

Abstract
BACKGROUND: Therapeutic strategies targeting immune checkpoint proteins have led to significant responses in patients with various tumor types. The success of these studies has led to the development of various antibodies/inhibitors for the different checkpoint proteins involved in immune evasion of the tumor. Adenosine present in high concentrations in the tumor microenvironment activates the immune checkpoint adenosine A2a receptor (A2aR), leading to the suppression of antitumor responses. Inhibition of this checkpoint has the potential to enhance antitumor T-cell responsiveness. METHODS: We developed a novel A2aR antagonist (PBF-509) and tested its antitumor response in vitro, in a mouse model, and in non-small cell lung cancer patient samples. RESULTS: Our studies showed that PBF-509 is highly specific to the A2aR as well as inhibitory of A2aR function in an in vitro model. In a mouse model, we found that lung metastasis was decreased after treatment with PBF-509 compared with its control. Furthermore, freshly resected tumor-infiltrating lymphocytes from lung cancer patients showed increased A2aR expression in CD4+ cells and variable expression in CD8+ cells. Ex vivo studies showed an increased responsiveness of human tumor-infiltrating lymphocytes when PBF-509 was combined with anti-PD-1 or anti-PD-L1. CONCLUSIONS: Our studies demonstrate that inhibition of the A2aR using the novel inhibitor PBF-509 could lead to novel immunotherapeutic strategies in non-small cell lung cancer.
Abbreviations: A2aR, adenosine A2a receptor; CAF, cancer-associated fibroblast; CHO, Chinese hamster ovary; IC50, concentration of compound that displaces the binding of radioligand by 50%; Kb, binding constant; Ki, inhibition constant; NECA, 5″-N-ethylcarboxamidoadenosine; NSCLC, non-small cell lung cancer; TIL, tumor-infiltrating lymphocyte
Introduction
Metastatic lung cancer remains an incurable disease. The standard treatment is chemotherapy, with targeted agents for patients who have actionable driver mutations. This has produced a survival benefit; however, median survival is only 12.3 months for patients with non-small cell lung cancer (NSCLC) [1]. Recently, immunotherapy with agents preventing the binding of the immune checkpoint PD-1 to its ligand PD-L1 was found to be efficacious in lung cancer. PD-1 is one of several T-cell surface receptors called immune checkpoint proteins, which delivers a negative signal to T-cells when it engages its ligand PD-L1 expressed on antigen-presenting cells [2]. This is a feedback mechanism whereby immune responses are dampened when no longer needed. This mechanism of control of T-cells can be co-opted by tumors to escape rejection by the immune system [3], [4]. A marker of malignancy is an inflamed tumor microenvironment [5], and inflammatory cytokines such as γ-interferon can induce PD-L1 expression on tumor cells [6]. Therefore, tumor antigen-specific T-cells activated in tumor-draining lymph nodes are shut down when they enter into the tumor microenvironment. This discovery has led to the development of a therapeutic strategy that involves monoclonal antibodies designed to prevent PD-1 binding to PD-L1. There are a number of different monoclonal antibodies specific for PD-1 or PD-L1 that are in clinical development for lung cancer, all having similar tumor response rates of 15% to 20% [7].
The adenosine A2a receptor (A2aR) is another T-cell surface immune checkpoint protein operational within the tumor microenvironment. It is known that the extracellular tumor microenvironment contains high levels of adenosine as a consequence of anaerobic glycolysis in hypoxic regions and preferential utilization of aerobic glycolysis for energy metabolism in non-hypoxic regions (the Warburg effect), producing a relative excess of AMP and tumor cell expression of the ectonucleotidase CD73, which catabolizes AMP to produce adenosine [8], [9]. Adenosine produced within the hypoxic microenvironment of inflamed tissue has been shown to limit the exuberance of inflammatory responses to reduce collateral damage of normal tissue by inflammatory cells and cytokines [10], [11]. This is due to a direct inhibitory effect of adenosine on T-cells that express adenosine A2aR, a T-cell surface immune checkpoint protein [12], leading to discovery that adenosine in the tumor microenvironment interferes with antitumor immunity [13] and suggesting that antagonism of the A2aR could be an effective cancer immunotherapeutic [14].
