DOI] [PubMed] [Google Scholar]
- 78.Takayama K, Horie-Inoue K, Katayama S, Suzuki T, Tsutsumi S, Ikeda K, et al. Androgen-responsive long noncoding RNA CTBP1-AS promotes prostate cancer. EMBO J. 2013 Jun 12;32(12):1665–80. doi: 10.1038/emboj.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Poser I, Golob M, Weidner M, Buettner R, Bosserhoff AK. Down-regulation of COOH-terminal binding protein expression in malignant melanomas leads to induction of MIA expression. Cancer Res. 2002 Oct 15;62(20):5962–6. [PubMed] [Google Scholar]
- 80.Winklmeier A, Poser I, Hoek KS, Bosserhoff AK. Loss of full length CtBP1 expression enhances the invasive potential of human melanoma. BMC Cancer. 2009;9:52. doi: 10.1186/1471-2407-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stossi F, Madak-Erdogan Z, Katzenellenbogen BS. Estrogen receptor alpha represses transcription of early target genes via p300 and CtBP1. Mol Cell Biol. 2009 Apr;29(7):1749–59. doi: 10.1128/MCB.01476-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lopez-Garcia J, Periyasamy M, Thomas RS, Christian M, Leao M, Jat P, et al. ZNF366 is an estrogen receptor corepressor that acts through CtBP and histone deacetylases. Nucleic Acids Res. 2006;34(21):6126–36. doi: 10.1093/nar/gkl875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim JH, Youn HD. C-terminal binding protein maintains mitochondrial activities. Cell Death Differ. 2009 Apr;16(4):584–92. doi: 10.1038/cdd.2008.186. [DOI] [PubMed] [Google Scholar]
- 84.Kovi RC, Paliwal S, Pande S, Grossman SR. An ARF/CtBP2 complex regulates BH3-only gene expression and p53-independent apoptosis. Cell Death Differ. 2010 Mar;17(3):513–21. doi: 10.1038/cdd.2009.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Paliwal S, Pande S, Kovi RC, Sharpless NE, Bardeesy N, Grossman SR. Targeting of C-terminal binding protein (CtBP) by ARF results in p53-independent apoptosis. Mol Cell Biol. 2006 Mar;26(6):2360–72. doi: 10.1128/MCB.26.6.2360-2372.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guadamillas MC, Cerezo A, Del Pozo MA. Overcoming anoikis--pathways to anchorage-independent growth in cancer. J Cell Sci. 2011 Oct 1;124(Pt 19):3189–97. doi: 10.1242/jcs.072165. [DOI] [PubMed] [Google Scholar]
- 87.Talbot LJ, Bhattacharya SD, Kuo PC. Epithelial-mesenchymal transition, the tumor microenvironment, and metastatic behavior of epithelial malignancies. Int J Biochem Mol Biol. 2012;3(2):117–36. [PMC free article] [PubMed] [Google Scholar]
- 88.Postigo AA, Depp JL, Taylor JJ, Kroll KL. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003 May 15;22(10):2453–62. doi: 10.1093/emboj/cdg226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Subramanian T, Chinnadurai G. Association of class I histone deacetylases with transcriptional corepressor CtBP. FEBS Lett. 2003 Apr 10;540(1–3):255–8. doi: 10.1016/s0014-5793(03)00275-8. [DOI] [PubMed] [Google Scholar]
- 90.Guaita S, Puig I, Franci C, Garrido M, Dominguez D, Batlle E, et al. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J Biol Chem. 2002 Oct 18;277(42):39209–16. doi: 10.1074/jbc.M206400200. [DOI] [PubMed] [Google Scholar]
- 91.Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, et al. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005 Mar 31;24(14):2375–85. doi: 10.1038/sj.onc.1208429. [DOI] [PubMed] [Google Scholar]
- 92.Pena C, Garcia JM, Silva J, Garcia V, Rodriguez R, Alonso I, et al. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: clinicopathological correlations. Hum Mol Genet. 2005 Nov 15;14(22):3361–70. doi: 10.1093/hmg/ddi366. [DOI] [PubMed] [Google Scholar]
- 93.Spoelstra NS, Manning NG, Higashi Y, Darling D, Singh M, Shroyer KR, et al. The transcription factor ZEB1 is aberrantly expressed in aggressive uterine cancers. Cancer Res. 2006 Apr 1;66(7):3893–902. doi: 10.1158/0008-5472.CAN-05-2881. [DOI] [PubMed] [Google Scholar]
- 94.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007 Jun;7(6):415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 95.Chen YW, Paliwal S, Draheim K, Grossman SR, Lewis BC. p19Arf inhibits the invasion of hepatocellular carcinoma cells by binding to C-terminal binding protein. Cancer Res. 2008 Jan 15;68(2):476–82. doi: 10.1158/0008-5472.CAN-07-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Paliwal S, Kovi RC, Nath B, Chen YW, Lewis BC, Grossman SR. The alternative reading frame tumor suppressor antagonizes hypoxia-induced cancer cell migration via interaction with the COOH-terminal binding protein corepressor. Cancer Res. 2007 Oct 1;67(19):9322–9. doi: 10.1158/0008-5472.CAN-07-1743. [DOI] [PubMed] [Google Scholar]
- 97.Hamada F, Bienz M. The APC tumor suppressor binds to C-terminal binding protein to divert nuclear beta-catenin from TCF. Dev Cell. 2004 Nov;7(5):677–85. doi: 10.1016/j.devcel.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 98.Sierra J, Yoshida T, Joazeiro CA, Jones KA. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 2006 Mar 1;20(5):586–600. doi: 10.1101/gad.1385806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mroz EA, Baird AH, Michaud WA, Rocco JW. COOH-terminal binding protein regulates expression of the p16INK4A tumor suppressor and senescence in primary human cells. Cancer Res. 2008 Aug 1;68(15):6049–53. doi: 10.1158/0008-5472.CAN-08-1279. [DOI] [PubMed] [Google Scholar]
