

Abstract
Diacylglycerol (DAG) is a fusogenic lipid that can be produced through phospholipase C activity on phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2], or through phosphatidic acid (PA) phosphatase activity. The fusion of Saccharomyces cerevisiae vacuoles requires DAG, PA and PI(4,5)P2, and the production of these lipids is thought to provide temporally specific stoichiometries that are critical for each stage of fusion. Furthermore, DAG and PA can be interconverted by the DAG kinase Dgk1 and the PA phosphatase Pah1. Previously we found that pah1Δ vacuoles were fragmented, blocked in SNARE priming and showed arrested endosomal maturation. In other pathways the effects of deleting PAH1 can be compensated for by additionally deleting DGK1, however deleting both genes did not rescue the pah1Δ vacuolar defects. Deleting DGK1 alone caused a marked increase in vacuole fusion that was attributed to elevated DAG levels. This was accompanied by a gain in resistance to the inhibitory effects of PA as well as inhibitors of Ypt7 activity. Together these data show that Dgk1 function can act as a negative regulator of vacuole fusion through the production of PA at the cost of depleting DAG and reducing Ypt7 activity.
Keywords: Diacylglycerol, Phosphatidic acid, Lipin 1, SNARE, Pah1, Membrane Fusion, Ypt7, Rab7, Vam7, Dgk1, Yck3, Mon1
INTRODUCTION
The movement of vesicles in eukaryotic cells is tightly regulated to prevent the missorting of cargo. Each route of membrane trafficking in the endocytic and secretory pathways is regulated by homologous sets of proteins that are conserved throughout eukarya. The final stage of cargo delivery occurs through the merger of two distinct membranes in a fusion reaction catalyzed by SNARE proteins.1 The fusion reaction pathway can be divided into stages that start with the disassembly of inactive cis-SNARE bundles. This process, known as priming, is executed by the SNARE chaperone NSF/Sec18 and its adaptor protein α-SNAP/Sec17.2 Membranes are subsequently brought into contact by the activity of Rab GTPases and their cognate tethering effectors.3 In Saccharomyces cerevisiae, vacuolar lysosomes undergo fusion with vesicles from the endocytic pathway, AP-3 coated membranes from the Golgi, autophagosomes, as well as with other vacuoles.4–7 Homotypic vacuole fusion requires the Rab Ypt7 and its effector complex HOPS for tethering.8,9 During docking, trans-SNARE complexes form between partner membranes leading to the release of luminal Ca2+ stores prior to fusion.10 Docked vacuoles become tightly apposed forming a flattened region where they are in contact that is referred to as the boundary domain. The proteins and lipids that drive fusion become enriched at the edge of the boundary termed the vertex ring microdomain.11–13 Fusion occurs around the vertex ring leading to the internalization of the boundary membrane and degradation of boundary-localized factors. DAG becomes enriched at the both the vertex and the boundary membrane leading to the potential degradation of this lipid after fusion occurs.
The regulation of membrane trafficking and fusion also involves a group of signaling lipids that play multiple roles in controlling these pathways. Yeast vacuole fusion requires diacylglycerol (DAG), phosphatidic acid (PA), as well as multiple phosphoinositides (PI) and ergosterol.11,14–20 During the docking stage of fusion, these lipids accumulate into membrane microdomains that organize the SNAREs, Ypt7, HOPS and actin at the site of fusion.11,16 The stoichiometry of these lipids is not static and requires the function of multiple lipid kinases, phosphatases and lipases at different time points to modify lipids to activate (or inactivate) their roles at each fusion stage. For instance, the PI 3-kinase Vps34 produces PI3P allowing the binding of Mon1, HOPS, and the soluble SNARE Vam7 leading to membrane tethering and docking.18,20–23 Similarly, the PA phosphatase Pah1 converts PA to DAG leading to the transfer of PA-bound Sec18 to cis-SNARE complexes for priming.19,24
Pah1 is the yeast Lipin1 orthologue and its activity is required for endoplasmic reticulum and nuclear envelope homeostasis as well as lipid drop formation.25,26 In addition, we have found that Pah1 activity is required for endosomal maturation. Deletion of Pah1 leads to the exclusion of Vps34, Mon1, Ypt7 and Vps39 from vacuolar membranes causing vacuole fragmentation and abolished fusion.18,19 Others have shown that the activity of Pah1 can be countered by the DAG kinase of Dgk1 to replenish PA.27,28 The morphological defect of the endoplasmic reticulum and nuclear envelope seen in pah1Δ cells is reversed by the concomitant deletion of DGK1, illustrating the effects of interconverting PA and DAG on organelle homeostasis. DAG produced by Pah1 is used for the synthesis of triacylglycerol and the formation of lipid droplets as well as feeding into the Kennedy pathway for the production of phosphatidylethanolamine and phosphatidylcholine.25,29,30 DAG is a fusogenic lipid due to the induction of negative curvature and destabilization of lipid bilayers.31,32 The fusogenicity of DAG is further demonstrated by its ability to promote the fusion of protein-free liposomes.33,34 These features are linked to its requirement for efficient vacuole fusion.
In this study we examined the role of Dgk1 in vacuole fusion. We first determined that deleting DGK1 in pah1Δ background strains did not restore the blocked endosomal maturation, fusogenicity, or the vacuole fragmentation phenotype caused by deleting PAH1 alone. This indicates that Pah1 and Dgk1 functions are not concurrent in vacuole homeostasis unlike their offsetting activities in other pathways. Importantly, the single dgk1Δ mutation led to an augmented fusion phenotype. We focused on finding the mechanism for the increased fusion of dgk1Δ vacuoles. These vacuoles contained elevated levels of the fusogenic lipid DAG and displayed an enhanced fusion phenotype. This was further characterized with in-creased resistance to the inhibitory effects of PA and elevated Ypt7-mediated vesicle tethering.
RESULTS
Vacuole morphology and Dgk1 localization
Vacuole fusion requires the production and turnover of numerous signaling lipids including PA and DAG.11,15,19 Multiple pathways feed into the production and interconversion of these lipids. DAG can be produced by the activity of Plc1 on PI(4,5)P2 35 or Pah1 on PA 26. PA can be produced by phospholipase D activity on PC or by Dgk1 on DAG.27,36 The production of DAG during vacuole fusion is important due to its fusogenic properties.37,38 DAG becomes enriched at the vertex ring and boundary membrane of docked vacuoles and regulates localization of SNAREs, Ypt7 and HOPS to the site of fusion.
We previously found that deletion or inactivation of PAH1 inhibited endosomal maturation that was characterized by vacuoles that lacked the late endosomal/lysosomal Rab Ypt7, the HOPS subunit Vps39, as well as the PI 3-kinase Vps34 and its product PI3P.19 The lack of Ypt7 was due to the exclusion of its guanine exchange factor Mon1-Ccz1, which is recruited to membranes in part through its interactions with PI3P.18,21 Because deletion of DGK1 restores the effects of deleting PAH1 in other pathways we began by testing whether the double deletion would restore the pah1Δ vacuolar defects. We first examined whether the double deletion would restore pah1Δ vacuole morphology. We found that dgk1Δ cells exhibited wild type vacuole morphology (Fig. 1A). Curiously, the dgk1Δ pah1Δ double deletion cells were fragmented in a manner similar to pah1Δ cells. Thus, deleting both enzymes did not rescue the pah1Δ vacuole fragmentation phenotype or blocked fusion. This was not in keeping with the effects seen in restoring nuclear/endoplasmic reticulum morphology in other pathways. Quantitation of vacuole fragmentation is shown in Figure 1B.
Figure 1. Dgk1 is localized to the vacuole.

(A) Vacuole morphology was examined using wild type, dgk1Δ, pah1Δ, and dgk1Δ pah1Δ yeast. Cells were incubated with 5 μM FM4-64 to label vacuoles. (B) Quantitation of vacuole fragmentation. The number of vacuoles per cell was determined for each strain (n>300 cells per strain). Cells with 8 or more vacuoles were combined into one population. (C) Dgk1-GFP expression in wild type and pah1Δ strains were double labeled with FM4-64 and used for fluorescence microscopy. (D) Immunoblotting of Dgk1-HA3 distribution in 10 μg samples of whole cell lysates, cytosol, vacuoles, endoplasmic reticulum and total membrane fraction using anti-HA antibody. Ypt7 and Sec61 were probed for as a controls for vacuole or endoplasmic reticulum enrichment. (E) Western blot analysis. Shown is the vacuolar enrichment of Dgk1 on vacuoles relative to contaminating membranes from the endoplasmic reticulum. Using densitometry, the relative amount of vacuolar Dgk1 was measured as a ratio to the amount of Sec61 in the vacuolar preparation and set to 1. The Dgk1/Sec61 ratio was also determined for the microsomal fraction in comparison to the vacuolar ratio. Densitometry was determined using BioRad ChimiDoc XRS+ System. Bar, 5μm.
Due to the lack of an effect of deleting DGK1 on the pah1Δ vacuole morphology phenotype we examined if Dgk1 was mislocalized in mutant cells. First, we visualized the distribution of Dgk1-GFP in wild type whole cells. Figure 1C shows widefield fluorescent images showing Dgk1-GFP localization in cells stained with FM4-64 to label vacuoles. Dgk1-GFP localized to ribbon-like structures of the endoplasmic reticulum similar to what has been previously reported.39 We also observed that the Dgk1-GFP ribbons were often adjacent to vacuoles and a fraction of Dgk1-GFP colocalized with FM4-64 suggesting that aside from the endoplasmic reticulum, vacuoles also harbor a portion of the enzyme. Dgk1-GFP localization was next examined in pah1Δ cells and we did not detect any appreciable differences in it localization relative to the wild type parent strain. Because some Dgk1-GFP appeared to colocalize with vacuoles we further examined Dgk1 distribution by cell fractionation and immunoblotting. Yeast cells expressing Dgk1-HA were fractionated into crude lysate, cytosol, vacuoles, a total membrane pool as well as microsomes containing endoplasmic reticulum. The blots showed that Dgk1-HA was present in microsomes and vacuoles (Fig. 1D). As a control, we probed for Dgk1-HA in the cytosolic fraction and found that this polytopic protein was absent. Ypt7 served as an enrichment marker for vacuoles. Sec61 served as a marker for resident endoplasmic reticulum components. Sec61 was detected in the vacuole fraction indicating that there was some contamination from the endoplasmic reticulum. Although this accounts for the majority of the Dgk1 detected in the vacuole fraction, it should also be noted that the ratio of Dgk1 to Sec61 is greater on vacuoles relative to the microsome fraction (Fig. 1E). Together with the Dgk1-GFP localization it is apparent that a subpopulation of Dgk1 does indeed reside on the vacuolar membrane.
Cells lacking DGK1 accumulate vacuolar DAG
To determine if deleting DGK1 would have an effect on vacuolar DAG levels we quantitated this lipid on vacuoles from wild type and dgk1Δ cells. Neutral lipids were extracted from wild type and dgk1Δ vacuoles and resolved by thin-layer chromatography as described in the Materials and Methods section. Purified DAG was used to generate a standard curve in the first six lanes of the plate (Fig. 2A). Three of the five trials run were loaded onto the plate shown. We found that dgk1Δ vacuoles consistently contained more DAG than wild type vacuoles. Figure 2B shows quantitation of five trials (p<0.05). The DAG content of wild type and dgk1Δ vacuoles was calculated using a standard curve of pure DAG and plotted as μg of DAG per 150 μg of vacuole protein. These data indicate that vacuoles will accumulate DAG in the absence of Dgk1. However, this does not indicate whether the difference in DAG content is due to Dgk1 activity on the vacuole or whether the lack of DAG kinase activity elsewhere affects the total amount of this lipid trafficked to vacuoles.
