

Abstract
Obsessive Compulsive Disorder (OCD) is a common neuropsychiatric disorder with unknown molecular underpinnings. Identification of genetic and non-genetic risk factors has largely been elusive, primarily because of a lack of power. In contrast, neuroimaging has consistently implicated the cortico-striatal-thalamo-cortical (CSTC) circuits in OCD. Pharmacological treatment studies also show specificity, with consistent response of OCD symptoms to chronic treatment with serotonin reuptake inhibitors; although most patients are left with residual impairment. In theory, animal models could provide a bridge from the neuroimaging and pharmacology data to an understanding of pathophysiology at the cellular and molecular level. Several mouse models have been proposed using genetic, immunological, pharmacological, and optogenetic tools. These experimental model systems allow testing of hypotheses about the origins of compulsive behavior. Several models have generated behavior that appears compulsive-like, particularly excessive grooming, and some have demonstrated response to chronic serotonin reuptake inhibitors, establishing both face validity and predictive validity. Construct validity is more difficult to establish in the context of a limited understanding of OCD risk factors. Our current models may help us to dissect the circuits and molecular pathways that can elicit OCD-relevant behavior in rodents. We can hope that this growing understanding, coupled with developing technology, will prepare us when robust OCD risk factors are better understood.
Keywords: Repetitive behavior, basal ganglia, striatum, optogenetic, autoimmune, autism
1. Introduction
Obsessive compulsive disorder (OCD) is a common, chronic condition characterized by persistent, intrusive obsessions, repetitive behavior, and anxiety (Calvocoressi et al., 1998). The disorder affects 1-3% of the population and is among the top ten causes of disability worldwide (Kessler et al., 2005, Koran et al., 2007, Michael S. Ritsner, 2007). First line forms of therapy include serotonin reuptake inhibitors (SRIs) and cognitive behavioral therapy; however, only 50-60% of patients show adequate response to available treatments (Koran et al., 2007). For example, a 20-40% decrease in OCD symptoms may result following SRI therapy (Dougherty et al., 2004), which leaves many with clinically significant residual symptoms. In some refractory OCD cases, deep brain stimulation (DBS) has also been used as a treatment alternative (Sturm et al., 2003, Goodman et al., 2010). Other augmentation therapies, such as antipsychotics and glutamatergic agents, are also being evaluated but have limited evidence for their utility (Arumugham and Reddy, 2013).
Ideally, more effective therapeutics would emerge from an understanding of the etiology of OCD. As with many neuropsychiatric conditions, the underlying causes of OCD are unknown and likely to involve both genetic and environmental factors. Hypotheses based on genetic and neuroimaging data have led researchers to create animal models that recapitulate the hallmarks of the disorder, with the aim of probing the underlying neurobiology. Here, we will discuss the growing number of proposed rodent models of OCD, including pharmacologically-induced, genetic, and optogenetic animal models, with a focus on assessment of the validity of the model in relation to knowledge of the human condition. Building upon the initial data on validity, we will describe the emerging understanding of neurobiological mechanisms in each model. Finally, we will discuss the approaches to take growing knowledge from these rodent models and translate it into novel treatments that can be applied in patients with OCD.
1.2 Genetic and non-genetic factors likely contribute to OCD risk
To understand the validity of rodent models, we must first understand the risk factors that may contribute to OCD. Abundant evidence for a heritable component of OCD stems from twin and family studies (Grados and Wilcox, 2007, Pauls, 2008). As reviewed elsewhere (Pauls, 2010), OCD symptoms are estimated to be 40-65% heritable in children and 27-47% heritable in adults (van Grootheest et al., 2005). Family studies indicate that OCD is twice as common in first-degree relatives of affected adults and ten times as likely in relatives of affected children (Pauls, 2008). These studies support the premise that OCD risk is derived from a complex combination of genetic and non-genetic factors.
Genetic linkage studies have yet to generate genome-wide significant findings for the core diagnosis of OCD, likely because of lack of statistical power in limited sample sizes (Hanna et al., 2002a, Mathews et al., 2012). The most promising linkage signal is on chromosome 9p24, based upon a suggestive linkage peak in the first genome-wide linkage study that was subsequently directly replicated in a study targeting only this region (Hanna et al., 2002a, Willour et al., 2004a). Another suggestive linkage peak on chromosome 15q14 was identified in two genome-wide linkage studies (Shugart et al., 2006). Genome-wide association studies (GWAS) have been similarly underpowered, to date, in OCD (Stewart et al., 2013b, Mattheisen et al., 2015). A single polymorphism near BTBD3 reached genome-wide significance in a family-based subset of one GWAS analysis, but was only suggestive in the overall sample (Stewart et al., 2013b).
Candidate genes for OCD have been identified based upon proximity to linkage peaks, biomarker findings, and relationship to pharmacological targets. The strongest candidate gene association findings in OCD focus on SLC1A1, which encodes the neuronal glutamate transporter, EAAT3. Multiple studies have reported significant evidence for association OCD with SLC1A1 polymorphisms, particularly in males with OCD and particularly with polymorphisms in the 3′ region of the gene (Arnold et al., 2006, Dickel et al., 2006, Stewart et al., 2007, Kwon et al., 2009, Shugart et al., 2009, Wendland et al., 2009b, Samuels et al., 2011, Stewart et al., 2013b, Wu et al., 2013); although a recent meta-analysis found only modest associations that were not significant after correcting for multiple testing (Stewart et al., 2013a). Interest in SLC1A1 stemmed from its location under the chromosome 9p24 linkage peak, as well as biomarker studies implicating the glutamate system in OCD. Elevated cerebrospinal fluid glutamate levels in two studies (Chakrabarty et al., 2005a, Bhattacharyya et al., 2009) are matched by increased glutamatergic signal in some magnetic resonance spectroscopy (MRS) studies in OCD (Moore et al., 1998, Rosenberg et al., 2000); although other MRS studies do not find significant differences (Brennan et al., 2013).
Other association studies have generated largely mixed results that are difficult to interpret in the context of a long history of psychiatric candidate genes that have failed to replicate consistently. Additional candidate genes in the glutamate system have also been studied, including GRIN2B, which shows evidence of association in some but not all studies (Arnold et al., 2004, Alonso et al., 2012, Cai et al., 2013). The other leading candidate gene is the serotonin transporter, SLC6A4, with both common and rare variants exhibiting association in some studies (Ozaki et al., 2003, Dickel et al., 2007, Grados et al., 2007, Saiz et al., 2008, Wendland et al., 2008), but not all (Taylor, 2013). The dopaminergic system (COMT and DRD4) has also been implicated via gene association studies; however these findings have yet to be replicated (Billett et al., 1998, Pooley et al., 2007). A lack of clear susceptibility genes contributes to the difficulty of modeling OCD in animals, though this issue is shared by most complex heterogeneous neuropsychiatric disorders.
Apart from genetics, autoimmunity has also received considerable attention as a potential risk factor for OCD. Observation of neuropsychiatric manifestations co-occuring with rheumatic fever and Sydenham's chorea lead to the description of Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal infections (PANDAS). As reviewed elsewhere (Williams and Swedo, 2015), children with this presentation may develop compulsions, tics, or other psychiatric symptoms very rapidly following an infection. Emerging data suggest that non-streptococcal infections may also serve as triggers for symptoms, leading to the more recent description of Pediatric Acute-onset Neuropsychiatric Syndrome (Chang et al., 2015). Anti-brain antibodies, including antibodies to the basal ganglia, have been identified in some cases of PANDAS, but with little evidence that antibodies consistently separate cases from controls or increase with symptom exacerbations (Pavone et al., 2004, Singer et al., 2004, Singer et al., 2005, Morris-Berry et al., 2013). In contrast, studies in the larger population of patients with idiopathic OCD have more consistently reported elevated anti-basal ganglia antibodies in serum, as supported by a recent meta-analysis (Pearlman et al., 2014), as well as one study in cerebrospinal fluid (Bhattacharyya et al., 2009). It is not clear, however, whether and how these antibodies might be causally connected to the pathophysiology of OCD.
1.3 Basal ganglia circuitry is implicated in OCD
Evidence from neuroimaging studies has consistently implicated the basal ganglia as being involved in the development of OCD, specifically the cortico-striatal-thalamo-cortical (CSTC) loop (Saxena and Rauch, 2000, Ting and Feng, 2011). The most consistently implicated basal ganglia subregions in OCD patients are the caudate nucleus and the putamen, which make up the human striatum (Rauch et al., 2001, Rosenberg et al., 2001). Structural neuroimaging studies have consistently demonstrated abnormalities in caudate volume in OCD (Radua and Mataix-Cols, 2009, Radua et al., 2010). Functional imaging studies have also identified hyperactivity in cortico-striatal circuits, both at baseline and after symptom provocation (Menzies et al., 2008). The caudate is extrinsically innervated primarily by excitatory inputs originating from the cerebral cortex, the thalamus, and the substatia nigra pars compacta (Parent and Hazrati, 1995). Many studies have found that increased activity within the cortex, specifically the anterior cingulate (ACC) and orbitofrontal cortex (OFC) are seen in OCD patients (Chamberlain et al., 2008, MacMaster et al., 2008, Menzies et al., 2008). These studies, along with further meta-analysis of functional neuroimaging data in OCD patients (Whiteside et al., 2004, Whiteside et al., 2006), suggest that there is a functional abnormality resulting in hyperactivity of the basal-ganglia loop. A common model of this increased CSTC signaling suggests that it could arise from hyperactivity of the direct-pathway or by hypoactivity of the indirect pathway resulting in repetitive and compulsive behaviors (Ting and Feng, 2011).
2. Validation of Mouse Models Related to OCD
Rodent models allow hypothesis testing via manipulation of OCD risk factors, as well as stimulation or inhibition of the circuitry implicated in OCD. The ability to perform such experimental manipulations is also accompanied by immediate access to the brain that allows for detailed probing of molecular and circuitry changes that may underlie the pathophysiology of OCD. Unfortunately, behavioral assessment in rodents is limited to compulsive-like behaviors, since obsessions are an internal experience that can only be measured via self-report. Animal studies aimed at eliciting OCD-relevant behaviors through the use of pharmacological, transgenic, immunologic, and optogenetic tools have resulted in a wide range of behavioral phenotypes with varying degrees of similarity to compulsive behaviors in the human population. Biochemical and electrophysiological analyses of the underlying cells and circuits believed to be involved in OCD are beginning to provide data that may enable researchers to develop novel therapeutics that lie closer to the pathophysiology of the disorder.
Model systems are necessary to test hypotheses and probe the underlying neurobiology, but models are inherently limited and cannot fully recapitulate the human disorder. To assess the potential utility of a given model, three criteria are typically used, as first described by Willner in 1991: 1) construct validity, 2) face validity, and 3) predictive validity (Willner, 1991). By using these criteria to assess how well an animal model mirrors the human condition, we can better contextualize the significance of resulting findings, potentially allowing us to target our attempts at translation to subgroups of the human population that may more clearly map onto the risk factor, behavioral profile, or circuitry implicated in the rodent.
2.1 Construct Validity
The first and likely most important criterion for assessing animal model validity is construct validity. This is best understood in the context of genetic models, where a putative OCD risk gene is manipulated in mice to observe its behavioral and neurobiological consequences. Similarly, an immunological trigger, such as the antibodies that have been detected in some cases of PANDAS or PANS, could allow direct transfer into an animal model (Swedo, 2002, Snider and Swedo, 2004). Drug-induced worsening of OCD symptoms, such has been reported with the anti-migraine 5-HT1B/D agonists (Koran et al., 2001a) and psychostimulants (Varley et al., 2001, Bloch et al., 2009), may also provide construct validity. Since these drugs seem to exacerbate but not cause OCD, this approach to validity may not work independently from another risk factor, as discussed in the predictive validity section below.
Since circuitry is better defined than risk factors, could circuits be a source of construct validity? Or does this represent face or predictive validity? Often, validation criterion may overlap, as is the case when thinking about brain circuitry. The differentiation between the validity criteria when it comes to brain circuits may come down to experimental design. If alteration of circuit activity is the foundation of the animal model, as is the case in some recent optogenetic work, then construct validity may be supported. Conversely, for genetic models that result in electrophysiological changes in brain regions implicated in OCD, the circuitry findings may be better classified as face validity, as described below.
2.2 Face Validity
The second criterion, face validity, is usually based on the emergence of observable behaviors reminiscent of compulsive behavior, since OCD is a behaviorally defined disorder. The most commonly observed rodent behavior that supports face validity is excessive grooming, which corresponds to an increase in a behavior that follows a stereotyped pattern even in wildtype mice (Welch et al., 2007, Shmelkov et al., 2010, Ahmari et al., 2013); although the stereotyped pattern itself may also be distorted in some models. In contrast, other models lead to a novel stereotyped, repetitive pattern of behavior, as is seen with dopaminergic (quinpirole) and serotonergic (8-OHDPAT) agonists that induce perseverative locomotor behavior in the Y-maze or open field chambers (Yadin et al., 1991, Szechtman et al., 1998). Other measures, such as reversal learning or prepulse inhibition deficits, parallel findings that are not explicitly diagnostic but are instead thought to underlie the symptoms of OCD, such as cognitive rigidity or motor inhibition deficits (Chamberlain et al., 2006, Remijnse et al., 2006, Gu et al., 2008, Valerius et al., 2008, Andersen et al., 2010, Ahmari et al., 2012, Bissonette and Powell, 2012, Brigman et al., 2012, Remijnse et al., 2013, Hatalova et al., 2014, Zhang et al., 2015).
Face validity may also be established in animal models by identifying alterations in putative biomarkers that mirror those previously identified in the human OCD population. As reviewed above, several such biomarkers could be used to assess a rodent model of OCD, such as abnormal caudate volume (Radua and Mataix-Cols, 2009, Radua et al., 2010), hyperactivity in the cortico-striatal circuit (Menzies et al., 2008), increased glutamate concentrations in the caudate (Moore et al., 1998, Whiteside et al., 2006, Starck et al., 2008), and elevated cerebrospinal fluid glutamate levels (Chakrabarty et al., 2005b). Identification of other abnormalities in previously hypothesized neurotransmitter system, such as serotonin, dopamine, or glutamate (Dickel et al., 2007, Taylor et al., 2010, Pittenger et al., 2011), could also be asserted to represent face validity, but we must be careful not to stretch too far in assessing what “looks like” our conception of OCD.