It has been shown that the use of an adenosine A2aR antagonist leads to synergistic tumor suppression with anti-PD-1 in murine models. Beavis and colleagues reported that an adenosine A2aR antagonist could protect mice from developing B16F10-CD73hi experimental metastases [15]. In a more recent paper, this group demonstrated that the combination of a blocking anti-PD-1 antibody and an A2aR antagonist were synergistic in the prevention of experimental and spontaneous metastases in two separate mouse models [16].
A2aR signaling in the brain contributes to neurologic conditions such as Parkinson disease, which has led to the development of A2aR antagonists that have been tested in randomized trials for these diseases [17]. As a class, these drugs are quite safe [18], [19]. Here, we describe the characteristics of PBF-509, a novel and potent A2aR antagonist that has already undergone two clinical trials to assess tolerability and safety (data has not been disclosed yet) [20], [21]. It enhances antitumor immune responses in murine and in human ex vivo models as a consequence of inhibiting the A2aR on T-cells where it functions as an immune checkpoint, contributing to immune evasion in the tumor microenvironment. Thus, PBF-509 may function as an anticancer immunotherapeutic agent in cancer patients.
Methods
Cell Lines
CHO-A1 and HEK-A2B cell lines were purchased from Euroscreen (now part of Perkin Elmer). Hela-A2A and Hela-A3 cells were obtained in house. These four cell lines were obtained more than 10 years ago and were characterized by means of radioligand binding saturation and competition with reference compound studies. This characterization was carried out each time a new batch of membranes was prepared for experiments. B16-CD73+ and MCA205 cells (used in the tumor model) were generously provided by Dr. Mark J. Smyth (Queensland University, Australia) and were not authenticated.
Radioligand Binding Competition Assay
A1, A2A, A2B, and A3 human receptors expressed in transfected Chinese hamster ovary (CHO; hA1), HeLa (hA2A and hA3), and HEK-293 (hA2B) cells were used. Concentration-response binding competition curves were carried out by assaying six different concentrations (range: between 10 nM and 100 μM). The inhibition constant (Ki) of each compound was calculated by the Cheng-Prusoff equation: Ki = IC50/(1 + [L]/Kd), where IC50 is the concentration of compound that displaces the binding of radioligand by 50%, [L] is the free concentration of radioligand, and Kd is the dissociation constant of each radioligand. IC50 values were obtained by fitting the data with nonlinear regression with the use of Prism 2.1 software.
cAMP Accumulation Inhibition Assay
These assays were performed with adenosine receptors transfected using a cAMP enzyme immunoassay kit (Amersham Biosciences). HEK-293 cells were seeded (10,000 cells/well) in 96-well culture plates and incubated at 37°C in an atmosphere with 5% CO2 in Eagle medium nutrient mixture F-12, containing 10% fetal calf serum and 1% l-glutamine. Cells were washed 3 times with 200 μl of assay medium (Eagle medium nutrient mixture F-12 and 25 mM HEPES; pH 7.4) and preincubated with assay medium containing 30 μM rolipram and test compounds at 37°C for 15 minutes. A second incubation step with 10 μM 5″-N-ethylcarboxamidoadenosine (NECA) was performed for 15 minutes at 37°C (total incubation time of 30 minutes). Reaction was stopped with lysis buffer supplied in the kit, and the enzyme immunoassay was carried out for detection of intracellular cAMP at 450 nm in an Ultra Evolution detector (Tecan). Data were fitted by non-linear regression using GraphPad Prism v2.01 (GraphPad Software). We calculated concentration-response curves by assaying six different concentrations (range from 10 nM to 100 μM). Data were expressed as binding constant (Kb) by following the formula reported by Leff and Dougall [22]: Kb = IC50/(2 + ([A]/[A50]n)(1/n) – 1, where IC50 is the concentration of compound that inhibits NECA by 50%; [A] is the concentration of NECA employed in the assay, [A50] is the NECA EC50 value, and n is the Hill slope of the curve.