Figure 2. DAG vacuolar content.
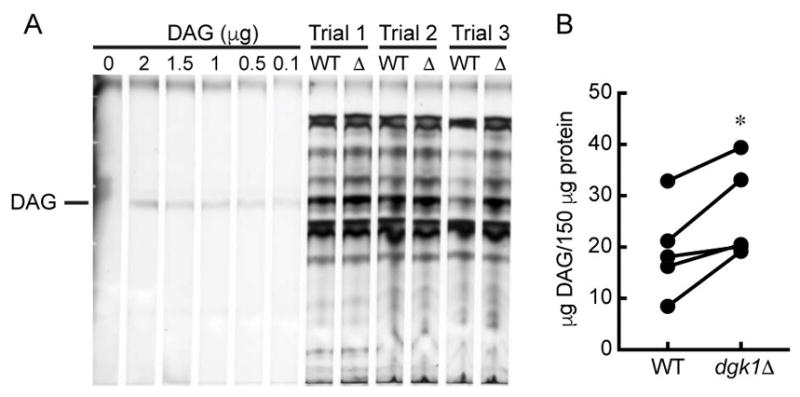
(A) Neutral lipids were extracted from wild type and dgk1Δ vacuoles and resolved by thin layer chromatography. Lipids were visualized by charring with copper (II) sulfate. (B) Quantitation of DAG from wild type and dgk1Δ vacuoles (150 μg of by protein). * P<0.05 (two-tailed paired t-test). n=5.
The absence of Dgk1 augments vacuole fusion
To further examine the consequence of deleting DGK1 on vacuole homeostasis we tested fusion efficiency. These experiments showed that dgk1Δ vacuole fusion was markedly increased by ~40% relative to wild type (Fig. 3A), suggesting that the conversion of DAG to PA by Dgk1 negatively regulates fusion. This is in keeping with the notion that DAG is fusogenic and that Dgk1 activity is needed to reduce unchecked fusion. We next asked if the rate of fusion itself was increased for dgk1Δ vacuoles. The slope of the fusion curve for dgk1Δ vacuoles appeared to be steeper in comparison to wild type. However, this appearance could be due to normalizing fusion to the wild type maximum. While this shows the overall increase in total fusion, it does not clearly show the initial rate of fusion. To better visualize the rate of dgk1Δ vacuole fusion we replotted the data as normalized to its own maximal fusion at the end of 90 min (Fig. 3A, red circles). This approach showed that the adjusted dgk1Δ fusion curve perfectly overlapped with that of wild type vacuoles, indicating that the rates of fusion were identical and that the augmented fusion of dgk1Δ vacuoles was due to other factors. We also asked whether the augmented fusion occurred during different rounds of fusion. To this aim we used a method shown to differentiate the first round of fusion from later events.40 Previously we used this method to determine that deleting NHX1 only affected the first round of fusion without affecting subsequent fusion cycles.41 To detect the first round of fusion, a 5:1 excess of effector vacuoles (PEP4 pho8Δ) was used. For later rounds of fusion reactions used a 4:2 excess of reporter vacuoles (pep4Δ PHO8). Using this method we found that dgk1Δ vacuoles showed enhanced fusion for both early and late rounds of fusion (Fig. 3B), suggesting that the effects of deleting DGK1 was consistent.
Figure 3. Dgk1 suppresses vacuole fusion.

(A) Fusion reactions were performed using wild type and dgk1Δ vacuoles. Reactions were incubated at 27°C for the indicated times and assayed for Pho8 activity. The red trace represents dgk1Δ fusion when normalized to its own maximum. This reveals the rate of dgk1Δ vacuole fusion versus wild type. (B) The ratio of effector and reporter vacuoles were altered to show early versus late rounds of fusion. To show early rounds of fusion an excess of effector vacuoles (5 μg PEP4 pho8Δ) were incubated with 1 μg of reporter vacuoles (pep4Δ PHO8). To show late rounds of fusion, an excess of reporter vacuoles (4 μg pep4Δ PHO8) were incubated with 2 μg of effector vacuoles (PEP4 pho8Δ). (C) Lipid mixing assay were performed with wild type or dgk1Δ vacuoles. Reporter vacuoles (2 μg) labeled with Rh-PE were incubated with 16 μg of unlabeled vacuoles. Fluorescence (λex=544 nm; λem=590 nm) was measured every 60 sec for 40 min. Shown is the average lipid mixing at 40 min. (D) Measurement of vacuole diameters after incubation (27°C, 90 min). Fusion reactions were treated with PS buffer, 12 μg/ml anti-Sec18, or 2 mM propranolol (Prop.). Measurements were plotted as a box plot representing median vacuoles with upper and lower quartile values. The lines extending from the boxes represent the minimum and maximum vacuoles in the data set. Outliers are displayed as points. (E–F) Fusion assays were performed with vacuoles from wild type, dgk1Δ, or dgk1Δ strains complemented with pDGK1 or pDGK1D177A. (G) Endpoint fusion efficiency was compared between wild type, dgk1Δ, pah1Δ, and pah1Δ dgk1Δ vacuoles. (H) Analysis of fusion components. Vacuoles were isolated from wild type BJ3505 and RFY17 (BJ3505 dgk1Δ) (5 μg protein) and immunoblotted with antibodies against the indicated proteins. To examine the effect of deleting DGK1 on Pep4 trafficking, vacuoles were examined from DKY6281 and RFY18 (DKY6281 dgk1Δ). Error bars represent S.E.M. (n≥3). *p<0.05, **p<0.01 (one-way ANOVA).
Although we have shown that the rate of full fusion and content mixing of dgk1Δ vacuoles matches that of wild type, it was possible that the elevated levels of DAG could destabilize the bilayers enough to promote faster hemifusion. Thus, we next measured the mixing of outer leaflet lipids as a reporter for hemifusion.16,42 Here, a population of vacuoles is loaded with rhodamine conjugated PE (Rh-PE) at levels that quench fluorescence. Rh-PE labeled vacuoles are then incubated with an 8-fold excess of unlabeled vacuoles. Upon lipid mixing of labeled and unlabeled vacuoles the Rh-PE is diluted and the fluorescence is de-quenched. This approach showed that dgk1Δ vacuoles underwent lipid mixing at the same rate versus wild type (Fig. 3C). Also shown are the negative controls of excluding ATP or blocking priming with anti-Sec17 antibody. These data suggest that the fusion phenotype of dgk1Δ vacuoles was not due to an increased rate of fusion of a set amount of vacuoles but perhaps is a refection that more vacuoles are initially participating in fusion.
Because changes in fusion, as a measure of Pho8 activity, could theoretically be affected indirectly by altered activity of the reporter enzyme itself, we verified that the increased fusion of dgk1Δ vacuoles was real by performing a visual assay to measure changes in vacuole diameter.40,43 Here, vacuole reactions were treated with buffer, anti-Sec18 IgG or propranolol. Both inhibitors potently inhibit fusion at the priming stage.2,19 Propranolol is a β-adrenergic receptor blocker that also inhibits PA phosphatase activity, which is required for Sec18-mediated SNARE priming to occur.24,44 After incubation (90 min, 27°C), fusion reactions were placed on ice and stained with FM4-64 for fluorescence microscopy evaluation. In accord with the content mixing assay, we found that the diameters of dgk1Δ vacuoles were significantly larger relative to wild type vacuoles (Fig. 3D, p<0.05). Incubating reactions on ice as well as treatment with anti-Sec18 or propranolol blocked the increase in vacuole diameter demonstrating that changes in vacuole size were due to fusion.
To determine whether the kinase activity of Dgk1 was linked to the fusion enhancement, dgk1Δ strains were complemented with plasmids encoding either wild type DGK or the inactivated kinase mutant DGKD177A.28 Complementation with pDGK1 partially restored fusion to near wild type levels (Fig. 3E). The lack of full wild type fusion restoration with complementation was thought to be due to plasmid loss during growth in non-selective medium for vacuole purification. Nevertheless, the difference in fusion between the dgk1Δ and complemented vacuoles was statistically significant (p<0.05). Moreover, the difference between wild type fusion and the complement was not significant (p=0.25). Complementation with the kinase-dead mutant pDGKD177A did not reverse the increased fusion seen with dgk1Δ vacuoles (Fig. 3F). These data demonstrate that the altered fusion seen with dgk1Δ vacuoles was directly due to the kinase activity of Dgk1.
In Figure 1A–B we showed that the dgk1Δ pah1Δ double deletion did not restore the vacuole fragmentation phenotype of pah1Δ alone. To further verify effect of deleting DGK1 on pah1Δ vacuole function we tested the mutations in content mixing assays. We found that the dgk1Δ pah1Δ double deletion strains exhibited attenuated fusion similar to what occurs with the single PAH1 deletion (Fig. 3G). As with the morphological examination of the double deletion strain, we concluded that the activities of these enzymes do not directly counterbalance each other with respect to vacuole homeostasis and fusion. For this reason we focused on the augmented fusion of dgk1Δ vacuoles for the remainder of the study.
Changes in fusion could be attributed to a variety of factors including changes in the concentration of regulatory proteins such as SNAREs. Others have shown that increased SNARE levels can enhance vacuole fusion.45 Deletion of the ABC transporter Ybt1 and the casein kinase Yck3 has also been shown to increase vacuole fusion through distinct mechanisms.43,46 To assess if the elevated fusion of dgk1Δ vacuoles was due to changes in the fusion machinery we performed immunoblotting analysis of known regulators. No differences were observed between dgk1Δ vacuoles and wild type (Fig. 3H). Taken together we conclude that Dgk1 activity suppresses maximal vacuole fusion and that the increased fusion seen with dgk1Δ vacuoles was not due to a fault in the reporter systems or caused by increased levels of the core fusion machinery proteins.
DGK1 deletion does not alter SNARE complex formation
Vacuole fusion can be augmented by the increase in the copies of SNAREs or by the improved efficiency of complex formation.16,45 As seen in Figure 3H, the amount of individual SNAREs was unaffected by deleting DGK1, however, their effectiveness is not apparent by immunoblotting alone. Thus we examined the efficiency of SNARE complex formation. This assay relies on blocking priming with anti-Sec17 IgG and its bypass by the addition of recombinant GST-Vam7 to restore fusion.47,48 First, we determined if the dosage required for the bypass was different for dgk1Δ vacuoles relative to wild type. Figure 4A shows the restoration of fusion upon addition of GST-Vam7 at the indicated concentrations. This showed that dgk1Δ vacuoles responded to the addition of GST-Vam7 at the same peak of efficacy compared to wild type.
Figure 4. SNARE complex formation and Ca2+ efflux are not affected on dgk1Δ vacuoles.

(A) Anti-Sec17 IgG bypass fusion. Fusion reactions were treated with anti-Sec17 IgG to block the priming stage of fusion. Recombinant GST-Vam7 was added at the indicated concentrations to stimulate fusion. (B) SNARE complex formation as described in the Materials and Methods section. (C) Quantitation of three GST-Vam7 pulldown assays. Protein band intensities were normalized to wild type at 27°C without inhibitors. (n=3). (D) Fusion reactions (2X) were prepared with 20 μg of wild type or dgk1Δ vacuoles in the presence of 150 nM low affinity Fluo-4 dextran in the presence or absence of ATP. A subset of reactions was treated with 2 μM Gyp1-46 to inhibit Ypt7 activity and block Ca2+ efflux. Fluo-4 fluorescence was normalized to the average fluorescence of wild type vacuoles without ATP. Error bars represent S.E.M. (n=3).
