

Abstract
Background
Detection of hepatitis C virus specific antibodies is the initial step in chronic HCV diagnosis. HCV NS4B is among the most immunogenic HCV antigens and has been widely used in commercial Enzyme Immunoassays (EIA). Additionally, NS4B, a key protein in the virus replication, can be an alternative target for antiviral therapy.
Objectives
Development of a new method for high-level expression and purification of NS4B coding region was the aim of the report.
Materials and Methods
Viral RNA was purified from the serum of an HCV positive patient and NS4B coding region was amplified using nested RT-PCR. PCR products were cloned into pET102/D-TOPO expression vector and transformed into E. coli BL21. Induction was performed by adding 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) to the culture medium. Immunoreactivity of the purified recombinant proteins was evaluated by immunoblotting and indirect enzymelinked immunosorbent assay (ELISA).
Results
The recombinant NS4B protein was expressed and its immunoreactivity was confirmed by ELISA and western blotting.
Conclusions
The directional TOPO cloning provides an efficient and easy platform for heterologous expression of immunoreactive HCV NS4B.
Keywords: Hepatitis C Virus, NS4B, Recombinant antigen, Serodiagnosis
1. Background
Hepatitis C is a liver disease caused by the hepatitis C virus. The virus is blood-borne and becomes contagious via unsafe injection practices, unsafe blood and blood product transfusions, and inadequate sterilization of medical equipment (1). Approximately 350,000 to 500,000 people die each year from hepatitis C-related liver diseases (2). Diagnosis of the chronic HCV infection is based on anti-HCV screening assays and a nucleic acid test for confirming chronic infection (1). The current standard treatment for hepatitis C is combination of antiviral therapy with interferon and ribavirin (1). There is currently no vaccine for the disease.
HCV is an RNA virus that is directly translated to a precursor polyprotein. The precursor is cleaved by host and viral proteases to produce four structural (core protein, E1, E2, and P7) and six non-structural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) viral proteins (3). Among these proteins, NS4B is studied relatively less than the others. A variety of functions have been suggested for NS4B, of which the most significant is providing a scaffold for the assembly of the HCV replication complex (4).
NS4B is among the HCV major immunogenic proteins in chronic infection (5). Some of the diagnostically important antigenic epitopes have been found to reside within NS4B (6-8). There are two FDA-approved commercially available enzyme immunoassays (EIA) for screening anti-HCV antibodies: Abbott HCV EIA 2.0 and ORTHO HCV Version 3.0 enzyme-linked immunosorbent assay (ELISA) Test System. Use of recombinant NS4B is a requirement in both EIAs. Hence, high level expression and purification of NS4B is essential for designing appropriate HCV serodiagnostic assay.
Although the HCV standard treatment with a combination of interferon and ribavirin is successful for many patient populations, this regimen has numerous limitations, including non-response, relapse, poor tolerability and long duration of treatment (9). To compensate for the limitations, pharmacological studies with the aim of finding new therapeutic targets are needed to be done. NS4B is among the key proteins in the viral life-cycle, and ironically it is among the least characterized of the HCV structural and nonstructural proteins (10). Therefore, pharmacological studies on NS4B are required to evaluate the effect of targeting NS4B as an alternative target for direct-acting antiviral agents. Meanwhile, high level expression and purification of NS4B could facilitate studies on drug design and development.
Here, a new method was described for substantial expression and purification of recombinant NS4B using pET102/D-TOPO expression vector and E. coli BL21 as the host. The immuno reactivity of the recombinant antigen was evaluated by western blot analysis and ELISA using HCV positive human sera.
2. Materials and Methods
2.1. Human Sera
HCV-positive (100 samples) and HCV-negative (50 samples) were collected from Iranian Blood Transfusion Organization. All sera were tested for the presence of anti-HCV antibody using thirdgeneration ELISA kit (Hepanostika HCV Ultra, BioMerieux, Marcy I’Etoile, France). Positive results of HCV antibody were confirmed by recombinant immunoblot assay (RIBA). Infecting HCV genotype was determined using HCV genotyping kit (Sacace, Como, Italy).