Experimental Tumor Model
Wild-type C57Bl/6 female mice were purchased from Charles River Laboratories and maintained at the Centre de Recherche du Centre Hospitalier de l'Université de Montréal (Montreal, Quebec, Canada). All experiments were carried out in accordance with guidelines set by the Animal Experimental Ethics Committee. C57Bl/6 mice were injected intravenously with 3 × 105 B16F10 tumor cells retrovirally gene-modified to express CD73 (herein referred to as B16-CD73+) [15] or 105 MCA205 cells. Mice were then treated daily with vehicle control or PBF-509 by oral gavage from day 0. Vehicle consisted of 0.1% Tween 80 and 0.5% sodium carboxymethylcellulose in water. At day 15 post-injection of tumor cells, mice were killed, lungs were harvested, and tumor nodules were counted under dissecting microscope.
Cell Culture
Primary human fibroblasts were isolated from portions of lung tumors resected from patients for clinically indicated reasons. The tumors were mechanically and enzymatically (collagenase, DNase, and protease inhibitors) digested, and the cells were cultured in DMEM-10% FBS (VWR), PenStrep (Gibco), and l-glutamine (Gibco) at 37°C. After 1 week of culture, tumor and immune cells died; however, the cancer-associated fibroblasts (CAFs) proliferated vigorously and survived for greater than 15 passages. Primary human tumor cells were isolated and cultured in F medium. The F medium consisted of DMEM/F12 (3:1) containing 5% serum, 0.4 μg hydrocortisone, 5 μg/ml insulin, 8.4 ng/ml cholera toxin, 10 ng/ml EGF, 24 μg/ml adenine, and penicillin–streptomycin. The isolated tumor cells were co-cultured with 3 T3 feeder cells (3000 cells/cm2) and 10 μM ROCK inhibitor Y-27632 (Enzo Life Sciences). Primary tumor-infiltrating lymphocytes (TILs) were isolated from portions of lung tumors. Pieces of the tumors were cultured in TIL medium. TIL medium consisted of RPMI, 10% hAB serum (Valley Biomedical), 10 mM HEPES (GE Life Sciences), PenStrep (Gibco), 50 mg/ml gentamycin (Gibco), and 55 μM β-mercaptoethanol (Gibco) in the presence of 6000 U/ml of rh-IL-2 (R&D Systems).
Ex Vivo Experiments
Tumor cells were isolated from portions of lung tumors resected from patients for clinically indicated reasons. The tumors were disaggregated for 2 hours in a collagenase-DNase solution in the presence of complete protease inhibitors (Roche). The disaggregated tumor cells were then counted and seeded in 96-well plates at 200,000 cells per well. PBF-509 (Palobiofarma; 1 μM), anti-PD-L1 (eBioscience; 10 μg/ml), anti-PD-1 (eBioscience; 10 μg/ml), and rh-IL-2 (6000 U/ml) were also added at the time of seeding. After 72-hour incubation, the supernatant was collected for γ-interferon ELISA (R&D Systems), which was performed in accordance with manufacturer's instructions.
Flow Cytometric Analysis
After tumor disaggregation, the cells were stained for Live/Dead NearIR (Life Technologies; L10119), CD3 (Fisher Scientific; BDB563546), CD4 (Fisher Scientific; BDB562970), CD8 (Fisher Scientific; BDB562282), and A2aR (Novus Biologicals; NBP1-39474FR) to determine the percentage of A2aR-positive cells in CD4+ and CD8+ cells. In addition, primary human CAFs and tumor cells were stained with anti-CD73 (BD Pharmigen; 550,257) to observe whether they expressed this 5′ ectonucleotidase. Fluorescence was measured using a LSRII flow cytometry system (BD Bioscience), and data were analyzed using FlowJo software (Treestar).
Statistical Analyses
Data are presented as means ± SEM. Statistical calculations were performed using Student t test. Statistical significance was accepted at P values of less than .05.