2.3 Predictive Validity
Predictive validity of animal models of OCD may be determined in a few ways. Unfortunately, there is no straightforward way to model the cognitive behavioral therapy that should be the first-line treatment in OCD; however compulsive-like behaviors may be rescued or protected against through the administration of SRIs, as these are the first line medications. Additionally, response to SRIs in OCD requires several weeks of administration, which could be used to add specificity, as has been suggested in a few models that show response to chronic or sub-chronic but not acute dosing. Adjunctive medications, such as atypical antipsychotics, which have some evidence of benefit in OCD, may also be expected to improve compulsive-like behavior, though this may be less specific since they also benefit tics in the human population. Invasive treatments, such capsulotomy or high frequency stimulation (HFS) protocols similar to deep brain stimulation (DBS), have shown benefit in SRI-resistant OCD and could be used to assess predictive validity with a clearer relationship to any observed changes in circuitry (Greenberg et al., 2006). From the opposite perspective, common anxiolytics or antidepressants that are ineffective in people suffering from OCD, including benzodiazepines and noradrenergic reuptake inhibitors (NRIs), should be expected to be ineffective in rescuing the behavioral phenotypes observed in a valid mouse model of OCD. One challenge for the field is that many people with OCD show only modest or even minimal response to available treatments, and lack of predictive validity could indicate that a model corresponds to this subset – the precise group that we would most like to help.
One question is how to conceptualize drugs that appear to worsen OCD symptoms. When administered to wildtype animals, these drugs may provide support for construct validity; although there is little evidence for drugs inducing persistent OCD symptoms once the drug has been stopped. In contrast, within an existing model of an OCD risk factor, a drug targeting the serotonin or dopamine system may be a way to reveal the worsened symptoms that have been reported with certain drugs in OCD patients, including clozapine, stimulants, and sumatriptan and its analogs (Broocks et al., 1998, Koran et al., 2001b, Gross-Isseroff et al., 2004, Bloch et al., 2009, Fonseka et al., 2014). Ultimately, assessing a given model does not necessarily require checking off each box in the three types of validity but instead assessing the overall strength of the data and carefully drawing boundaries around the human population that it models. For example, if clozapine were used to induce repetitive behavior and CSTC circuit abnormalities in a particular inbred strain of mouse, then the initial step in translation would target the human population that experiences treatment-emergent compulsions while taking clozapine.
3. Genetic Mouse Models of OCD
3.1 Sapap3 Null Mice
At the time of the initial description of OCD-related phenotypes in mice lacking Sapap3 (Sap90/PSD-95 associated protein 3) (Welch et al., 2007, Burguiere et al., 2013), no genetic data were available to examine its potential involvement in OCD. A subsequent study identified rare amino acid variants in the human Sapap3 ortholog, DLGAP3 (Discs, Large (Drosophila) Homolog-Associated Protein 3), in OCD and trichotillomania populations; although the degree to which these variants contribute to OCD risk remains somewhat unclear (Zuchner et al., 2009). Another association study found nominally significant association with a number of common polymorphisms and haplotypes in individuals with a grooming disorder (trichotillomania, excoriation, or nail-biting) in addition to OCD, but not in OCD alone (Bienvenu et al., 2009). Additionally, the largest genome-wide association study in OCD found some suggestive but not significant evidence of association with another DLGAP family member, DLGAP1 (Stewart et al., 2013b). Collectively, these individual pieces of data provide plausible hints of construct validity without replicated evidence of involvement of DLGAP3 in human OCD.
The face validity and predictive validity of the Sapap3 null mouse is quite appealing and provided an example for subsequent studies of putatively compulsive-like behavior. Between 4-6 months, Sapap3-null mice exhibit a perseverative grooming phenotype that results in open skin wounds, as well as increased anxiety-like behavior, which together parallel the common co-occurrence of maladaptive repetitive behavior and anxiety in human OCD. It is also possible, however, to argue that increased, self-injurious grooming in the mouse maps less well onto the cleaning symptoms commonly seen in OCD, such as compulsive hand-washing, and more neatly onto the OC-spectrum conditions of trichotillomania or excoriation/skin-picking. Without access to “obsessions” that may drive the grooming behavior, it is impossible to clearly separate these possibilities. It may be most useful, then, to conceptualize this mouse as a model of OC-spectrum behavior. Importantly, the excessive grooming is alleviated by sub-chronic (6 days), but not acute administration of fluoxetine, an SRI. It is important to note that chronic administration of SRIs for a month or more is necessary to achieve efficacy in the human OCD population; however differences in brain circuitry in mice compared to humans could underlie this difference.
SAPAP3 is a post-synaptic scaffolding protein that interacts with Post Synaptic Density Protein 95 and Shank protein families (Takeuchi et al., 1997), and is highly expressed within glutamatergic synapses of the striatum, providing plausibility for its involvement in OCD risk (Saxena and Rauch, 2000, Welch et al., 2007, Menzies et al., 2008, Ting and Feng, 2011). The initial characterization of Sapap3 null mice demonstrated cortico-striatal glutamatergic transmission abnormalities, including defective synaptic transmission and reduction in in N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) glutamate receptor function (Welch et al., 2007). Further electrophysiological work with this model has revealed that loss of SAPAP3 enhances AMPA receptor endocytosis via an glutamate metabotropic receptor mGlu5 dependent mechanism (Wan et al., 2011). Additional defects in inhibitory neurotransmission of Sapap3-null mice have also been identified, including reductions of parvalbumin-positive fast spiking interneurons, resulting in abnormalities in a learning related behavioral task that will be discussed in future sections (Burguiere et al., 2013), further supporting the hypothesis of dysfunctional neurotransmission within the cortico-striatal circuit in this model.
Surprisingly, crossing the Sapap3 null mouse with a line of animals with either constitutive or induced deletion of Mc4r, the gene coding for the melanocortin 4 receptor (MC4R), was able to rescue the elevated grooming phenotype (Xu et al., 2013). Prior work showed that stimulation of MC4R resulted in a compulsive-like grooming phenotype (Alvaro et al., 2003), leading to the hypothesis that deletion of Mc4r within Sapap3-null mice could diminish the grooming phenotype. Sapap3 null mice and Mc4r mice have opposite electrophysiology findings at corticostriatal synapses, with the double null animals landing in the middle, indistinguishable from wildtype animals. Remarkably, the cross with Sapap3 null animals also rescued the hyperphagia and obesity that are the primary phenotype associated with loss of Mc4r (Farooqi, 2008). The molecular mechanisms underlying this rescue remain unknown, but it suggests the corticostriatal signaling abnormalities as the key target for rescuing the compulsive-like grooming behavior in the Sapap3 null mice. It remains to be seen whether drugs that block MC4R show similar effects, but these surprising findings point to the potential for mouse models to yield completely novel hypotheses about OCD treatment. As the first genetic mouse model to be described with SRI-responsive, striatally-dependent repetitive behavior, the Sapap3 null mouse has already begun to yield insights that could potentially translate into human OC-spectrum disorders. In the absence of clear genetic data, however, it remains difficult to assess whether findings in this mouse offer a direct parallel to human OCD.
3.2 Slitrk5 Null Mice
Interest in the SLITRK (Slit- and Trk-like) gene family initially emerged from a chromosomal translocation and follow-up rare variant association study linking SLITRK1 and Tourette's syndrome (Abelson et al., 2005), with support in some but not all subsequent studies (Keen-Kim et al., 2006, Scharf et al., 2008, Miranda et al., 2009, O'Roak et al., 2010, Karagiannidis et al., 2012, Ozomaro et al., 2013). SLITRKs are single-transmembrane proteins containing two extracellular leucine-rich repeat domains, similar to SLIT proteins, and a carboxy-terminal domain similar to tyrosine kinase receptors (Proenca et al., 2011). Like SLIT proteins, SLITRK proteins are widely expressed throughout the brain and are believed to direct neurite outgrowth. Tic-like or compulsive-like behavior has not been described in Slitrk1-null mice, but these animals do exhibit increased anxiety-like behaviors and elevated norepinephrine levels in the prefrontal cortex and nucleus accumbens (Katayama et al., 2010). On the other hand, knockout of another SLITRK family member, Slitrk5, resulted in a phenotype similar to that previously described in the Sapap3 null mice. It is unclear whether genetic association data at SLITRK1 should be seen as contributing to construct validity of Slitrk5 null mice as a model of OC-spectrum behavior. Similarly, a recent genome-wide study in OCD found suggestive association near PTPRD (Protein tyrosine phosphatase, receptor type D), which encodes a member of the receptor protein tyrosine phosphatase family that interacts with SLITRK3, providing more circumstantial evidence for involvement of this protein family, if not SLITRK5 itself. Recent work from Song and colleagues (2015), indicates that SLITRK5 acts in conjunction with the TrkB receptor to positively modulate the sensitivity of neurons to brain-derived neurotrophic factor (BDNF) (Song et al., 2015).
Shmelkov and colleagues (2010) developed and characterized a Slitrk5-null mouse that displayed face-validity in the form of elevated grooming resulting in facial hair loss and skin lesion by three months of age (Shmelkov et al., 2010). It is also of note that hemizygous Slitrk5 mice developed similar lesions but with a delayed onset at seven to nine months of age, which has not been reported in the Sapap3 heterozygous animals. Slitrk5-null mice also displayed increased anxiety-like behaviors in the elevated plus maze and open-field test. With regards to predictive-validity, chronic treatment with the SRI, fluoxetine, reversed the elevated grooming phenotype in Slitrk5-nulls; although the consequences of acute treatment were not shown.
The Slitrk5 null animals also showed biochemical and structural findings in the CSTC circuit implicated in OCD. Increased FosB immunoreactivity in the orbitofrontal cortex in these animals may parallel functional imaging finding in OCD (Chamberlain et al., 2008). Similarly, they show decreased striatal volume that may parallel some reports of reduced striatal volume relative to whole-brain measurements in OCD patients (Rosenberg et al., 1997); however other studies suggest that putamen volume may actually be increased in OCD (Radua et al., 2010, de Wit et al., 2014). As with the Sapap3 null, defective neurotransmission was measured in striatal slice preparations from Slitrk5 null mice, linking abnormalities in brain regions previously implicated in OCD to this model. As an almost direct parallel to the established pattern of validation in the Sapap3 null mouse, the Slitrk5 null mouse model shows SRI-sensitive, excessive grooming behavior in the context of altered corticostriatal structure and function. Again, more genetic data will be needed to evaluate whether this provides a direct model of OCD or OC-spectrum risk; however it is possible that insights from this model will translate to SRI-dependent repetitive behavior across species, regardless of whether this particular genetic mechanism underlies OCD itself.
3.3 HoxB8 Null Mice
HOXB8 is a component of the mammalian Hox (Homeobox-containing) complex, a family of 39 transcription factors important in determining the anteroposterior axis during development (Capecchi, 1997). In contrast to the above models, where some genetic data, albeit limited, point to potential genetic involvement in OCD or OC-spectrum disorders, no data implicate HOXB8 or its gene family in the human disorder. On the other hand, Greer and Capecchi (2002) noted that HoxB8-null mice show excessive grooming resulting in extensive hair loss and skin lesions (Greer and Capecchi, 2002). HoxB8-null animals self-groom excessively and also have increased social grooming of control animals. A subsequent report from another group using a different Hoxb8 null mouse line suggested that deficits in spinal cord function led to changes in sensitivity to pain, potentially underlying the excessive, self-injurious grooming behavior (Holstege et al., 2008). A follow-up study conducted by the Capecchi group in the original Hoxb8 null mice, however, clearly demonstrated that central nervous system function was responsible for excessive grooming in these animals (Chen et al., 2010). Remarkably, targeted knockout of Hoxb8 in the hematopoietic system alone was found to induce excessive grooming in these mice, and bone marrow transplant resulted in rescue of the grooming phenotype in Hoxb8 null animals. Taken together, these findings suggest that dysfunction resulting from peripherally-derived microglia accounts for compulsive-like grooming in these animals.
While the latest study on targeted Hoxb8 knockout ruled out peripheral nervous system (PNS) defects as the cause of the grooming phenotype, the broad expression of Hoxb8 in microglia of the brain prevents straightforward localization of the implicated brain region(s). As noted above, this model lacks clear construct validity, and predictive validity has not been assessed with an SRI rescue experiment. Despite this, the novelty of microglial involvement in excessive grooming provides an interesting new direction for potential treatment of compulsive-like behavior. As noted above, immune or autoimmune mechanisms have been explored in OCD; although without conclusive data tying a particular mechanism to emerging symptoms (Swedo, 2002, Martino et al., 2009). Intravenous immunoglobulin or plasmopheresis have been explored for treatment of Streptococcal infection-associated compulsive behaviors (Snider and Swedo, 2004); however, no current treatments have targeted the monocyte lineages leading to microglia. The Hoxb8 null model, then, may be an example of a genetic mouse model limited to face validity that nevertheless generates a very novel hypothesis about OCD risk or treatment. Without construct or predictive validity at the outset, however, translational studies targeting peripherally-derived microglia seem quite risky and should likely be reserved for truly refractory OCD unless more data emerge implicating this mechanism in a subgroup of patients with OCD.
3.4 Slc1a1 null mice
As a counter example to these three mouse models that show compulsive-like behavior but without strong construct validity, the neuronal glutamate transporter gene SLC1A1 (Solute Carrier Family 1 Member 1) has the most genetic evidence to date, but behavioral assessment of mice lacking Slc1a1 has not suggested altered compulsive-like behavior, to date. As noted above, both linkage and association data point to SLC1A1 in OCD, particularly in males with early-onset symptoms (Veenstra-VanderWeele et al., 2001, Hanna et al., 2002b, Willour et al., 2004b, Arnold et al., 2006, Dickel et al., 2006, Stewart et al., 2007, Shugart et al., 2009, Wendland et al., 2009a, Samuels et al., 2011). Adding circumstantial evidence, a recent study of 3 patients with dicarboxylic aminoaciduria identified rare loss-of-function mutations in SLC1A1, with one of the probands reporting long-standing checking and hand-washing behaviors that could be OCD-related but were not formally evaluated (Bailey et al., 2011). Slc1a1 mRNA and its corresponding protein, EAAT3 (Excitatory Amino Acid Transporter 3), are strongly expressed in the cortico-striatal regions implicated in OCD (Nieoullon et al., 2006). EAAT3 is also involved in mediating neuronal cysteine transport, an essential rate-limiting step in the synthesis of the antioxidant glutathione (Aoyama et al., 2006, Aoyama and Nakaki, 2013).
Initial publication of an Slc1a1/EAAT3 null mouse suggested little behavioral relevance to OCD (Peghini et al., 1997), with the only significant change being decreased spontaneous open-field locomotor activity. Subsequent studies with the EAAT3 null mouse focused on the potential neurodegenerative effects associated with the loss of neuronal cysteine transport. Aoyama et al. (2006) reported reduced neuronal glutathione levels and age-dependent neurodegeneration, shown by cortical thinning and enlarged ventricles. As they aged, EAAT3 null mice were observed to show increased aggression and to appear disheveled. At eleven months of age, but not at two months of age, EAAT3 null mice showed hippocampal atrophy and declines in learning on the Morris water maze (Aoyama et al., 2006). The available data, then, certainly do not suggest face validity for the EAAT3 null animals in relation to OCD. This is not very surprising because the majority of the genetic evidence suggests that increased SLC1A1/EAAT3 expression, rather than decreased expression, as in the null animals, would be predicted from the OCD-associated polymorphisms (Wendland et al., 2009a). The Slc1a1/EAAT3 null mouse would then, if anything, model decreased susceptibility to OCD; although a knockout mouse also provides an opportunity to understand the role of this protein in the circuitry underlying OCD. Further work to evaluate the impact of Slc1a1/EAAT3 should focus on modeling increased expression. Alternatively, face validity could potentially be demonstrated by attenuation of pharmacologically- or genetically-mediated compulsive-like behaviors in mice with diminished EAAT3 expression.
