

Preface
To maintain protein homeostasis, AAA+ proteolytic machines degrade damaged and unneeded proteins in bacteria, archaea and eukaryotes. This process involves ATP-dependent unfolding of a target protein and subsequent translocation into a self-compartmentalized proteolytic chamber. Related AAA+ enzymes also disaggregate and remodel proteins. Recent structural and biochemical studies, in combination with direct visualization of unfolding and translocation in single-molecule experiments, have illuminated molecular mechanisms and suggest how remodelling of macromolecular complexes by AAA+ enzymes could occur without global denaturation. In this Review, we discuss the structural and mechanistic features of AAA+ proteases and remodeling machines, focusing on bacterial ClpXP and ClpX as paradigms. We also consider the potential of these enzymes as antibacterial targets and outline future challenges for the field.
Graphical Abstract
AAA+ proteolytic machines unfold and degrade damaged and unneeded proteins in all domains of life. In this Review, Olivares and colleagues discuss the molecular mechanisms and structures of bacterial AAA+ machines, focusing on recent studies of ClpXP as a paradigm.
Introduction
In all domains of life, cellular compartments are packed with proteins, many of which are in the process of folding, are intrinsically disordered, or contain both natively folded and unstructured regions1. Because the peptide bonds in an unstructured polypeptide are highly sensitive to proteolytic cleavage, the cytoplasm of bacteria and archaea, and most eukaryotic cellular compartments, do not contain indiscriminate proteases. Instead, specific proteins in these intracellular environments are degraded by proteolytic machines that sequester the active sites for peptide-bond cleavage within a protected chamber. These enzymes are known as AAA+ proteases, owing to the presence of a AAA+ unfoldase that recognizes specific substrates and uses the chemical energy of ATP hydrolysis to mechanically unfold the target protein and then translocate it into the degradation chamber2–4.
AAA+ proteases present in bacteria, mitochondria, and chloroplasts include ClpXP, ClpAP, ClpCP, HslUV, Lon and FtsH2. Other proteases in the AAA+ family consist of the 20S peptidase, which is found in all three domains of life, in combination with different AAA+ unfoldase partners, such as Mpa (bacteria), PAN or Cdc48/p97 (archaea) or the Rpt1–6 ring of the 26S proteasome (eukaryotic cytosol and nucleus)3–5. These AAA+ proteases enforce protein quality control by recognizing and destroying proteins that have been damaged by oxidation and heat stress6,7 and protein fragments that have been generated by endoproteolytic cleavage or failures in translation8–10. Cellular processes can also be controlled by AAA+ proteases that degrade regulatory proteins, including the bacterial stationary-phase sigma factor11,12, cell-division checkpoint inhibitors of the DNA-damage response13, and proteins that regulate cell-cycle progression14. For example, DNA damage in Escherichia coli results in synthesis of SulA, a cell-division inhibitor that must be degraded by the Lon protease before growth can resume13, and ClpXP degradation of CtrA, a master regulator of transcription in Caulobacter crescentus, is required for cell-cycle progression and initiation of chromosomal replication14. Developmental transitions frequently involve degradation of specific proteins by AAA+ proteases, altering the composition of the intracellular proteome. ATP-dependent degradation of specific target proteins can also facilitate development of genetic competence, sporulation, virulence and biofilm formation15,16.
In this Review, we discuss recent work that has shed light on the molecular mechanisms of bacterial AAA+ proteases. In particular, we highlight biochemical, structural, and single-molecule studies of protein degradation by E. coli ClpXP that illuminate the principles and dynamic interactions that enable the unfolding, translocation and degradation of a wide variety of structurally diverse protein substrates. Related principles explain how AAA+ enzymes can also function to remodel macromolecular complexes. We also examine the diversity of AAA+ proteases present in the bacterial domain and the potential of some of these enzymes as targets for antibacterial therapy. Finally, we outline future challenges for the field and the technological advances that will be needed to address them.
Bacterial AAA+ proteases
Most bacterial phyla utilize ClpXP, ClpAP or ClpCP, HslUV, Lon and FtsH to execute ATP-dependent protein degradation, whereas Actinobacteria also employ the Mpa•20S proteasome2,3. Mycoplasma, which have the smallest bacterial genomes, typically encode only the Lon and FtsH proteases17,18.
ClpXP, a paradigm for AAA+ proteases
ClpXP, the best characterized AAA+ protease, consists of the ClpX unfoldase and ClpP peptidase19. Each ClpX subunit contains a large AAA+ domain and a small AAA+ domain, which together form the ATP-hydrolysis and motor module. In the ClpX hexamer, the AAA+ domains pack together to form a ring with an axial channel or pore that serves to initially engage a portion of the target protein, has an active role in unfolding, and is the conduit for translocation into the degradation chamber of ClpP19. ClpX also contains a family-specific N domain required for efficient recognition of adaptors and auxiliary signals in some substrates20,21. Like ClpX, subunits of the HslU, Mpa, PAN, Lon and FtsH unfolding enzymes contain a single ATP-hydrolysis and motor module, whereas subunits of the ClpA, ClpC, and Cdc48 enzymes have two ATP-hydrolysis and motor modules, which form discrete stacked rings in the hexamer2.
ClpP consists of two heptameric rings that enclose a chamber containing the active sites for peptide-bond cleavage (Fig. 1a)22–24. A portal at the centre of each ring controls access to the degradation chamber and allows entry only to small peptides when ClpP is not bound to a AAA+ unfoldase partner25. The peptidases of other AAA+ proteases differ from ClpP in subunit structure, in the number of subunits in each ring, in the number of rings, and in the chemistry of the active-site residues that catalyse peptide-bond cleavage (Figs. 1b–f)2. Nevertheless, each of these peptidases also sequesters its active sites within a self-compartmentalized chamber, with access controlled by a AAA+ unfoldase partner, and thus uses the same strategy as ClpP to limit non-specific degradation.
Figure 1. Self-compartmentalized peptidases are the degradation components of AAA+ proteases.

In each panel, a single representative structure is shown. (a) The ClpP peptidase from E. coli (pdb 1TYF) consists of two heptameric rings, uses a Ser–His–Asp catalytic triad for peptide-bond cleavage, and functions with one of three homohexameric AAA+ partners (ClpX, ClpA, or ClpC). In different species, ClpP can consist of 14 identical subunits, distinct homomeric rings or a mixture of subunits in each ring. (b) The 20S proteasome from Thermoplasma acidophilum (pdb 1PMA) has an α7β7β7α7 structure, uses a Thr nucleophile for peptide-bond cleavage, and partners with homohexameric Mpa in bacteria, homohexameric PAN or Cdc48/p97 in archaea, or the heterohexameric Rpt1–6 ring in the eukaryotic 26S proteasome. The α and β rings have seven identical subunits in bacteria and archaea and seven distinct α or β subunits in eukaryotes. (c) The HslV peptidase from Haemophilus influenzae (pdb 1G3I) consists of two homohexameric rings (each subunit is homologous to a β subunit of the 20S proteasome), uses a Thr nucleophile for peptide-bond cleavage, and partners with an HslU homohexamer. (d) The homohexameric Lon protease from Thermococcus onnurineus (pdb 3K1J) is assembled from subunits in which the AAA+ module is fused to the peptidase domain and uses a Ser–Lys dyad for peptide-bond cleavage. (e) Two E. coli Lon hexamers can combine to form a dodecamer, which is stabilized by N-domain interactions that form portals of ~45 Å into the enzyme lumen. The panel shows the E. coli 3LJC and B. subtilis 3M6A structures modeled into a low-resolution electron-density map91. (f) The homohexameric Thermotoga maritima FtsH protease (pdb 3KDS) also assembles from subunits in which the AAA+ module is fused to the peptidase domain. FtsH uses an Asp–Zn++ active site for peptide-bond hydrolysis.
ClpX rings can bind to one or both ClpP rings to form singly or doubly capped complexes in which the portals of ClpP are aligned with the axial channels of ClpX, allowing substrates translocated through the ClpX channel to enter ClpP26. Assembly of these proteolytic complexes, which have an inherent symmetry mismatch owing to different numbers of subunits in the ClpX and ClpP rings, is stabilised by flexible loops from one face of a hexameric ClpX ring that dock into clefts on a heptameric ClpP ring20,27,28. Mismatched docking of a hexameric AAA+ ring with a heptameric proteolytic ring also occurs in ClpAP, ClpCP, Mpa•20S, PAN•20S, Cdc48•20S and the 26S proteasome (Figs. 1a–b). In HslUV, Lon and FtsH, by contrast, the AAA+ ring and proteolytic ring are both hexamers (Figs. 1c–f).