Results
Binding Assays Against Adenosine Receptors
The affinity of PBF-509 for the four human adenosine receptors was tested using standard radioligand binding competition assays in vitro using A1, A2a, A2b, and A3 human receptors expressed in transfected CHO (hA1), HeLa (hA2a and hA3), and HEK-293 (hA2b) cells. We calculated concentration-response binding competition curves for 6 different concentrations (range: from 10 nM to 100 μM). Figure 1A shows that the inhibition constant against the human adenosine receptors were as follows: 12 nM for A2aR, with values for other adenosine receptors of 2500 nM for A1 (selectivity of 208-fold), 1000 nM for A2b (selectivity of 83-fold), and 5000 nM for A3 (selectivity of 416-fold). To evaluate the influence of the plasma protein binding in the PBF-509 affinity against the adenosine A2a receptor, a binding assay was performed in the presence of 100% human plasma. We observed a three-fold Ki shift for PBF-509, from 12 nM to 32 nM, versus the assay in plasma (Figure 1B). These results show the specificity of the PBF-509 compound to A2aR at low doses.
Figure 1.

PBF-509 is specific for the A2aR. (A) Dose response curves of PBF-509 versus 4 adenosine receptors in transfected CHO (hA1), HeLa (hA2a and hA3), and HEK-293 (hA2b) cells. (B) Representative dose–response curves of PBF-509 in a binding assay against the human A2aR in buffer (median Ki = 12 nM) and in human plasma (median Ki = 32 nM).
PBF-509 Inhibits cAMP Accumulation in Cells
The A2aR is a G-coupled receptor that activates via Gs signal transduction proteins, resulting in increased intracellular levels of cAMP [13]. To determine the functional potency of PBF-509, we treated CHO cells transfected with the adenosine A2aR [23] with PBF-509. We determined the ability of PBF-509 to reduce cAMP elevation (cAMP produced by the full adenosine A2a agonist NECA), with a typical dose–response curve of PBF-509 shown in Figure 2. The concentration of PBF-509 that reduced by 50% the total cAMP elevation induced by NECA (IC50) was determined to be 25 nM. Our results thus show that PBF-509 inhibits A2aR function in an in vitro model.
Figure 2.
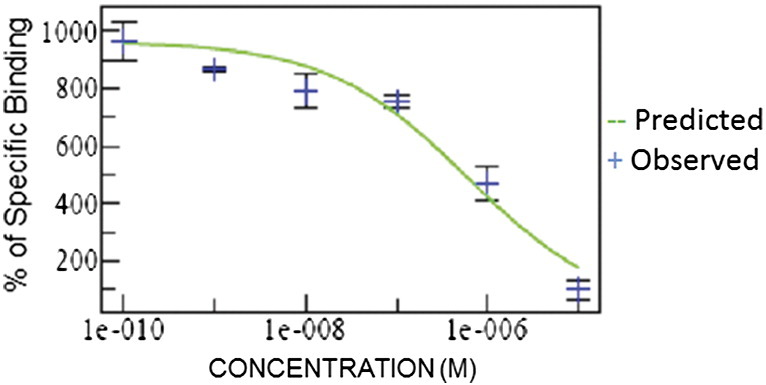
PBF-509 inhibits accumulation of cAMP. CHO cells transfected with the A2aR were used to determine the concentration of PBF-509 to reduce the concentration of cAMP by 50%.
Secondary Pharmacology
PBF-509 was investigated in a diverse panel of 21 different enzymes (selected from known pharmacologically relevant enzyme families, including kinases, phosphodiesterases, and proteases), 5 transporters for amino acids and neurotransmitters (such as dopamine, serotonin, and the adenosine transporter), 8 ion channels, and 55 different receptors (which included the main neurotransmitter receptors dopamine, serotonin, GABA, NMDA, adenosine, and muscarinic receptors). A concentration of 10 μM was used for an initial screen. Apart from the expected activity against the adenosine receptors, PBF-509 did not show any relevant effect in the secondary profile analyses (data not shown).
PBF-509 Reduces Lung Metastasis Formation in Mouse Models
We next investigated the antitumor activity of orally administered PBF-509 in two syngeneic mouse models of cancer. Mice were injected intravenously with B16F10 tumor cells retrovirally gene-modified to express CD73 (B16-CD73+ cells) or with MCA205 cells, which endogenously express high levels of CD73, and treated daily with vehicle control or PBF-509 for 15 days. As shown in Figure 3, oral administration of PBF-509 significantly reduced tumor burden of mice injected intravenously with B16-CD73+ or MCA205 tumor cells.
Figure 3.