3.5 Other genetic manipulations also lead to increased grooming
Excessive grooming is a common behavioral phenotype described in these mouse models of compulsive-like behavior, but it is not exclusive to OCD-like mouse models. The D1CT transgenic mouse was an early model created to mimic hyperactivity of the direct basal ganglia pathways postulated to underlie obsessive and compulsive behavior in humans (Campbell et al., 1999). These mice express intracellular cholera toxin (CT) in dopamine receptor 1 (Drd1) expressing neurons, resulting in potentiation of CSTC circuitry. This manipulation results in increased repetitive behavior, including repetitive leaping (Campbell et al., 1999), as well as increased anxiety-like behavior (McGrath et al., 1999). Even in the absence of clear construct validity, the face validity resulting from manipulation of OCD-related circuitry in this model is intriguing; although rescue with serotonin reuptake inhibitors was not assessed to allow evaluation of predictive validity. Notably, this model is a precursor, in some ways, to the use of optogenetics to affect neuronal activity in implicated circuitry, as discussed below. Many other genetic manipulations similarly generate increased levels of grooming, especially those related to autism spectrum disorders (ASD), where one hallmark of the disorder is excessive repetitive behavior that may or may not be OCD-like (Jacob et al., 2009). ASD-related models, including the Cntnap2 null, Neurexin1 null, Shank3 null, and Integrin beta 3 null, are primarily based on genes that have been associated with ASD and other neurodevelopmental disorders (Etherton et al., 2009, Carter et al., 2011, Peca et al., 2011, Penagarikano et al., 2011). The Cntnap2 null and the Shank3 null mice also show altered striatal structure or function, supporting relevance to OCD despite construct validity primarily related to ASD. Although the construct validity of these models specifically to OCD may be tenuous, the above genetic models thought to be most relevant to OCD are also lacking in either construct or face validity. Importantly, none of the ASD genetic models have been evaluated for response to SRIs, which limits evaluation of predictive validity. On the other hand, the Cntnap2 null mouse does show decreased grooming with a non-sedating dose of risperidone, an atypical antipsychotic medication frequently used in ASD but also used as an adjunctive medication in OCD (Bloch et al., 2006). Overall, the emerging pattern of increased grooming in genetic mouse models suggests that this species-typical repetitive behavior may be a useful readout for altered corticostriatal signaling, regardless of whether the model is most relevant to OCD or ASD (Kalueff et al., 2016).
4. Immunological Models
Alternative models aimed at understanding the potential role of the immune system and the induction abnormal neuropsychiatric behavior may also be a fruitful path in relation to OCD. One model with potential construct validity is based on the observation that streptococcal infections may induce obsessive-compulsive behavior and tics in children, as noted above (Swedo et al., 1998). Mouse models intended to mimic PANDAS have shown mixed results. The primary hypothesis of these investigations is that immune response to streptococcal infection would alter neuronal function and lead to the induction of repetitive behavior reminiscent of those reported in PANDAS. Since rodents are not naturally infected with group A beta-hemolytic streptococcus pyogenes (GABHS) it is challenging to develop a direct model of the human pathophysiology. An alternative would be to transfer antibodies or sera from human patients into mice; although this also has challenges due to the differences between protein antigens across species.
4.1 Mouse model of Streptococcal antigen exposure
The first attempt at a PANDAS model used repeated injection of a GABHS homogenate together with Freund's adjuvant to generate an immune response (Hoffman et al., 2004). In comparison to animals injected with adjuvant alone, sera from these GABHS-exposed mice showed greater immunoreactivity to the globus pallidus and thalamus with the CSTC loop implicated in OCD; however the strongest finding was greater immunoreactivity to the deep cerebellar nuclei (DCN) (Hoffman et al., 2004). Animals with enhanced immunoreactivity to the DCN exhibited increased rearing behavior in the open field arena and in hole board tests compared to controls and to GABHS-immunized mice without increased DCN immunoreactivity. A follow-up study from the same group tested if passive transfer of antibodies from GABHS-immunized animals to non-immunized animals was capable of inducing similar behavioral disturbances (Yaddanapudi et al., 2010). Increased rearing behavior was again observed in the open-field, and a more extensive behavioral evaluation also demonstrated reduced aggression and social interaction in resident-intruder behavioral tasks, as well as motor coordination deficits. In theory, the rearing behavior observed in this model could represent a repetitive, compulsive-like behavior, but rearing is also an exploratory behavior that may indicate decreased anxiety-like behavior or increased exploratory drive. Motor coordination deficits and broad changes in emotional reactivity are sometimes reported in PANDAS, but this seems less relevant to OCD symptoms in particular (Swedo et al., 1998). Evaluating predictive validity in a PANDAS model is a little challenging, since serotonin reuptake inhibitors have not been explicitly tested in this subpopulation of OCD.
4.2 Rat model of Streptococcal antigen exposure
Recent work has also explored a potential rat model of PANDAS using Group A streptococcal (GABHS) antigen, again paired with Freund's adjuvant (Brimberg et al., 2012). These animals showed impaired motor symptoms, including beam walking deficits and ratings of food pellet manipulation. After being misted with water, they also showed increased grooming. The food manipulation phenotype improved after an acute administration of the dopamine D2 receptor antagonist haloperidol, which the authors argue parallels antipsychotic drug treatment in Sydenham's chorea. Offering some evidence of predictive validity for OCD, three days of treatment with the SRI paroxetine diminished water spray-induced grooming; although this falls short of the duration of chronic SRI treatment usually thought to parallel OCD treatment. (Brimberg et al., 2012). In GABHS antigen-exposed rats, antibody deposition was seen in the striatum as well as the thalamus and cortex. Interestingly, dopamine levels were elevated in the cortex of exposed rats, and sera from exposed rats reacted with human dopamine D1 and D2 receptors. To evaluate this as a potential mechanism in the human condition, they examined cross-reactivity of sera from PANDAS patients, finding a similar pattern of binding to D1 and D2 receptors.
A follow-up study from the same group tested the impact of intra-striatal transfer of isolated antibodies of GABHS-exposed rats (GAS-I). Paralleling the motor abnormalities in immunized animals, rats striatally infused with IgG took longer to traverse a narrow beam; however no induced-grooming phenotypes emerged (Lotan et al., 2014). Interestingly, the GAS-I rats did exhibit increased marble burying behavior compared to control animals, which had been previously observed in GABHS-exposed rats. Face validity of elevated marble burying for OCD is uncertain due to benzodiazepines being able to attenuate the behavior in wild-type mice (Nicolas et al., 2006), despite their ineffectiveness in the human OCD population (Crockett et al., 2004). Taken together, this rat model has some appealing parallels to PANDAS in the human population, including behavioral disruption from passive transfer of antibodies into a specific brain region implicated in OCD, but it is somewhat difficult to evaluate the construct validity given the lack of pathogenicity of Streptococcus in rodents.
4.3 Direct injection of PANDAS sera into mice
Injecting sera from human patients with PANDAS into rodents may be a more valid approach, given that mice are not typically infected with GABHS; although species differences could also distort patterns of immunoreactivity. One study infused sera isolated from patients with PANDAS or Tourette syndrome into either the ventral or ventrolateral striatum of rats. They found no behavioral abnormalities and concluded that antibody reactivity in the striatum was unlikely to mediate the behaviors observed in PANDAS (Loiselle et al., 2004). The body of literature on immunological models of OCD is quite small and difficult to evaluate at this point because of the limitation of studies using GABHS in rodents. On the other hand, interest in immune-mediated mechanisms continues to grow in light of the growing literature on PANDAS and PANS as well as the rescue of the excessive grooming behavior in the Hoxb8 null mouse by bone marrow transplantation.
5. Pharmacological Models
Pharmacological models of OCD-relevant behavior are based on drug-induced behavioral phenotypes reminiscent of OCD behaviors observed in human patients (e.g., perseveration, compulsive checking). In the models evaluated, drugs that affect previously implicated neurotransmitter systems drive the OCD-relevant behavior. Due to numerous reports of alterations of both the dopaminergic and serotonergic system in people with OCD (Denys et al., 2004, Fineberg et al., 2012), it is reasonable to believe that manipulation of these neurotransmitter systems could lead to OCD-relevant behaviors in rodents. It is unclear, however, exactly how dopamine and serotonin are involved in the underlying pathogenesis of the disorder, thus the validity of each model must be carefully evaluated when making conclusions from these data, with predictive validity becoming even more important. In contrast, two drug classes have been reported to exacerbate or trigger OCD in the human population. One of these, the 5-HT1B agonist drugs in the triptan class, has led to a pharmacological model, as described below. The other, atypical antipsychotic drugs, with clozapine as the primary offender (Fonseka et al., 2014), has not yet been examined as a potential trigger of compulsive-like behavior in rodents.
5.1 8-OH-DPAT
One of the initial drugs to be studied in relation to compulsive-like behavior was 8-hydroxy-2-(din-propylamino)-tetraline (8-OH-DPAT), a serotonin 5-HT1A receptor agonist that also has some activity at 5-HT7 (Yadin et al., 1991). Of note, no compulsive-like response has been described in humans in response to 5-HT1A agonists, such as buspirone, so construct validity is limited to the general implication of the serotonin system in OCD. Acute administration of 8-OH-DPAT to rats results in a decrease in spontaneous alternation behavior in the T-maze. Instead of spontaneously switching the arm that is entered, rats who receive the drug tend to return repeatedly to the same arm, which may parallel the perseverative behavior often observed in OCD but could also indicate defects in working memory (Dek et al., 2015). Chronic fluoxetine prevents this drug-induced behavior, potentially providing predictive validity; whereas desipramine does not, providing evidence of specificity (Fernandez-Guasti et al., 2003). Importantly, however, diminution of serotonin receptor-mediated behavior with chronic administration of a serotonin reuptake inhibitor is expected due to receptor down-regulation in the context of tonic increases in extracellular serotonin levels. It is difficult, therefore, to know whether this truly indicates predictive validity or whether it simply represents an expected homeostatic mechanism. An alternative approach to predictive validity would be to parallel neuromodulatory treatments in OCD. Both bilateral lesions and bilateral low-frequency stimulation of the thalamic reticular nucleus diminished perseverative response to 8-OH-DPAT; although high-frequency stimulation, which more clearly parallels the response to deep brain stimulation in OCD, had no effect (Andrade et al., 2009, Andrade et al., 2010). More recent work suggests that 8-OH-DPAT may also induce perseverative behavior in other contexts, including returning repeatedly to the same location in an open field arena, which has been described as “compulsive checking” behavior (Alkhatib et al., 2013). Overall, despite interesting data, this model is challenged by difficulties in establishing clear validation in any of the three domains.
5.2 Quinpirole
Chronic administration of the D2/3 receptor agonist quinpirole to rats results in a perseverative exploration phenotype also described as “compulsive checking” behavior (Szechtman et al., 1998). This model has some degree of construct validity with regard to the dopamine system generally, since stimulant drugs and dopamine agonists can lead to repetitive behaviors, or frank compulsive behavior, in some contexts, such as with treatment of Parkinson's disease or attention deficit hyperactivity disorder (Borcherding et al., 1990, Djamshidian et al., 2011, Weintraub et al., 2015). On the other hand, stimulants do not appear to be a common cause of worsening in OCD patients themselves and may even lead to improvements in some patients (Insel et al., 1983). The quinpirole model uses an open field chamber containing four small cubes, and rats receiving 5 or more weeks of drug treatment show more rapid and excessive returns to preferred objects, as well as ritual-like motor behaviors, in comparison to rats receiving vehicle injections (Szechtman et al., 1998). Chronic treatment with clomipramine, an SRI in the tricyclic class, delayed but did not eliminate this behavior. High frequency stimulation and temporary inactivation of the subthalamic nucleus (STN) was able to attenuate this compulsive-like behavior (Winter et al., 2008), paralleling one of the most common neuromodulatory targets in OCD (Mallet et al., 2002, Fontaine et al., 2004). Similarly, high-frequency stimulation of the OCD-linked nucleus accumbens, another common target in OCD (Sturm et al., 2003, Greenberg et al., 2006, Rauch et al., 2006), also reduced quinpirole-induced checking (Mundt et al., 2009). In sum, the predictive validity of this model is impressive, with interesting behavioral parallels to OCD as well, but the construct validity is more difficult to evaluate.
5.3 RU24969
Serotonin 5-HT1B receptor agonist drugs, such as sumatriptan, are used to treat migraines and have been reported to worsen OCD in some patients (Koran et al., 2001a, Gross-Isseroff et al., 2004). Based upon this observation, treatment with the 5-HT1B agonist RU24969, which also has some activity at 5-HT1A, has been examined as a possible construct valid pharmacological model of OCD. Acute treatment with RU24969 in mice results in a dramatic increase in activity, and this activity is dominated by perseverative circling around the perimeter of an open field chamber (Shanahan et al., 2009). Direct injection of a 5-HT1B antagonist demonstrated that 5-HT1B in the orbitofrontal cortex (OFC) is necessary for this perseverative circling response (Shanahan et al., 2009, Shanahan et al., 2011), paralleling neuroimaging data implicating the OFC in OCD (Menzies et al., 2008). Furthermore, RU24969 treatment results in prepulse inhibition (PPI) deficits that may parallel data in OCD, as well as in a number of other neuropsychiatric conditions' (Swerdlow et al., 1993, Shanahan et al., 2009, Ahmari et al., 2012). Chronic treatment but not acute treatment with fluoxetine, but not desipramine, rescued both the repetitive circling behavior and the PPI deficits (Shanahan et al., 2009). This provides some degree of predictive validity but with the same challenge of circularity when examining 5-HT receptor response after chronically increasing extracellular 5-HT with an SRI. Considering all of the evidence, RU24969 treatment is one of the most convincing pharmacologically-induced models due to the most impressive construct validity, coupled with some evidence for both face and predictive validity.
5.3 Neonatal clomipramine
Neonatal exposure to fluoxetine has previously been described to lead to long-term increases in anxiety-like behavior in mice (Ansorge et al., 2004), supporting the possibility that such exposure could also lead to risk of compulsive-like behavior. No epidemiological studies have yet examined this hypothesis in human studies, but the Andersen lab administered clomipramine, an SRI in the tricyclic class, to rat pups from postnatal day 9-16, followed by assessment of relevant behaviors in adulthood (Andersen et al., 2010). Exposed rats showed increased anxiety-like behavior on the elevated plus maze, increased marble burying, and increased “hoarding” of food pellets at the bottom of the cage, as well as deficits in OCD-relevant cognitive tasks (spontaneous alternation, reversal learning, and working memory-related tasks). All of the above behaviors are evocative of symptoms or cognitive deficits observed in OCD patients (Krebs and Heyman, 2015). They also reported increased 5-HT2C receptor mRNA in the OFC and dopamine D2 receptor mRNA in the striatum of experimental rats compared to controls, providing some parallel to neuroimaging studies in OCD (Remijnse et al., 2006, Menzies et al., 2008). This developmental model shows the broadest range of OCD-relevant phenotypes with perhaps greater face validity than any other model described to date, but it lacks construct and predictive validity at this point. These intriguing findings, however, raise the possibility that early life programming, either via pharmacological exposure or via genetics, may result in adaptive changes in OCD-relevant brain regions that lead to later emergence of compulsive-like behavior.