Next, we performed Vam7 bypass experiments to examine the formation of SNARE complexes and HOPS binding. We found that GST-Vam7 interacted with its cognate SNAREs Nyv1 and Vam3 with equal efficiency on either wild type or dgk1Δ vacuoles (Fig. 4B–C). Hence, it appears that the altered fusion seen with dgk1Δ vacuoles was not due to increased SNARE complex formation. To verify that SNARE complexes were similarly active on both vacuole types we examined the efflux of luminal Ca2+ stores that are released upon trans-SNARE complex formation.10 Previously we have found that deletion of the ABC transporter Ybt1 strikingly delayed the kinetics of Ca2+ efflux, which was linked to increased vacuole fusion.43 However, there was no difference in the kinetics or amplitude of Ca2+ release between dgk1Δ and wild type vacuoles (Fig. 4D). Together these data indicated that increased fusion of dgk1Δ vacuoles was not directly due to altered SNARE complex formation.
Deletion of DGK1 confers resistance to PA
Previous studies showed that reducing, or blocking, DAG inhibited the enrichment of regulatory lipids at the vertex ring assembly and blocked fusion.11,15 We next asked if increasing DAG levels through the deletion of DGK1 would alter the role of regulatory lipids in fusion. To this aim we examined the effects of blocking regulatory signaling lipids on dgk1Δ vacuoles. We found that blocking the function of PI3P, PI4P, PI(4,5)P2, and ergosterol with the ligands FYVE, FappPH, ENTH, and filipin respectively inhibited dgk1Δ vacuole fusion indistinguishably from wild type (Fig. 5A). There was also no difference when PI3P and PI(4,5)P2 were modified by the phosphatases MTM-1 and SigD, respectively. This indicated that the enhanced fusion phenotype was not due to additional changes in the core fusion machinery or due to off pathway mechanisms. Because Dgk1 activity altered DAG levels on isolated vacuoles we next tested if dgk1Δ vacuoles were less sensitive to the DAG ligand C1b. We found that C1b inhibited dgk1Δ vacuole fusion at the same concentration needed to block wild type fusion (Fig. 5B). This suggests that the change in DAG does not directly affect a protein-lipid interaction such as the binding of Protein kinase C (PKC) to DAG on other membranes.49 Instead it is likely that the augmented fusion is a result of an indirect effect such as changes in membrane curvature and fluidity. It is also possible that a population of DAG was inaccessible to C1b by accumulating in the boundary membrane or by translocating across the bilayer.11,50
Figure 5. Deletion of DGK1 confers resistance to PA.
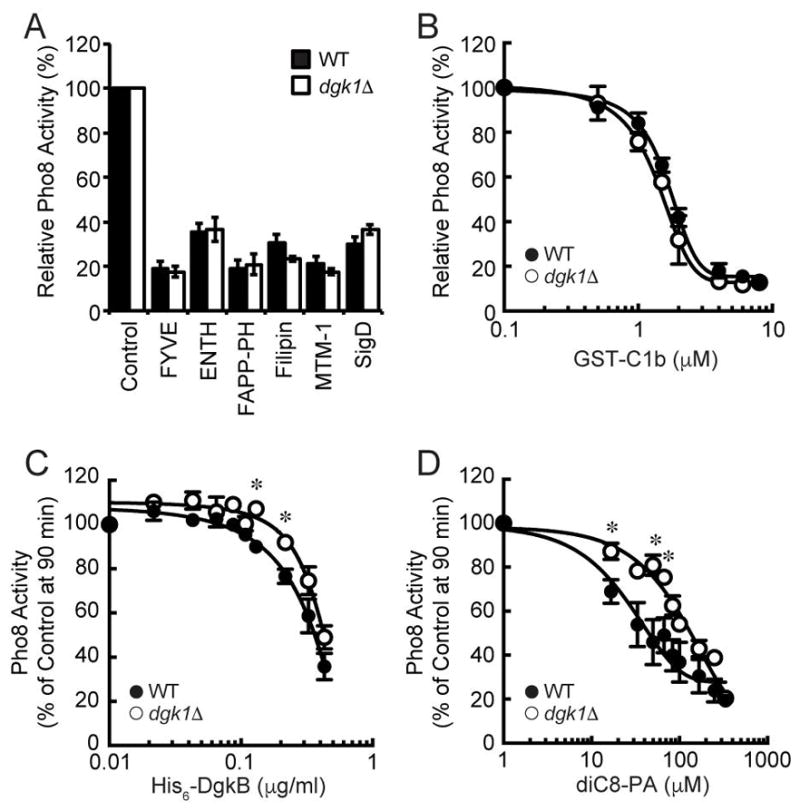
(A) Fusion reactions were performed using wild type or dgk1Δ vacuoles. Fusion reactions containing wild type or dgk1Δ vacuoles treated with PS buffer, 2 μM GST-FYVE, 20 μM GST-ENTH, 10 μM MBP-FappPH, 20 μM Filipin, 1 μM His6-MTM1 or 1.2 μM SigD. (B–D) Dose response curves of GST-C1b, His6-DgkB and diC8-PA at the indicated concentrations. Error bars represent S.E.M. (n=3). * p<0.05 (one-way ANOVA).
As is now evident, the conversion of PA to DAG by Pah1 is necessary for vacuolar SNARE priming.24 Increasing PA concentrations through inactivation of Pah1 or through the addition of short-chain PA (diC8-PA) potently blocked fusion by sequestering Sec18 away from SNAREs in a PA-dependent manner. Thus, we asked if the increase in DAG on dgk1Δ vacuoles would shift the DAG:PA ratio in a way that would alter the sensitivity of fusion towards PA. To answer this we tested the effect of converting DAG to PA with recombinant His6-DgkB, the soluble DAG kinase from Staphylococcus aureus.51 We found that DgkB potently inhibited wild type vacuole fusion as well as fusion by dgk1Δ vacuoles (Fig. 5C). However, there was a right-shift in the inhibition curves for dgk1Δ vacuole fusion that was statistically significant at 130 and 220 μg/ml (p<0.05). Although the shift was not large it was consistent with needing more DgkB for the elevated level of DAG at the beginning of the reaction.
To further test the effects of increasing PA on fusion we added diC8-PA to fusion assays. As previously shown, the addition of diC8-PA had a marked inhibitory effect on the fusion of wild type vacuoles (Fig. 5D). Importantly, the inhibitory effect of excess PA was clearly reduced with dgk1Δ vacuoles. The IC50 of diC8-PA on wild type vacuoles was 31.2±5.1 μM whereas the IC50 was 122.8±15.2 μM for dgk1Δ vacuoles (*p<0.05). The increased resistance of dgk1Δ vacuole fusion to diC8-PA suggests that the elevated levels of DAG lead to a requirement of more diC8-PA to fully inhibit fusion. Together these data indicate that the ratio of DAG to PA is a critical component of regulating vacuole fusion.
Augmented dgk1Δ vacuole fusion is linked to enhanced Ypt7 activity
To test if the augmented fusion of dgk1Δ vacuoles was on pathway and regulated by the known machinery, we tested a panel of inhibitors that targeted the proteins that regulate vacuole fusion. Figure 6A shows that antibodies against the SNAREs Nyv1, Vam3, or the HOPS subunit Vps33 inhibited dgk1Δ vacuole fusion with similar effects compared to wild type. The relative fusion endpoints for both wild type and dgk1Δ vacuoles were normalized to 100% (Fig. 6A). Interestingly, dgk1Δ vacuole fusion showed reduced sensitivity to anti-Ypt7 IgG. To further examine the change in sensitivity of dgk1Δ vacuoles to anti-Ypt7 we performed dose response assays to calculate the IC50. This showed that dgk1Δ fusion was significantly less sensitive to anti-Ypt7 with an IC50 of 124.9Δ10.7 μg/ml while the IC50 for wild type was 61.2±8.1 μg/ml (Fig. 6B, *p<0.05). This suggests that Ypt7 could be more active in dgk1Δ vacuoles. It is also possible that changes in the membrane curvature and fluidity could allosterically induce conformational changes in Ypt7 leading to decreased binding by the antibody. Based on the effects of anti-Ypt7 IgG we also examined the efficacy of the GTPase activating protein Gyp1 and the Rab-GDP dissociation inhibitor GDI. We found that both proteins had right-shifted inhibition curves with dgk1Δ vacuole fusion. Figure 6C shows that dgk1Δ vacuole were less sensitive to Gyp1-46 with an IC50 of 2.17±0.06 μM compared to wild type (IC50 = 1.54±0.046 μM, *p<0.05). Similarly, dgk1Δ vacuole fusion was more resistant to GDI versus wild type. The IC50 for GDI with dgk1Δ vacuoles was 112.7±8.7 nM, while the IC50 with wild type was 209.2±14.1 nM (Fig. 6D, *p<0.05). Together these data suggest that Ypt7 function is enhanced in dgk1Δ vacuoles. That said, these assays do not distinguish whether dgk1Δ vacuoles have increased activity per individual Ypt7 molecule or if there are simply more numerous activated Ypt7 copies per vacuole. Alternatively, it is possible the dgk1Δ require less Ypt7 to promote fusion.
Figure 6. The augmented fusion of dgk1Δ vacuoles is linked to Ypt7 function.
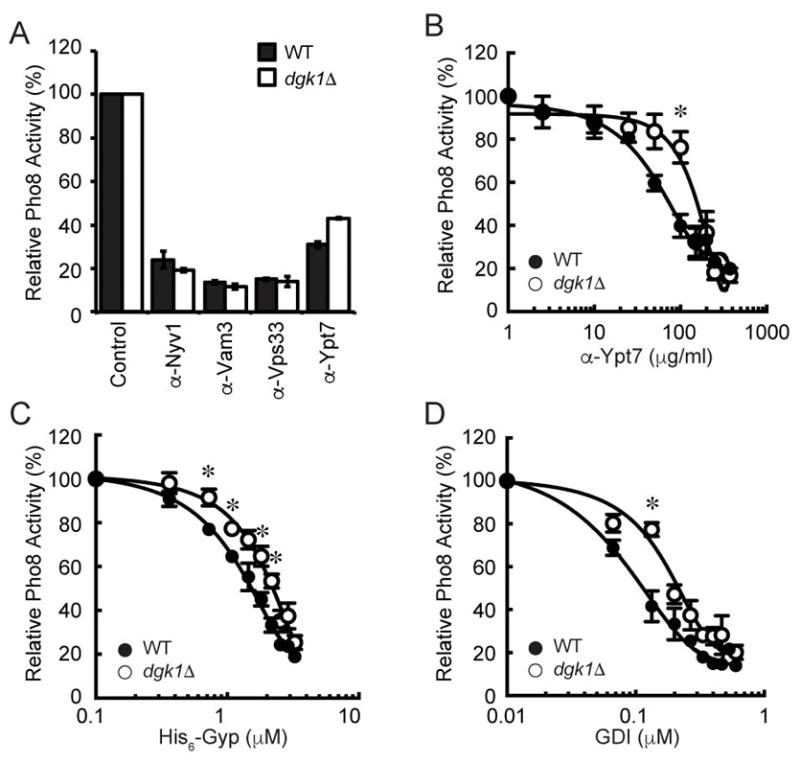
(A) Fusion reactions were performed using wild type or dgk1Δ vacuoles. Individually, reactions were treated with PS buffer (Control), or antibodies against Nyv1 (17 μg/ml), Vam3 (27 μg/ml), Vps33 (5 μg/ml) or Ypt7 (8 μg/ml). (B–D) Dose response curves of anti-Ypt7 IgG (B), Gyp1-46 (C), and GDI (D). Reactions containing wild type of dgk1Δ vacuoles were incubated for 90 min at 27°C and tested for fusion. Error bars represent S.E.M. (n=3). *P<0.05 (one-way ANOVA).