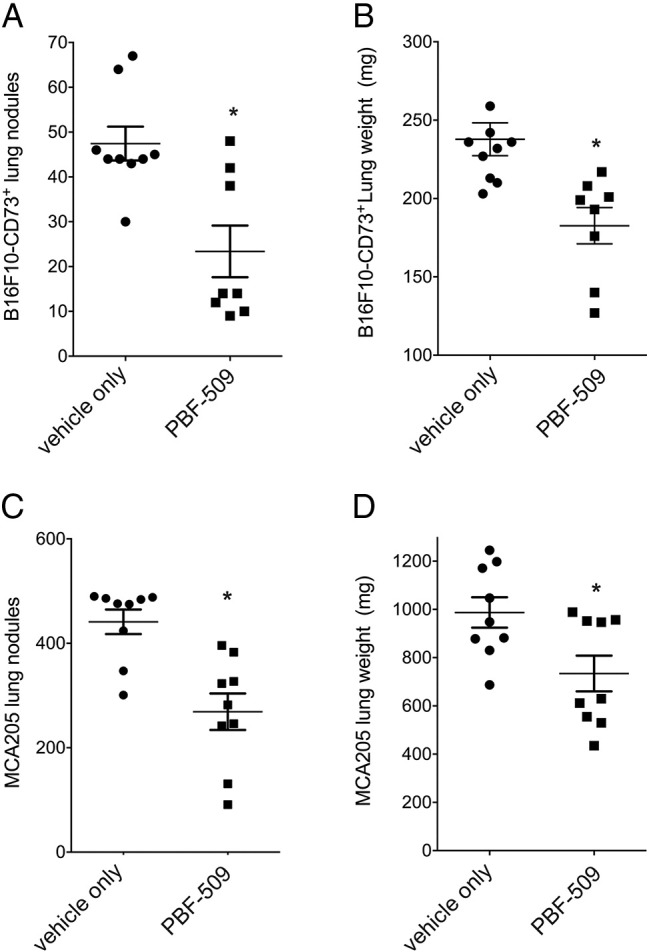
Antitumor activity of PBF-509 in mice. Syngeneic C57Bl/6 mice were injected with 3 × 105 B16-CD73+ tumor cells intravenously and treated daily for 15 days with vehicle control or PBF-509 at 15 mg/kg/day by oral gavage (A and B) or injected with 2 × 105 MCA205 tumor cells intravenously and treated daily for 7 days with vehicle control or PBF-509 at 30 mg/kg/day by oral gavage (C and D). Mean numbers of lung nodules and mean lung weights of 9–10 mice/group ± standard errors are shown. *P < .05 (Student t test).
CD73 Levels are Frequently Elevated in Human Lung Cancers
We established long-term human primary CAFs and human primary tumor cell lines (using a ROCK1 inhibitor and 3T3 feeder cells [24]) from 12 NSCLC tissue samples and used flow cytometry to observe the expression of CD73, a 5′-ectonucleotidase that cleaves AMP and produces adenosine. As shown in Figure 4, A and B, the cell lines (CAF as well as tumor cells) constitutively expressed high levels of CD73.
Figure 4.

Human primary tumor, CAF, and TIL cell lines express CD73 and A2aR. Tumor (Tm), cancer-associated fibroblast (CAF), and tumor infiltrating lymphocyte (TIL) cell lines were established from 12 resected NSCLC tumors. (A) Tumor cells or CAFs were stained with anti-CD73 and subjected to flow cytometry. Most cells from nearly all of the cell lines (both tumor cells and CAFs) expressed high levels of CD73. (B) Representative experiment of tumor cells stained with anti-CD73 (blue) or isotype control (green). (C) CD4+ or CD8+ TIL cells were stained with anti-A2aR and subjected to flow cytometric analysis showing that high levels of A2aR were being expressed in CD4+ cells, with variable expression in CD8+ cells.