6. Optogenetic Mouse Models of OCD
As both human neuroimaging data and electrophysiology data from genetic mouse models continue to refine our understanding of the underlying circuitry, optogenetic approaches can be used to test these hypotheses by directly stimulating or inhibiting components of the cortico-striatal-thalamo-cortical network. By taking an alternative approach from targeting genes identified from OCD genetic studies, the optogenetic approach may point to circuitry-based treatment options such as repetitive transcranial stimulation or deep brain stimulation, rather than focusing on a molecular understanding or potential pharmacological treatments. These techniques permit alteration of neural activity with brain region, genetic, and temporal precision. Optogenetic technology has recently been used to in two separate studies in an attempt to 1) rescue OC-relevant behaviors in a previously studied genetic mouse model via stimulation of neurons projecting from the lateral OFC to striatum, and 2) induce compulsive-like behaviors using optogenetic stimulation of neurons projecting from the orbitofrontal cortex to the ventral medial striatum. Whether used in conjunction with previously validated transgenic models or on their own, optogenetic tools may revolutionize the study of disease-relevant circuits in animal models of OCD.
6.1 Optogenetic induction of excessive grooming behavior
Ahmari and colleagues used optogenetics to test circuitry-based hypotheses about compulsive behavior (Ahmari et al., 2013). Rather than focusing on a genetic or immune hypothesis of OCD risk, they focused on the intermediate phenotype of hyperactivition of the orbitofrontal cortex and ventral medial striatum (VMS) in OCD (Insel and Winslow, 1992, Rosenberg and Hanna, 2000, Pauls et al., 2014), matched by the observation that deep brain stimulation of the ventral striatum is effective in reducing OCD symptoms in some patients (Rodriguez-Romaguera et al., 2012). Based upon these lines of evidence in humans with OCD, they hypothesized that increasing signaling from the OFC to the VMS would result in increased compulsive-like behavior in mice. The optogenetic approach is very different from genetic manipulations that attempt to establish construct validity by linking to the most proximal cause of OCD risk. Instead, this type of experiment may be thought to primarily examine the plausibility of the cortico-striatal-thalamo-cortical circuit hypothesis of OCD. Remarkably, however, whereas the results may not directly inform our understanding of the proximal risk factors contributing to OCD risk, the results suggest that manipulation of this circuit could be used to understand the dysfunction that generates compulsive-like behavior, and potentially to develop new treatments based upon this understanding.
Ahmari and colleagues introduced the light-sensitive cation channel, Channelrhodopsin via viral injection to the OFC. They then applied light stimulation in the VMS to establish specificity for the OFC-VMS circuit. Contrary to the initial hypothesis, acute OFC-VMS hyperstimulation did not increase repetitive behavior. In contrast, repeated hyperstimulation of the OFC-VMS projection generated a progressive increase in grooming, which persisted for two weeks after the stimulation was stopped. No self-injury was described in these animals, but the increase in grooming suggests that the OFC projection to the VMS can alter this species-typical stereotyped behavior that is also increased in genetic models with face and predictive validity for OCD. Importantly, electrophysiologic measurements were also taken and a progressive increase in light-evoked firing was observed with repeated stimulation, paralleling the increase in repetitive grooming behavior.
Moving beyond testing the plausibility of the CSTC hypothesis, they then examined whether an SRI could rescue the induced grooming. Remarkably, treatment with two weeks of fluoxetine following a seven-day optogenetic stimulation protocol was able to normalize light-evoked neuronal activity and attenuate grooming behavior, providing evidence of predictive validity. Taken as a whole, these data move beyond establishing plausibility for altered corticostriatal signaling as an intermediate phenotype in compulsive-like behavior. The increase in VMS response to light-induced activity with repeated hyperstimulation suggests that plasticity at OFC-VMS synapses induces long-lasting alterations that prime the OFC-VMS synapses, resulting in a reduced activation threshold during subsequent bouts of stimulation. Increased OFC-VMS activity may transmit information through the CSTC circuit, leading to multiple downstream events that generate a repetitive pattern of behavior. The combination of construct, face, and predictive validity make this model very appealing for further dissecting the impact on local microcircuits in the VMS, downstream effects on CSTC signaling, as well as the molecular events underlying the observed synaptic plasticity, with the possibility that other manipulations, beyond SRIs, could also rescue the altered corticostriatal signaling and compulsive-like behavior.
6.2 Optogenetic rescue in Sapap3 null mice
Burguire and colleagues (2013) used optogenetics to take the opposite approach. Rather than using optogenetics to induce compulsive-like behavior, they evaluated the ability of circuit manipulation to rescue compulsive-like behavior in the Sapap3 null mouse model. They first designed a novel delayed conditioning task to achieve temporal and conditioned control over the previously described excessive grooming phenotype. They presented a tone followed by an unconditioned stimulus of a water drop delivered to the nose, which induced grooming behavior in both the wildtype and the Sapap3 null mice. Probe trials, with the tone presented alone without the water drop, were interspersed with conditioning trials. In mid-training, both wildtype and Sapap3 null mice showed grooming response to the tone alone, but later in training this was suppressed in the wildtype but not the mutant mice. This suggests that Sapap3 null mice are unable to inhibit their conditioned grooming response to the tone, even after observing only intermittent presentation of the unconditioned water drop stimulus. In parallel, they found that medium spiny neuron activity was higher in the Sapap3 null animals late in the training period when the response pattern diverged. Interestingly, they noted a decrease in striatal parvalbumin positive (PV+) interneurons in the Sapap3 null animals, suggesting that defective inhibition could account for the electrophysiological and behavioral defects.
To assess whether cortical input could be used to rescue inhibition deficits in the striatum, Burguire and colleagues used optogenetics to stimulate axon terminals of the lateral orbitofrontal cortex (l-OFC) within the striatum, based on previous findings implicating these regions in OCD (Chamberlain et al., 2005, Remijnse et al., 2006, Chamberlain et al., 2008). Remarkably, activation of l-OFC input to the striatum in the Sapap3 null mice was able to attenuate their elevated MSN activity late in training, presumably by enhanced feed-forward inhibition driven by cortical activation of fast-spiking striatal interneurons. Beyond the electrophysiological rescue, they also found that optogenetic stimulation rescued the tone-response inhibition deficits observed in the mutants. Even outside the context of the delayed conditioning task, they found that optogenetic activation of l-OFC input to the striatum decreased compulsive-like grooming in the mutant mice. Importantly, this optogenetic rescue makes use of l-OFC input to striatum, despite no clear evidence that l-OFC activity is defective in the Sapap3 null mice (Burguiere et al., 2013). Such an approach could be paralleled by deep brain stimulation in the human population, where the manipulation depends upon an understanding of the circuits implicated in OCD, without knowing specifically what node in the circuit may be dysfunctional in an individual patient. This idea could be further tested by extending this optogenetic paradigm to other mouse models with compulsive-like grooming behavior due to other genetic manipulations, such as the Slitrk5 or Hoxb8 null mice.
At first glance, these two optogenetic studies seem to have conflicting results. One is able to rescue elicit compulsive-like behavior, whereas the other rescues it, despite activating similar brain regions. It is important to note that there are some fundamental differences. Burguire and colleagues (2013) took advantage of a genetic knockout mouse model with established repetitive behavioral phenotypes that result from abnormal neural activity resulting from the genetic manipulation. In contrast, Ahmari and colleagues (2013) used wild-type mice that had undergone normal neural development prior to introduction of the channelrhodopsin and stimulation of the OFC-VMS circuit. Initial work characterizing the Sapap3-null mouse showed abnormal cortico-striatal function that may be a result of abnormal neural development (Welch et al., 2007), suggesting that the circuits in the two studies may be fundamentally different at the onset. Secondly, the stimulation protocols are actually quite different, with different stimulation durations in either the lateral OFC and primarily dorsal striatum (Burguire) or the medial OFC and ventral striatum (Ahmari). These are distinctly separate cell populations that were previously posited to have alternative roles in the pathophysiology of the disorder (Milad and Rauch, 2012). Clearly, more work will be needed to better understand the stimulus pattern, subregion, and cell type specificity in relation to compulsive-like behavior.
7. Discussion and Future Directions
The primary limitation across rodent models of OCD is the lack of clear risk factors that would support construct validity. At least one model in each of the four major categories reviewed here shows both face and predictive validity, but construct validity is considerably more challenging. The genetic category is likely the easiest domain to establish construct validity, but the field is limited by the lack of power in OCD linkage and genome-wide association studies. The published genetic models with increased grooming that responds to SRI treatment are limited to circumstantial genetic evidence supporting their potential involvement in OCD in the human population. Conversely, those genes with the strongest support in the OCD literature have not yet been adequately evaluated in rodent models, either for compulsive-like behavior or even in relation to their function in the circuits implicated in OCD. Beyond genetic linkage and association studies, no postmortem studies have been published to date in OCD, leaving us without the RNA, protein, or cellular profiles that have guided development of animal models in schizophrenia or autism spectrum disorder (Berg et al., 2015, Brown et al., 2015).
Construct validity is harder to establish for immunological, pharmacological, or optogenetic studies. PANDAS and PANS are exciting clinical syndromes that are poorly understood and offer technical challenges for rodent models. Our knowledge of pharmacological triggers is primarily limited to triptans and atypical antipsychotics, but these are not thought to cause OCD itself but typically to worsen symptoms, and even then only in a subset of patients. The circuitry-based models are perhaps the most exciting and can offer us an opportunity to test hypotheses based upon intermediate phenotypes in OCD, rather than requiring a clear understanding of the original factors that contributed to risk in an individual. It may be difficult to assess construct validity in these models, but they seem to offer substantial promise for understanding how the circuitry implicated in OCD works, even if they do not directly model the disorder itself. We must be careful, however, to not over-interpret models based upon expressing a foreign, light-sensitive protein in an ectopic pattern and then stimulating neurons in a temporal pattern that would not typically occur in the brain.
The difficulty in establishing construct validity also complicates our understanding of the most valid behavioral or cognitive tests that are relevant to OCD. If we had several genes clearly implicated in OCD with mouse models that mimic the human risk variant, we would be able to survey the phenotypes in those animals to understand the true range of “OCD-like” behavior in the rodent. Just as OCD is often difficult to diagnose in the human population, some of those animals would likely be engaging in subtle rituals or compulsive-like behaviors that we may not anticipate or detect without extensive characterization. In the absence of construct valid models to serve as examples, we are restricted to behaviors that fit our expectations for how compulsivity might present in a rodent. In this context, a broad assessment that includes both spontaneous behaviors, such as grooming, as well as cognitive measures reported to be abnormal in OCD (Dittrich and Johansen, 2013), is important to better understand the strengths and limitations of a given model. None of the genetic models have had a significant assessment of cognitive function reported to date; although the conditioned-grooming assay in the Sapap3 model begins to move toward an understanding of cognitive function in these animals, albeit only in the context of the already identified grooming phenotype. Some intermediate phenotypes identified in OCD, such as reversal learning deficits or impaired prepulse inhibition, can be assessed across species; although no single cognitive or sensory gating measure is specific for OCD to the exclusion of other neuropsychiatric disorders.
In contrast to the challenges in understanding the risk factors and molecular etiology of OCD, neuroimaging has made significant progress in identifying affected brain regions through the use of continually advancing techniques. Likewise, the use of deep brain stimulation points to brain regions or tracts that are likely to at least reflect downstream consequences of the pathophysiology of OCD. This understanding of brain regions and circuits has become one of the key arguments for the validity of particular genetic or pharmacological models, rather than the construct being modeled. Abnormalities in the OFC or the striatum, or corticostriatal signaling specifically, are reassuring findings across models, but we should be particularly excited to see more specific findings arise, such as the decrease in fast-spiking striatal interneurons in the Sapap3 null mice. Even in the absence of construct or predictive validity, the sheer novelty of the Hoxb8 microglia rescue raises the possibility that an animal model will transform our understanding of OCD pathophysiology, even if just in a small subset of the overall population.
When assessing the available models, a few conclusions can be drawn. First, no single model is likely to be all-encompassing, with each model likely corresponding to a subset of the disorder and potentially to a particular symptom type or cognitive deficit. Second, excessive grooming is a species-appropriate, stereotyped behavior that seems particularly sensitive to perturbations that affect the CSTC circuit implicated in OCD and may be a very useful readout across models. Importantly, rodent grooming also increases in novel or stressful environments (Hammamieh et al., 2012, Xu et al., 2015), and anxiolytic response to SRIs may therefore complicate assessment of predictive validity specific to compulsive-like behavior. Third, emerging techniques to manipulate specific circuits offer the possibility of working back and forth between genetic, immunological, and pharmacological models that perturb specific circuits, and optogenetic models that can evaluate whether an observed change in circuit function is necessary and specific to generate the observed compulsive-like behavior, with optogenetic rescue of the Sapap3 null mice as the pioneering example. The rapid advance in technologies to develop and study rodent models is moving forward at an astonishing pace, and we may expect more genetic models to emerge that may force us to test construct validity after observing a rodent phenotype, as in the Sapap3 and Slitrk5 null mice. We can also expect that optogenetic findings will direct the attention of human neuroimaging studies to specific circuits or nodes that may otherwise have been ignored. Ideally, however, we will also continue to pursue models with explicit construct validity in order to maintain a focus on factors that impact upon OCD risk in humans, rather than the myriad of manipulations that may generate compulsive-like behavior in rodents.
Figure 1.