2.2. HCV RNA Extraction and cDNA Synthesis
HCV RNA was extracted from the serum of a patient infected with HCV genotype 1a usingHighPure Viral RNA MiniKit (Roche Applied Biosystems, Frankfurt, Germany) according to the manufacturer’s instructions. The extracted RNA was reverse transcribed into cDNA using reverse transcriptase enzymes l Maloney Maurine Leukemia Virus ((MMLV)(New England Biolabs, Ipswich, MA, USA)) (NEB#M0253USA) according to the manufacturer’s protocol.
2.3. Nested PCR for Amplification of NS4B Coding Region
Coding region of NS4B was amplified by nested PCR. Designed forward and reverse outer primers were 5'GAGGTCCTCTACCGGGAGTTCG3' and 5'CCA GTCCCAGATGTCCCTTAGCCA3'. Designed forward and reverse inner primers were 5'CACCTCCC AGCACTTACCGTAC3' and 5'GACACACGTGTTGCA GTCTATCA3'. To enable insertion of blunt end PCR product into TOPO cloning vector in the right orientation, the inner forward primer contained the sequence CACC at the 5'end. The first PCR reaction was assembled in a final volume of 25 μL using the following reagents: 0.7 μL Pfu DNA polymerase (2.5 U.μL-1; Stratagene, La Jolla, CA, USA), 1 μL (100 ng.μL-1) of each of the forward and reverse outer primers, 2.5 μL of 10× reaction buffer, 2 μL dNTPs (10 μM) and 3 μL template (cDNA 100 ng.μL-1). The second PCR reaction was performed on the product of the first PCR (3 μL template) using inner primers as described for the first PCR. Thermal cycling conditions except for the annealing step were similar for the first and the second PCR and as follows: initial denaturation (95°C for 3 min), 35 cycles of denaturation (95°C for 45 seconds), annealing (the first PCR: 58°C for 30 seconds and the second PCR: 62°C for 30 seconds), extension (72°C for 1 min), and the final extension (72°C for 10 min).
2.4. Construction of NS4B Plasmid (TOPO Cloning)
After agarose gel electrophoresis, PCR products with correct size were extracted from the gel using High Pure PCR Product Purification Kit (Roche, Frankfurt, Germany) according to the manufacturer’s protocol. The purified PCR products were ligated into pET102/D-TOPO expression vector (Invitrogen, Montreal, Canada) according to the manufacturer’s instructions. The ligation products were transformed into E. coli BL21 (DE3) strain (Invitrogen, Montreal, Canada) and cultured in Luria Bertani (LB) agar plate containing 100 μg.mL-1 of ampicillin. Plasmids were extracted from colonies on agar plate using High Pure Plasmid Isolation Kit (Roche, Frankfurt, Germany) according to the manufacturer’s instructions. To confirm the accuracy of the construct, colony PCR and DNA sequencing (Bioneer, Daejeon, South Korea) were performed on extracted plasmids.
2.5. Expression of Recombinant NS4B
E. coli BL21 (DE3) cells carrying recombinant plasmid were grown in 10 μL LB broth containing 100 μg.μL-1 ampicillin at 37°C with gentle shaking (150 rpm) overnight. Once the optical density (OD) of the culture reached 0.6 at 600 nm, induction was performed using 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) (Invitrogen, Frankfurt, Germany). The culture was left to grow at 37°C with gentle shake for 4 h. Cells were harvested by centrifugation at 5,000 rpm at 4°C for 10 min.