A2aR Checkpoint Protein is Frequently Expressed on TILs in Human NSCLC Tumors
To assess the relative potential contribution of the various known T-cell immune checkpoints in human lung cancers, we surveyed a series of resected NSCLC tumors. Immediately after resection, the tumors were mechanically and enzymatically (collagenase, DNase, and protease inhibitors) disaggregated, stained with antibodies specific for CD3, CD4, CD8, LAG3, PD-1, BTLA, TIM3, and A2aR and analyzed using flow cytometry. The TIL cell populations of interest were identified using a gating strategy of morphology, live cells, CD3, and CD4 or CD8. We observed significant heterogeneity of expression of LAG3 and PD-1 among the tumors but uniformly low levels of expression of TIM3 and CTLA-4 (data not shown). Interestingly, most of the tumors contained CD4 TILs that were nearly 100% positive for A2aR, with greater heterogeneity of expression on the CD8 TILs among the tumors (Figure 4C).
PBF-509 and Anti-PD-L1 Restore Immune Responsiveness in Human TILs Ex Vivo
To determine the functional significance of A2aR expression on TILs and CD73 expression on tumor cells and CAF cells, we performed short-term ex vivo assays of freshly disaggregated resected human NSCLC tumors. After 3 days of culture, the supernatants were assayed for γ-interferon as a measure of T-cell reactivity to autologous tumor cells. As was expected with no manipulation or with the addition of IL-2, the T-cells displayed little to no activity. The addition of either anti-PD-L1 or the A2aR antagonist PBF-509 partially restored TIL reactivity in some of the samples, and the combination improved TIL function in an additive fashion (Figure 5, A and B). In other tumors, the addition of anti-PD-L1, anti-PD-1, or PBF-509 had no effect on TIL function, but the combination was synergistically capable of restoring TIL function (Figure 5, C and D).
Figure 5.

Anti-PD-L1 and PBF-509 restore immune responsiveness of TILs. Four different resected NSCLC tumors were disaggregated and cultured with or without anti-PD-L1 (aPDL1; 10 mg/ml), anti-PD-1 (aPD1; 10 μg/ml), or the A2aR antagonist PBF-509 (1 μM). After 3 days in culture, the supernatants were assayed for the presence of γ-interferon using an ELISA.
Discussion
The numerous immune-evasive mechanisms co-opted by tumors in the tumor microenvironment perhaps explain primary resistance to the PD-1/PD-L1 blocking of immunotherapeutic agents. One of these mechanisms is the induced expression of the immune checkpoint protein, the adenosine A2aR on the surface of T-cells. Here, we showed that the novel A2aR antagonist PBF-509 has an effect in in vivo models as well as ex vivo models of lung cancer.
Currently, various A2aR antagonists are being studied in clinical trials, mainly in patients with Parkinson disease, where it has provided motor function-related benefits in patients [25]. Our studies focused on one of these A2aR antagonists called PBF-509. PBF-509 is a new A2aR antagonist that is currently the only A2aR antagonist being studied in cancer patients in a clinical trial [26]. We were able to demonstrate that PBF-509 has a high affinity and potency for A2aR at low doses (12 nM and 25 nM, respectively) in in vitro experiments. In addition, no activity was observed in the presence of a panel of receptors and enzymes, which reiterated the specificity of the drug.
Increased adenosine in the tumor microenvironment has been shown to have negative effects on T-cell activation and effector activity, specifically through the A2aR [11], [14], [27], [28], [29]. Thus, inhibition of this receptor has been studied as a possible novel therapeutic approach in cancer. Various groups have studied the effects of A2aR antagonists in vivo as a single agent or in combination with various immune checkpoint inhibitors [13], [15], [16], [30], [31], [32]. These studies have shown that inhibition of the A2aR with antagonists such as ZM241385 and SCH58261 alone or in combination with anti-PD-1 and anti-CTLA4 leads to decreased tumor burden. Our studies are the first to show that the A2aR antagonist PBF-509 significantly decreased tumor burden in mice treated with the inhibitor compared with control results in two different cell lines that express high levels of CD73. CD73 is an ectonucleotidase that, in combination with CD39, generates adenosine from ATP leading to the activation of the A2aR [33].