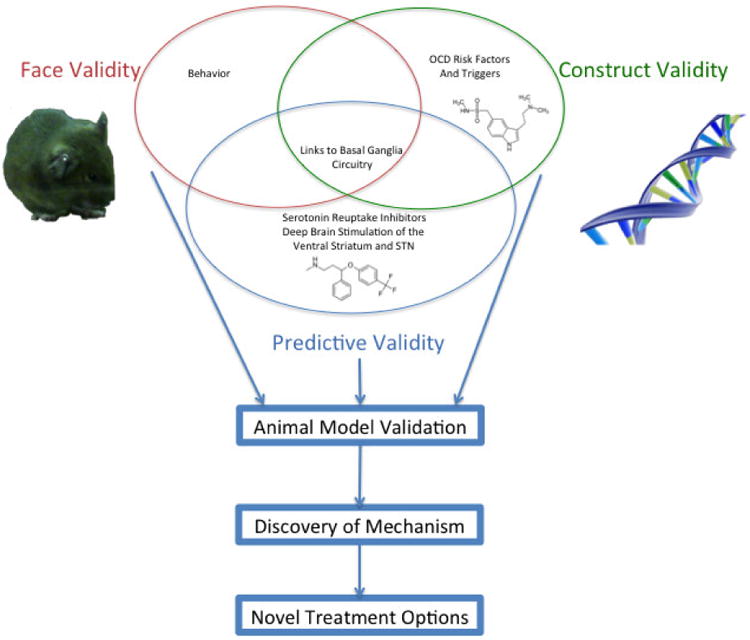
The most commonly used criteria to evaluate rodent models of Obsessive Compulsive Disorder are: 1) construct validity, 2) face validity, and 3) predictive validity. Each criterion contains exclusive components (grooming behavior for face validity or behavioral response to serotonin reuptake inhibitors for predictive validity). Some findings, such as the implication of cortico-striatal circuitry, may contribute to validation in any one of the three domains, depending upon context. For example, an optogenetic mouse model was constructed via chronic stimulation of cortical inputs to the striatum (Ahmari et al., 2013). Abnormal corticostriatal signaling is seen in a couple of genetic mouse models (Welch et al., 2007, Shmelkov et al., 2010), a form of face validity paralleling human neuroimaging findings. Finally, a different optogenetic paradigm seeks predictive validity by rescuing compulsive-like behavior by stimulating a cortico-striatal circuit (Burguiere et al., 2013).
Highlights.
Genetic and environmental risk factors for obsessive compulsive disorder (OCD) have been difficult to identify.
Neuroimaging studies implicate cortico-striatal-thalamo-cortical circuitry in OCD.
Chronic administration of a serotonin reuptake inhibitor is the only effective medication treatment in OCD.
Rodent models have been proposed based upon genetic, pharmacological, immune, and circuit approaches.
No single model fully captures the human disorder, but multiple models converge on abnormal cortico-striatal signaling.
Acknowledgments
Support for this work was provided by NIH MH096200, New York State Psychiatric Institute, the Mortimer D. Sackler, M.D., Chair Fund, and the Columbia University Department of Psychiatry.
Footnotes
Biomedical conflicts of interest: Dr. Veenstra-VanderWeele has consulted with Roche Pharmaceuticals, Novartis, and SynapDx and has had research funding from Roche Pharmaceuticals, Novartis, SynapDx, Seaside Therapeutics, Forest, and Sunovion. He receives an honorarium for editorial work from Springer and John Wiley and Sons.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abelson JF, Kwan KY, O'Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, Davis NR, Ercan-Sencicek AG, Guez DH, Spertus JA, Leckman JF, Dure LSt, Kurlan R, Singer HS, Gilbert DL, Farhi A, Louvi A, Lifton RP, Sestan N, State MW. Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science. 2005;310:317–320. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- Ahmari SE, Risbrough VB, Geyer MA, Simpson HB. Impaired sensorimotor gating in unmedicated adults with obsessive-compulsive disorder. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2012;37:1216–1223. doi: 10.1038/npp.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmari SE, Spellman T, Douglass NL, Kheirbek MA, Simpson HB, Deisseroth K, Gordon JA, Hen R. Repeated cortico-striatal stimulation generates persistent OCD-like behavior. Science. 2013;340:1234–1239. doi: 10.1126/science.1234733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhatib AH, Dvorkin-Gheva A, Szechtman H. Quinpirole and 8-OH-DPAT induce compulsive checking behavior in male rats by acting on different functional parts of an OCD neurocircuit. Behav Pharmacol. 2013;24:65–73. doi: 10.1097/FBP.0b013e32835d5b7a. [DOI] [PubMed] [Google Scholar]
- Alonso P, Gratacos M, Segalas C, Escaramis G, Real E, Bayes M, Labad J, Lopez-Sola C, Estivill X, Menchon JM. Association between the NMDA glutamate receptor GRIN2B gene and obsessive-compulsive disorder. J Psychiatry Neurosci. 2012;37:273–281. doi: 10.1503/jpn.110109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvaro JD, Taylor JR, Duman RS. Molecular and behavioral interactions between central melanocortins and cocaine. The Journal of pharmacology and experimental therapeutics. 2003;304:391–399. doi: 10.1124/jpet.102.040311. [DOI] [PubMed] [Google Scholar]
- Andersen SL, Greene-Colozzi EA, Sonntag KC. A novel, multiple symptom model of obsessive-compulsive-like behaviors in animals. Biol Psychiatry. 2010;68:741–747. doi: 10.1016/j.biopsych.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Andrade P, Carrillo-Ruiz JD, Ramirez Y, Jimenez F. Effects of Thalamic Reticular Nucleus Electrical Stimulation in Rats in a T-maze Perseverative Behavior Model Induced by 8-OH-DPAT. Neuromodulation. 2010;13:2–9. doi: 10.1111/j.1525-1403.2009.00242.x. [DOI] [PubMed] [Google Scholar]
- Andrade P, Fernandez-Guasti A, Carrillo-Ruiz JD, Ulloa RE, Ramirez Y, Reyes R, Jimenez F. Effects of bilateral lesions in thalamic reticular nucleus and orbitofrontal cortex in a T-maze perseverative model produced by 8-OH-DPAT in rats. Behav Brain Res. 2009;203:108–112. doi: 10.1016/j.bbr.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Ansorge MS, Zhou M, Lira A, Hen R, Gingrich JA. Early-life blockade of the 5-HT transporter alters emotional behavior in adult mice. Science. 2004;306:879–881. doi: 10.1126/science.1101678. [DOI] [PubMed] [Google Scholar]
- Aoyama K, Nakaki T. Neuroprotective properties of the excitatory amino acid carrier 1 (EAAC1) Amino acids. 2013;45:133–142. doi: 10.1007/s00726-013-1481-5. [DOI] [PubMed] [Google Scholar]
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nature neuroscience. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Arnold PD, Rosenberg DR, Mundo E, Tharmalingam S, Kennedy JL, Richter MA. Association of a glutamate (NMDA) subunit receptor gene (GRIN2B) with obsessive-compulsive disorder: a preliminary study. Psychopharmacology (Berl) 2004;174:530–538. doi: 10.1007/s00213-004-1847-1. [DOI] [PubMed] [Google Scholar]
- Arnold PD, Sicard T, Burroughs E, Richter MA, Kennedy JL. Glutamate transporter gene SLC1A1 associated with obsessive-compulsive disorder. Archives of general psychiatry. 2006;63:769–776. doi: 10.1001/archpsyc.63.7.769. [DOI] [PubMed] [Google Scholar]
- Arumugham SS, Reddy JY. Augmentation strategies in obsessive-compulsive disorder. Expert Rev Neurother. 2013;13:187–202. doi: 10.1586/ern.12.160. quiz 203. [DOI] [PubMed] [Google Scholar]
- Bailey CG, Ryan RM, Thoeng AD, Ng C, King K, Vanslambrouck JM, Auray-Blais C, Vandenberg RJ, Broer S, Rasko JE. Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria. J Clin Invest. 2011;121:446–453. doi: 10.1172/JCI44474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JM, Lee C, Chen L, Galvan L, Cepeda C, Chen JY, Penagarikano O, Stein JL, Li A, Oguro-Ando A, Miller JA, Vashisht AA, Starks ME, Kite EP, Tam E, Gdalyahu A, Al-Sharif NB, Burkett ZD, White SA, Fears SC, Levine MS, Wohlschlegel JA, Geschwind DH. JAKMIP1, a Novel Regulator of Neuronal Translation, Modulates Synaptic Function and Autistic-like Behaviors in Mouse. Neuron. 2015;88:1173–1191. doi: 10.1016/j.neuron.2015.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Khanna S, Chakrabarty K, Mahadevan A, Christopher R, Shankar SK. Anti-brain autoantibodies and altered excitatory neurotransmitters in obsessive-compulsive disorder. Neuropsychopharmacology. 2009;34:2489–2496. doi: 10.1038/npp.2009.77. [DOI] [PubMed] [Google Scholar]
- Bienvenu OJ, Wang Y, Shugart YY, Welch JM, Grados MA, Fyer AJ, Rauch SL, McCracken JT, Rasmussen SA, Murphy DL, Cullen B, Valle D, Hoehn-Saric R, Greenberg BD, Pinto A, Knowles JA, Piacentini J, Pauls DL, Liang KY, Willour VL, Riddle M, Samuels JF, Feng G, Nestadt G. Sapap3 and pathological grooming in humans: Results from the OCD collaborative genetics study. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:710–720. doi: 10.1002/ajmg.b.30897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billett EA, Richter MA, Sam F, Swinson RP, Dai XY, King N, Badri F, Sasaki T, Buchanan JA, Kennedy JL. Investigation of dopamine system genes in obsessive-compulsive disorder. Psychiatric genetics. 1998;8:163–169. doi: 10.1097/00041444-199800830-00005. [DOI] [PubMed] [Google Scholar]
- Bissonette GB, Powell EM. Reversal learning and attentional set-shifting in mice. Neuropharmacology. 2012;62:1168–1174. doi: 10.1016/j.neuropharm.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch MH, Landeros-Weisenberger A, Kelmendi B, Coric V, Bracken MB, Leckman JF. A systematic review: antipsychotic augmentation with treatment refractory obsessive-compulsive disorder. Mol Psychiatry. 2006;11:622–632. doi: 10.1038/sj.mp.4001823. [DOI] [PubMed] [Google Scholar]
- Bloch MH, Panza KE, Landeros-Weisenberger A, Leckman JF. Meta-analysis: treatment of attention-deficit/hyperactivity disorder in children with comorbid tic disorders. J Am Acad Child Adolesc Psychiatry. 2009;48:884–893. doi: 10.1097/CHI.0b013e3181b26e9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borcherding BG, Keysor CS, Rapoport JL, Elia J, Amass J. Motor/vocal tics and compulsive behaviors on stimulant drugs: is there a common vulnerability? Psychiatry Res. 1990;33:83–94. doi: 10.1016/0165-1781(90)90151-t. [DOI] [PubMed] [Google Scholar]
- Brennan BP, Rauch SL, Jensen JE, Pope HG., Jr A critical review of magnetic resonance spectroscopy studies of obsessive-compulsive disorder. Biol Psychiatry. 2013;73:24–31. doi: 10.1016/j.biopsych.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigman JL, Powell EM, Mittleman G, Young JW. Examining the genetic and neural components of cognitive flexibility using mice. Physiol Behav. 2012;107:666–669. doi: 10.1016/j.physbeh.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimberg L, Benhar I, Mascaro-Blanco A, Alvarez K, Lotan D, Winter C, Klein J, Moses AE, Somnier FE, Leckman JF, Swedo SE, Cunningham MW, Joel D. Behavioral, pharmacological, and immunological abnormalities after streptococcal exposure: a novel rat model of Sydenham chorea and related neuropsychiatric disorders. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2012;37:2076–2087. doi: 10.1038/npp.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broocks A, Pigott TA, Hill JL, Canter S, Grady TA, L'Heureux F, Murphy DL. Acute intravenous administration of ondansetron and m-CPP, alone and in combination, in patients with obsessive-compulsive disorder (OCD): behavioral and biological results. Psychiatry Res. 1998;79:11–20. doi: 10.1016/s0165-1781(98)00029-8. [DOI] [PubMed] [Google Scholar]
- Brown JA, Ramikie TS, Schmidt MJ, Baldi R, Garbett K, Everheart MG, Warren LE, Gellert L, Horvath S, Patel S, Mirnics K. Inhibition of parvalbumin-expressing interneurons results in complex behavioral changes. Mol Psychiatry. 2015;20:1499–1507. doi: 10.1038/mp.2014.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguiere E, Monteiro P, Feng G, Graybiel AM. Optogenetic stimulation of lateral orbitofronto-striatal pathway suppresses compulsive behaviors. Science. 2013;340:1243–1246. doi: 10.1126/science.1232380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Zhang W, Yi Z, Lu W, Wu Z, Chen J, Yu S, Fang Y, Zhang C. Influence of polymorphisms in genes SLC1A1, GRIN2B, and GRIK2 on clozapine-induced obsessive-compulsive symptoms. Psychopharmacology (Berl) 2013;230:49–55. doi: 10.1007/s00213-013-3137-2. [DOI] [PubMed] [Google Scholar]
- Calvocoressi L, Libman D, Vegso SJ, McDougle CJ, Price LH. Global functioning of inpatients with obsessive-compulsive disorder, schizophrenia, and major depression. Psychiatric services. 1998;49:379–381. doi: 10.1176/ps.49.3.379. [DOI] [PubMed] [Google Scholar]
- Capecchi MR. Hox genes and mammalian development. Cold Spring Harb Symp Quant Biol. 1997;62:273–281. [PubMed] [Google Scholar]
- Carter MD, Shah CR, Muller CL, Crawley JN, Carneiro AM, Veenstra-VanderWeele J. Absence of preference for social novelty and increased grooming in integrin beta3 knockout mice: initial studies and future directions. Autism Res. 2011;4:57–67. doi: 10.1002/aur.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty K, Bhattacharyya S, Christopher R, Khanna S. Glutamatergic dysfunction in OCD. Neuropsychopharmacology. 2005a;30:1735–1740. doi: 10.1038/sj.npp.1300733. [DOI] [PubMed] [Google Scholar]
- Chakrabarty K, Bhattacharyya S, Christopher R, Khanna S. Glutamatergic dysfunction in OCD. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2005b;30:1735–1740. doi: 10.1038/sj.npp.1300733. [DOI] [PubMed] [Google Scholar]
- Chamberlain SR, Blackwell AD, Fineberg NA, Robbins TW, Sahakian BJ. The neuropsychology of obsessive compulsive disorder: the importance of failures in cognitive and behavioural inhibition as candidate endophenotypic markers. Neurosci Biobehav Rev. 2005;29:399–419. doi: 10.1016/j.neubiorev.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Chamberlain SR, Fineberg NA, Blackwell AD, Robbins TW, Sahakian BJ. Motor inhibition and cognitive flexibility in obsessive-compulsive disorder and trichotillomania. Am J Psychiatry. 2006;163:1282–1284. doi: 10.1176/ajp.2006.163.7.1282. [DOI] [PubMed] [Google Scholar]
- Chamberlain SR, Menzies L, Hampshire A, Suckling J, Fineberg NA, del Campo N, Aitken M, Craig K, Owen AM, Bullmore ET, Robbins TW, Sahakian BJ. Orbitofrontal dysfunction in patients with obsessive-compulsive disorder and their unaffected relatives. Science (New York, NY) 2008;321:421–422. doi: 10.1126/science.1154433. [DOI] [PubMed] [Google Scholar]