E. coli ClpP consists of 14 identical subunits, as do most ClpP enzymes in other bacteria. However, Actinobacteria, Cyanobacteria and some species from other phyla contain multiple ClpP paralogues, which are often differentially expressed23,24. In Mycobacterium tuberculosis, discrete ClpP1 and ClpP2 rings form the proteolytic barrel29,30, whereas in some Cyanobacteria and plants, each heptameric ring contains multiple paralogous subunits31,32. Potential biological advantages of using more than one type of ClpP subunit to form active tetradecamers include altering specificity for different AAA+ unfoldase partners, developmental regulation of activity, enhancing the diversity of peptide-bond cleavage specificity and increasing resistance to antibiotics that target ClpP29,33. In many species, ClpP assembles into active tetradecamers without assistance, but interactions with a AAA+ unfoldase partner and substrate delivery can also be required to stabilize active double-ring barrels28,34,35. Crystal structures of inactive and active ClpP tetradecamers reveal alterations in the geometry of the active sites and substrate-binding pockets as a consequence of different packing arrangements at the ring–ring interface23,24. It has been suggested that peptide products exit the ClpP chamber through ring–ring interface windows that open transiently36.
Both ClpP and its AAA+ unfoldase partners are targets for antibacterial drugs, including small molecules that prevent partner binding and open the portals into the ClpP chamber, suicide inhibitors of the peptidase active sites and cyclic peptides that act via the AAA+ enzymes33,35,37–45 (Box 1).
Box 1. Clp proteases as drug targets.
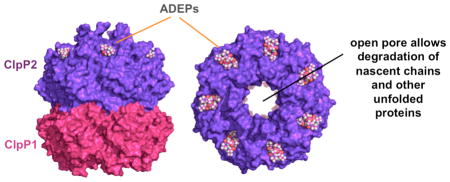
Clp-family proteases have emerged as promising antibacterial targets over the past decade. Brötz-Oesterhelt and colleagues first discovered that Gram-positive bacteria could be killed by small-molecule acyldepsipeptides (ADEPs)37, which activate ClpP degradation of proteins with little stable native structure in the absence of a AAA+ partner. These results suggested that ADEP killing of bacterial cells results from uncontrolled proteolysis of unfolded proteins and newly synthesized nascent chains. As shown in the figure, ADEPs bind in the ClpP clefts that normally serve as partner-docking sites, preventing binding of AAA+ unfoldases, and also widen the axial entry portal into the degradation chamber, allowing unfolded proteins to enter and be degraded35,38–40. As expected, clpP null mutations confer ADEP resistance in bacteria, like Escherichia coli, in which ClpP is not essential37. Although ClpP is also not essential in Staphylococcus aureus, combining an ADEP variant with rifampicin, an inhibitor of RNA polymerase, eradicated S. aureus biofilms in a chronic infection mouse model42. ADEP derivatives with enhanced antibacterial potency have been synthesized44, but their ultimate use as therapies remains to be determined.
In Mycobacterium tuberculosis and related actinobacteria, ClpP, ClpX and ClpC (a homologue of ClpA) are all essential enzymes107,108. Thus, ADEP-induced lethality might, in principle, result from inhibition of ClpXP or ClpCP proteolysis as well as from uncontrolled ClpP degradation35. Suicide inhibitors that react specifically with the active-site serine of ClpP also kill M. tuberculosis and other bacteria in which ClpP function is essential33,41. Finally, natural cyclic peptides can kill strains of M. tuberculosis that are otherwise drug resistant by binding to the N-terminal domain of ClpC, hyper-activating ATP hydrolysis and uncoupling this hydrolysis from ClpCP degradation43,45,93.
Principles of ATP-fuelled proteolysis
ClpXP and other AAA+ proteases share a common basic mechanism (Fig. 2a)2. ClpX or another AAA+ unfoldase ring hexamer engages a recognition tag attached to a target protein that is too large in its native state to pass through the pore. Cycles of ATP binding and hydrolysis power conformational changes in the AAA+ unfoldase ring that pull on the native substrate, typically resulting in failed unfolding and/or substrate release but occasionally causing unfolding. Once unfolding is successful, the polypeptide is translocated through the pore and into the chamber of the associated self-compartmentalized peptidase, where it is cleaved into peptides, typically 5–15 amino acids in length46,47.
Figure 2. Substrate recognition and degradation.

(a) Minimal model for recognition, unfolding, translocation and degradation of a single-domain protein by a AAA+ protease. Reaction steps in the forward direction are ATP dependent. In the initial recognition step, a disordered engagement tag in the native protein substrate is bound in the axial pore of the AAA+ ring hexamer. ATP-fuelled conformational changes in the ring then pull on the substrate, which can result either in failed unfolding, substrate release or substrate denaturation. The probability of each of these outcomes depends on substrate stability, as a very stable protein might be bound and released many times resulting in unproductive hydrolysis of a substantial amount of ATP. Following forced unfolding, the denatured polypeptide is processively translocated through the pore and into the peptidase chamber for degradation. (b) Efficient recognition of some protein substrates requires secondary recognition signals, which are substrate sequences that bind to the AAA+ enzyme either directly or via adaptor proteins. In principle, these secondary signals might affect any of the pre-unfolding steps shown in panel a.
The specificity of degradation by AAA+ proteases is controlled in several ways. In the simplest case, binding sites in the pore of the AAA+ unfoldase ring recognize an aminoacid sequence in the unstructured engagement tag of the substrate (Fig. 2a), although tag access can also be regulated by unfolding, dissociation or post-translational modification reactions2. Specificity can also depend on additional recognition sequences in the protein substrate or by ubiquitination or pupylation48,49. For example, pupylation and degradation of pupylated substrates by Mpa•20S allows Mycobacterium smegmatis to survive nitrogen starvation50. Secondary recognition signals on substrates typically bind to auxiliary domains on the AAA+ unfolding ring either directly or via adaptor proteins (Fig. 2b). Such interactions can mediate recognition by increasing the effective concentration of the engagement tag with respect to the pore, improving the probability of engagement, or by strengthening affinity, which may help keep substrates bound after failed unfolding attempts2. These secondary recognition signals can also be used to direct the unfoldase to a specific oligomeric or conformational state of the substrate, and thus serve a regulatory function. Adaptor-mediated degradation can also be influenced by “anti-adaptor” proteins or post-translational modification to enhance or inhibit protease recognition51–53.
ClpX ring structure and function
Crystal structures of the AAA+ ring of single-chain variants of E. coli ClpX have been solved in different nucleotide states54,55 (Fig. 3a). Differences in the orientations of the large and small AAA+ domains generate subunits with a “loadable” (L) conformation, which have a nucleotide-binding pocket in a cleft between the large and small domains, and subunits with an “unloadable” (U) conformation, which do not have a binding pocket (Fig. 3b). Subunits in most crystal structures are arranged in a pseudo-symmetric L-L-U-L-L-U pattern (Fig. 3a), which is unlikely to represent the major configuration of the working ring, as conformational assays support an asymmetric 5:1 ratio of L:U subunits (Fig. 3c)55. A 5:1 arrangement has also been observed by electron microscopy for the Rpt1–6 AAA+ ring of the 26S proteasome56.
Figure 3. ClpX ring structure.

(a) Views of the AAA+ ring of E. coli ClpX (pdb 3HWS). In each subunit, the large AAA+ domain is coloured dark or light grey and the small AAA+ domain is coloured purple. Hinges between the large and small domains of each subunit are coloured red. (b) Subunits can adopt either a loadable (L) or an unloadable (U) conformation. In L subunits, ATP binds in a cleft between the large and small AAA+ domains. In U subunits, rotation of the small domain destroys the binding pocket. (c) Cartoon of a 5L:1U ClpX ring showing how six rigid-body units connected by six hinges are created by packing between the small AAA+ domain of a subunit (coloured blue) and the large AAA+ domain of a neighbouring subunit (dark grey for L subunits; light grey for U subunits). Because the ring is topologically closed, changes in the conformation of any single hinge — caused by ATP binding, ATP hydrolysis or product release — propagates around the ring.
Dynamic switching of subunits between the L and U conformations is required for robust ClpX function. For example, locking one subunit in either conformation by crosslinking permits ATP hydrolysis in the ring but largely uncouples ATP hydrolysis from mechanical activity55,57. Different L subunits in the ring bind ATP in an asymmetric pattern with a wide range of affinities, presumably owing to structural variations. L⇔U conformational switching may prevent machine stalling when power strokes fail to unfold a stable protein by resetting the functional roles of subunits55,57. It is also possible that subunit switching is a way to avoid ring malfunction when ADP fails to dissociate or binds to an empty subunit instead of ATP.