Although our murine data showed the importance of the A2aR antagonist PBF-509, we wanted to show similar results in a more clinically relevant model. For this reason, we utilized an ex vivo model to study the TILs from freshly resected lung tumors, as well as derive primary CAF and tumor cell lines from the same patients. Our data showed that most of the tumor and CAF cell lines had high expression of CD73. In addition, the TILs from the same patients showed high expression of A2aR in CD4+ cells and variable expression in CD8+ cells. This demonstrated that, in the tumor microenvironment of lung cancer patients, the expression levels of these two immunosuppressive molecules are high and the use of an inhibitor for the A2aR could lead to improved therapies. To further demonstrate the importance of the A2aR and of the new A2aR antagonist PBF-509, an ex vivo model was utilized to determine its effectiveness in combination with known immune-based therapies. Using 4 different patient samples, we were able to show that PBF-509 combined with anti-PD-1 or anti-PD-L1 restores immune responsiveness in NSCLC tumors.
Conclusions
The A2aR antagonist PBF-509 has potential as a novel therapeutic approach in NSCLC. Our data showed the safety of the drug, its capability of reducing the tumor burden in in vivo models, its competence to restore immune responsiveness ex vivo, and the association of high A2aR expression in lung cancer patients with poor prognosis.
Ethics Approval and Consent to Participate
All experiments were carried out in accordance with guidelines set by the Animal Experimental Ethics Committee. The study was exempted from human subjects’ activities; therefore, the Institutional Review Board declared that no approval was required.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
Competing Interests
JS is a paid consultant and science advisory board member for Surface Oncology Inc. and received research grants from Surface Oncology Inc., MedImmune LLC, and Palobiofarma. All other authors declare that they have no competing interests.
Funding
This work has been supported in part by the Flow Cytometry Core Facility and Tissue Core Facility at the H. Lee Moffitt Cancer Center & Research Institute, a comprehensive cancer center designated by the National Cancer Institute (P30-CA76292–14).
Authors' Contributions
MM-V, JC, AC, DV, DCH, and JS developed study concepts and design and performed data analysis and interpretation. MM-V, AC, DN, and DCH performed flow cytometric analysis, ex vivo assays, and drafted the manuscript. JC performed binding assays. JS performed in vivo studies. SA, JC, and JS conceived of the study, participated in hypothesis generation and testing, study design, data analysis, interpretation, and manuscript preparation. All authors read and approved the final manuscript.
Acknowledgements
We thank Rasa Hamilton (Moffitt Cancer Center) for editorial assistance.
References
- 1.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 2.Yao S, Zhu Y, Chen L. Advances in targeting cell surface signalling molecules for immune modulation. Nat Rev Drug Discov. 2013;12:130–146. doi: 10.1038/nrd3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3 - potential mechanisms of action. Nat Rev Immunol. 2014;15:45–56. doi: 10.1038/nri3790. [DOI] [PubMed] [Google Scholar]
- 4.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, Oh S, Shin JG, Yao S, Chen L. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274) FEBS Lett. 2006;580:755–762. doi: 10.1016/j.febslet.2005.12.093. [DOI] [PubMed] [Google Scholar]
- 7.Philips GK, Atkins M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int Immunol. 2015;27:39–46. doi: 10.1093/intimm/dxu095. [DOI] [PubMed] [Google Scholar]
- 8.Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, Shin T, Curiel TJ, Zhang B. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 2010;70:2245–2255. doi: 10.1158/0008-5472.CAN-09-3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29:5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- 10.Di Paola R, Melani A, Esposito E, Mazzon E, Paterniti I, Bramanti P, Pedata F, Cuzzocrea S. Adenosine A2A receptor-selective stimulation reduces signaling pathways involved in the development of intestine ischemia and reperfusion injury. Shock. 2010;33:541–551. doi: 10.1097/SHK.0b013e3181c997dd. [DOI] [PubMed] [Google Scholar]