- Chang K, Frankovich J, Cooperstock M, Cunningham MW, Latimer ME, Murphy TK, Pasternack M, Thienemann M, Williams K, Walter J, Swedo SE, Consortium PC Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol. 2015;25:3–13. doi: 10.1089/cap.2014.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SK, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, Capecchi MR. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell. 2010;141:775–785. doi: 10.1016/j.cell.2010.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam with sertraline in obsessive-compulsive disorder. Ann Clin Psychiatry. 2004;16:127–132. doi: 10.1080/10401230490486972. [DOI] [PubMed] [Google Scholar]
- de Wit SJ, Alonso P, Schweren L, Mataix-Cols D, Lochner C, Menchon JM, Stein DJ, Fouche JP, Soriano-Mas C, Sato JR, Hoexter MQ, Denys D, Nakamae T, Nishida S, Kwon JS, Jang JH, Busatto GF, Cardoner N, Cath DC, Fukui K, Jung WH, Kim SN, Miguel EC, Narumoto J, Phillips ML, Pujol J, Remijnse PL, Sakai Y, Shin NY, Yamada K, Veltman DJ, van den Heuvel OA. Multicenter voxel-based morphometry mega-analysis of structural brain scans in obsessive-compulsive disorder. Am J Psychiatry. 2014;171:340–349. doi: 10.1176/appi.ajp.2013.13040574. [DOI] [PubMed] [Google Scholar]
- Dek EC, van den Hout MA, Engelhard IM, Giele CL, Cath DC. Perseveration causes automatization of checking behavior in obsessive-compulsive disorder. Behav Res Ther. 2015;71:1–9. doi: 10.1016/j.brat.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Denys D, Zohar J, Westenberg HG. The role of dopamine in obsessive-compulsive disorder: preclinical and clinical evidence. J Clin Psychiatry. 2004;65(14):11–17. [PubMed] [Google Scholar]
- Dickel DE, Veenstra-VanderWeele J, Bivens NC, Wu X, Fischer DJ, Van Etten-Lee M, Himle JA, Leventhal BL, Cook EH, Jr, Hanna GL. Association studies of serotonin system candidate genes in early-onset obsessive-compulsive disorder. Biological psychiatry. 2007;61:322–329. doi: 10.1016/j.biopsych.2006.09.030. [DOI] [PubMed] [Google Scholar]
- Dickel DE, Veenstra-VanderWeele J, Cox NJ, Wu X, Fischer DJ, Van Etten-Lee M, Himle JA, Leventhal BL, Cook EH, Jr, Hanna GL. Association testing of the positional and functional candidate gene SLC1A1/EAAC1 in early-onset obsessive-compulsive disorder. Arch Gen Psychiatry. 2006;63:778–785. doi: 10.1001/archpsyc.63.7.778. [DOI] [PubMed] [Google Scholar]
- Dittrich WH, Johansen T. Cognitive deficits of executive functions and decision-making in obsessive-compulsive disorder. Scand J Psychol. 2013;54:393–400. doi: 10.1111/sjop.12066. [DOI] [PubMed] [Google Scholar]
- Djamshidian A, Averbeck BB, Lees AJ, O'Sullivan SS. Clinical aspects of impulsive compulsive behaviours in Parkinson's disease. J Neurol Sci. 2011;310:183–188. doi: 10.1016/j.jns.2011.07.031. [DOI] [PubMed] [Google Scholar]
- Dougherty DD, Rauch SL, Jenike MA. Pharmacotherapy for obsessive-compulsive disorder. Journal of clinical psychology. 2004;60:1195–1202. doi: 10.1002/jclp.20083. [DOI] [PubMed] [Google Scholar]
- Etherton MR, Blaiss CA, Powell CM, Sudhof TC. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:17998–18003. doi: 10.1073/pnas.0910297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi IS. Monogenic human obesity. Front Horm Res. 2008;36:1–11. doi: 10.1159/000115333. [DOI] [PubMed] [Google Scholar]
- Fernandez-Guasti A, Ulloa RE, Nicolini H. Age differences in the sensitivity to clomipramine in an animal model of obsessive-compulsive disorder. Psychopharmacology (Berl) 2003;166:195–201. doi: 10.1007/s00213-002-1301-1. [DOI] [PubMed] [Google Scholar]
- Fineberg NA, Reghunandanan S, Brown A, Pampaloni I. Pharmacotherapy of obsessive-compulsive disorder: Evidence-based treatment and beyond. The Australian and New Zealand journal of psychiatry. 2012 doi: 10.1177/0004867412461958. [DOI] [PubMed] [Google Scholar]
- Fonseka TM, Richter MA, Muller DJ. Second generation antipsychotic-induced obsessive-compulsive symptoms in schizophrenia: a review of the experimental literature. Curr Psychiatry Rep. 2014;16:510. doi: 10.1007/s11920-014-0510-8. [DOI] [PubMed] [Google Scholar]
- Fontaine D, Mattei V, Borg M, von Langsdorff D, Magnie MN, Chanalet S, Robert P, Paquis P. Effect of subthalamic nucleus stimulation on obsessive-compulsive disorder in a patient with Parkinson disease. Case report. J Neurosurg. 2004;100:1084–1086. doi: 10.3171/jns.2004.100.6.1084. [DOI] [PubMed] [Google Scholar]
- Goodman WK, Foote KD, Greenberg BD, Ricciuti N, Bauer R, Ward H, Shapira NA, Wu SS, Hill CL, Rasmussen SA, Okun MS. Deep brain stimulation for intractable obsessive compulsive disorder: pilot study using a blinded, staggered-onset design. Biol Psychiatry. 2010;67:535–542. doi: 10.1016/j.biopsych.2009.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grados M, Wilcox HC. Genetics of obsessive-compulsive disorder: a research update. Expert review of neurotherapeutics. 2007;7:967–980. doi: 10.1586/14737175.7.8.967. [DOI] [PubMed] [Google Scholar]
- Grados MA, Samuels J, Shugart YY, Willour VL, Wang Y, Cullen B, Bienvenu OJ, Hoehn-Saric R, Valle D, Liang KY, Riddle MA, Wendland JR, Murphy DL, Nestadt G, Detera-Wadleigh S. Rare plus common SERT variants in obsessive-compulsive disorder. Mol Psychiatry. 2007;12:422–423. doi: 10.1038/sj.mp.4001970. [DOI] [PubMed] [Google Scholar]
- Greenberg BD, Malone DA, Friehs GM, Rezai AR, Kubu CS, Malloy PF, Salloway SP, Okun MS, Goodman WK, Rasmussen SA. Three-year outcomes in deep brain stimulation for highly resistant obsessive-compulsive disorder. Neuropsychopharmacology. 2006;31:2384–2393. doi: 10.1038/sj.npp.1301165. [DOI] [PubMed] [Google Scholar]
- Greer JM, Capecchi MR. Hoxb8 is required for normal grooming behavior in mice. Neuron. 2002;33:23–34. doi: 10.1016/s0896-6273(01)00564-5. [DOI] [PubMed] [Google Scholar]
- Gross-Isseroff R, Cohen R, Sasson Y, Voet H, Zohar J. Serotonergic dissection of obsessive compulsive symptoms: a challenge study with m-chlorophenylpiperazine and sumatriptan. Neuropsychobiology. 2004;50:200–205. doi: 10.1159/000079970. [DOI] [PubMed] [Google Scholar]
- Gu BM, Park JY, Kang DH, Lee SJ, Yoo SY, Jo HJ, Choi CH, Lee JM, Kwon JS. Neural correlates of cognitive inflexibility during task-switching in obsessive-compulsive disorder. Brain. 2008;131:155–164. doi: 10.1093/brain/awm277. [DOI] [PubMed] [Google Scholar]
- Hammamieh R, Chakraborty N, De Lima TC, Meyerhoff J, Gautam A, Muhie S, D'Arpa P, Lumley L, Carroll E, Jett M. Murine model of repeated exposures to conspecific trained aggressors simulates features of post-traumatic stress disorder. Behav Brain Res. 2012;235:55–66. doi: 10.1016/j.bbr.2012.07.022. [DOI] [PubMed] [Google Scholar]
- Hanna G, Veenstra-Vander Weele J, Cox N, Boehnke M, Himle J, Curtis G, Leventhal B, Cook E. Genome-wide linkage analysis of families with obsessive-compulsive disorder ascertained through pediatric probands. American Journal of Medical Genetics (Neuropsychiatric Genetics) 2002a;114:541–552. doi: 10.1002/ajmg.10519. [DOI] [PubMed] [Google Scholar]
- Hanna GL, Veenstra-VanderWeele J, Cox NJ, Boehnke M, Himle JA, Curtis GC, Leventhal BL, Cook EH. Genome-wide linkage analysis of families with obsessive-compulsive disorder ascertained through pediatric probands. American journal of medical genetics. 2002b;114:541–552. doi: 10.1002/ajmg.10519. [DOI] [PubMed] [Google Scholar]
- Hatalova H, Radostova D, Pistikova A, Vales K, Stuchlik A. Spatial reversal learning in chronically sensitized rats and in undrugged sensitized rats with dopamine d2-like receptor agonist quinpirole. Frontiers in behavioral neuroscience. 2014;8:122. doi: 10.3389/fnbeh.2014.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman KL, Hornig M, Yaddanapudi K, Jabado O, Lipkin WI. A murine model for neuropsychiatric disorders associated with group A beta-hemolytic streptococcal infection. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:1780–1791. doi: 10.1523/JNEUROSCI.0887-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege JC, de Graaff W, Hossaini M, Cardona Cano S, Jaarsma D, van den Akker E, Deschamps J. Loss of Hoxb8 alters spinal dorsal laminae and sensory responses in mice. Proc Natl Acad Sci U S A. 2008;105:6338–6343. doi: 10.1073/pnas.0802176105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR, Hamilton JA, Guttmacher LB, Murphy DL. D-amphetamine in obsessive-compulsive disorder. Psychopharmacology (Berl) 1983;80:231–235. doi: 10.1007/BF00436159. [DOI] [PubMed] [Google Scholar]
- Insel TR, Winslow JT. Neurobiology of obsessive compulsive disorder. Psychiatr Clin North Am. 1992;15:813–824. [PubMed] [Google Scholar]
- Jacob S, Landeros-Weisenberger A, Leckman JF. Autism spectrum and obsessive-compulsive disorders: OC behaviors, phenotypes and genetics. Autism Res. 2009;2:293–311. doi: 10.1002/aur.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalueff AV, Stewart AM, Song C, Berridge KC, Graybiel AM, Fentress JC. Neurobiology of rodent self-grooming and its value for translational neuroscience. Nat Rev Neurosci. 2016;17:45–59. doi: 10.1038/nrn.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannidis I, Rizzo R, Tarnok Z, Wolanczyk T, Hebebrand J, Nothen MM, Lehmkuhl G, Farkas L, Nagy P, Barta C, Szymanska U, Panteloglou G, Miranda DM, Feng Y, Sandor P, Barr C, TsgeneSee. Paschou P. Replication of association between a SLITRK1 haplotype and Tourette Syndrome in a large sample of families. Mol Psychiatry. 2012;17:665–668. doi: 10.1038/mp.2011.151. [DOI] [PubMed] [Google Scholar]
- Katayama K, Yamada K, Ornthanalai VG, Inoue T, Ota M, Murphy NP, Aruga J. Slitrk1-deficient mice display elevated anxiety-like behavior and noradrenergic abnormalities. Mol Psychiatry. 2010;15:177–184. doi: 10.1038/mp.2008.97. [DOI] [PubMed] [Google Scholar]
- Keen-Kim D, Mathews CA, Reus VI, Lowe TL, Herrera LD, Budman CL, Gross-Tsur V, Pulver AE, Bruun RD, Erenberg G, Naarden A, Sabatti C, Freimer NB. Overrepresentation of rare variants in a specific ethnic group may confuse interpretation of association analyses. Hum Mol Genet. 2006;15:3324–3328. doi: 10.1093/hmg/ddl408. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koran LM, Hanna GL, Hollander E, Nestadt G, Simpson HB. Practice guideline for the treatment of patients with obsessive-compulsive disorder. The American journal of psychiatry. 2007;164:5–53. [PubMed] [Google Scholar]
- Koran LM, Pallanti S, Quercioli L. Sumatriptan, 5-HT(1D) receptors and obsessive-compulsive disorder. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology. 2001a;11:169–172. doi: 10.1016/s0924-977x(01)00082-7. [DOI] [PubMed] [Google Scholar]
- Koran LM, Pallanti S, Quercioli L. Sumatriptan, 5-HT(1D) receptors and obsessive-compulsive disorder. Eur Neuropsychopharmacol. 2001b;11:169–172. doi: 10.1016/s0924-977x(01)00082-7. [DOI] [PubMed] [Google Scholar]
- Krebs G, Heyman I. Obsessive-compulsive disorder in children and adolescents. Arch Dis Child. 2015;100:495–499. doi: 10.1136/archdischild-2014-306934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JS, Joo YH, Nam HJ, Lim M, Cho EY, Jung MH, Choi JS, Kim B, Kang DH, Oh S, Park T, Hong KS. Association of the glutamate transporter gene SLC1A1 with atypical antipsychotics-induced obsessive-compulsive symptoms. Archives of general psychiatry. 2009;66:1233–1241. doi: 10.1001/archgenpsychiatry.2009.155. [DOI] [PubMed] [Google Scholar]
- Loiselle CR, Lee O, Moran TH, Singer HS. Striatal microinfusion of Tourette syndrome and PANDAS sera: failure to induce behavioral changes. Mov Disord. 2004;19:390–396. doi: 10.1002/mds.10522. [DOI] [PubMed] [Google Scholar]
- Lotan D, Benhar I, Alvarez K, Mascaro-Blanco A, Brimberg L, Frenkel D, Cunningham MW, Joel D. Behavioral and neural effects of intra-striatal infusion of anti-streptococcal antibodies in rats. Brain Behav Immun. 2014;38:249–262. doi: 10.1016/j.bbi.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMaster FP, O'Neill J, Rosenberg DR. Brain imaging in pediatric obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2008;47:1262–1272. doi: 10.1097/CHI.0b013e318185d2be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet L, Mesnage V, Houeto JL, Pelissolo A, Yelnik J, Behar C, Gargiulo M, Welter ML, Bonnet AM, Pillon B, Cornu P, Dormont D, Pidoux B, Allilaire JF, Agid Y. Compulsions, Parkinson's disease, and stimulation. Lancet. 2002;360:1302–1304. doi: 10.1016/S0140-6736(02)11339-0. [DOI] [PubMed] [Google Scholar]
- Martino D, Defazio G, Giovannoni G. The PANDAS subgroup of tic disorders and childhood-onset obsessive-compulsive disorder. J Psychosom Res. 2009;67:547–557. doi: 10.1016/j.jpsychores.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Mathews CA, Badner JA, Andresen JM, Sheppard B, Himle JA, Grant JE, Williams KA, Chavira DA, Azzam A, Schwartz M, Reus VI, Kim SW, Cook EH, Hanna GL. Genome-wide linkage analysis of obsessive-compulsive disorder implicates chromosome 1p36. Biol Psychiatry. 2012;72:629–636. doi: 10.1016/j.biopsych.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattheisen M, Samuels JF, Wang Y, Greenberg BD, Fyer AJ, McCracken JT, Geller DA, Murphy DL, Knowles JA, Grados MA, Riddle MA, Rasmussen SA, McLaughlin NC, Nurmi EL, Askland KD, Qin HD, Cullen BA, Piacentini J, Pauls DL, Bienvenu OJ, Stewart SE, Liang KY, Goes FS, Maher B, Pulver AE, Shugart YY, Valle D, Lange C, Nestadt G. Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Mol Psychiatry. 2015;20:337–344. doi: 10.1038/mp.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath MJ, Campbell KM, Veldman MB, Burton FH. Anxiety in a transgenic mouse model of cortical-limbic neuro-potentiated compulsive behavior. Behav Pharmacol. 1999;10:435–443. doi: 10.1097/00008877-199909000-00001. [DOI] [PubMed] [Google Scholar]