In crystal structures of the ClpX hexamer, each small AAA+ domain packs against the large AAA+ domain of the neighbouring subunit in the same way, forming a ring with six identical rigid-body units connected by short hinges between the large and small AAA+ domains of each subunit (Fig. 3c)54,55. Engineering disulfide bonds across each rigid-body interface between subunits in the hexamer results in a topologically closed ring that is highly active in ATP-dependent protein unfolding and degradation58. Thus, ClpX function does not require an open ring or lock-washer conformation, and all functional ring conformations can be accessed by changing the hinge conformations.
How does ATP hydrolysis drive changes in the conformation of the ClpX ring? Bound ATP contacts both the AAA+ domains and the hinge, and thus sets the hinge conformation. When ATP is hydrolysed, and ADP and Pi are subsequently released, the hinge conformation changes again. Thus, changes in the orientations of the large and small AAA+ domains in one subunit – caused by ATP binding, hydrolysis or product release – propagate conformational changes around the ring to drive substrate unfolding and/or translocation. In addition, altering the conformation of any single hinge will change the hinge conformations of flanking subunits to maintain a closed ring. The precise nature of these conformational changes is not yet known, but structures in different nucleotide states show movements of ~1 nm of the axial pore loops that are thought to drive unfolding and translocation54.
Finally, one ATP hydrolysis event is sufficient to generate a power stroke and mechanical function, as a ClpX ring with a single hydrolytically active subunit supports protein degradation59. This result rules out mechanochemical models in which multiple ClpX subunits hydrolyse ATP synchronously or individual subunits hydrolyse ATP in a strictly coordinated sequence60,61.
Watching unfolding and translocation
Single-molecule force spectroscopy has recently been used to visualize ClpX mechanical activity directly. Attachment of ClpXP to one laser-trapped bead and attachment of a multi-domain protein substrate to another bead enables the substrate to be captured by the protease, creating a tether. The positions and power of the lasers in the optical trap are set so that a small and constant force would pull the beads apart in the absence of this tether. As the degradation reaction progresses, ATP-fuelled unfolding and subsequent translocation of each domain changes the bead-to-bead distance, providing a direct readout of ClpXP mechanical activity (Fig. 4)62,63. For example, unfolding of the structure of a domain causes a sudden increase in distance, which is followed by a slower decrease as the unfolded polypeptide is spooled through ClpX and into ClpP for degradation62,63. As substrate domains are translocated against resisting tension in these experiments, ClpXP must itself generate mechanical force. After completing translocation of one unfolded domain, ClpXP attempts to unfold the next domain, and no substantial change in distance is observed until unfolding occurs62,63.
Figure 4. Single-molecule force spectroscopy of ClpXP.

(a) Cartoon of an optical-trapping experiment. Micron-sized beads, trapped by infrared lasers, are tethered to either ClpXP or a multi-domain substrate via a DNA linker. When ClpXP engages the substrate, ATP-fuelled mechanical activity can be monitored by measuring bead movements relative to the centre of laser focus (dotted lines). (b) ClpXP unfolding of an individual substrate domain (panels a2 and a5) results in an increase in bead-to-bead distance. Translocation of the substrate (panels a3 and a6) results in a decrease in bead-to-bead distance that corresponds to the length of the translocated domain. Periods of no movement are dwells (panels a1 and a4) in which ClpXP tries to unfold the next native domain in the substrate.
ClpXP unfolding of individual protein domains typically occurs in a cooperative all-or-none fashion. However, intermediates in unfolding are observed when ClpXP extraction of one structural element of a domain does not cause global denaturation62–65. As ClpXP attempts to unfold a domain in the optical trap, the length of the pre-unfolding dwell time provides information important for understanding how unfolding occurs. As expected for a reaction with a single rate-limiting step, these dwell times are exponentially distributed for a specific protein domain with a time constant that reflects its mechanical stability65. For rapidly unfolded domains, the unfolding time constant matches the time constant for ATP hydrolysis, indicating that one hydrolysis event and associated power stroke can drive unfolding62. For domains that unfold more slowly, many ATPs are hydrolysed during the pre-unfolding dwell time62–65, a result consistent with biochemical studies showing that ClpXP hydrolyses hundreds of ATPs during degradation of a single protein substrate66. In these cases, most power strokes do not force unfolding and only an occasional power stroke succeeds. Nevertheless, the exponential distribution of unfolding dwell times implies that every power stroke has the same probability of causing unfolding, as a lag in the kinetics would be expected if repeated power strokes were needed to weaken the structure to the point of failure or multiple ATP-hydrolysis events were required to build mechanical tension in the enzyme. Importantly, the average number of power strokes required for unfolding increases as the local stability of structural elements adjacent to the site of enzyme-mediated pulling increases65–67. Transient fraying of this local structure, caused by random thermal motions, would result in a subpopulation vulnerable to unfolding by a single power stroke. By taking advantage of the stochastic local dynamics of a substrate domain, enzymatic pulling by ClpXP and other AAA+ machines could enhance an unfolding pathway that relatively few protein molecules would normally follow in solution67.
Following unfolding, ClpX translocates the denatured polypeptide into ClpP, resulting in decreased bead-to-bead distance in the optical trap. The average length of the smallest translocation steps corresponds to movement of ~5 amino acids of the substrate through the ClpX pore during a power stroke. However, steps ~2, ~3 and ~4 times longer than the smallest step are also observed, which appear to result from kinetic bursts of power strokes64,65. Interestingly, steps of different lengths do not occur in any specific pattern, indicating that some aspect of ClpX ring function is inherently probabilistic. One model to explain random stepping behaviour posits that step size depends on the number of ATP molecules bound to the ring and subsequently hydrolysed64. Another model proposes that an initial ATP hydrolysis event can occur in any one of the L subunits in an ATP-saturated ring, followed by a burst of ATP hydrolysis and translocation steps in neighbouring subunits until a U subunit is reached65.
Substrate gripping and release
In ClpX and related AAA+ machines, conformational changes in the ring, which are powered by cycles of ATP hydrolysis, are analogous to the movements within the engine of a motor vehicle. However, additional mechanisms are still needed to grip the protein substrate, apply force and perform mechanical work. Loops with a highly conserved aromatic–hydrophobic–glycine motif in the axial pore of ClpX have crucial roles in substrate gripping during protein unfolding. These loops can be crosslinked to substrates, severe loop mutations eliminate mechanical activity, and reducing the number of wild-type loops in the ClpX hexamer slows unfolding, resulting in an increased ATP cost for degradation (Fig. 5a)68–71. Intriguingly, the relationship between unfolding activity and the number of wild-type pore loops suggests that five or six pore loops combine to grip the substrate and apply an unfolding force as a consequence of ATP hydrolysis in a single ClpX subunit70,71. Pore loops with very similar aromatic–hydrophobic–glycine motifs are found in all hexameric AAA+ machines that unfold or remodel proteins, implying that the role of these loops in the function of AAA+ unfoldases is highly conserved2.
Figure 5. Factors influencing the energetic cost of degradation.

The average number of ATPs hydrolysed by the ClpXP protease during degradation of a single protein substrate depends upon the protein’s stability and how well it is gripped by ClpX. (a) The axial pore of ClpX contains a loop from each subunit that grips the substrate during protein unfolding. Mutating one or two loops decreases the maximal rate of degradation of a GFP substrate and increases the ATP cost70. Thus, maximal unfolding and degradation efficiency requires the combined gripping action of five or six pore loops. (b) Structure of the I27 domain of the human muscle protein titin (pdb 1TIT) and the locations of two mutations (V13P and V15P) that destabilize this domain by removing or disrupting hydrogen bonds. Also shown is an engagement tag at the C-terminus that allows ClpX to recognize and pull on titinI27 variants. The bar graph shows the average ATP cost of ClpXP degradation for the wild-type (WT), V15P and V13P titinI27 domains65,66. ATP hydrolysed during translocation, terminal unfolding attempts and unfolding attempts that result in substrate release are indicated.
Experiments with peptide substrates show that ClpX does not recognize specific side-chain features or the regular spacing of peptide bonds in translocating polypeptides72. Translocation can proceed in an N→C or C→N direction, depending on the location of the engagement tag67,73,74, and multiple polypeptides can be concurrently translocated through the axial pore75,76. How might gripping occur if ClpX does not recognize specific chemical features of a translocating polypeptide? One possibility is that the axial pore is elastic and can close tightly around one or multiple polypeptides, with the gripping mechanism dependent on nonspecific van der Waals interactions54.