The dgk1Δ fusion effect is in parallel to the function of Yck3
In addition to the effect of deleting DGK1 on Ypt7 function shown here, Ungermann et al. found that deleting the type I casein kinase Yck3 resulted in fusion that was resistant to Gdi1 and Gyp7-47.52 This was linked to augmented vacuole fusion. Yck3 activity reduces fusion by phosphorylating Vps41, Vam3 and Mon1.18,52,53 The similar phenotypes of elevated fusion and increased Ypt7 activity in both yck3Δ and dgk1Δ vacuoles led us to ask if they shared a common pathway. We previously reported that the Ypt7 GEF Mon1-Ccz1 was phosphorylated by the casein kinase Yck3 leading to its release from the membrane.18 Deletion of YCK3 or mutation of Mon1 phosphorylation sites prevented Mon1 from being released from the membrane during fusion. The influence of Yck3 on fusion is further illustrated through the inhibitory activity of adding exogenous recombinant His6-Yck3.46 Together, the data suggest that Dgk1 and Yck3 lie on the same pathway of regulating vacuole fusion.
The experiments in Figure 7 begin addressing the connection between Dgk1 and Yck3. First, we immunoblotted for Yck3 on wild type and dgk1Δ vacuoles and found that the kinase was present at equal levels on membranes from both strains (Fig. 7A–B). This shows that Yck3 does not require Dgk1 for its recruitment to vacuoles. Next we examined if Yck3-dependent Mon1 release occurred on dgk1Δ vacuoles. Figure 7C shows that Mon1 was released at the same rate on both sets of vacuoles, indicating that Yck3 function was normal on dgk1Δ membranes. Figure 7D shows quantitation of Mon1 retention over time. Although Mon1 release was not affected on dgk1Δ we went on to test if the addition of recombinant Yck3 would differentially affect the two types of vacuoles. We found that added His6-Yck3 inhibited the fusion of wild type and dgk1Δ vacuoles with identical dose responses (Fig. 7E). Thus far, it appears that Yck3 and Dgk1 functions are likely in parallel pathways. To further determine if the underlying mechanisms of the dgk1Δ and yck3Δ fusion phenotypes are linked, we constructed yck3Δ dgk1Δ double deletion strains. We hypothesized that if the two functions were in parallel we would observed additive increases in vacuole fusion relative to either yck3Δ or dgk1Δ single deletions. The lack of further augmented fusion would suggest that the Dgk1 and Yck3 pathways overlap. We found that both yck3Δ and dgk1Δ single deletions had enhanced fusion as seen previously. Unexpectedly, the double yck3Δ dgk1Δ double deletion fused to a similar extent the wild type parent strains (Fig. 7F, p>0.05). The loss of augmented fusion suggests that Yck3 and Dgk1 function in different pathways. Although evidence is lacking, it is not unlikely that the altered function of Ypt7 due to the lack of Dgk1 was negatively affected by the retention of unphosphorylated Mon1 due to the lack of Yck3 resulting in a return to normal overall function. That said, further studies would be required to explore this possibility, which go beyond the scope of this study.
Figure 7. The dgk1Δ fusion effect is in parallel to the function of Yck3.

(A–B) Immunoblotting and quantitation of Yck3 content on vacuoles from wild type and dgk1Δ cells. (C) Immunoblotting of Mon1 release from vacuoles during the fusion reaction. Fusion reactions were incubated for 0, 30, or 60 min after which membranes (bound) and supernatants (unbound) were separated by centrifugation (13,000 g, 15 min, 4°C). (D) Quantitation of bound Mon1 at the indicated times. (E) Fusion reactions containing were treated with indicated concentrations of recombinant His6-Yck3. (F) Fusion of wild type, dgk1Δ, yck3Δ and dgk1Δ/yck3Δ vacuoles. Fusion reactions were incubated for 90 min at 27°C. Error bars represent S.E.M. (n=3).
Exogenously added DAG enhances fusion of wild type vacuoles
Because dgk1Δ vacuoles contain increased levels of DAG, we next asked if adding the lipid exogenously to wild type vacuoles would alter fusion. Previously we found that while vacuole fusion was inhibited with short chain phosphatidic acid (diC8-PA), fusion was not affected by diC8-DAG.24 The effect of diC8-PA was attributed to the interaction of the PA headgroup with Sec18 to prevent SNARE priming. DAG on the other hand is directly fusogenic and transmits its effects on membranes through its acyl chains. Thus, the lack of an effect by diC8-DAG was attributed to the inability of short acyl chains to influence membrane curvature or bilayer stability. To better determine the effect of exogenous addition of this lipid we used long chain (16:0, 18:1) DAG to wild type and dgk1Δ vacuole fusion reactions. We found that adding DAG enhanced the fusion wild type vacuoles by ~17% at 5 nM while it had no effect on dgk1Δ vacuoles (Fig. 8A, *p<0.05). Higher levels of DAG did not further enhance fusion (not shown). Although the addition of DAG did not fully mimic the effect of deleting DGK1, it does support the notion that DAG levels alone can modulate the efficacy of fusion. Similarly, the addition of 5 nM DAG increased lipid mixing by approximately 20% (Fig. 8B, **p<0.01).
Figure 8. Fusion of dgk1Δ vacuoles and resistance to diC8-PA.
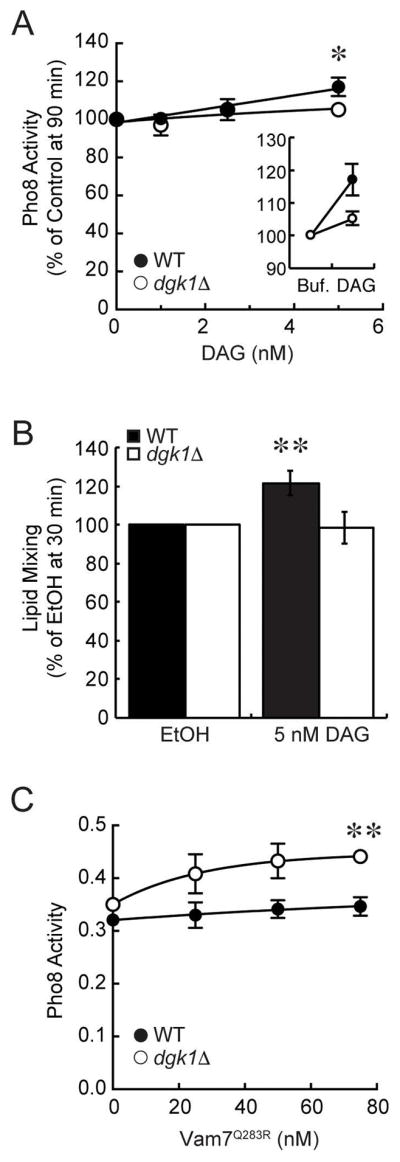
(A) Exogenous addition of DAG at the indicated concentrations to fusion reactions containing wild type or dgk1Δ vacuoles. Inset shows the direct comparison of 5 nM DAG and buffer with an equivalent volume of carrier (EtOH). (B) Lipid mixing using wild type and dgk1Δ vacuoles in the presence of 5 nM DAG or buffer with an equivalent volume of EtOH. (C) Anti-Sec17 IgG bypass fusion reactions using wild type or dgk1Δ vacuoles. Individual fusion reactions were treated with recombinant GST-Vam7Q283R to stimulate fusion. Fusion reactions were incubated for 90 min at 27°C. Error bars rep-resent S.E.M. (n=3). *p<0.05, **p<0.01 (one-way ANOVA).
The deletion of DGK1 restores the fusogenicity of mutant Vam7Q283R
In previous studies we found that mutating the ionic zero layer of the Vam7 SNARE motif (Q283R) prevented anti-Sec17 bypass fusion.48,54 Fusion reactions containing Vam7Q283R were arrested at a hemifusion intermediate. Fusion could be restored by altering the fluidity and curvature of the vacuole with the cationic amphiphile chlorpromazine. We hypothesized that mutant SNARE complexes containing Vam7Q283R transferred insufficient energy to the membrane to trigger fusion. Thus, the effect of chlorpromazine was thought to be due to lowering the energy barrier for fusion to a level where Vam7Q283R could function. Due to the increased DAG concentration in dgk1Δ vacuoles we asked if fluidity and curvature was sufficiently altered to promote Vam7Q283R mediated fusion. Fusion reactions blocked with anti-Sec17 IgG were treated with recombinant Vam7Q283R at the indicated concentrations. We found that Vam7Q283R was able to promote a modest level of fusion in dgk1Δ vacuoles whereas wild type vacuole fusion were unresponsive (Fig. 8C, **p<0.01). This result is consistent with the notion that the increase in DAG on dgk1Δ lowers the energy barrier for fusion in a similar manner to what was seen with chlorpromazine. It is important to consider that exogenous Vam7 rescues anti-Sec17 blocked fusion also bypasses the role of Ypt7 dependent tethering, which requires Sec17 mediated SNARE priming. Thus, the enhanced Ypt7 activity likely did not play a role in this experiment.
DISCUSSION
It has been well established that most membrane fusion is catalyzed by SNARE proteins, yet the interdependence of the fusion machinery and the membrane itself remains unclear. Among the lipids that compose the membrane is a group that is subject to modification by specific kinases, phosphatases, and lipases to regulate membrane trafficking. The most studied of these lipids are the phosphoinositides that can be differentially phosphorylated on their inositol head groups to yield a panel of bioactive molecules that help orchestrate the fusion machinery through direct interactions with protein ligands. Specific PIs are necessary at various stages of fusion. For instance, vacuole fusion requires PI(4,5)P2 during priming as well as after docking.55,56 PI(4,5)P2 is turned over by Plc1 to yield IP3 and DAG, a fusogenic lipid required for vacuole content mixing.15 DAG can also be produced through the activity of the PA phosphatase Pah1, a pathway that is complemented by the activity of Dgk1 on DAG to restore PA levels.28,57 PA plays important roles in various membrane trafficking pathways including sporulation, mitochondrial fusion, and Glut4 trafficking.58–60 Like DAG, PA induces negative curvature.61 However the monophosphoester head group of PA adds a negative charge to the membrane that can play a role in binding proteins, including NSF/Sec18.24,62 Moreover, Pah1 activity is required during Sec18-mediated priming.19,24 Deletion of Pah1 leads to broad cellular defects including the enlargement of the endoplasmic reticulum and nuclear membranes, blocked lipid droplet synthesis, and inhibited endosomal maturation.18,19,25,26
In other pathways, such as lipid droplet formation, the effects caused by deleting PAH1 can be countered by concurrent deletion of DGK1 illustrating that the interconversion of PA and DAG by Pah1 and Dgk1 respectively can negate the effects of inactivating one of the enzymes.25,27 Importantly, it shows that the balance struck by these two enzymes is consistent during the homeostasis of some pathways. Unlike other pathways deleting DGK1 had no effect on the pah1Δ vacuolar phenotype (Fig. 9). This suggested one of two things. First, it is likely that the defect seen in pah1Δ vacuoles was not simply due to a steady-state imbalance of PA and DAG. It is possible that the PA:DAG ratio varies throughout the fusion pathway. For example, Pah1 activity is needed during priming, indicating that a reduction of PA and increase in DAG is necessary.19 However, inhibiting Pah1 activity after priming has no effect on fusion, demonstrating that PA levels no longer need to be reduced. Our previous findings show that DAG does not play a role during priming but is instead required after the formation of trans-SNARE complexes.15,16,47,48 This leads us to propose that the activities of Dgk1 and Pah1 are separated temporally on the fusion pathway making it unlikely to balance activities at any time. A second possibility is that DAG generated by Pah1 is fed into lipid synthesis pathways to generate phosphatidylethanolamine and phosphatidylcholine through the Kennedy pathway, or into the formation of triacylglycerol for storage.25,63 However, the latter is unlikely as lipid synthesis occurs at the endoplasmic reticulum and not the vacuole. Nevertheless, changes in lipid synthesis can propagate throughout the cell via membrane trafficking.