- 100.Guan C, Shi H, Wang H, Zhang J, Ni W, Chen B, et al. CtBP2 contributes to malignant development of human esophageal squamous cell carcinoma by regulation of p16INK4A. J Cell Biochem. 2013 Jun;114(6):1343–54. doi: 10.1002/jcb.24475. [DOI] [PubMed] [Google Scholar]
- 101.Madison DL, Lundblad JR. C-terminal binding protein and poly(ADP)ribose polymerase 1 contribute to repression of the p21(waf1/cip1) promoter. Oncogene. 2010 Nov 11;29(45):6027–39. doi: 10.1038/onc.2010.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000 Feb 18;100(4):387–90. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 103.Escriva M, Peiro S, Herranz N, Villagrasa P, Dave N, Montserrat-Sentis B, et al. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol Cell Biol. 2008 Mar;28(5):1528–40. doi: 10.1128/MCB.02061-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roussel MF. The INK4 family of cell cycle inhibitors in cancer. Oncogene. 1999 Sep 20;18(38):5311–7. doi: 10.1038/sj.onc.1202998. [DOI] [PubMed] [Google Scholar]
- 105.Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34(5):1416–26. doi: 10.1093/nar/gkl010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Di LJ, Fernandez AG, De Siervi A, Longo DL, Gardner K. Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat Struct Mol Biol. 2010 Dec;17(12):1406–13. doi: 10.1038/nsmb.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Deng Y, Liu J, Han G, Lu SL, Wang SY, Malkoski S, et al. Redox-dependent Brca1 transcriptional regulation by an NADH-sensor CtBP1. Oncogene. 2010 Dec 16;29(50):6603–8. doi: 10.1038/onc.2010.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004 Oct;4(10):814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 109.Tripathi MK, Misra S, Khedkar SV, Hamilton N, Irvin-Wilson C, Sharan C, et al. Regulation of BRCA2 gene expression by the SLUG repressor protein in human breast cells. J Biol Chem. 2005 Apr 29;280(17):17163–71. doi: 10.1074/jbc.M501375200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995 Jan 20;267(5196):383–6. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 111.Blevins MA, Kouznetsova J, Krueger AB, King R, Griner LM, Hu X, et al. Small Molecule, NSC95397, Inhibits the CtBP1-Protein Partner Interaction and CtBP1-Mediated Transcriptional Repression. J Biomol Screen. 2014 Dec 4; doi: 10.1177/1087057114561400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lazo JS, Nemoto K, Pestell KE, Cooley K, Southwick EC, Mitchell DA, et al. Identification of a potent and selective pharmacophore for Cdc25 dual specificity phosphatase inhibitors. Mol Pharmacol. 2002 Apr;61(4):720–8. doi: 10.1124/mol.61.4.720. [DOI] [PubMed] [Google Scholar]
- 113.Berg MG, Wan L, Younis I, Diem MD, Soo M, Wang C, et al. A quantitative high-throughput in vitro splicing assay identifies inhibitors of spliceosome catalysis. Mol Cell Biol. 2012 Apr;32(7):1271–83. doi: 10.1128/MCB.05788-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pandey RN, Wang TS, Tadjuidje E, McDonald MG, Rettie AE, Hegde RS. Structure-activity relationships of benzbromarone metabolites and derivatives as EYA inhibitory anti-angiogenic agents. PLoS One. 2013;8(12):e84582. doi: 10.1371/journal.pone.0084582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tadjuidje E, Wang TS, Pandey RN, Sumanas S, Lang RA, Hegde RS. The EYA tyrosine phosphatase activity is pro-angiogenic and is inhibited by benzbromarone. PLoS One. 2012;7(4):e34806. doi: 10.1371/journal.pone.0034806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Straza MW, Paliwal S, Kovi RC, Rajeshkumar B, Trenh P, Parker D, et al. Therapeutic targeting of C-terminal binding protein in human cancer. Cell Cycle. 2010 Sep 15;9(18):3740–50. doi: 10.4161/cc.9.18.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bergman LM, Morris L, Darley M, Mirnezami AH, Gunatilake SC, Blaydes JP. Role of the unique N-terminal domain of CtBP2 in determining the subcellular localisation of CtBP family proteins. BMC Cell Biol. 2006;7:35. doi: 10.1186/1471-2121-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Park H, Jung SK, Yu KR, Kim JH, Kim YS, Ko JH, et al. Structure-based virtual screening approach to the discovery of novel inhibitors of eyes absent 2 phosphatase with various metal chelating moieties. Chem Biol Drug Des. 2011 Oct;78(4):642–50. doi: 10.1111/j.1747-0285.2011.01192.x. [DOI] [PubMed] [Google Scholar]
- 119.Hubler D, Rankovic M, Richter K, Lazarevic V, Altrock WD, Fischer KD, et al. Differential spatial expression and subcellular localization of CtBP family members in rodent brain. PLoS One. 2012;7(6):e39710. doi: 10.1371/journal.pone.0039710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, et al. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev. 2008 May 15;22(10):1397–409. doi: 10.1101/gad.1666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang Y, Foncea R, Deis JA, Guo H, Bernlohr DA, Chen X. Lipocalin 2 expression and secretion is highly regulated by metabolic stress, cytokines, and nutrients in adipocytes. PLoS One. 2014;9(5):e96997. doi: 10.1371/journal.pone.0096997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010 Jul;38(Web Server issue):W695–9. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]