During the degradation of multi-domain substrates, ClpXP can release partially degraded protein fragments, which may regulate protein function in some cases67,73,77–79. Release of partially degraded protein fragments occurs when one or more domains have been unfolded and degraded, and another stable domain is encountered. Experiments with multi-domain substrates show that ClpXP has some chance of degrading each stable domain and some chance of releasing a partially degraded protein with an unstructured tail of ~40 residues, which would span the distance from the top of the ClpX pore to the active sites of ClpP. These released fragments escape further degradation if their tails do not contain engagement tags for ClpXP. Recent studies show that this mechanism allows ClpXP to partially degrade the C. crescentus DnaX clamp loader but then release a truncated fragment with a new activity when it encounters a glycine-rich sequence before a stable domain79. Earlier work demonstrated generation of specific truncation products of a few eukaryotic substrates by the 26S proteasome80–82.
ClpXP degrades some protein substrates at a rate proportional to the ATP-hydrolysis rate but ceases to degrade other substrates when hydrolysis falls below a critical threshold value as a consequence of low ATP concentrations or ClpX mutations12,77,83. This ATP dependence provides a mechanism for specific regulation of cellular processes by ClpXP. For example, E. coli σS, which regulates entry into stationary phase, is constitutively degraded by ClpXP during exponential growth when ATP levels are high but is not degraded when ATP levels fall, signalling a need to change the transcriptional program to deal with a low-energy environment12. Why does ClpXP degradation of some but not all substrates stall at low ATP concentrations? A likely possibility is that grip on the substrate is reduced when the ClpX ring is only partially saturated with ATP, which differentially affects more mechanically stable proteins that require a stronger grip to initiate unfolding70.
Degradation energetics
Because cleavage of a peptide bond is energetically favourable, the cost of ClpXP degradation depends on how much ATP must be hydrolysed to ensure unfolding and translocation of a given protein substrate. This value can vary substantially for closely related substrates. For example, ClpXP degradation of a titinI27 domain, a model substrate, consumes ~600 ATPs, whereas degradation of the less stable V15P and V13P titinI27 variants consumes ~230 and ~120 ATPs, respectively (Fig. 5b, 5c)66. To place these costs into context, synthesis of a titin domain of ~100 amino acids requires an energetic investment comparable to hydrolysis of ~400 ATPs. As translocation of a titin domain by ClpXP requires ~20 ATPs65, the vast majority of ATP hydrolysis during ClpXP degradation of titinI27 occurs as the ClpXP engages and attempts to unfold the substrate. Comparison of single-molecule and solution-biochemical experiments shows that ~25% of the ATP required for eventual unfolding is hydrolysed while ClpXP remains bound to the substrate65. Another ~75% is consumed in reactions in which the substrate is bound but eventually released after unsuccessful unfolding66,73.
ClpA, a double-ring AAA+ unfoldase
In the ClpAP protease, the ClpP peptidase partners with the AAA+ ClpA unfoldase to degrade protein substrates2,3. Because ClpAP and ClpXP use different unfoldases, they have different functional properties, as discussed beow. Each ClpA subunit contains two AAA+ modules, D1 and D2, which form distinct stacked rings in the hexamer26. The D2 ring, which is proximal to ClpP, seems to play the major role in unfolding and translocation, as inactivation of ATP hydrolysis in this ring abrogates robust degradation of stable native substrates84, and axial pore loops in this ring contact the engagement tags of substrates85. By contrast, ClpA retains substantial degradation activity when the D1 ring cannot hydrolyse ATP, suggesting that this ring has a secondary role in force generation84,86.
In optical-trap assays of mechanochemical activity, ClpAP translocates unfolded polypeptides more slowly than ClpXP, largely because the average steps are smaller, corresponding to movement of ~5 or ~10 amino acids per step87. Despite slower translocation than ClpXP, ClpAP unfolds most protein domains substantially faster78,87,88. This enhanced unfolding does not occur because ClpAP applies more force than ClpXP87. However, because the double-ring architecture of ClpA creates a longer axial pore, loops from both the D1 and D2 rings could cooperate in gripping substrates more tightly, resulting in more efficient transfer of force to the substrate during a power stroke. Alternatively, as ClpAP and ClpXP pull on a native protein, differences in the enzyme surfaces that the substrate contacts may allow better unfolding of certain classes of proteins by one enzyme compared with the other. For example, ClpAP might be more efficient than ClpXP at initial extraction of beta strands from proteins. Whether evolution has matched degradation of a given substrate to a specific proteolytic machine that can unfold the protein more rapidly or at lower energetic costs remains to be determined, as information about natural ClpXP and ClpAP substrates is currently limited. Interestingly, ClpX unfolds proteins with roughly the same efficiency as ClpXP, whereas ClpA alone is a substantially poorer unfoldase than ClpAP86. This difference occurs, in part, because ClpP binding doubles the rate of ATP hydrolysis by ClpA but modestly suppresses the ATPase activity of ClpX84,86. It is unclear if these opposing effects of ClpP on ATP hydrolysis by its AAA+ unfoldase partners are biologically significant.
AAA+ protease diversity
Depending on genome size, bacteria typically have two to five AAA+ proteases. Similarly, eukaryotic organelles of bacterial origin often have multiple AAA+ proteases. By contrast, ATP-dependent degradation in the cytosol and nucleus of eukaryotic cells depends exclusively on the 26S proteasome, perhaps because the ubiquitin system with its highly diverse E3 ligases is largely responsible for substrate identification in these compartments4,56. The roles of specific AAA+ proteases can change between organisms. In most bacteria, for example, ClpXP degrades ssrA-tagged proteins produced by tmRNA-mediated rescue of stalled ribosomes, whereas Lon serves this function in Mesoplasma florum, Mycoplasma pneumoniae and other Mycoplasma spp.9,10,17,18.
The use of multiple AAA+ proteases by bacterial cells or eukaryotic organelles may, in part, be related to the diverse subcellular locations of substrates. For example, bacterial FtsH and its mitochondrial homologues are membrane bound, and some of their substrates are peripheral or integral membrane proteins89,90. In archaea, LonB-family proteases are also membrane bound, whereas most Lon orthologues in bacteria are cytoplasmic and those in mitochondria function in the matrix7. FtsH and Lon are unique among AAA+ proteases because they consist of a single polypeptide in which the AAA+ subunit is fused directly to a peptidase subunit. It is unclear whether this fused architecture has advantages in comparison with complexes that have distinct ClpP, HslV or 20S peptidases that operate with different AAA+ unfoldase partners. Interestingly, the architecture of Lon hexamers allows them to assemble into a dodecamer in which access to the engagement pores may be restricted by portals at the hexamer–hexamer interface (Fig. 1e)91. If dodecamer assembly alters the substrate repertoire, increased cellular Lon concentrations would increase the proportion of dodecamers and thus influence which proteins are degraded. Lon substrates also have a more active role in controlling AAA+ unfoldase ring activity than substrates of other AAA+ proteases92. As natural antibiotics can target both the peptidase and unfoldase components of AAA+ proteases37,45,93, using multiple proteases with different peptidase active site architectures and distinct AAA+ unfoldase enzymes may minimize susceptibility to any single inhibitor. In a given cell, different AAA+ proteases are also likely to function optimally under different conditions. For instance, E. coli HslUV is overexpressed under heat-shock conditions and has a temperature optimum for substrate degradation of ~50 °C in vitro, whereas ClpXP is not under heat-shock control and is optimally active at ~30 °C94. Furthermore, specific protein substrates are often degraded by more than one AAA+ protease in bacteria. For example, the SulA inhibitor of cell division in E. coli is a substrate of both Lon and HslUV, which may ensure proper degradation following DNA damage over a range of temperatures13,95.
As discussed above, certain AAA+ proteases are likely to be better at unfolding and degrading specific cellular substrates. Moreover, almost all AAA+ proteases have family-specific auxiliary domains that function directly in substrate recognition or bind adaptors that alter activity and/or substrate preference, enabling diversity in the evolution and control of degradation2,96. For example, both ClpXP and ClpAP degrade ssrA-tagged proteins efficiently in vitro. In E. coli, by contrast, the SspB adaptor enhances ClpXP degradation of ssrA-tagged substrates, whereas the ClpS adaptor represses ClpAP degradation of ssrA-tagged substrates but facilitates degradation of N-end-rule substrates, which typically begin with Leu, Phe, Tyr, or Trp2.