Figure 9. Schematic of role of PA and DAG production and modification on vacuole fusion.
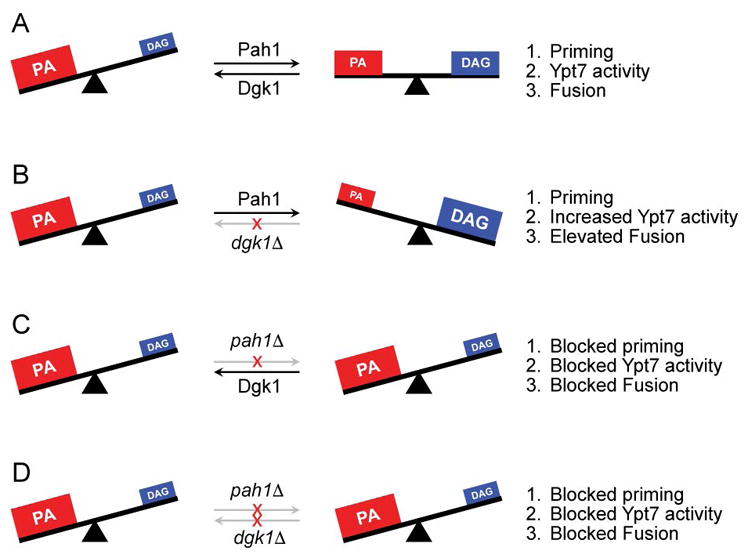
(A) Wild type fusion (PAH1 DGK1) leads to a reduction of PA by Pah1 and increase in DAG at the start of the pathway to promote SNARE priming, Ypt7 activation and support fusion. (B) In the absence of Dgk1 there is an accumulation of DAG leading to increased Ypt7 activity and augmented fusion. (C) In the absence of Pah1 high PA levels are maintained that sequester Sec18 from SNARE complexes and preventing priming. Consequently, Ypt7 activity is blocked and fusion is abrogated. (D) The double deletion of PAH1 and DGK1 does not restore fusion and phenocopies the pah1Δ single deletion.
Although both PA and DAG are essential for vacuole fusion, their exact function remains to be fully elucidated. DAG is a fusogenic lipid due in part to its intrinsic negative curvature in a membrane and ability to destabilize lipid bilayers and even trigger the fusion of protein-free vesicles.31–34 DAG can also bind fusion regulators such as protein kinase C (PKC) through its C1 domain.64,65 PKC has been shown to affect membrane fusion by several pathways including the phosphorylation of NSF to regulate neurotransmitter release, or phosphorylating the SNARE Syntaxin4 to control thrombin release from platelets.62,66 That said, it has not been determined whether PKC activity occurs on isolated vacuoles.
Another potential reason for the enhanced fusion of dgk1Δ vacuoles is that skewing the lipid balance to have less PA versus DAG can have an effect on priming. In a separate study we found that PA binds Sec18 to prevent cis-SNARE complex association.24 Conversion of PA to DAG by Pah1 releases Sec18 allowing it to engage cis-SNARE bundles. An excess of PA or inhibiting Pah1 activity results in the retention of Sec18 on PA domains. Thus we theorized that the converse could occur in the absence of Dgk1 and a likely reduced pool of PA would facilitate the recruitment of Sec18 to cis-SNARE complexes and lead to more efficient priming. Enhanced priming could result in a functional increase in active SNAREs and would be in keeping with other studies showing that overexpression of SNAREs augments vacuole fusion. In our hands however, we did not observe a difference in priming activity on dgk1Δ vacuoles (not shown). Thus, we conclude that the effects of deleting DGK1 are mostly due to the physical changes caused by elevated DAG. This notion is consistent with bilayer destabilization by the non-lamellar lipid DAG and lowering the energy barrier needed for SNAREs to trigger fusion.
One of the striking findings of this study was that fusion showed decreased sensitivity to reagents that target Ypt7 activity. Our results showed that fusion gained varying levels of resistance to anti-Ypt7 antibody, the GAP Gyp1-46, and GDI. One interpretation of these results is that Ypt7 is more active on dgk1Δ vacuoles and that increased activity would manifest in augmented tethering. Even though measuring tethering efficiency is difficult to quantitate using light microscopy, we propose a model where the lipid composition of the vacuole modulates Rab activity. Alternatively, these results could reflect a reduced need of Ypt7 for dgk1Δ vacuole fusion.
How would an increase in DAG levels affect Ypt7 function? To date there is no direct evidence of membrane fluidity affecting Rab proteins. One can imagine that the membrane affects the rotation or angle of insertion of the Ypt7 lipid anchor leading to an altered conformational state that strengthens its interactions with the HOPS complex, or a yet to be identified binding partner. Alternatively, the increase in DAG could lead to the enhanced recruitment of a Rab interacting protein. For example, in mammalian renal cortical tubule and mesangial cells the production of DAG promotes the interactions of hmunc13 with Rab34.67 This was due to the DAG recruitment of hmunc13 to membranes containing Rab34. The latter is unlikely here as the IC50 of C1b was unaltered on dgk1Δ vacuoles, suggesting that there was no change in competition for a protein ligand. Therefore, we favor a model where the physical properties of the membrane modulate Ypt7 activity. Although evidence linking Rab activity to the physical properties of a membrane is lacking, there are some clues that come from cholesterol storage defects. For instance, cholesterol accumulation prevents the mammalian Ypt7 homologue Rab7 from recruiting its effector protein RILP.68 This is linked with the inhibition of fusion between lysosomes and phagosomes and is likely due to decreased membrane fluidity caused by the increase in cholesterol as seen Niemann-Pick type C disease cells.69 Decreased fluidity due to excess cholesterol in Niemann-Pick type C disease cells has also been shown to reduce the ability of GDI to extract Rab7.70 The authors subsequently drew the conclusion cholesterol plays a role in the Rab7 cycle. These studies are in keeping with our proposed model where membrane fluidity modulates Rab activity.
In summary, our observations indicate that the PA-to-DAG ratio, regulated in part by the non-concurrent, yet complementary functions of Pah1 and Dgk1, can modulate vacuole fusion. The effects of increased DAG in the absence of Dgk1 led to improved Ypt7 activity as shown by the increased resistance to Gyp1-46, GDI and anti-Ypt7 antibody. Finally, the changes in the physical properties of dgk1Δ vacuole membranes restored the ability of Vam7Q283R to stimulate fusion.
Materials and Methods
Reagents
Reagents were dissolved in PS buffer (20 mM PIPES-KOH, pH 6.8, 200 mM sorbitol). Recombinant GST-FYVE 71, His6-MTM-1 72, GST-C1b 65, His6-SigD 73, GST-ENTH 74, MBP-FAPP1-PH 75, His6-DgkB 51, GST-Vam7 and GST-Vam7Q283R 48, Gdi1 45, His6-Gyp1-46 76, His6-Yck3 46 and Pbi2 77 were prepared as described and stored in PS buffer with 125 mM KCl. The plasmid for expressing His6-DgkB was a gift from Dr. C. Rock (St. Jude Children’s Research Hospital, Memphis, TN). Antibodies against Sec17 2, Sec18 78, Vam3 79, Nyv1 80, Vps33 9, Ypt7 8 were prepared as described. diC8-PA (1,2-dioctanoyl-sn-glycero-3-phosphate), diC8-DAG (1,2-diC8-sn-glycerol) and POG/DAG (palmitoyl-2-oleoyl-sn-glycerol) were purchased from Avanti Polar Lipids as chloroform stock solutions and stored at −20ºC. Filipin III was dissolved in DMSO at a stock concentration of 10 mM. Propranolol (Sigma) was dissolved in PS buffer.
Strains
Vacuoles from BJ3505 and DKY6281 were used for fusion assays (Table 1).81 Strains lacking PAH1 or YCK3 were described previously.19,82 DGK1 was deleted by homologous recombination using PCR products amplified using 5′-DGK1-KO and 3′-DGK1-KO primers with homology flanking the DGK1 coding sequence (Table 2). The PCR product was transformed into yeast by standard lithium acetate methods and plated on YPD media containing G418 (250 μg/μl) to generate BJ3505 dgk1Δ::kanMX6 (RFY60) and DKY6281 dgk1Δ::kanMX6 (RFY61). DGK1 was deleted from pah1Δ strains using PCR products amplified from pFA6A-hphMX4 and plated on YPD media containing hygromycin to generate RFY62 and RFY63. For complementation of dgk1Δ, RFY60 and RFY61 were transformed with pRS416 plasmids encoding DGK1 or DGK1D177A inserted between the EcoRI and NotI sites to generate RFY64-67. The DGK1 open reading frame was cloned using 5′-DGK1 and 3′-DGK1 primers using genomic DNA as a template. Point mutations generated using 5′-DGK1D177A and 3′-DGK1D177A primers. Transformants were selected on complete synthetic media lacking uracil. For cellular distribution analysis, DGK1 was fused in frame to GFP. DKY6281 was transformed with a PCR product amplified from pFA6a-GFP(S65T)-KanMX6 with homology flanking the stop codon of DGK1 with 5′-DGK-GFP and 3′DGK-GFP primers. Transformants were plated on YPD medium containing G418 to generate RFY68 83. PAH1 was deleted from RFY68 using PCR products amplified with 5′-PAH1-KO and 3′-PAH1-KO primers from pAG32 with homology flanking the PAH1 coding sequence to generate RFY71. For immunoblotting DGK1 was fused in frame to HA3. BJ3505 was transformed with a PCR products amplified from pFA6a-3HA-kanMX6 with homology flanking the stop codon to generate RFY69.84 Using the 5′-YCK3-KO and 3′-YCK3-KO primers YCK3 was deleted from dgk1Δ strains to generate RFY72-73.
Table 1.
Yeast strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| BJ3505 | MATa pep4::HIS3 prb1-Δ1.6R his3–200 lys2–801 trp1Δ101 (gal3) ura3–52 gal2 can1 | 90 |
| DKY6281 | MATα pho8::TRP1 leu2–3 leu 2–112 ura3–52 his3-Δ200 trp1-Δ901 lys2–801 | 82 |
| RFY17 | BJ3505, pah1Δ::kanMX6 | 19 |
| RFY18 | DKY6281, pahΔ::kanMX6 | 19 |
| CHY53 | BJ3505, yck3Δ::hphMX4 | 46 |
| CHY54 | DKY6281, yck3Δ::hphMX4 | 46 |
| RFY60 | BJ3505, dgk1Δ::hphMX4 | This study |
| RFY61 | DKY6281, dgk1Δ::hphMX4 | This study |
| RFY62 | RFY17, dgk1Δ:: hphMX4 | This study |
| RFY63 | RFY18, dgk1Δ:: hphMX4 | This study |
| RFY64 | RFY60, pRS416-DGK1 | This study |
| RFY65 | RFY61, pRS416-DGK1 | This study |
| RFY66 | RFY60, pRS416-DGK1D177A | This study |
| RFY67 | RFY61, pRS416-DGK1D177A | This study |
| RFY68 | BJ3505, DGK1::GFP-kanMX6 | This study |
| RFY69 | BJ3505, DGK1::3XHA-kanMX6 | This study |
| RFY71 | RFY68, pah1Δ:: hphMX4 | This study |
| RFY72 | RFY60, yck3Δ:: kanMX6 | This study |
| RFY73 | RFY61, yck3Δ:: kanMX6 | This study |
Table 2.