2.6. Purification of Recombinant NS4B
Ni-NTA Purification System (Invitrogen, Frankfurt, Germany) was used to purify recombinant NS4B according to the user manual. The harvested cells were subjected to cell lysis under denaturing conditions. The cell pellet from a 50 μL culture was resuspended in 8 μL Guanidinium Lysis Buffer (containing 6 M guanidine hydrochloride, 20 mM sodium phosphate, and 0.5 M NaCl) for 10 min on a shaker. The cell lysate was sonicated on ice with three 5-second pulses at high intensity. The lysate was centrifuged at 3,000 ×g for 15 min and the supernatant was purified using Ni-NTA purification system. A volume of 8 μL of the lysate was applied to the purification column and incubated for 30 min with gentle agitation. The column was centrifuged and the supernatant was removed. The column was washed twice with 4 μL Denaturing Binding Buffer pH 6.0. Then the column was washed twice with 4 μL Denaturing Wash Buffer pH 5.3. The recombinant protein was eluted with 5 μL Denaturing Elution Buffer. The fractions were subsequently quantified by Micro BCA protein assay kit (Pierce, Rockford, IL, USA) according to the user’s guide.
2.7. Western Blot
An amount of 25 μg of protein was separated on 10% SDS-PAGE gel and transferred onto PVDF membrane (Amersham, London, United Kingdom). The membrane was blocked for 1.5 h with 5% skim milk in TBST buffer (Tris-Buffered Saline containing 0.1% v/v Tween 20), the membrane was incubated at 4ºC overnight with a pool of HCV positive sera (containing antibodies to 1a, 1b, and 3a HCV genotypes) diluted 1:15000 in 3% skim milk in TBS buffer. The membrane was washed for 15min wash and three 5-min washes with TBST buffer. Membrane was incubated withHRP-conjugated anti-human IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1:100,000 in 3% skim milk in TBS buffer for 1 h at room temperature. After washing (as described previously), the signals were detected by chemiluminescence using the ECL detection reagent (GE Healthcare, Piscataway, NJ, USA).
In a parallel experiment, western blot analysis was performed using anti-His tag monoclonal antibody (Invitrogen, Frankfurt, Germany) diluted 1:2000 in 3% skim milk in TBS buffer. This antibody specifically binds to poly-histidine tag of the recombinant protein and confirms the presence of the recombinant NS4B.
2.8. ELISA
Each ELISA well was incubated with 100 μL of the purified recombinant NS4B in 0.1 M sodium carbonate buffer pH 9.6 (40 μg.μL-1), at 4°C overnight. Wells were incubated with blocking buffer (1% BSA in phosphate buffer saline) at 22°C for 1 h. After 3 washes with wash buffer (phosphate buffer saline containing 0.1% Tween-20), 40 HCV positive sera fromdifferent HCV genotypes (1a, 1b, and 3a), each separately diluted 1:1000 in PBS containing 0.1% (v/v) Tween-20 and 0.01% (w/v) BSA, was added to the wells and incubated at 37°C for 30 min. Sera from 40 healthy individuals were similarly used as negative control. After 3 washes, 100 μL of HRP-conjugated rabbit anti-human IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1:100,000 was added to each well and incubated at 37°C for 30 min. After 3 washes, 100 μL of TMB substrate (Roche Diagnostics, Sussex, UK) was added to each well and incubated for 15 min at 22°C. The reaction was stopped by adding 50 μL of 0.2 M H2SO4 and the absorbance was measured at 450 nm with ELISA reader (Biochrome, Cambridge, UK).
3. Results
3.1. Gene Amplification, TOPO Cloning and Transformation
Amplification of HCV NS4B protein coding sequence was performed successfully by nested RT-PCR. Electrophoresis of the PCR products showed a single clear band equal to 800 bp (Figure 1). The accuracy of the recombinant plasmid was confirmed by colony PCR and sequencing. The efficiency of bacterial transformation was approved by colony formation on LB agar plate containing 100 mg.μL-1 ampicillin.
Figure 1 .