AAA+ remodelling machines
Some AAA+ enzymes that function in proteolysis also remodel protein structures and complexes. For instance, E. coli ClpX disassembles a tetramer of the bacteriophage MuA transposase bound to recombined DNA by unfolding at least one protein subunit in the complex97. In this case, target recognition involves multiple MuA sequence elements, some of which bind in the ClpX pore and others to the N-terminal domain98. Bacteria and fungi also use double-ring AAA+ ClpB, ClpV and Hsp104 hexamers, which do not have peptidase partners but function as dedicated remodelling enzymes, sometimes to resolubilize aggregates or fragment amyloid fibers99,100. ClpV, for example, disassembles and recycles components of a contractile injection apparatus important for delivery of effector proteins in the type VI secretion system of Vibrio cholerae and other Gram-negative bacteria99. In eukaryotes, the single-ring AAA+ katanin and spastin enzymes sever microtubules by extracting tubulin subunits101. During cell division in bacteria, ClpX performs a similar function by disassembling the Z-ring, which is composed of tubulin-like subunits102. In each of these cases, it is likely that the AAA+ unfoldase engages and unfolds one or more subunits in the ring or polymer (Fig. 6a), leading to disassembly.
Figure 6. AAA+ remodelling of proteins and protein complexes.

(a) A AAA+ remodeling machine breaks a polymer into two pieces by unfolding an interior subunit. Remodelling of the polymer but not the monomer is possible if signals for AAA+ recognition are only properly arranged in the polymer. Related mechanisms may explain severing of microtubules, cell-division rings and amyloid fibers by AAA+ enzymes. (b) Model in which a AAA+ machine enhances the rate of cofactor insertion into a metabolic enzyme by inducing a conformational change in the cofactor binding site. This mechanism appears to be used by mitochondrial ClpX to catalyse incorporation of a cofactor into a haem biosynthesis enzyme103. (c) Multi-point binding of a complex to a AAA+ ring, followed by a major conformational change in the ring, might pull the complex apart. This mechanism may account for ClpB remodeling of some bacterial substrates and for disassembly of complexes required for membrane fusion in eukaryotic cells.
Some AAA+ machines appear to remodel substrates by a mechanism that does not require complete translocation of the target subunit through the axial pore. For example, ClpX orthologs in mitochondria, some of which lack ClpP partners, catalyse incorporation of a cofactor into a haem biosynthesis enzyme103, possibly by pulling on it to populate a more open insertion-competent structure (Fig. 6b). Similar functions have yet to be documented in bacteria but seem likely to emerge given the evolutionary relationship between mitochondria and α-proteobacteria. In other cases, disassembly of a protein or protein-DNA complex might occur simply by coupling multipoint substrate binding to a large change in conformation in the AAA+ unfoldase ring (Fig. 6c). In eukaryotes, disassembly of soluble NSF attachment protein receptor (SNARE) complexes during membrane fusion by the double-ring N-ethylmaleimide sensitive factor (NSF) enzyme seems to occur by this mechanism. Structural characterization of NSF-SNARE complexes by cryo-electron microscopy (cryo-EM) 104 and single-molecule experiments suggest a spring-loaded mechanism of disassembly105. ClpB may remodel some bacterial substrates by a similar mechanism, as a recent study suggests that it dissociates from an unfolded substrate after only one or two translocation steps106.
Future challenges
A combination of structural, biochemical and single-molecule studies have outlined the basic mechanisms of several bacterial AAA+ enzymes that unfold and/or remodel proteins. In most cases, however, it is not understood at a detailed structural level how target proteins are initially bound or how conformational changes coupled to ATP binding, hydrolysis and product release drive unfolding, translocation or remodelling. High-resolution cryo-EM or crystal structures may answer some of these questions. The development and application of single-molecule assays that simultaneously monitor ATP binding, conformational changes in the AAA+ unfoldase ring and mechanical activity may be needed to address other questions. What dictates whether AAA+ proteases processively degrade or release partially processed substrates is incompletely understood. Moreover, it is not known if tension-relief mechanisms, such as that proposed for NSF, are fundamentally different than power-stroke mechanisms, and whether a single AAA+ unfoldase enzyme is limited to using one mechanism or the other. Finally, our knowledge of the repertoire of natural substrates and degrons recognized by the different bacterial AAA+ proteases and remodelling machines is limited, as is our understanding of machine and target regulation by adaptors or modification reactions. Thus, there is ample room for continued discovery and exciting progress in our understanding of these fascinating molecular machines.
Key points.
In ATP-dependent proteases, a ring-shaped AAA+ machine harnesses the chemical energy of ATP binding and hydrolysis to mechanically unfold target proteins by translocating them through an axial pore and into the degradation chamber of a compartmental peptidase.
Recognition of specific target proteins involves direct binding of amino-acid sequences to the axial pore of the AAA+ ring, binding of sequences to auxiliary domains, and/or binding mediated by adaptor proteins. Degron sequences can be revealed or added to substrates by protein-modification reactions.
Novel antibiotics kill some bacteria by binding to the ClpP compartmental peptidase and transforming it into a rogue enzyme that indiscriminately degrades nascent polypeptides and unstructured cellular proteins.
Single-molecule optical trapping has directly visualized the unfolding and translocation activities of the ClpXP and ClpAP AAA+ proteases. These experiments and solution studies support a probabilistic model of AAA+ ring function and show that each power stroke has a constant and typically low probability of unfolding a stable protein domain.
Although protein degradation by AAA+ proteases is typically highly processive, multi-domain substrates are sometimes partially proteolyzed, with the released products having new biological functions.
AAA+ enzymes can function independently to solubilize aggregated proteins, disassemble macromolecular complexes, and catalyze incorporation of cofactors into enzymes.
Acknowledgments
We thank members of the Baker and Sauer labs and P. Chien, E. Gur, and M. Laub for helpful discussions. Work from our labs was supported by NIH grants AI-16892, GM-101988, GM-49224, and the Howard Hughes Medical Institute. T.A.B. is an employee of the Howard Hughes Medical Institute.
Glossary
- AAA+ enzyme
AAA+ (pronounced “triple A plus”) enzymes use the chemical energy of ATP binding, hydrolysis and product release to perform mechanical work in cells.
- Adaptor
A protein that binds a substrate and delivers it to a AAA+ protease for degradation.
- Acyldepsipeptides (ADEPs)
Antibacterial compounds that kill bacteria by activating non-specific ClpP degradation of unfolded proteins and nascent polypeptide chains.
- Cryo-electron microscopy (cryo-EM)
A technique to visualize single macromolecules in a thin layer of vitreous ice. Recent advances in direct electron detectors and data-processing algorithms now allow some structures to be determined at atomic resolution.
- Degradation chamber
The interior of a barrel-like compartment that contains the active sites for peptide-bond cleavage in a AAA+ protease.
- Degron
Any sequence or structural element, including the engagement tag, that is required for recognition and degradation of a substrate by a AAA+ protease.
- E3 ligases
Eukaryotic enzymes responsible for the addition of ubiquitin chains, which in some cases target proteins for degradation by the proteasome.
- Engagement tag
A specific but intrinsically disordered sequence that binds in the pore of the AAA+ unfoldase ring and allows the enzyme to pull on an attached native protein to unfold it.
- Loadable subunit
A subunit of ClpX or any other AAA+ unfoldase ring that can bind ATP and other nucleotides.
- Lock-washer conformation
AAA+ unfoldase ring with the subunits arranged in a helical conformation, creating an open interface between the first and last subunits.
- N-ethylmaleimide sensitive factor (NSF)
AAA+ remodelling machine that disassembles soluble NSF attachment protein receptor (SNARE) complexes, which are required for vesicle fusion.
- Persisters
Microbial cells in a non-dividing dormant state that escape killing by conventional antibiotics but are not inherently antibiotic resistant.
- Power stroke
A conformational movement of the AAA+ ring – generated by ATP binding, hydrolysis or product release – that pulls on or propels a peptide segment of a substrate through the axial channel.
- Proteasome
AAA+ protease that uses the 20S peptidase for degradation.
- Pupylation
A post-translational modification that attaches a Pup (prokaryotic ubiquitin-like protein) to a lysine in a protein to direct its degradation by the Mpa•20S proteasome.
- Rigid-body unit
A structure that shows little structural variation formed by packing of the small AAA+ domain of one ClpX subunit against the large AAA+ domain of a neighbouring subunit.