Primers used in this study
| Oligonucleotide | Sequence |
|---|---|
| 5′-DGK1-KO | 5′-GTCGTAGAGCAGTTGAACATTACGTAAACAGATATCTACGATAGGCCACTAGTGGATCTG–3′ |
| 3′-DGK1-KO | 5′-GAGGTGCTGGATCGTTAACCACAGATAAACTGAATCGCGCTC AGCTGAAGCTTCGTACGC–3′ |
| 5′-DGK1 | 5′-GTACTCGAATTCCTCTTACAAGAGACCATC-3′ |
| 3′-DGK1 | 5′-GTACTCGCGGCCGCGCGCTTGTTGGCACATAT-3′ |
| 5′-DGK1D177A | 5′-TTGGTCCGCTACAGCCGCCGCAACTATTGGAAGAAAGTAT-3′ |
| 3′-DGK1D177A | 5′-CGGCTGTAGCGGACCAACTTAGCAAAAATAACGATATTAAGG-3′ |
| 5′-DGK1-GFP/HA | 5′-ATCACTTTTTATGAACGCAGTAATCAAAACATTCAAGAAACGGAT CCCCGGGTTAATTAA-3′ |
| 3′-DGK1-GFP/HA | 5′-CGAAGAATATAAAACACTCCTGTTTTTGGTATATATGCTTGAATT CGAGCTCGTTTAAAC-3′ |
| 5′-PAH1-KO | 5′-ACAGGGAAGAAATTACTGAAGATAGACACATCGGTCGATTCTGTTTAGCTTGCCTTGTCC-3′ |
| 3′-PAH1-KO | 5′-AGTATGGATCGTTATAAATAATATTCGGCTACAAGAATCTGACACTGGATGGCGGCGTTA-3′ |
Vacuole isolation, cell fractionation, and in vitro vacuole fusion
Vacuoles were isolated as described.81 In vitro fusion reactions (30 μl) contained 3 μg each of vacuoles from BJ3505 and DKY6281 backgrounds, reaction buffer (20 mM PIPES-KOH pH 6.8, 200 mM sorbitol, 125 mM KCl, 5 mM MgCl2), ATP regenerating system (1 mM ATP, 0.1 mg/ml creatine kinase, 29 mM creatine phosphate), 10 μM CoA, and 283 nM Pbi2 (Protease B inhibitor). Reactions were incubated at 27°C and Pho8 activity was assayed in 250 mM Tris-Cl, pH 8.5, 0.4% Triton X-100, 10 mM MgCl2, 1 mM p-nitrophenylphosphate. Fusion units were measured by determining the p-nitrophenolate produced and absorbance was detected at 400 nm.
For Dgk1 localization by immunoblotting, yeast cells were fractionated into a total lysate, a total endomembrane fraction (Membranes), cytosol, endoplasmic reticulum (microsomes), and vacuoles as previously described.41,85 For the total membrane preparation, yeast cells were resuspended in bead buffer (10 mM Tris/HCl, pH 7.4, 0.3 M sorbitol, 0.1 M NaCl, 5 mM MgCl2) containing 1 mM PMSF at a density of 200 A600 units/ml. Cells were disrupted with glass beads by vortexing in five 1-min pulses, with intermittent chilling on ice. Lysates were centrifuged in a table-top centrifuge at 100 g for 5 min to pellet large debris and the supernatants carefully withdrawn and transferred to ultracentrifuge tubes. The total membrane fraction was collected by centrifugation (100,000 g, 1 h, 4°C) and resuspended in PS buffer (20 mM PIPES-KOH, pH 6.8, 200 mM sorbitol). The supernatant was saved as the cytosolic fraction. Endoplasmic reticulum derived microsomes were prepared as described.86 Briefly, yeast cells were treated with lyticase to produce spheroplasts as performed for vacuole preparations. Spheroplasts were resuspended in buffer (5 mM MES, pH 5.0, 0.5 mM EDTA, 0.6 M sorbitol, 1 mM PMSF) and centrifuged (20,000 g, 0.5 h, 4°C) using a JA-20 rotor. Supernatants were collected and microsomes were pelleted by centrifugation (SW-41 rotor, 60,000 g, 0.5 h, 4°C). The pellet was resuspended in 100 μl of buffer.
Quantification of Vacuolar Diacylglycerol by Thin-Layer Chromatography
Lipids were extracted from purified DKY6281 and dgk1Δ vacuoles (200 μg protein content) by the method of Bligh and Dyer then dried overnight under vacuum.87 Purified lipids were resuspended in chloroform:methanol (2:1) before being resolved by neutral lipid thin-layer chromatography as previously described.88 Briefly, Whatman Partisil® LK6D Silica Gel Plates (60 Å) were pre-washed with methanol/ethyl acetate (6:4) for 30 minutes. Individual channels were loaded with the indicated amount of diacylglycerol standard (Avanti Lipids) or vacuolar lipids. Plates were run twice with CH2Cl2/ethyl acetate/acetone (80:16:4) to 40 and 55 mm then hexanes/ethyl acetate to 68 mm (90:10), 80 mm (95:5), and 90 mm (100:0). Plates were then sprayed with a solution of 10% copper (II) sulfate in 8% phosphoric acid and dried for 10 minutes before charring at 145º C for 10 minutes. Imaging of plates was performed using a BioRad ChemiDoc™ MP Visualization System (605/50 filter, Green Epi illumination). Densitometry values were measured using Image Lab 4.0.1 software.
GST-Vam7 SNARE complex isolation and bypass fusion
SNARE complex isolation was performed as described previously using GST-Vam7.23,47,48 Briefly, 6X fusion reactions were incubated with 85 μg/ml anti-Sec17 IgG to block priming. After 15 min, 2 μM Gyp1-56 or 2 mM propranolol were added to selected reactions and incubated for an additional 5 min before adding 150 nM GST-Vam7. After a total of 90 min, reactions were placed on ice and 30 μl aliquots were removed to measure Pho8 activity. The remaining 150 μl reactions were sedimented (11,000 g, 10 min, 4°C), and the supernatants were discarded before extracting vacuoles with solubilization buffer (SB: 20 mM HEPES-KOH, pH 7.4, 100 mM NaCl, 2 mM EDTA, 20% glycerol, 0.5% Triton X-100, 1 mM DTT) with protease inhibitors (1 mM PMSF, 10 μM Pefabloc-SC, 5 μM pepstatin A, and 1 μM leupeptin). Vacuole pellets were overlaid with 100 μl SB and resuspended gently. An additional 100 μl SB was added, gently mixed, and incubated on ice for 20 min. Insoluble debris was sedimented (16,000 g, 10 min, 4°C) and 176 μl of supernatants were removed and placed in chilled tubes. Next, 16 μl was removed from each reaction as 10% total samples, mixed with 8 μl of 3X SDS loading buffer and heated (95°C, 5 min). Equilibrated glutathione beads (30 μl) were incubated with the remaining extracts (15 h, 4°C, nutation). Beads were sedimented and washed 5X with 1 ml SB (735 g, 2 min, 4°C), and bound material was eluted with 40 μl 1X SDS loading buffer. Protein complexes were examined by Western blotting.
Ca2+ efflux assay
Vacuole lumen Ca2+ efflux was measured as described.43 Fusion reactions (2X) contained 20 μg of vacuoles isolated from BJ3505 backgrounds, fusion reaction buffer and the fluorescent Ca2+ probe Fluo-4 dextran at 150 nM (Invitrogen). Reaction mixtures were transferred to a black 96-well plate. ATP regenerating system, or buffer was added and reactions were incubated at 27°C while monitoring Fluo-4 fluorescence (λex=488 nm; λem=520 nm).
Lipid mixing
Lipid Mixing assays were conducted using rhodamine B DHPE (Rh-PE; Thermo Fisher) as described.41 BJ3505 vacuoles (300 μg) were isolated and then incubated in 400 μl of PS buffer containing 150 μM Rh-PE (10 min, 4°C, nutating). Next, 800 μl of 15% Ficoll was added and then transferred to an 11 × 60 mm pollyalomar ultracentrifuge tube, overlaid with 1.2 ml of 8% and 4%, and 0.5 ml of PS buffer. Labeled vacuoles were isolated by centrifugation (105,200 X g, 25 min, 4°C, SW-60 Ti rotor) and recovered from the 0–4% Ficoll interface. Lipid mixing assays (90 μl) contained 2 μg of labeled vacuoles and 16 μg of unlabeled vacuoles in fusion buffer. Reaction mixtures were transferred to a black, half volume 96-well flat-bottom microtiter plate on ice. The plate was transferred to a fluorescence plate reader at 27°C to start the reactions. Measurements were taken every 60 sec for 40 min yielding fluorescence values (λex=544 nm; λem=590 nm) at the onset (F0) and during the reaction (Ft). After 40 min 0.45% (vol/vol) Triton X-100 was added and the final 10 measurements were averaged to give the value of fluorescence after infinite dilution (FTX100). The relative fluorescence change ΔFt/FTX100 = (Ft − F0)/FTX100 − F0 was calculated.
Microscopy
Vacuole morphology was monitored by incubating yeast with YPD broth containing the vital dye FM4-64.89 Cultures were grown to saturation, diluted to ~0.2 OD600 in YPD containing 5 μM FM4-64, grown for 1 h in 30°C shaker, washed with PBS, and resuspended in dye-free YPD to chase the dye for 3 h at 30°C. Cells were washed in PBS, mixed with 0.6% agarose, and mounted on glass slides for observation. Images were acquired using a Zeiss Axio Observer Z1 microscope equipped with an X-Cite 120XL light source, Plan Apochromat 63X oil objective (NA 1.4), and an AxioCam CCD camera.
Statistical analysis
Statistical analysis was calculated using one-way ANOVA or two-tailed paired t-test. P values of ≤0.05 were considered significant. The half maximal inhibitory concentrations (IC50) were determined using Origin software (OriginPro 9.1 software, OriginLab).
Synopsis.
In this study we found that the yeast diacylglycerol kinase Dgk1 acts as a negative regulator of vacuole fusion. Deletion of DGK1 leads to the accumulation of DAG on isolated vacuoles resulting in augmented fusion efficiency. This was thought to be caused by the destabilization of lipid bilayers and increased fluidity and membrane curvature that lowers the energy barrier needed for SNARE-triggered fusion. In addition, there was increased Ypt7 activity and vesicle tethering.
Acknowledgments
We thank Drs. William Wickner, Randy Schekman, Christian Ungermann, George Carman and Charles Rock for generous gifts of strains, antisera and plasmids. We also thank Dr. Terry Sasser, Mr. Colin Stoy, Mr. Alan Weisgerber, and Mr. Albert Chang for their technical assistance. This research was supported by a grant from the National Institutes of Health (R01-GM101132) to RAF. MLS was supported by a NIGMS-NIH Chemistry-Biology Interface Training Grant (5T32-GM070421).
Footnotes
Conflict of Interest
The authors declare no potential conflict of interest.