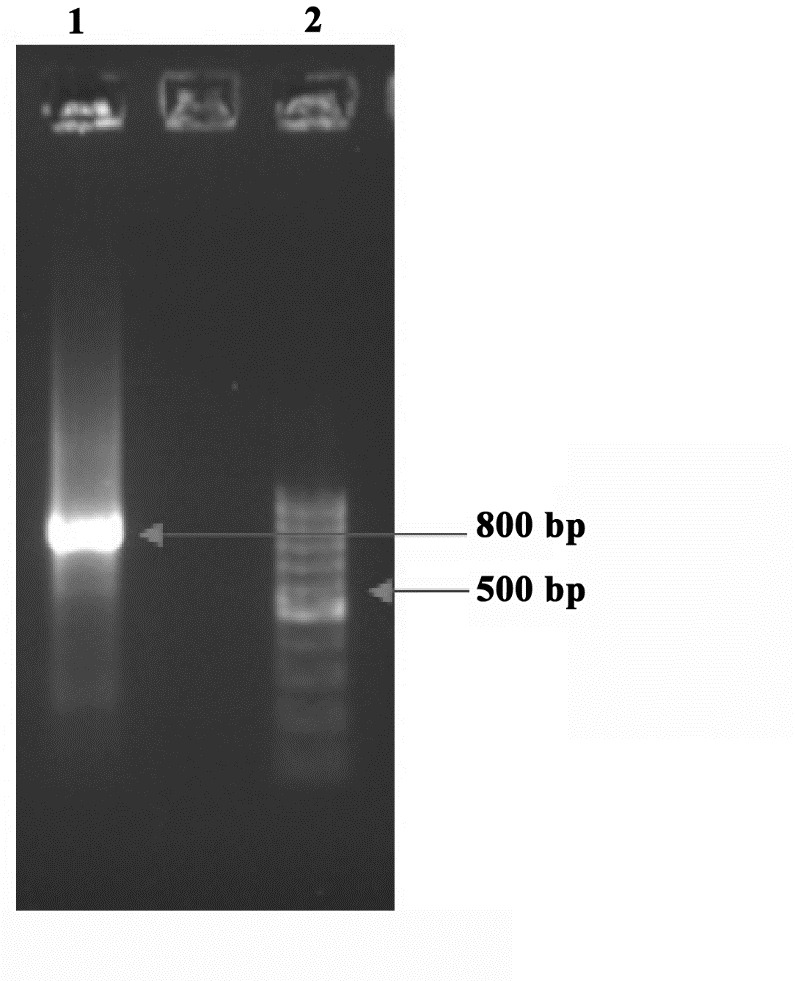
PCR amplification of HCV NS4B coding sequence. Lane 1: the amplified product of HCV NS4B by pfuDNA polymerase (800 bp); lane 2: DNA marker (100 bp)
3.2. Analysis of NS4B Expression by SDS-PAGE, Protein Assay and Western Blot
SDS-PAGE showed successful expression of the recombinant NS4B after IPTG induction (Figure 2). It was determined that the molecular weight of the recombinant NS4B was about 44 kDa. Protein purification using Ni-NTA purification system yielded highly pure fractions of recombinant protein near to homogeneity, as observed by SDS-PAGE (data not shown) and the recombinant protein concentrations in the eluted fractions were as high as 48 μg.μL-1. Western blot analysis using anti-His antibody confirmed the authenticity of the recombinant protein (Figure 3) and Western blot analysis using human sera from HCV-infected patients confirmed the immunoreactivity of the recombinant protein (Figure 4).
Figure 2 .