- Self-compartmentalized peptidase
An enzyme in which multiple subunits assemble to form a barrel-shaped structure in which the active sites for peptide-bind cleavage are located in an internal chamber.
- Single-molecule force spectroscopy
A biophysical method that uses optical tweezers, magnetic tweezers or atomic-force microscopy to study the behavior of a single macromolecule under force.
- Suicide inhibitors
Substrate analogues that cause irreversible inhibition of an enzyme by forming an irreversible covalent bond with the active site of an enzyme.
- Time constant
The reciprocal of the kinetic rate constant for any reaction. For example, if the rate constant for ATP hydrolysis by a single ClpX enzyme is 4 s−1, then the time constant is 0.25 s.
- TitinI27 domain
One of the many related β-sheet immunoglobulin-like domains, each comprising ~100 amino acids, in the titin protein, which is responsible for muscle elasticity. The titinI27 domain has been used for many biochemical and biophysical studies of protein folding.
- tmRNA
A bacterial RNA molecule that acts both like a tRNA and an mRNA molecule to rescue ribosomes stalled during translation and to add the ssrA degradation tag to the partially synthesized nascent polypeptide.
- van der Waals interactions
Electrostatic interactions that occur between atoms of all types and have an attractive component, resulting from transient induced dipoles (typically maximal at inter-atomic distances of 2–3.5 Å), and a repulsive component at closer distances where the electron shells of the two atoms overlap.
Biographies
Adrian O. Olivares is a research scientist at MIT, Cambridge, USA. His graduate work at Yale, New Haven, USA, focused on the molecular mechanism of myosin motors. At MIT, he has spearheaded a multi-laboratory effort to apply single-molecule optical trapping to understanding how the ClpXP and ClpAP proteases unfold and translocate specific protein substrates.
Tania A. Baker is Professor of Biology and an investigator of the Howard Hughes Medical Institute, at MIT, Cambridge, USA. Her laboratory studies mechanisms of direct and adaptor-mediated substrate recognition of protein substrates by AAA+ proteases and protein-remodelling chaperones.
Robert T. Sauer is Professor of Biology at MIT, Cambridge, USA. His lab studies the structures and molecular mechanisms of protein unfolding, translocation and degradation by AAA+ proteases and also studies PDZ proteases involved in signalling and protein quality control.
Footnotes
Competing interests statement
The authors declare no competing interests.
Contributor Information
Adrian O. Olivares, Dept. of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139 USA
Tania A. Baker, Dept. of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139 USA. Howard Hughes Medical Institute, M.I.T., Cambridge, MA 02139 USA
Robert T. Sauer, Dept. of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139 USA
References
- 1.Oldfield CJ, Dunker AK. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem. 2014;83:553–584. doi: 10.1146/annurev-biochem-072711-164947. [DOI] [PubMed] [Google Scholar]
- 2.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 3.Striebel F, Kress W, Weber-Ban E. Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr Opin Struct Biol. 2009;19:209–217. doi: 10.1016/j.sbi.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Matyskiela ME, Martin A. Design principles of a universal protein degradation machine. J Mol Biol. 2013;425:199–213. doi: 10.1016/j.jmb.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barthelme D, Sauer RT. Identification of the Cdc48•20S proteasome as an ancient AAA+ proteolytic machine. Science. 2012;337:843–846. doi: 10.1126/science.1224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyer AS, Baker TA. Proteolysis in the Escherichia coli heat shock response: a player at many levels. Curr Opin Microbiol. 2011;14:194–199. doi: 10.1016/j.mib.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gur E. The Lon AAA+ protease. Subcell Biochem. 2013;66:35–51. doi: 10.1007/978-94-007-5940-4_2. [DOI] [PubMed] [Google Scholar]
- 8.Flynn JM, Levchenko I, Sauer RT, Baker TA. Modulating substrate choice: the SspB adaptor delivers a regulator of the extracytoplasmic-stress response to the AAA+ protease ClpXP for degradation. Genes Dev. 2004;18:2292–2301. doi: 10.1101/gad.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore SD, Sauer RT. The tmRNA system for translational surveillance and ribosome rescue. Annu Rev Biochem. 2007;76:101–124. doi: 10.1146/annurev.biochem.75.103004.142733. [DOI] [PubMed] [Google Scholar]
- 10.Keiler KC. Mechanisms of ribosome rescue in bacteria. Nat Rev Microbiol. 2015;13:285–297. doi: 10.1038/nrmicro3438. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Y, Gottesman S. Regulation of proteolysis of the stationary-phase sigma factor RpoS. J Bacteriol. 1998;180:1154–1158. doi: 10.1128/jb.180.5.1154-1158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peterson CN, Levchenko I, Rabinowitz JD, Baker TA, Silhavy TJ. RpoS proteolysis is controlled directly by ATP levels in Escherichia coli. Genes Dev. 2012;26:548–553. doi: 10.1101/gad.183517.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizusawa S, Gottesman S. Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein. Proc Natl Acad Sci USA. 1983;80:358–362. doi: 10.1073/pnas.80.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenal U, Fuchs T. An essential protease involved in bacterial cell-cycle control. EMBO J. 1998;17:5658–5669. doi: 10.1093/emboj/17.19.5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ingmer H, Brøndsted L. Proteases in bacterial pathogenesis. Res Microbiol. 2009;160:704–710. doi: 10.1016/j.resmic.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 16.Konovalova A, Søgaard-Andersen L, Kroos L. Regulated proteolysis in bacterial development. FEMS Microbiol Rev. 2014;38:493–522. doi: 10.1111/1574-6976.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gur E, Sauer RT. Evolution of the ssrA degradation tag in Mycoplasma: specificity switch to a different protease. Proc Natl Acad Sci USA. 2008;105:16113–16118. doi: 10.1073/pnas.0808802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ge Z, Karzai AW. Co-evolution of multipartite interactions between an extended tmRNA tag and a robust Lon protease in Mycoplasma. Mol Microbiol. 2009;74:1083–1099. doi: 10.1111/j.1365-2958.2009.06923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker TA, Sauer RT. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta. 2012;1823:15–28. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh SK, et al. Functional domains of the ClpA and ClpX molecular chaperones identified by limited proteolysis and deletion analysis. J Biol Chem. 2001;276:29420–29429. doi: 10.1074/jbc.M103489200. [DOI] [PubMed] [Google Scholar]
- 21.Wojtyra UA, Thibault G, Tuite A, Houry WA. The N-terminal zinc binding domain of ClpX is a dimerization domain that modulates the chaperone function. J Biol Chem. 2003;278:48981–48990. doi: 10.1074/jbc.M307825200. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Hartling JA, Flanagan JM. The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell. 1997;91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- 23.Yu AYH, Houry WA. ClpP: a distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 2007;581:3749–3757. doi: 10.1016/j.febslet.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 24.Alexopoulos JA, Guarné A, Ortega J. ClpP: a structurally dynamic protease regulated by AAA+ proteins. J Struct Biol. 2012;179:202–210. doi: 10.1016/j.jsb.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Lee ME, Baker TA, Sauer RT. Control of substrate gating and translocation into ClpP by channel residues and ClpX binding. J Mol Biol. 2010;399:707–718. doi: 10.1016/j.jmb.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimaud R, Kessel M, Beuron F, Steven AC, Maurizi MR. Enzymatic and structural similarities between the Escherichia coli ATP-dependent proteases, ClpXP and ClpAP. J Biol Chem. 1998;273:12476–12481. doi: 10.1074/jbc.273.20.12476. [DOI] [PubMed] [Google Scholar]
- 27.Kim YI, et al. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat Struct Biol. 2001;8:268–271. doi: 10.1038/84967. [DOI] [PubMed] [Google Scholar]
- 28.Martin A, Baker TA, Sauer RT. Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol Cell. 2007;27:41–52. doi: 10.1016/j.molcel.2007.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akopian T, et al. The active ClpP protease from M tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 2012;31:1529–1541. doi: 10.1038/emboj.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmitz KR, Sauer RT. Substrate delivery by the AAA+ ClpX and ClpC1 unfoldases activates the mycobacterial ClpP1P2 peptidase. Mol Microbiol. 2014;93:617–628. doi: 10.1111/mmi.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stanne TM, Pojidaeva E, Andersson FI, Clarke AK. Distinctive types of ATP-dependent Clp proteases in cyanobacteria. J Biol Chem. 2007;282:14394–14402. doi: 10.1074/jbc.M700275200. [DOI] [PubMed] [Google Scholar]