References
- 1.Jahn R, Scheller RH. SNAREs - engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7(9):631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 2.Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85(1):83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 3.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 4.Piper RC, Bryant NJ, Stevens TH. The membrane protein alkaline phosphatase is delivered to the vacuole by a route that is distinct from the VPS-dependent pathway. J Cell Biol. 1997;138(3):531–545. doi: 10.1083/jcb.138.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishihara N, Hamasaki M, Yokota S, Suzuki K, Kamada Y, Kihara A, Yoshimori T, Noda T, Ohsumi Y. Auto-phagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell. 2001;12(11):3690–3702. doi: 10.1091/mbc.12.11.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cabrera M, Langemeyer L, Mari M, Rethmeier R, Orban I, Perz A, Brocker C, Griffith J, Klose D, Steinhoff HJ, Reggiori F, Engelbrecht-Vandre S, Ungermann C. Phosphorylation of a membrane curvature-sensing motif switches function of the HOPS subunit Vps41 in membrane tethering. J Cell Biol. 2010;191(4):845–859. doi: 10.1083/jcb.201004092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wickner W. Membrane Fusion: Five Lipids, Four SNAREs, Three Chaperones, Two Nucleotides, and a Rab, All Dancing in a Ring on Yeast Vacuoles. Annu Rev Cell Dev Biol. 2010:26115–136. doi: 10.1146/annurev-cellbio-100109-104131. [DOI] [PubMed] [Google Scholar]
- 8.Mayer A, Wickner W. Docking of yeast vacuoles is catalyzed by the Ras-like GTPase Ypt7p after symmetric priming by Sec18p (NSF) J Cell Biol. 1997;136(2):307–17. doi: 10.1083/jcb.136.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seals DF, Eitzen G, Margolis N, Wickner WT, Price A. A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc Natl Acad Sci U S A. 2000;97(17):9402–7. doi: 10.1073/pnas.97.17.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merz AJ, Wickner W. Trans-SNARE interactions elicit Ca2+ efflux from the yeast vacuole lumen. J Cell Biol. 2004:164195–206. doi: 10.1083/jcb.200310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fratti RA, Jun Y, Merz AJ, Margolis N, Wickner W. Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles. J Cell Biol. 2004;167(6):1087–1098. doi: 10.1083/jcb.200409068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Seeley ES, Wickner W, Merz AJ. Vacuole Fusion at a Ring of Vertex Docking Sites Leaves Membrane Fragments within the Organelle. Cell. 2002;108(3):357–69. doi: 10.1016/s0092-8674(02)00632-3. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Merz AJ, Collins KM, Wickner W. Hierarchy of protein assembly at the vertex ring domain for yeast vacuole docking and fusion. J Cell Biol. 2003;160(3):365–374. doi: 10.1083/jcb.200209095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boeddinghaus C, Merz AJ, Laage R, Ungermann C. A cycle of Vam7p release from and PtdIns 3-P-dependent rebinding to the yeast vacuole is required for homotypic vacuole fusion. J Cell Biol. 2002;157(1):79–89. doi: 10.1083/jcb.200112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jun Y, Fratti RA, Wickner W. Diacylglycerol and its formation by Phospholipase C regulate Rab- and SNARE- dependent yeast vacuole fusion. J Biol Chem. 2004:27953186–53195. doi: 10.1074/jbc.M411363200. [DOI] [PubMed] [Google Scholar]
- 16.Karunakaran S, Fratti R. The Lipid Composition and Physical Properties of the Yeast Vacuole Affect the Hemifusion-Fusion Transition. Traffic. 2013;14(6):650–662. doi: 10.1111/tra.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato M, Wickner W. Ergosterol is required for the Sec18/ATP-dependent priming step of homotypic vacuole fusion. EMBO J. 2001;20(15):4035–40. doi: 10.1093/emboj/20.15.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence G, Brown CC, Flood BA, Karunakaran S, Cabrera M, Nordmann M, Ungermann C, Fratti RA. Dynamic association of the PI3P-interacting Mon1-Ccz1 GEF with vacuoles is controlled through its phosphorylation by the type-1 casein kinase Yck3. Mol Biol Cell. 2014;25(10):1608–1619. doi: 10.1091/mbc.E13-08-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasser T, Qiu QS, Karunakaran S, Padolina M, Reyes A, Flood B, Smith S, Gonzales C, Fratti RA. Yeast lipin 1 orthologue pah1p regulates vacuole homeostasis and membrane fusion. J Biol Chem. 2012;287(3):2221–2236. doi: 10.1074/jbc.M111.317420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stroupe C, Collins KM, Fratti RA, Wickner W. Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J. 2006;25(8):1579–1589. doi: 10.1038/sj.emboj.7601051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cabrera M, Nordmann M, Perz A, Schmedt D, Gerondopoulos A, Barr F, Piehler J, Engelbrecht-Vandre S, Ungermann C. The Mon1-Ccz1 GEF activates the Rab7 GTPase Ypt7 via a longin fold-Rab interface and association with PI-3-P-positive membranes. J Cell Sci. 2014 doi: 10.1242/jcs.140921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheever ML, Sato TK, de Beer T, Kutateladze TG, Emr SD, Overduin M. Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat Cell Biol. 2001;3(7):613–8. doi: 10.1038/35083000. [DOI] [PubMed] [Google Scholar]
- 23.Miner GE, Starr ML, Hurst LR, Sparks RP, Padolina M, Fratti RA. The Central Polybasic Region of the Soluble SNARE (Soluble N-Ethylmaleimide-sensitive Factor Attachment Protein Receptor) Vam7 Affects Binding to Phosphatidylinositol 3-Phosphate by the PX (Phox Homology) Domain. J Biol Chem. 2016;291(34):17651–17663. doi: 10.1074/jbc.M116.725366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Starr ML, Hurst LR, Fratti RA. Phosphatidic acid sequesters Sec18p from cis-SNARE complexes to inhibit priming. Traffic. 2016;17(10):1091–1109. doi: 10.1111/tra.12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adeyo O, Horn PJ, Lee S, Binns DD, Chandrahas A, Chapman KD, Goodman JM. The yeast lipin orthologue Pah1p is important for biogenesis of lipid droplets. J Cell Biol. 2011;192(6):1043–1055. doi: 10.1083/jcb.201010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han GS, Siniossoglou S, Carman GM. The cellular functions of the yeast lipin homolog PAH1p are dependent on its phosphatidate phosphatase activity. J Biol Chem. 2007;282(51):37026–37035. doi: 10.1074/jbc.M705777200. [DOI] [PubMed] [Google Scholar]
- 27.Han GS, O’Hara L, Carman GM, Siniossoglou S. An unconventional diacylglycerol kinase that regulates phospholipid synthesis and nuclear membrane growth. J Biol Chem. 2008:28320433–20442. doi: 10.1074/jbc.M802903200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han GS, LOH, Siniossoglou S, Carman GM. Characterization of the yeast DGK1-encoded CTP-dependent diacylglycerol kinase. J Biol Chem. 2008:28320443–20453. doi: 10.1074/jbc.M802866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karanasios E, Han GS, Xu Z, Carman GM, Siniossoglou S. A phosphorylation-regulated amphipathic helix controls the membrane translocation and function of the yeast phosphatidate phosphatase. Proc Natl Acad Sci U S A. 2010;107(41):17539–17544. doi: 10.1073/pnas.1007974107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi HS, Su WM, Morgan JM, Han GS, Xu Z, Karanasios E, Siniossoglou S, Carman GM. Phosphorylation of phosphatidate phosphatase regulates its membrane association and physiological functions in Saccharomyces cerevisiae: identification of SER(602), THR(723), AND SER(744) as the sites phosphorylated by CDC28 (CDK1)-encoded cyclin-dependent kinase. J Biol Chem. 2011;286(2):1486–1498. doi: 10.1074/jbc.M110.155598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das S, Rand RP. Diacylglycerol causes major structural transitions in phospholipid bilayer membranes. Biochem Biophys Res Commun. 1984;124(2):491–496. doi: 10.1016/0006-291x(84)91580-8. [DOI] [PubMed] [Google Scholar]
- 32.Seddon JM. An inverse face-centered cubic phase formed by diacylglycerol-phosphatidylcholine mixtures. Biochemistry. 1990;29(34):7997–8002. doi: 10.1021/bi00486a031. [DOI] [PubMed] [Google Scholar]
- 33.Sánchez-Migallón MP, Aranda FJ, Gómez-Fernández JC. The dissimilar effect of diacylglycerols on Ca(2+)-induced phosphatidylserine vesicle fusion. Biophys J. 1995;68(2):558–566. doi: 10.1016/S0006-3495(95)80217-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villar AV, Alonso A, Goñi FM. Leaky vesicle fusion induced by phosphatidylinositol-specific phospholipase C: observation of mixing of vesicular inner monolayers. Biochemistry. 2000;39(46):14012–14018. doi: 10.1021/bi992515c. [DOI] [PubMed] [Google Scholar]
- 35.Flick JS, Thorner J. Genetic and biochemical characterization of a phosphatidylinositol-specific phospholipase C in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13(9):5861–5876. doi: 10.1128/mcb.13.9.5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waksman M, Eli Y, Liscovitch M, Gerst JE. Identification and characterization of a gene encoding phospholipase D activity in yeast. J Biol Chem. 1996;271(5):2361–2364. doi: 10.1074/jbc.271.5.2361. [DOI] [PubMed] [Google Scholar]
- 37.Goñi FM, Alonso A. Structure and functional properties of diacylglycerols in membranes. Prog Lipid Res. 1999;38(1):1–48. doi: 10.1016/s0163-7827(98)00021-6. [DOI] [PubMed] [Google Scholar]
- 38.Chernomordik LV, Kozlov MM. Protein-lipid interplay in fusion and fission of biological membranes. Annu Rev Biochem. 2003:72175–207. doi: 10.1146/annurev.biochem.72.121801.161504. [DOI] [PubMed] [Google Scholar]
- 39.Wolinski H, Hofbauer HF, Hellauer K, Cristobal-Sarramian A, Kolb D, Radulovic M, Knittelfelder OL, Rechberger GN, Kohlwein SD. Seipin is involved in the regulation of phosphatidic acid metabolism at a subdomain of the nuclear envelope in yeast. Biochim Biophys Acta. 2015;1851(11):1450–1464. doi: 10.1016/j.bbalip.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Merz AJ, Wickner WT. Resolution of organelle docking and fusion kinetics in a cell-free assay. Proc Natl Acad Sci U S A. 2004;101(32):11548–11553. doi: 10.1073/pnas.0404583101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu QS, Fratti RA. The Na+/H+ exchanger Nhx1p regulates the initiation of Saccharomyces cerevisiae vacuole fusion. J Cell Sci. 2010;123(Pt 19):3266–3275. doi: 10.1242/jcs.067637. [DOI] [PubMed] [Google Scholar]
- 42.Reese C, Mayer A. Transition from hemifusion to pore opening is rate limiting for vacuole membrane fusion. J Cell Biol. 2005;171(6):981–990. doi: 10.1083/jcb.200510018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sasser TL, Padolina M, Fratti RA. The Yeast Vacuolar ABC Transporter Ybt1p Regulates Membrane Fusion Through Ca2+ Transport Modulation. Biochem J. 2012;448(3):365–372. doi: 10.1042/BJ20120847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meier KE, Gause KC, Wisehart-Johnson AE, Gore AC, Finley EL, Jones LG, Bradshaw CD, McNair AF, Ella KM. Effects of propranolol on phosphatidate phosphohydrolase and mitogen-activated protein kinase activities in A7r5 vascular smooth muscle cells. Cell Signal. 1998;10(6):415–426. doi: 10.1016/s0898-6568(97)00140-x. [DOI] [PubMed] [Google Scholar]