- Menzies L, Chamberlain SR, Laird AR, Thelen SM, Sahakian BJ, Bullmore ET. Integrating evidence from neuroimaging and neuropsychological studies of obsessive-compulsive disorder: the orbitofronto-striatal model revisited. Neuroscience and biobehavioral reviews. 2008;32:525–549. doi: 10.1016/j.neubiorev.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael S, Ritsner AGA. Quality of Life Impairment in Schizophrenia, Mood and Anxiety Disorders. New Perspectives on Research and Treatment 2007 [Google Scholar]
- Milad MR, Rauch SL. Obsessive-compulsive disorder: beyond segregated cortico-striatal pathways. Trends Cogn Sci. 2012;16:43–51. doi: 10.1016/j.tics.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda DM, Wigg K, Kabia EM, Feng Y, Sandor P, Barr CL. Association of SLITRK1 to Gilles de la Tourette Syndrome. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:483–486. doi: 10.1002/ajmg.b.30840. [DOI] [PubMed] [Google Scholar]
- Moore GJ, MacMaster FP, Stewart C, Rosenberg DR. Case study: caudate glutamatergic changes with paroxetine therapy for pediatric obsessive-compulsive disorder. Journal of the American Academy of Child and Adolescent Psychiatry. 1998;37:663–667. doi: 10.1097/00004583-199806000-00017. [DOI] [PubMed] [Google Scholar]
- Morris-Berry CM, Pollard M, Gao S, Thompson C, Tourette Syndrome Study G. Singer HS. Anti-streptococcal, tubulin, and dopamine receptor 2 antibodies in children with PANDAS and Tourette syndrome: single-point and longitudinal assessments. J Neuroimmunol. 2013;264:106–113. doi: 10.1016/j.jneuroim.2013.09.010. [DOI] [PubMed] [Google Scholar]
- Mundt A, Klein J, Joel D, Heinz A, Djodari-Irani A, Harnack D, Kupsch A, Orawa H, Juckel G, Morgenstern R, Winter C. High-frequency stimulation of the nucleus accumbens core and shell reduces quinpirole-induced compulsive checking in rats. The European journal of neuroscience. 2009;29:2401–2412. doi: 10.1111/j.1460-9568.2009.06777.x. [DOI] [PubMed] [Google Scholar]
- Nicolas LB, Kolb Y, Prinssen EP. A combined marble burying-locomotor activity test in mice: a practical screening test with sensitivity to different classes of anxiolytics and antidepressants. Eur J Pharmacol. 2006;547:106–115. doi: 10.1016/j.ejphar.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Nieoullon A, Canolle B, Masmejean F, Guillet B, Pisano P, Lortet S. The neuronal excitatory amino acid transporter EAAC1/EAAT3: does it represent a major actor at the brain excitatory synapse? Journal of neurochemistry. 2006;98:1007–1018. doi: 10.1111/j.1471-4159.2006.03978.x. [DOI] [PubMed] [Google Scholar]
- O'Roak BJ, Morgan TM, Fishman DO, Saus E, Alonso P, Gratacos M, Estivill X, Teltsh O, Kohn Y, Kidd KK, Cho J, Lifton RP, State MW. Additional support for the association of SLITRK1 var321 and Tourette syndrome. Mol Psychiatry. 2010;15:447–450. doi: 10.1038/mp.2009.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki N, Goldman D, Kaye WH, Plotnicov K, Greenberg BD, Lappalainen J, Rudnick G, Murphy DL. Serotonin transporter missense mutation associated with a complex neuropsychiatric phenotype. Mol Psychiatry. 2003;8:933–936. doi: 10.1038/sj.mp.4001365. [DOI] [PubMed] [Google Scholar]
- Ozomaro U, Cai G, Kajiwara Y, Yoon S, Makarov V, Delorme R, Betancur C, Ruhrmann S, Falkai P, Grabe HJ, Maier W, Wagner M, Lennertz L, Moessner R, Murphy DL, Buxbaum JD, Zuchner S, Grice DE. Characterization of SLITRK1 variation in obsessive-compulsive disorder. PLoS One. 2013;8:e70376. doi: 10.1371/journal.pone.0070376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent A, Hazrati LN. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res Brain Res Rev. 1995;20:91–127. doi: 10.1016/0165-0173(94)00007-c. [DOI] [PubMed] [Google Scholar]
- Pauls DL. The genetics of obsessive compulsive disorder: a review of the evidence. American journal of medical genetics Part C, Seminars in medical genetics. 2008;148C:133–139. doi: 10.1002/ajmg.c.30168. [DOI] [PubMed] [Google Scholar]
- Pauls DL. The genetics of obsessive-compulsive disorder: a review. Dialogues Clin Neurosci. 2010;12:149–163. doi: 10.31887/DCNS.2010.12.2/dpauls. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauls DL, Abramovitch A, Rauch SL, Geller DA. Obsessive-compulsive disorder: an integrative genetic and neurobiological perspective. Nat Rev Neurosci. 2014;15:410–424. doi: 10.1038/nrn3746. [DOI] [PubMed] [Google Scholar]
- Pavone P, Bianchini R, Parano E, Incorpora G, Rizzo R, Mazzone L, Trifiletti RR. Anti-brain antibodies in PANDAS versus uncomplicated streptococcal infection. Pediatr Neurol. 2004;30:107–110. doi: 10.1016/S0887-8994(03)00413-2. [DOI] [PubMed] [Google Scholar]
- Pearlman DM, Vora HS, Marquis BG, Najjar S, Dudley LA. Anti-basal ganglia antibodies in primary obsessive-compulsive disorder: systematic review and meta-analysis. Br J Psychiatry. 2014;205:8–16. doi: 10.1192/bjp.bp.113.137018. [DOI] [PubMed] [Google Scholar]
- Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peghini P, Janzen J, Stoffel W. Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. Embo J. 1997;16:3822–3832. doi: 10.1093/emboj/16.13.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger C, Bloch MH, Williams K. Glutamate abnormalities in obsessive compulsive disorder: neurobiology, pathophysiology, and treatment. Pharmacology & therapeutics. 2011;132:314–332. doi: 10.1016/j.pharmthera.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooley EC, Fineberg N, Harrison PJ. The met(158) allele of catechol-O-methyltransferase (COMT) is associated with obsessive-compulsive disorder in men: case-control study and meta-analysis. Mol Psychiatry. 2007;12:556–561. doi: 10.1038/sj.mp.4001951. [DOI] [PubMed] [Google Scholar]
- Proenca CC, Gao KP, Shmelkov SV, Rafii S, Lee FS. Slitrks as emerging candidate genes involved in neuropsychiatric disorders. Trends Neurosci. 2011;34:143–153. doi: 10.1016/j.tins.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radua J, Mataix-Cols D. Voxel-wise meta-analysis of grey matter changes in obsessive-compulsive disorder. The British journal of psychiatry : the journal of mental science. 2009;195:393–402. doi: 10.1192/bjp.bp.108.055046. [DOI] [PubMed] [Google Scholar]
- Radua J, van den Heuvel OA, Surguladze S, Mataix-Cols D. Meta-analytical comparison of voxel-based morphometry studies in obsessive-compulsive disorder vs other anxiety disorders. Archives of general psychiatry. 2010;67:701–711. doi: 10.1001/archgenpsychiatry.2010.70. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Dougherty DD, Malone D, Rezai A, Friehs G, Fischman AJ, Alpert NM, Haber SN, Stypulkowski PH, Rise MT, Rasmussen SA, Greenberg BD. A functional neuroimaging investigation of deep brain stimulation in patients with obsessive-compulsive disorder. J Neurosurg. 2006;104:558–565. doi: 10.3171/jns.2006.104.4.558. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Whalen PJ, Curran T, Shin LM, Coffey BJ, Savage CR, McInerney SC, Baer L, Jenike MA. Probing striato-thalamic function in obsessive-compulsive disorder and Tourette syndrome using neuroimaging methods. Adv Neurol. 2001;85:207–224. [PubMed] [Google Scholar]
- Remijnse PL, Nielen MM, van Balkom AJ, Cath DC, van Oppen P, Uylings HB, Veltman DJ. Reduced orbitofrontal-striatal activity on a reversal learning task in obsessive-compulsive disorder. Arch Gen Psychiatry. 2006;63:1225–1236. doi: 10.1001/archpsyc.63.11.1225. [DOI] [PubMed] [Google Scholar]
- Remijnse PL, van den Heuvel OA, Nielen MM, Vriend C, Hendriks GJ, Hoogendijk WJ, Uylings HB, Veltman DJ. Cognitive inflexibility in obsessive-compulsive disorder and major depression is associated with distinct neural correlates. PLoS One. 2013;8:e59600. doi: 10.1371/journal.pone.0059600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Romaguera J, Do Monte FH, Quirk GJ. Deep brain stimulation of the ventral striatum enhances extinction of conditioned fear. Proc Natl Acad Sci U S A. 2012;109:8764–8769. doi: 10.1073/pnas.1200782109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg DR, Hanna GL. Genetic and imaging strategies in obsessive-compulsive disorder: potential implications for treatment development. Biological psychiatry. 2000;48:1210–1222. doi: 10.1016/s0006-3223(00)01073-8. [DOI] [PubMed] [Google Scholar]
- Rosenberg DR, Keshavan MS, O'Hearn KM, Dick EL, Bagwell WW, Seymour AB, Montrose DM, Pierri JN, Birmaher B. Frontostriatal measurement in treatment-naive children with obsessive-compulsive disorder. Archives of general psychiatry. 1997;54:824–830. doi: 10.1001/archpsyc.1997.01830210068007. [DOI] [PubMed] [Google Scholar]
- Rosenberg DR, MacMaster FP, Keshavan MS, Fitzgerald KD, Stewart CM, Moore GJ. Decrease in caudate glutamatergic concentrations in pediatric obsessive-compulsive disorder patients taking paroxetine. Journal of the American Academy of Child and Adolescent Psychiatry. 2000;39:1096–1103. doi: 10.1097/00004583-200009000-00008. [DOI] [PubMed] [Google Scholar]
- Rosenberg DR, MacMillan SN, Moore GJ. Brain anatomy and chemistry may predict treatment response in paediatric obsessive--compulsive disorder. Int J Neuropsychopharmacol. 2001;4:179–190. doi: 10.1017/S1461145701002401. [DOI] [PubMed] [Google Scholar]
- Saiz PA, Garcia-Portilla MP, Arango C, Morales B, Bascaran MT, Martinez-Barrondo S, Florez G, Sotomayor E, Paredes B, Alvarez C, San Narciso G, Carreno E, Bombin I, Alvarez V, Coto E, Fernandez JM, Bousono M, Bobes J. Association study between obsessive-compulsive disorder and serotonergic candidate genes. Progress in neuro-psychopharmacology & biological psychiatry. 2008;32:765–770. doi: 10.1016/j.pnpbp.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Samuels J, Wang Y, Riddle MA, Greenberg BD, Fyer AJ, McCracken JT, Rauch SL, Murphy DL, Grados MA, Knowles JA, Piacentini J, Cullen B, Bienvenu OJ, Rasmussen SA, Geller D, Pauls DL, Liang KY, Shugart YY, Nestadt G. Comprehensive family-based association study of the glutamate transporter gene SLC1A1 in obsessive-compulsive disorder. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics. 2011;156B:472–477. doi: 10.1002/ajmg.b.31184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Rauch SL. Functional neuroimaging and the neuroanatomy of obsessive-compulsive disorder. The Psychiatric clinics of North America. 2000;23:563–586. doi: 10.1016/s0193-953x(05)70181-7. [DOI] [PubMed] [Google Scholar]
- Scharf JM, Moorjani P, Fagerness J, Platko JV, Illmann C, Galloway B, Jenike E, Stewart SE, Pauls DL Tourette Syndrome International Consortium for G. Lack of association between SLITRK1var321 and Tourette syndrome in a large family-based sample. Neurology. 2008;70:1495–1496. doi: 10.1212/01.wnl.0000296833.25484.bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan NA, Holick Pierz KA, Masten VL, Waeber C, Ansorge M, Gingrich JA, Geyer MA, Hen R, Dulawa SC. Chronic reductions in serotonin transporter function prevent 5-HT1B-induced behavioral effects in mice. Biol Psychiatry. 2009;65:401–408. doi: 10.1016/j.biopsych.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan NA, Velez LP, Masten VL, Dulawa SC. Essential role for orbitofrontal serotonin 1B receptors in obsessive-compulsive disorder-like behavior and serotonin reuptake inhibitor response in mice. Biological psychiatry. 2011;70:1039–1048. doi: 10.1016/j.biopsych.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmelkov SV, Hormigo A, Jing D, Proenca CC, Bath KG, Milde T, Shmelkov E, Kushner JS, Baljevic M, Dincheva I, Murphy AJ, Valenzuela DM, Gale NW, Yancopoulos GD, Ninan I, Lee FS, Rafii S. Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nat Med. 2010;16:598–602. doi: 10.1038/nm.2125. 591p following 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugart YY, Samuels J, Willour VL, Grados MA, Greenberg BD, Knowles JA, McCracken JT, Rauch SL, Murphy DL, Wang Y, Pinto A, Fyer AJ, Piacentini J, Pauls DL, Cullen B, Page J, Rasmussen SA, Bienvenu OJ, Hoehn-Saric R, Valle D, Liang KY, Riddle MA, Nestadt G. Genomewide linkage scan for obsessive-compulsive disorder: evidence for susceptibility loci on chromosomes 3q, 7p, 1q, 15q, and 6q. Mol Psychiatry. 2006;11:763–770. doi: 10.1038/sj.mp.4001847. [DOI] [PubMed] [Google Scholar]
- Shugart YY, Wang Y, Samuels JF, Grados MA, Greenberg BD, Knowles JA, McCracken JT, Rauch SL, Murphy DL, Rasmussen SA, Cullen B, Hoehn-Saric R, Pinto A, Fyer AJ, Piacentini J, Pauls DL, Bienvenu OJ, Riddle MA, Liang KY, Nestadt G. A family-based association study of the glutamate transporter gene SLC1A1 in obsessive-compulsive disorder in 378 families. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics. 2009;150B:886–892. doi: 10.1002/ajmg.b.30914. [DOI] [PubMed] [Google Scholar]