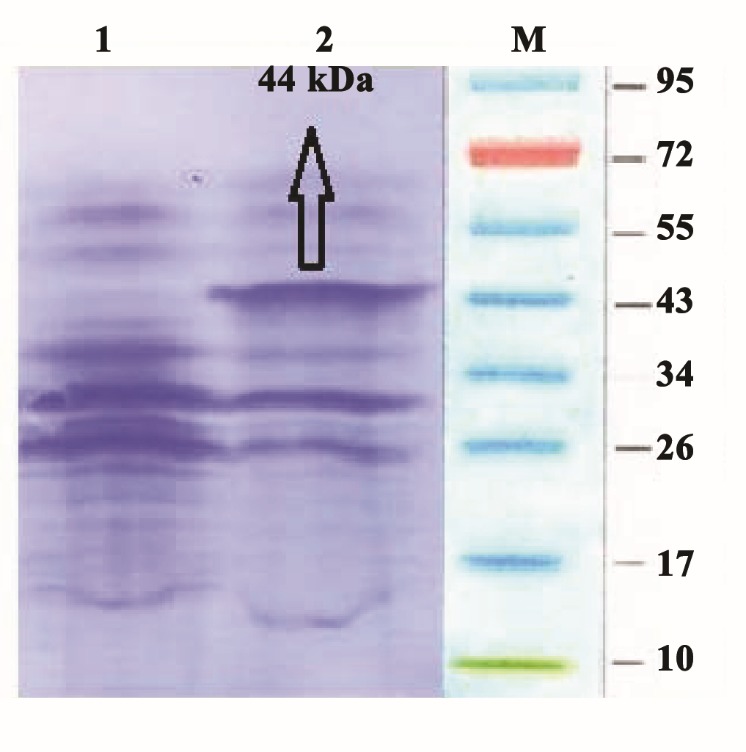
SDS-PAGE analysis of recombinant-NS4B fusion protein. Lane M: Protein marker; lane1: E. coli BL21 cell lysate harboring recombinant plasmid before induction with IPTG; lane 2: E. coli BL21 cell lysate harboring recombinant plasmid after induction with IPTG (44 kDa)
Figure 3 .
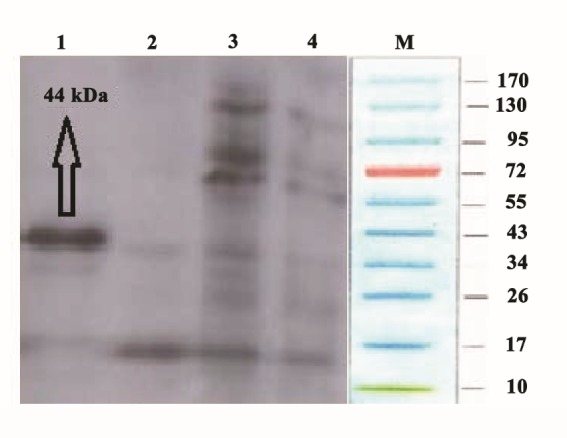
Western blot analysis of the recombinant NS4B protein with mouse anti-HIS antibodies. Lane M: Protein marker; lane 1: purified NS4B (44 kDa); lane 2: fraction from wash step of Ni-NTApurification; lane 3: total protein of E. coli BL21carrying pET102-NS3 without IPTG induction; lane 4: Total protein of Escherichia coli BL21 as negative control
Figure 4 .
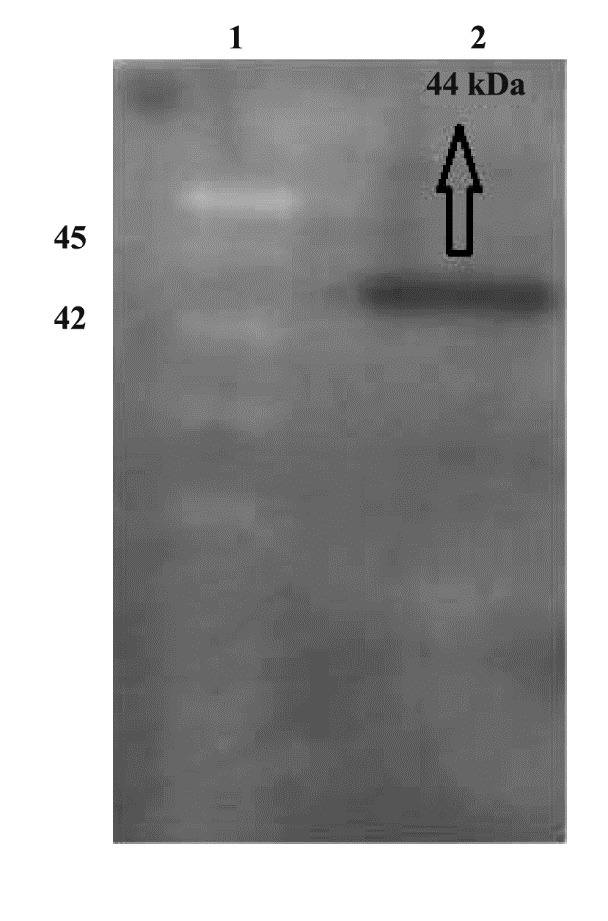
Western blot analysis of the recombinant NS4B protein with HCV-positive human sera. Lane 1: Protein marker; lane 2: purified NS4B (44 kDa)
3.3. ELISA
ELISA was performed on 40 HCV negative sera and 40 HCV positive sera from different HCV genotypes (1a, 1b, and 3a). The optimum cutoff value (0.78) was calculated as the mean OD value of negative samples plus three times the standard deviation. The ELISA showed 85% sensitivity and 80% specificity.