- 45.Starai VJ, Jun Y, Wickner W. Excess vacuolar SNAREs drive lysis and Rab bypass fusion. Proc Natl Acad Sci U S A. 2007;104(34):13551–13558. doi: 10.1073/pnas.0704741104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hickey CM, Stroupe C, Wickner W. The major role of the Rab Ypt7p in vacuole fusion is supporting HOPS membrane association. J Biol Chem. 2009;284(24):16118–16125. doi: 10.1074/jbc.M109.000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fratti RA, Wickner W. Distinct Targeting and Fusion Functions of the PX and SNARE Domains of Yeast Vacuolar Vam7p. J Biol Chem. 2007;282(17):13133–13138. doi: 10.1074/jbc.M700584200. [DOI] [PubMed] [Google Scholar]
- 48.Fratti RA, Collins KM, Hickey CM, Wickner W. Stringent 3Q: 1R composition of the SNARE 0-layer can be by-passed for fusion by compensatory SNARE mutation or by lipid bilayer modification. J Biol Chem. 2007;282(20):14861–14867. doi: 10.1074/jbc.M700971200. [DOI] [PubMed] [Google Scholar]
- 49.Stahelin RV, Digman MA, Medkova M, Ananthanarayanan B, Rafter JD, Melowic HR, Cho W. Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Cdelta. J Biol Chem. 2004;279(28):29501–29512. doi: 10.1074/jbc.M403191200. [DOI] [PubMed] [Google Scholar]
- 50.Bai J, Pagano RE. Measurement of spontaneous transfer and transbilayer movement of BODIPY-labeled lipids in lipid vesicles. Biochemistry. 1997;36(29):8840–8848. doi: 10.1021/bi970145r. [DOI] [PubMed] [Google Scholar]
- 51.Miller DJ, Jerga A, Rock CO, White SW. Analysis of the Staphylococcus aureus DgkB structure reveals a common catalytic mechanism for the soluble diacylglycerol kinases. Structure. 2008;16(7):1036–1046. doi: 10.1016/j.str.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.LaGrassa TJ, Ungermann C. The vacuolar kinase Yck3 maintains organelle fragmentation by regulating the HOPS tethering complex. J Cell Biol. 2005;168(3):401–414. doi: 10.1083/jcb.200407141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brett CL, Plemel RL, Lobingier BT, Vignali M, Fields S, Merz AJ. Efficient termination of vacuolar Rab GTPase signaling requires coordinated action by a GAP and a protein kinase. J Cell Biol. 2008;182(6):1141–1151. doi: 10.1083/jcb.200801001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karunakaran V, Wickner W. Fusion proteins and select lipids cooperate as membrane receptors for the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) Vam7p. J Biol Chem. 2013;288(40):28557–28566. doi: 10.1074/jbc.M113.484410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Collins KM, Thorngren NL, Fratti RA, Wickner WT. Sec17p and HOPS, in distinct SNARE complexes, mediate SNARE complex disruption or assembly for fusion. EMBO J. 2005;24(10):1775–1786. doi: 10.1038/sj.emboj.7600658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mayer A, Scheglmann D, Dove S, Glatz A, Wickner W, Haas A. Phosphatidylinositol 4,5-bisphosphate regulates two steps of homotypic vacuole fusion. Mol Biol Cell. 2000;11(3):807–17. doi: 10.1091/mbc.11.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han GS, Wu WI, Carman GM. The Saccharomyces cerevisiae Lipin homolog is a Mg2+-dependent phosphatidate phosphatase enzyme. J Biol Chem. 2006;281(14):9210–9218. doi: 10.1074/jbc.M600425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. 2006;8(11):1255–1262. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- 59.Nakanishi H, Morishita M, Schwartz CL, Coluccio A, Engebrecht J, Neiman AM. Phospholipase D and the SNARE Sso1p are necessary for vesicle fusion during sporulation in yeast. J Cell Sci. 2006;119(Pt 7):1406–1415. doi: 10.1242/jcs.02841. [DOI] [PubMed] [Google Scholar]
- 60.Vicogne J, Vollenweider D, Smith JR, Huang P, Frohman MA, Pessin JE. Asymmetric phospholipid distribution drives in vitro reconstituted SNARE-dependent membrane fusion. Proc Natl Acad Sci U S A. 2006;103(40):14761–14766. doi: 10.1073/pnas.0606881103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kooijman EE, Burger KN. Biophysics and function of phosphatidic acid: a molecular perspective. Biochim Biophys Acta. 2009;1791(9):881–888. doi: 10.1016/j.bbalip.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 62.Matveeva EA, Whiteheart SW, Vanaman TC, Slevin JT. Phosphorylation of the N-ethylmaleimide-sensitive factor is associated with depolarization-dependent neurotransmitter release from synaptosomes. J Biol Chem. 2001;276(15):12174–12181. doi: 10.1074/jbc.M007394200. [DOI] [PubMed] [Google Scholar]
- 63.Kennedy EP, Weiss SB. The function of cytidine coenzymes in the biosynthesis of phospholipides. J Biol Chem. 1956;222(1):193–214. [PubMed] [Google Scholar]
- 64.Dries DR, Newton AC. Kinetic analysis of the interaction of the C1 domain of protein kinase C with lipid membranes by stopped-flow spectroscopy. J Biol Chem. 2008;283(12):7885–7893. doi: 10.1074/jbc.M709943200. [DOI] [PubMed] [Google Scholar]
- 65.Johnson JE, Giorgione J, Newton AC. The C1 and C2 domains of protein kinase C are independent membrane targeting modules, with specificity for phosphatidylserine conferred by the C1 domain. Biochemistry. 2000;39(37):11360–11369. doi: 10.1021/bi000902c. [DOI] [PubMed] [Google Scholar]
- 66.Chung SH, Polgar J, Reed GL. Protein kinase C phosphorylation of syntaxin 4 in thrombin-activated human plate-lets. J Biol Chem. 2000;275(33):25286–25291. doi: 10.1074/jbc.M004204200. [DOI] [PubMed] [Google Scholar]
- 67.Speight P, Silverman M. Diacylglycerol-activated Hmunc13 serves as an effector of the GTPase Rab34. Traffic. 2005;6(10):858–865. doi: 10.1111/j.1600-0854.2005.00321.x. [DOI] [PubMed] [Google Scholar]
- 68.Huynh KK, Gershenzon E, Grinstein S. Cholesterol accumulation by macrophages impairs phagosome maturation. J Biol Chem. 2008;283(51):35745–35755. doi: 10.1074/jbc.M806232200. [DOI] [PubMed] [Google Scholar]
- 69.Koike T, Ishida G, Taniguchi M, Higaki K, Ayaki Y, Saito M, Sakakihara Y, Iwamori M, Ohno K. Decreased membrane fluidity and unsaturated fatty acids in Niemann-Pick disease type C fibroblasts. Biochim Biophys Acta. 1998;1406(3):327–335. doi: 10.1016/s0925-4439(98)00019-2. [DOI] [PubMed] [Google Scholar]
- 70.Lebrand C, Corti M, Goodson H, Cosson P, Cavalli V, Mayran N, Fauré J, Gruenberg J. Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J. 2002;21(6):1289–1300. doi: 10.1093/emboj/21.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, Parton RG, Stenmark H. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 2000;19(17):4577–88. doi: 10.1093/emboj/19.17.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taylor GS, Maehama T, Dixon JE. Inaugural article: myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc Natl Acad Sci U S A. 2000;97(16):8910–8915. doi: 10.1073/pnas.160255697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marcus SL, Wenk MR, Steele-Mortimer O, Finlay BB. A synaptojanin-homologous region of Salmonella typhimurium SigD is essential for inositol phosphatase activity and Akt activation. FEBS Lett. 2001;494(3):201–207. doi: 10.1016/s0014-5793(01)02356-0. [DOI] [PubMed] [Google Scholar]
- 74.Rosenthal JA, Chen H, Slepnev VI, Pellegrini L, Salcini AE, Di Fiore PP, De Camilli P. The epsins define a family of proteins that interact with components of the clathrin coat and contain a new protein module. J Biol Chem. 1999;274(48):33959–33965. doi: 10.1074/jbc.274.48.33959. [DOI] [PubMed] [Google Scholar]
- 75.Dowler S, Currie RA, Campbell DG, Deak M, Kular G, Downes CP, Alessi DR. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem J. 2000;351(Pt 1):19–31. doi: 10.1042/0264-6021:3510019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang CW, Stromhaug PE, Kauffman EJ, Weisman LS, Klionsky DJ. Yeast homotypic vacuole fusion requires the Ccz1-Mon1 complex during the tethering/docking stage. J Cell Biol. 2003;163(5):973–985. doi: 10.1083/jcb.200308071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Slusarewicz P, Xu Z, Seefeld K, Haas A, Wickner WT. I2B is a small cytosolic protein that participates in vacuole fusion. Proc Natl Acad Sci U S A. 1997;94(11):5582–7. doi: 10.1073/pnas.94.11.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haas A, Wickner W. Homotypic vacuole fusion requires Sec17p (yeast alpha-SNAP) and Sec18p (yeast NSF) EMBO J. 1996;15(13):3296–305. [PMC free article] [PubMed] [Google Scholar]
- 79.Nichols BJ, Ungermann C, Pelham HR, Wickner WT, Haas A. Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature. 1997;387(6629):199–202. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- 80.Ungermann C, Nichols BJ, Pelham HR, Wickner W. A vacuolar v-t-SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J Cell Biol. 1998;140(1):61–9. doi: 10.1083/jcb.140.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haas A, Scheglmann D, Lazar T, Gallwitz D, Wickner W. The GTPase Ypt7p of Saccharomyces cerevisiae is required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. EMBO J. 1995;14(21):5258–70. doi: 10.1002/j.1460-2075.1995.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Haas A, Conradt B, Wickner W. G-protein ligands inhibit in vitro reactions of vacuole inheritance. J Cell Biol. 1994;126(1):87–97. doi: 10.1083/jcb.126.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wach A, Brachat A, Alberti-Segui C, Rebischung C, Philippsen P. Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast. 1997;13(11):1065–1075. doi: 10.1002/(SICI)1097-0061(19970915)13:11<1065::AID-YEA159>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 84.Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14(10):953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 85.Sorin A, Rosas G, Rao R. PMR1, a Ca2+-ATPase in yeast Golgi, has properties distinct from sarco/endoplasmic reticulum and plasma membrane calcium pumps. J Biol Chem. 1997;272(15):9895–9901. doi: 10.1074/jbc.272.15.9895. [DOI] [PubMed] [Google Scholar]
- 86.Grillitsch K, Connerth M, Köfeler H, Arrey TN, Rietschel B, Wagner B, Karas M, Daum G. Lipid particles/droplets of the yeast Saccharomyces cerevisiae revisited: lipidome meets proteome. Biochim Biophys Acta. 2011;1811(12):1165–1176. doi: 10.1016/j.bbalip.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 88.Churchward MA, Rogasevskaia T, Brandman DM, Khosravani H, Nava P, Atkinson JK, Coorssen JR. Specific lipids supply critical negative spontaneous curvature--an essential component of native Ca2+-triggered membrane fusion. Biophys J. 2008;94(10):3976–3986. doi: 10.1529/biophysj.107.123984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128(5):779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jones EW, Zubenko GS, Parker RR. PEP4 gene function is required for expression of several vacuolar hydrolases in Saccharomyces cerevisiae. Genetics. 1982;102(4):665–677. doi: 10.1093/genetics/102.4.665. [DOI] [PMC free article] [PubMed] [Google Scholar]