- 32.Olinares PDB, Kim J, Davis JI, van Wijk KJ. Subunit stoichiometry, evolution, and functional implications of an asymmetric plant plastid ClpP/R protease complex in Arabidopsis. Plant Cell. 2011;23:2348–2361. doi: 10.1105/tpc.111.086454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Compton CL, Schmitz KR, Sauer RT, Sello JK. Antibacterial activity of and resistance to small molecule inhibitors of the ClpP peptidase. ACS Chem Biol. 2013;8:2669–2677. doi: 10.1021/cb400577b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang SG, Dimitrova MN, Ortega J, Ginsburg A, Maurizi MR. Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J Biol Chem. 2005;280:35424–35432. doi: 10.1074/jbc.M507240200. [DOI] [PubMed] [Google Scholar]
- 35.Schmitz KR, Carney DW, Sello JK, Sauer RT. Crystal structure of Mycobacterium tuberculosis ClpP1P2 suggests a model for peptidase activation by AAA+ partner binding and substrate delivery. Proc Natl Acad Sci USA. 2014;111:E4587–E4595. doi: 10.1073/pnas.1417120111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sprangers R, Gribun A, Hwang PM, Houry WA, Kay LE. Quantitative NMR spectroscopy of supramolecular complexes: dynamic side pores in ClpP are important for product release. Proc Natl Acad Sci USA. 2005;102:16678–16683. doi: 10.1073/pnas.0507370102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brötz-Oesterhelt H, et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med. 2005;11:1082–1087. doi: 10.1038/nm1306. Discovery that ADEPs kill bacteria by targeting the ClpP peptidase and activating degradation of unstructured proteins in the absence of a AAA+ unfoldase partner. [DOI] [PubMed] [Google Scholar]
- 38.Kirstein J, et al. The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol Med. 2009;1:37–49. doi: 10.1002/emmm.200900002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee BG, et al. Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat Struct Mol Biol. 2010;17:471–478. doi: 10.1038/nsmb.1787. [DOI] [PubMed] [Google Scholar]
- 40.Li DH, et al. Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: A model for the ClpX/ClpA-bound state of ClpP. Chem Biol. 2010;17:959–969. doi: 10.1016/j.chembiol.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeiler E, Korotkov VS, Lorenz-Baath K, Böttcher T, Sieber SA. Development and characterization of improved β-lactone-based anti-virulence drugs targeting ClpP. Bioorg Med Chem. 2012;20:583–591. doi: 10.1016/j.bmc.2011.07.047. [DOI] [PubMed] [Google Scholar]
- 42.Conlon BP, et al. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature. 2013;503:365–370. doi: 10.1038/nature12790. This study demonstrates that ADEPs, in combination with a traditional antibiotic, can effectively eliminate dormant persister cells in biofilms, which are responsible for many drug-resistant chronic infections. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vasudevan D, Rao SPS, Noble CG. Structural basis of mycobacterial inhibition by cyclomarin A. J Biol Chem. 2013;288:30883–30891. doi: 10.1074/jbc.M113.493767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carney DW, Schmitz KR, Truong JV, Sauer RT, Sello JK. Restriction of the conformational dynamics of the cyclic acyldepsipeptide antibiotics improves their antibacterial activity. J Am Chem Soc. 2014;136:1922–1929. doi: 10.1021/ja410385c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gavrish E, et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem Biol. 2014;21:509–518. doi: 10.1016/j.chembiol.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson MW, Singh SK, Maurizi MR. Processive degradation of proteins by the ATP-dependent Clp protease from Escherichia coli Requirement for the multiple array of active sites in ClpP but not ATP hydrolysis. J Biol Chem. 1994;269:18209–18215. [PubMed] [Google Scholar]
- 47.Jennings LD, Lun DS, Médard M, Licht S. ClpP hydrolyzes a protein substrate processively in the absence of the ClpA ATPase: mechanistic studies of ATP-independent proteolysis. Biochem. 2008;47:11536–11546. doi: 10.1021/bi801101p. [DOI] [PubMed] [Google Scholar]
- 48.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Striebel F, Imkamp F, Özcelik D, Weber-Ban E. Pupylation as a signal for proteasomal degradation in bacteria. Biochim Biophys Acta. 2014;1843:103–113. doi: 10.1016/j.bbamcr.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 50.Elharar Y, et al. Survival of mycobacteria depends on proteasome-mediated amino acid recycling under nutrient limitation. EMBO J. 2014;33:1802–1814. doi: 10.15252/embj.201387076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bougdour A, Cunning C, Baptiste PJ, Elliott T, Gottesman S. Multiple pathways for regulation of σS (RpoS) stability in Escherichia coli via the action of multiple anti-adaptors. Mol Microbiol. 2008;68:298–313. doi: 10.1111/j.1365-2958.2008.06146.x. [DOI] [PubMed] [Google Scholar]
- 52.Abel S, et al. Regulatory cohesion of cell cycle and cell differentiation through interlinked phosphorylation and second messenger networks. Mol Cell. 2011;43:550–560. doi: 10.1016/j.molcel.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rood KL, Clark NE, Stoddard PR, Garman SC, Chien P. Adaptor-dependent degradation of a cell-cycle regulator uses a unique substrate architecture. Structure. 2012;20:1223–1232. doi: 10.1016/j.str.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell. 2009;139:744–756. doi: 10.1016/j.cell.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stinson BM, et al. Nucleotide binding and conformational switching in the hexameric ring of a AAA+ machine. Cell. 2013;153:628–639. doi: 10.1016/j.cell.2013.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matyskiela ME, Lander GC, Martin A. Conformational switching of the 26S proteasome enables substrate degradation. Nat Struct Mol Biol. 2013;20:781–788. doi: 10.1038/nsmb.2616. This paper reports a cryo-EM structure of the 26S proteasome, which provides important mechanistic insights into substrate recognition, deubiquitination, unfolding and translocation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stinson BM, Baytshtok V, Schmitz KR, Baker TA, Sauer RT. Subunit asymmetry and roles of conformational switching in the hexameric AAA+ ring of ClpX. Nat Struct Mol Biol. 2015;22:411–416. doi: 10.1038/nsmb.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glynn SE, Nager AR, Baker TA, Sauer RT. Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat Struct Mol Biol. 2012;19:616–622. doi: 10.1038/nsmb.2288. This study probes ClpX using structure-guided crosslinking across the rigid-body interfaces, which reveals that a topologically closed ring is mechanically active and assumes different conformations by altering the geometry of the hinges between the large and small AAA+ domains of each subunit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin A, Baker TA, Sauer RT. Rebuilt AAA+ motors reveal operating principles for ATP-fuelled machines. Nature. 2005;437:1115–1120. doi: 10.1038/nature04031. In this study, engineering and characterization of single-chain ClpX hexamers with different combinations of active and inactive subunits supports a probabilistic model of AAA+ ring function in which ATP hydrolysis in a single subunit generates a power stroke. [DOI] [PubMed] [Google Scholar]
- 60.Gai D, Zhao R, Li D, Finkielstein CV, Chen XS. Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell. 2004;119:47–60. doi: 10.1016/j.cell.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 61.Smith D, Fraga H, Reis C, Kafri G. ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell. 2011;144:526–538. doi: 10.1016/j.cell.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aubin-Tam ME, Olivares AO, Sauer RT, Baker TA, Lang MJ. Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell. 2011;145:257–267. doi: 10.1016/j.cell.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maillard RA, et al. ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell. 2011;145:459–469. doi: 10.1016/j.cell.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sen M, et al. The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell. 2013;155:636–646. doi: 10.1016/j.cell.2013.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cordova JC, et al. Stochastic but highly coordinated protein unfolding and translocation by the ClpXP proteolytic machine. Cell. 2014;158:647–658. doi: 10.1016/j.cell.2014.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kenniston JA, Baker TA, Fernandez JM, Sauer RT. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell. 2003;114:511–520. doi: 10.1016/s0092-8674(03)00612-3. [DOI] [PubMed] [Google Scholar]
- 67.Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A. ATP-Dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell. 2001;7:627–637. doi: 10.1016/s1097-2765(01)00209-x. The first demonstration that AAA+ proteases catalyse unfolding by processively unravelling substrates from the engagement tag, with the stability of adjacent local secondary structure having an important role in degradation susceptibility. [DOI] [PubMed] [Google Scholar]