4. Discussion
The HCV non-structural 4B (NS4B) protein is a 27 kDa hydrophobic protein localized in the endoplasmic reticulum membrane (11) and it is mainly involved in HCV replication (4). NS4B is amongthe HCV major immunogenic proteins and it is widely used in anti-HCV antibody detection in diagnostic serologic tests (6-8). Furthermore, since NS4B is a key protein in virus life cycle, it provides new opportunities for antiviral intervention against a leading cause of liver diseases worldwide (12). Therefore, rapid, high yield, and economical production of recombinant HCV NS4B is essential for producing HCV diagnostic kits. Moreover, biochemical characterization of the protein, and development of anti-HCV drugs are all dependent on having enough protein that can be obtained via heterologous expression.
NS4B has often been expressed in insect cells using baculovirus vector (5,13-15). In the present work, we described the expression and purification of recombinant HCV NS4B coding region in E. coli BL21 using pET102/D-TOPO expression vector and evaluated its immunoreactivity. To the best of our knowledge, this is the first time that HCV NS4B has been produced using directional TOPO cloning.
Directional TOPO vectors in general provide a fast and easy method for cloning and expression of recombinant proteins. These vectors take advantage from T7lac promoter for high-level, IPTG-inducible expression of the gene of interest. This is essential for expressing NS4B because the basal expression of this protease may lead to plasmid instability and cell death. In particular, PET102/D-TOPO provides N-terminal His-Patch thioredoxin for increased translation efficiency and solubility of heterologous proteins. It also provides one-step purification of NS4B in native buffers without urea and chaotic agents in order to provide the possibility of refolding the recombinant protein which may play an important role in the immunoreactivity of NS4B due to natural conformation of the epitopes. BL21 strain of E. coli (DE3) permits high-yield expression of the recombinant protein because BL21 Star (DE3) carries a mutated rne gene that encodes a truncated RNase E resulting in an increase in mRNA stability.
Most papers did not emphasize on the high yield of the recombinant protein. However, we obtained approximately 1 mg purified NS4B from 1 liter of IPTG induced cell culture. Furthermore, most reports did not analyze the immunoreactivity of the recombinant NS4B. To our knowledge, only Sillanpaa et al. (2009) analyzed the immunoreactivity of three structural and six non-structural HCV proteins including NS4B produced in Sf9 insect cells by using the baculovirus expression system (5). Based on western blot analyses, this study reported that NS4B was recognized in 86% of patient sera, which is consistent with 85% sensitivity obtained from ELISA here.
We have previously expressed and purified hepatitis C virus NS3 protease, NS5A protein and NS3 helicase fragment using directional TOPO vector pET102/D-TOPO (16,17).
In conclusion, the directional TOPO cloning provides an efficient and easy method for cloning and expression of recombinant HCV NS4B. The directional TOPO cloning should be evaluated for production of other viral proteins.