- Singer HS, Hong JJ, Yoon DY, Williams PN. Serum autoantibodies do not differentiate PANDAS and Tourette syndrome from controls. Neurology. 2005;65:1701–1707. doi: 10.1212/01.wnl.0000183223.69946.f1. [DOI] [PubMed] [Google Scholar]
- Singer HS, Loiselle CR, Lee O, Minzer K, Swedo S, Grus FH. Anti-basal ganglia antibodies in PANDAS. Mov Disord. 2004;19:406–415. doi: 10.1002/mds.20052. [DOI] [PubMed] [Google Scholar]
- Snider LA, Swedo SE. PANDAS: current status and directions for research. Mol Psychiatry. 2004;9:900–907. doi: 10.1038/sj.mp.4001542. [DOI] [PubMed] [Google Scholar]
- Song M, Giza J, Proenca CC, Jing D, Elliott M, Dincheva I, Shmelkov SV, Kim J, Schreiner R, Huang SH, Castren E, Prekeris R, Hempstead BL, Chao MV, Dictenberg JB, Rafii S, Chen ZY, Rodriguez-Boulan E, Lee FS. Slitrk5 Mediates BDNF-Dependent TrkB Receptor Trafficking and Signaling. Dev Cell. 2015;33:690–702. doi: 10.1016/j.devcel.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starck G, Ljungberg M, Nilsson M, Jönsson L, Lundberg S, Ivarsson T, Ribbelin S, Ekholm S, Carlsson A, Forssell-Aronsson E, Carlsson ML. A 1H magnetic resonance spectroscopy study in adults with obsessive compulsive disorder: relationship between metabolite concentrations and symptom severity. Journal of neural transmission (Vienna, Austria : 1996) 2008;115:1051–1062. doi: 10.1007/s00702-008-0045-4. [DOI] [PubMed] [Google Scholar]
- Stewart SE, Fagerness JA, Platko J, Smoller JW, Scharf JM, Illmann C, Jenike E, Chabane N, Leboyer M, Delorme R, Jenike MA, Pauls DL. Association of the SLC1A1 glutamate transporter gene and obsessive-compulsive disorder. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics. 2007;144B:1027–1033. doi: 10.1002/ajmg.b.30533. [DOI] [PubMed] [Google Scholar]
- Stewart SE, Mayerfeld C, Arnold PD, Crane JR, O'Dushlaine C, Fagerness JA, Yu D, Scharf JM, Chan E, Kassam F, Moya PR, Wendland JR, Delorme R, Richter MA, Kennedy JL, Veenstra-VanderWeele J, Samuels J, Greenberg BD, McCracken JT, Knowles JA, Fyer AJ, Rauch SL, Riddle MA, Grados MA, Bienvenu OJ, Cullen B, Wang Y, Shugart YY, Piacentini J, Rasmussen S, Nestadt G, Murphy DL, Jenike MA, Cook EH, Pauls DL, Hanna GL, Mathews CA. Meta-analysis of association between obsessive-compulsive disorder and the 3′ region of neuronal glutamate transporter gene SLC1A1. Am J Med Genet B Neuropsychiatr Genet. 2013a;162B:367–379. doi: 10.1002/ajmg.b.32137. [DOI] [PubMed] [Google Scholar]
- Stewart SE, Yu D, Scharf JM, Neale BM, Fagerness JA, Mathews CA, Arnold PD, Evans PD, Gamazon ER, Davis LK, Osiecki L, McGrath L, Haddad S, Crane J, Hezel D, Illman C, Mayerfeld C, Konkashbaev A, Liu C, Pluzhnikov A, Tikhomirov A, Edlund CK, Rauch SL, Moessner R, Falkai P, Maier W, Ruhrmann S, Grabe HJ, Lennertz L, Wagner M, Bellodi L, Cavallini MC, Richter MA, Cook EH, Jr, Kennedy JL, Rosenberg D, Stein DJ, Hemmings SM, Lochner C, Azzam A, Chavira DA, Fournier E, Garrido H, Sheppard B, Umana P, Murphy DL, Wendland JR, Veenstra-VanderWeele J, Denys D, Blom R, Deforce D, Van Nieuwerburgh F, Westenberg HG, Walitza S, Egberts K, Renner T, Miguel EC, Cappi C, Hounie AG, Conceicao do Rosario M, Sampaio AS, Vallada H, Nicolini H, Lanzagorta N, Camarena B, Delorme R, Leboyer M, Pato CN, Pato MT, Voyiaziakis E, Heutink P, Cath DC, Posthuma D, Smit JH, Samuels J, Bienvenu OJ, Cullen B, Fyer AJ, Grados MA, Greenberg BD, McCracken JT, Riddle MA, Wang Y, Coric V, Leckman JF, Bloch M, Pittenger C, Eapen V, Black DW, Ophoff RA, Strengman E, Cusi D, Turiel M, Frau F, Macciardi F, Gibbs JR, Cookson MR, Singleton A, North American Brain Expression C. Hardy J, Database UKBE. Crenshaw AT, Parkin MA, Mirel DB, Conti DV, Purcell S, Nestadt G, Hanna GL, Jenike MA, Knowles JA, Cox N, Pauls DL. Genome-wide association study of obsessive-compulsive disorder. Mol Psychiatry. 2013b;18:788–798. doi: 10.1038/mp.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm V, Lenartz D, Koulousakis A, Treuer H, Herholz K, Klein JC, Klosterkotter J. The nucleus accumbens: a target for deep brain stimulation in obsessive-compulsive- and anxiety-disorders. J Chem Neuroanat. 2003;26:293–299. doi: 10.1016/j.jchemneu.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Swedo SE. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) Mol Psychiatry. 2002;7(2):S24–25. doi: 10.1038/sj.mp.4001170. [DOI] [PubMed] [Google Scholar]
- Swedo SE, Leonard HL, Garvey M, Mittleman B, Allen AJ, Perlmutter S, Lougee L, Dow S, Zamkoff J, Dubbert BK. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: clinical description of the first 50 cases. The American journal of psychiatry. 1998;155:264–271. doi: 10.1176/ajp.155.2.264. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Benbow CH, Zisook S, Geyer MA, Braff DL. A preliminary assessment of sensorimotor gating in patients with obsessive compulsive disorder. Biological psychiatry. 1993;33:298–301. doi: 10.1016/0006-3223(93)90300-3. [DOI] [PubMed] [Google Scholar]
- Szechtman H, Sulis W, Eilam D. Quinpirole induces compulsive checking behavior in rats: a potential animal model of obsessive-compulsive disorder (OCD) Behav Neurosci. 1998;112:1475–1485. doi: 10.1037//0735-7044.112.6.1475. [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Hata Y, Hirao K, Toyoda A, Irie M, Takai Y. SAPAPs. A family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J Biol Chem. 1997;272:11943–11951. doi: 10.1074/jbc.272.18.11943. [DOI] [PubMed] [Google Scholar]
- Taylor JL, Rajbhandari AK, Berridge KC, Aldridge JW. Dopamine receptor modulation of repetitive grooming actions in the rat: potential relevance for Tourette syndrome. Brain research. 2010;1322:92–101. doi: 10.1016/j.brainres.2010.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S. Molecular genetics of obsessive-compulsive disorder: a comprehensive meta-analysis of genetic association studies. Mol Psychiatry. 2013;18:799–805. doi: 10.1038/mp.2012.76. [DOI] [PubMed] [Google Scholar]
- Ting JT, Feng G. Neurobiology of obsessive-compulsive disorder: insights into neural circuitry dysfunction through mouse genetics. Curr Opin Neurobiol. 2011;21:842–848. doi: 10.1016/j.conb.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerius G, Lumpp A, Kuelz AK, Freyer T, Voderholzer U. Reversal learning as a neuropsychological indicator for the neuropathology of obsessive compulsive disorder? A behavioral study. The Journal of neuropsychiatry and clinical neurosciences. 2008;20:210–218. doi: 10.1176/jnp.2008.20.2.210. [DOI] [PubMed] [Google Scholar]
- van Grootheest DS, Cath DC, Beekman AT, Boomsma DI. Twin studies on obsessive-compulsive disorder: a review. Twin Res Hum Genet. 2005;8:450–458. doi: 10.1375/183242705774310060. [DOI] [PubMed] [Google Scholar]
- Varley CK, Vincent J, Varley P, Calderon R. Emergence of tics in children with attention deficit hyperactivity disorder treated with stimulant medications. Comprehensive psychiatry. 2001;42:228–233. doi: 10.1053/comp.2001.23145. [DOI] [PubMed] [Google Scholar]
- Veenstra-VanderWeele J, Kim SJ, Gonen D, Hanna GL, Leventhal BL, Cook EH. Genomic organization of the SLC1A1/EAAC1 gene and mutation screening in early-onset obsessive-compulsive disorder. Molecular psychiatry. 2001;6:160–167. doi: 10.1038/sj.mp.4000806. [DOI] [PubMed] [Google Scholar]
- Wan Y, Feng G, Calakos N. Sapap3 deletion causes mGluR5-dependent silencing of AMPAR synapses. J Neurosci. 2011;31:16685–16691. doi: 10.1523/JNEUROSCI.2533-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub D, David AS, Evans AH, Grant JE, Stacy M. Clinical spectrum of impulse control disorders in Parkinson's disease. Mov Disord. 2015;30:121–127. doi: 10.1002/mds.26016. [DOI] [PubMed] [Google Scholar]
- Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding JD, Feliciano C, Chen M, Adams JP, Luo J, Dudek SM, Weinberg RJ, Calakos N, Wetsel WC, Feng G. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448:894–900. doi: 10.1038/nature06104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland JR, Moya PR, Kruse MR, Ren-Patterson RF, Jensen CL, Timpano KR, Murphy DL. A novel, putative gain-of-function haplotype at SLC6A4 associates with obsessive-compulsive disorder. Human molecular genetics. 2008;17:717–723. doi: 10.1093/hmg/ddm343. [DOI] [PubMed] [Google Scholar]
- Wendland JR, Moya PR, Timpano KR, Anavitarte AP, Kruse MR, Wheaton MG, Ren-Patterson RF, Murphy DL. A haplotype containing quantitative trait loci for SLC1A1 gene expression and its association with obsessive-compulsive disorder. Arch Gen Psychiatry. 2009a;66:408–416. doi: 10.1001/archgenpsychiatry.2009.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland JR, Moya PR, Timpano KR, Anavitarte AP, Kruse MR, Wheaton MG, Ren-Patterson RF, Murphy DL. A haplotype containing quantitative trait loci for SLC1A1 gene expression and its association with obsessive-compulsive disorder. Archives of general psychiatry. 2009b;66:408–416. doi: 10.1001/archgenpsychiatry.2009.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteside SP, Port JD, Abramowitz JS. A meta-analysis of functional neuroimaging in obsessive-compulsive disorder. Psychiatry Res. 2004;132:69–79. doi: 10.1016/j.pscychresns.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Whiteside SP, Port JD, Deacon BJ, Abramowitz JS. A magnetic resonance spectroscopy investigation of obsessive-compulsive disorder and anxiety. Psychiatry research. 2006;146:137–147. doi: 10.1016/j.pscychresns.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Williams KA, Swedo SE. Post-infectious autoimmune disorders: Sydenham's chorea, PANDAS and beyond. Brain Res. 2015;1617:144–154. doi: 10.1016/j.brainres.2014.09.071. [DOI] [PubMed] [Google Scholar]
- Willner P. Methods for Assessing the Validity of Animal Models of Human Psychopathology. In: Boulton A, et al., editors. Animal Models in Psychiatry, I. Vol. 18. Humana Press; 1991. pp. 1–23. [Google Scholar]
- Willour VL, Yao Shugart Y, Samuels J, Grados M, Cullen B, Bienvenu OJ, 3rd, Wang Y, Liang KY, Valle D, Hoehn-Saric R, Riddle M, Nestadt G. Replication study supports evidence for linkage to 9p24 in obsessive-compulsive disorder. Am J Hum Genet. 2004a;75:508–513. doi: 10.1086/423899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willour VL, Yao Shugart Y, Samuels J, Grados M, Cullen B, Bienvenu OJ, Wang Y, Liang KY, Valle D, Hoehn-Saric R, Riddle M, Nestadt G. Replication study supports evidence for linkage to 9p24 in obsessive-compulsive disorder. American journal of human genetics. 2004b;75:508–513. doi: 10.1086/423899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter C, Mundt A, Jalali R, Joel D, Harnack D, Morgenstern R, Juckel G, Kupsch A. High frequency stimulation and temporary inactivation of the subthalamic nucleus reduce quinpirole-induced compulsive checking behavior in rats. Exp Neurol. 2008;210:217–228. doi: 10.1016/j.expneurol.2007.10.020. [DOI] [PubMed] [Google Scholar]
- Wu H, Wang X, Xiao Z, Yu S, Zhu L, Wang D, Jiang K, Wang Z, Zhang T, Fralick D. Association between SLC1A1 gene and early-onset OCD in the Han Chinese population: a case-control study. J Mol Neurosci. 2013;50:353–359. doi: 10.1007/s12031-013-9995-6. [DOI] [PubMed] [Google Scholar]
- Xu M, Li L, Ohtsu H, Pittenger C. Histidine decarboxylase knockout mice, a genetic model of Tourette syndrome, show repetitive grooming after induced fear. Neuroscience letters. 2015;595:50–53. doi: 10.1016/j.neulet.2015.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Grueter BA, Britt JK, McDaniel L, Huntington PJ, Hodge R, Tran S, Mason BL, Lee C, Vong L, Lowell BB, Malenka RC, Lutter M, Pieper AA. Double deletion of melanocortin 4 receptors and SAPAP3 corrects compulsive behavior and obesity in mice. Proc Natl Acad Sci U S A. 2013;110:10759–10764. doi: 10.1073/pnas.1308195110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaddanapudi K, Hornig M, Serge R, De Miranda J, Baghban A, Villar G, Lipkin WI. Passive transfer of streptococcus-induced antibodies reproduces behavioral disturbances in a mouse model of pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection. Molecular psychiatry. 2010;15:712–726. doi: 10.1038/mp.2009.77. [DOI] [PubMed] [Google Scholar]
- Yadin E, Friedman E, Bridger WH. Spontaneous alternation behavior: an animal model for obsessive-compulsive disorder? Pharmacol Biochem Behav. 1991;40:311–315. doi: 10.1016/0091-3057(91)90559-k. [DOI] [PubMed] [Google Scholar]
- Zhang J, Yang X, Yang Q. Neuropsychological dysfunction in adults with early-onset obsessive-compulsive disorder: the search for a cognitive endophenotype. Rev Bras Psiquiatr. 2015;37:126–132. doi: 10.1590/1516-4446-2014-1518. [DOI] [PubMed] [Google Scholar]
- Zuchner S, Wendland JR, Ashley-Koch AE, Collins AL, Tran-Viet KN, Quinn K, Timpano KC, Cuccaro ML, Pericak-Vance MA, Steffens DC, Krishnan KR, Feng G, Murphy DL. Multiple rare SAPAP3 missense variants in trichotillomania and OCD. Mol Psychiatry. 2009;14:6–9. doi: 10.1038/mp.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]