- 68.Martin A, Baker TA, Sauer RT. Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of ssrA-tagged substrates. Mol Cell. 2008;29:441–450. doi: 10.1016/j.molcel.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martin A, Baker TA, Sauer RT. Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat Struct Mol Biol. 2008;15:1147–1151. doi: 10.1038/nsmb.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iosefson O, Nager AR, Baker TA, Sauer RT. Coordinated gripping of substrate by subunits of an AAA+ proteolytic machine. Nat Chem Biol. 2015;11:201–206. doi: 10.1038/nchembio.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iosefson O, Olivares AO, Baker TA, Sauer RT. Dissection of axial-pore loop function during unfolding and translocation by a AAA+ proteolytic machine. Cell Reports. 2015;12:1–10. doi: 10.1016/j.celrep.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barkow SR, Levchenko I, Baker TA, Sauer RT. Polypeptide translocation by the AAA+ ClpXP protease machine. Chem Biol. 2009;16:605–612. doi: 10.1016/j.chembiol.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kenniston JA, Baker TA, Sauer RT. Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci USA. 2005;102:1390–1395. doi: 10.1073/pnas.0409634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell. 2003;11:671–683. doi: 10.1016/s1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 75.Burton RE, Siddiqui SM, Kim YI, Baker TA, Sauer RT. Effects of protein stability and structure on substrate processing by the ClpXP unfolding and degradation machine. EMBO J. 2001;20:3092–3100. doi: 10.1093/emboj/20.12.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bolon DN, Grant RA, Baker TA, Sauer RT. Nucleotide-dependent substrate handoff from the SspB adaptor to the AAA+ ClpXP protease. Mol Cell. 2004;16:343–350. doi: 10.1016/j.molcel.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 77.Martin A, Baker TA, Sauer RT. Protein unfolding by a AAA+ protease is dependent on ATP-hydrolysis rates and substrate energy landscapes. Nat Struct Mol Biol. 2008;15:139–145. doi: 10.1038/nsmb.1380. [DOI] [PubMed] [Google Scholar]
- 78.Koodathingal P, et al. ATP-dependent proteases differ substantially in their ability to unfold globular proteins. J Biol Chem. 2009;284:18674–18684. doi: 10.1074/jbc.M900783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vass RH, Chien P. Critical clamp loader processing by an essential AAA+ protease in Caulobacter crescentus. Proc Natl Acad Sci USA. 2013;110:18138–18143. doi: 10.1073/pnas.1311302110. Discovery of an unexpected mode of partial proteolytic processing by ClpXP that generates DNA-clamp loader isoforms required for C. crescentus viability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 81.Hoppe T, et al. Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell. 2000;102:577–586. doi: 10.1016/s0092-8674(00)00080-5. [DOI] [PubMed] [Google Scholar]
- 82.Tian L, Holmgren RA, Matouschek A. A conserved processing mechanism regulates the activity of transcription factors Cubitus interruptus and NF-kappaB. Nat Struct Mol Biol. 2005;12:1045–1053. doi: 10.1038/nsmb1018. [DOI] [PubMed] [Google Scholar]
- 83.Nager AR, Baker TA, Sauer RT. Stepwise unfolding of a β barrel protein by the AAA+ ClpXP protease. J Mol Biol. 2011;413:4–16. doi: 10.1016/j.jmb.2011.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kress W, Mutschler H, Weber-Ban E. Both ATPase domains of ClpA are critical for processing of stable protein structures. J Biol Chem. 2009;284:31441–31452. doi: 10.1074/jbc.M109.022319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hinnerwisch J, et al. Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell. 2005;121:1029–1041. doi: 10.1016/j.cell.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 86.Baytshtok V, Baker TA, Sauer RT. Assaying the kinetics of protein denaturation catalyzed by AAA+ unfolding machines and proteases. Proc Natl Acad Sci USA. 2015;112:5377–5382. doi: 10.1073/pnas.1505881112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Olivares AO, Nager AR, Iosefson O, Sauer RT, Baker TA. Mechanochemical basis of protein degradation by a double-ring AAA+ machine. Nat Struct Mol Biol. 2014;21:871–875. doi: 10.1038/nsmb.2885. This paper uses optical trapping experiments to reveal similarities and differences in the mechanical unfolding and translocation activities of single-ring and double-ring AAA+ partners of ClpP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Flynn JM, et al. Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis. Proc Natl Acad Sci USA. 2001;98:10584–10589. doi: 10.1073/pnas.191375298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ito K, Akiyama Y. Cellular functions, mechanism of action, and regulation of FtsH protease. Annu Rev Microbiol. 2005;59:211–231. doi: 10.1146/annurev.micro.59.030804.121316. [DOI] [PubMed] [Google Scholar]
- 90.Gerdes F, Tatsuta T, Langer T. Mitochondrial AAA proteases--towards a molecular understanding of membrane-bound proteolytic machines. Biochim Biophys Acta. 2012;1823:49–55. doi: 10.1016/j.bbamcr.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 91.Vieux EF, Wohlever ML, Chen JZ, Sauer RT, Baker TA. Distinct quaternary structures of the AAA+ Lon protease control substrate degradation. Proc Natl Acad Sci USA. 2013;110:E2002–E2008. doi: 10.1073/pnas.1307066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gur E, Sauer RT. Degrons in protein substrates program the speed and operating efficiency of the AAA+ Lon proteolytic machine. Proc Natl Acad Sci USA. 2009;106:18503–18508. doi: 10.1073/pnas.0910392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schmitt EK, et al. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew Chemie Int Ed. 2011;50:5889–5891. doi: 10.1002/anie.201101740. [DOI] [PubMed] [Google Scholar]
- 94.Burton RE, Baker TA, Sauer RT. Nucleotide-dependent substrate recognition by the AAA+ HslUV protease. Nat Struct Mol Biol. 2005;12:245–251. doi: 10.1038/nsmb898. [DOI] [PubMed] [Google Scholar]
- 95.Wu WF, Zhou Y, Gottesman S. Redundant in vivo proteolytic activities of Escherichia coli Lon and the ClpYQ (HslUV) protease. J Bacteriol. 1999;181:3681–3687. doi: 10.1128/jb.181.12.3681-3687.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schrader EK, Harstad KG, Matouschek A. Targeting proteins for degradation. Nat Chem Biol. 2009;5:815–822. doi: 10.1038/nchembio.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Burton BM, Baker TA. Remodeling protein complexes: insights from the AAA+ unfoldase ClpX and Mu transposase. Protein Sci. 2005;14:1945–1954. doi: 10.1110/ps.051417505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ling L, Montaño SP, Sauer RT, Rice PA, Baker TA. Deciphering the roles of multicomponent recognition signals by the AAA+ unfoldase ClpX. J Mol Biol. 2015 doi: 10.1016/j.jmb.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kapitein N, et al. ClpV recycles VipA/VipB tubules and prevents non-productive tubule formation to ensure efficient type VI protein secretion. Mol Microbiol. 2013;87:1013–1028. doi: 10.1111/mmi.12147. [DOI] [PubMed] [Google Scholar]
- 100.Winkler J, Tyedmers J, Bukau B, Mogk A. Chaperone networks in protein disaggregation and prion propagation. J Struct Biol. 2012;179:152–160. doi: 10.1016/j.jsb.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 101.Roll-Mecak A, McNally FJ. Microtubule-severing enzymes. Curr Opin Cell Biol. 2010;22:96–103. doi: 10.1016/j.ceb.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Camberg JL, Viola MG, Rea L, Hoskins JR, Wickner S. Location of dual sites in E coli FtsZ important for degradation by ClpXP; one at the C-terminus and one in the disordered linker. PLoS One. 2014;9:e94964. doi: 10.1371/journal.pone.0094964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kardon JR, et al. Mitochondrial ClpX activates a key enzyme for heme biosynthesis and erythropoiesis. Cell. 2015;161:858–867. doi: 10.1016/j.cell.2015.04.017. Discovery that mitochondrial ClpX remodels an enzyme required for haem biosynthesis to accelerate the rate of cofactor insertion and regulate activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao M, et al. Mechanistic insights into the recycling machine of the SNARE complex. Nature. 2015;518:61–67. doi: 10.1038/nature14148. A paper reporting cryo-EM structures of NSF in different nucleotide states bound to SNARE complexes that reveal dramatic changes in conformation that may explain SNARE disassembly. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ryu JK, et al. Spring-loaded unraveling of a single SNARE complex by NSF in one round of ATP turnover. Science. 2015;347:1485–1489. doi: 10.1126/science.aaa5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li T, et al. E. coliClpB is a non-processive polypeptide translocase. Biochem J. doi: 10.1042/BJ20141457. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 108.Raju RM, et al. Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 2012;8:e1002511. doi: 10.1371/journal.ppat.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]