Acknowledgments
The authors would like to thank Dr. Keyvan from Tehran University of Medical Sciences, Department of Virology for providing HCV positive sera samples. We are also grateful to Mrs. N. Saemi for technical assistance. This work was supported by grant no. P-1087 of Tehran University of Medical Sciences.
References
- 1. Organization WH. Hepatitis C [updated April 2014]. Available from: http://www.who.int/mediacentre/factsheets/fs164/en/.
- 2.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet (London, England) 2012;380(9859):2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436(7053):933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 4.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5(6):453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 5.Sillanpaa M, Melen K, Porkka P, Fagerlund R, Nevalainen K, Lappalainen M. et al. Hepatitis C virus core, NS3, NS4B and NS5A are the major immunogenic proteins in humoral immunity in chronic HCV infection. Virol J. 2009;6:84. doi: 10.1186/1743-422X-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang JC, Seidel C, Ofenloch B, Jue DL, Fields HA, Khudyakov YE. Antigenic heterogeneity of the hepatitis C virus NS4 protein as modeled with synthetic peptides. Virology. 1999;257(1):177–190. doi: 10.1006/viro.1999.9612. [DOI] [PubMed] [Google Scholar]
- 7.Masalova OV, Lakina EI, Abdulmedzhidova AG, Atanadze SN, Semiletov YA, Shkurko TV. et al. Characterization of monoclonal antibodies and epitope mapping of the NS4 protein of hepatitis C virus. Immunol Lett. 2002;83(3):187–196. doi: 10.1016/S0165-2478(02)00081-0. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez-Lopez M, Riezu-Boj JI, Ruiz M, Berasain C, Civeira MP, Prieto J. et al. Immunogenicity of variable regions of hepatitis C virus proteins: selection and modification of peptide epitopes to assess hepatitis C virus genotypes by ELISA. J Gen Virol. 1999;80(Pt3):727–738. doi: 10.1099/0022-1317-80-3-727. [DOI] [PubMed] [Google Scholar]
- 9.Rai R, Deval J. New opportunities in anti-hepatitis C virus drug discovery: targeting NS4B. Antivir Res. 2011;90(2):93–101. doi: 10.1016/j.antiviral.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Dvory-Sobol H, Pang PS, Glenn JS. The Future of HCV Therapy: NS4B as an Antiviral Target. Viruses. 2010;2(11):2481–2492. doi: 10.3390/v2112481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hugle T, Fehrmann F, Bieck E, Kohara M, Krausslich HG, Rice CM. et al. The hepatitis C virus nonstructural protein 4B is an integral endoplasmic reticulum membrane protein. Virology. 2001;284(1):70–81. doi: 10.1006/viro.2001.0873. [DOI] [PubMed] [Google Scholar]
- 12.Gouttenoire J, Penin F, Moradpour D. Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev Med Virol. 2010;20(2):117–129. doi: 10.1002/rmv.640. [DOI] [PubMed] [Google Scholar]
- 13.Grakoui A, Wychowski C, Lin C, Feinstone SM, Rice CM. Expression and identification of hepatitis C virus polyprotein cleavage products. J Virol. 1993;67(3):1385–1395. doi: 10.1128/jvi.67.3.1385-1395.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirowatari Y, Hijikata M, Tanji Y, Shimotohno K. Expression and processing of putative nonstructural proteins of hepatitis C virus in insect cells using baculovirus vector. Virus Res. 1995;35(1):43–61. doi: 10.1016/0168-1702(94)00078-Q. [DOI] [PubMed] [Google Scholar]
- 15.Thompson AA, Zou A, Yan J, Duggal R, Hao W, Molina D. et al. Biochemical characterization of recombinant hepatitis C virus nonstructural protein 4B: evidence for ATP/GTP hydrolysis and adenylate kinase activity. Biochemistry. 2009;48(5):906–916. doi: 10.1021/bi801747p. [DOI] [PubMed] [Google Scholar]