- 11.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 12.Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, Drake CG, Powell JD. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008;111:251–259. doi: 10.1182/blood-2007-03-081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A. 2006;103:13132–13137. doi: 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Discov. 2014;4:879–888. doi: 10.1158/2159-8290.CD-14-0341. [DOI] [PubMed] [Google Scholar]
- 15.Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, Dwyer K, Stagg J, Smyth MJ, Darcy PK. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci U S A. 2013;110:14711–14716. doi: 10.1073/pnas.1308209110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mittal D, Young A, Stannard K, Yong M, Teng MW, Allard B, Stagg J, Smyth MJ. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res. 2014;74:3652–3658. doi: 10.1158/0008-5472.CAN-14-0957. [DOI] [PubMed] [Google Scholar]
- 17.Dungo R, Deeks ED. Istradefylline: first global approval. Drugs. 2013;73:875–882. doi: 10.1007/s40265-013-0066-7. [DOI] [PubMed] [Google Scholar]
- 18.Hauser RA, Olanow CW, Kieburtz KD, Pourcher E, Docu-Axelerad A, Lew M, Kozyolkin O, Neale A, Resburg C, Meya U. Tozadenant (SYN115) in patients with Parkinson's disease who have motor fluctuations on levodopa: a phase 2b, double-blind, randomised trial. Lancet Neurol. 2014;13:767–776. doi: 10.1016/S1474-4422(14)70148-6. [DOI] [PubMed] [Google Scholar]
- 19.Pinna A. Adenosine A2A receptor antagonists in Parkinson's disease: progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs. 2014;28:455–474. doi: 10.1007/s40263-014-0161-7. [DOI] [PubMed] [Google Scholar]
- 20.ClinicalTrials.gov Study To Assess the Safety and Tolerability of PBF-509 in Male Healthy Volunteers. https://clinicaltrials.gov/ct2/show/NCT01691924 Available at:
- 21.ClinicalTrials.gov Study to Assess the Safety, Tolerability and Pharmacokinetic Profile of PBF −509 (80 mg, 160 mg and 240 mg) "After Multiple Oral Doses" in Healthy Volunteers. https://clinicaltrials.gov/ct2/show/NCT02111330 Available at: [Accessed June 20, 2016]
- 22.Leff P, Dougall IG. Further concerns over Cheng-Prusoff analysis. Trends Pharmacol Sci. 1993;14:110–112. doi: 10.1016/0165-6147(93)90080-4. [DOI] [PubMed] [Google Scholar]
- 23.Vu CB, Pan D, Peng B, Kumaravel G, Smits G, Jin X, Phadke D, Engber T, Huang C, Reilly J. Novel diamino derivatives of [1,2,4]triazolo[1,5-a][1,3,5]triazine as potent and selective adenosine A2a receptor antagonists. J Med Chem. 2005;48:2009–2018. doi: 10.1021/jm0498396. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180:599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jenner P. Istradefylline, a novel adenosine A2A receptor antagonist, for the treatment of Parkinson's disease. Expert Opin Investig Drugs. 2005;14:729–738. doi: 10.1517/13543784.14.6.729. [DOI] [PubMed] [Google Scholar]
- 26.ClinicalTrials.gov. Trial of PBF-509 and PDR001 in Patients With Advanced Non-small Cell Lung Cancer (NSCLC).
- 27.Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014;74:7250–7259. doi: 10.1158/0008-5472.CAN-13-3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cekic C, Linden J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res. 2014;74:7239–7249. doi: 10.1158/0008-5472.CAN-13-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muller-Haegele S, Muller L, Whiteside TL. Immunoregulatory activity of adenosine and its role in human cancer progression. Expert Rev Clin Immunol. 2014;10:897–914. doi: 10.1586/1744666X.2014.915739. [DOI] [PubMed] [Google Scholar]
- 30.Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, Kershaw MH, Stagg J, Darcy PK. Adenosine Receptor 2A Blockade Increases the Efficacy of Anti-PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol Res. 2015;3:506–517. doi: 10.1158/2326-6066.CIR-14-0211. [DOI] [PubMed] [Google Scholar]
- 31.Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am J Cancer Res. 2014;4:172–181. [PMC free article] [PubMed] [Google Scholar]
- 32.Mediavilla-Varela M, Luddy K, Noyes D, Khalil FK, Neuger AM, Soliman H, Antonia SJ. Antagonism of adenosine A2A receptor expressed by lung adenocarcinoma tumor cells and cancer associated fibroblasts inhibits their growth. Cancer Biol Ther. 2013;14:860–868. doi: 10.4161/cbt.25643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang B. CD73: a novel target for cancer immunotherapy. Cancer Res. 2010;70:6407–6411. doi: 10.1158/0008-5472.CAN-10-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.