

Abstract
Background
Quinazoline-based α1-adrenoceptor antagonists suppress tumor growth by inducing apoptosis via an α1-adrenoceptor-independent action. Anoikis is a unique mode of apoptosis consequential to insufficient cell-matrix interactions.
Objective
This study investigated the apoptotic effect of novel quinazoline-based compounds on human renal cancer cells.
Design, setting, and participants
Two cell lines were used: renal cell carcinoma (RCC) 786-0, harboring a von Hippel-Lindau (VHL) tumor-suppressor gene mutation with a highly angiogenic phenotype, and Caki cells (no VHL mutation).
Measurements
The lead compound DZ-50 (10 μM) led to significant inhibition of tumor-cell adhesion, migration, and invasion at a lower dose than doxazosin (25 μM) in both RCC lines.
Results and limitations
Doxazosin induced death-receptor-mediated apoptosis, while DZ-50 led to anoikis via targeting of the focal adhesion complex and AKT signaling that subsequently increased RCC susceptibility to caspase-8-mediated apoptosis. Both quinazoline compounds, doxazosin and DZ-50, significantly reduced RCC metastatic potential in vivo.
Conclusions
Quinazoline-based drugs trigger anoikis in RCC by targeting the focal adhesion survival signaling. This potent antitumor action against human RCC suggests a novel quinazoline-based therapy targeting renal cancer.
Keywords: Renal cancer, Invasion, Metastasis, Akt signaling, Focal adhesion complex
1. Introduction
Renal cell carcinoma (RCC) accounts for an estimated of 58 000 new cases of cancer and about 13 000 deathsin the United States annually. The incidence of RCC has increased by >30% in the last decade [1], with an annual mortality-to-incidence ratio significantly higher than for other urologic tumors. Approximately 30% of RCC patients have metastasis at the time of diagnosis; the 5-yr survival rate for patients with metastasis is <10% and the overall 5-yr survival rate is 60% [2,3]. Significantly, 60% of RCC cases harbor a von Hippel-Lindau (VHL) tumor-suppressor gene mutation that mediates the hypoxia-inducing factor (HIF)–related, highly angiogenic phenotype [2,3]. Combination regimens of recombinant cytokines with interferon-α (IFN-α) or interleukin-2 exhibit a modest response of 10–20% with no impact on survival [3]. In the era of targeted therapy, sunitinib, a potent antiangiogenic tyrosine kinase inhibitor approved for the first- and second-line treatment of patients with metastatic RCC, demonstrates longer overall survival compared with IFN-α plus improved response and progression-free survival [4].
Quinazoline-based α1-adrenoreceptor antagonists are clinically effective for the relief of symptoms associated with benign prostatic hyperplasia [5]. We previously reported the ability of quinazoline α1-adrenoceptor antagonists, but not sulfonamide-based α1-antagonists, to induce apoptosis in prostate epithelial and endothelial cells [5]. This apoptotic effect is an α1-adrenoceptor-independent action, engaging the death receptor Fas-associated death domain (FADD)-mediated caspase-8 activation, Smad activation of transforming growth factor (TGF)-ß1 signaling, and targeting the Akt survival mechanism [6–8]. Drug optimization approaches led to the development of novel quinazoline-based compounds, the lead analogue of which, DZ-50, exerts a potent antiangiogenic effect targeting the prostate tumor microenvironment [9].
Tumor epithelial and endothelial cells require attachment to the extracellular matrix (ECM) for survival; on loss of adhesion, they undergo anoikis [10,11], a process that plays a key role in tumor angiogenesis and metastasis [12]. During metastatic progression, cells lack firm attachment to the ECM and are susceptible to anoikis [12]. Integrin-linked kinase (ILK) is a serine/threonine protein kinase that interacts with the cytoplasmic domain of β1-integrin and β3-integrin [13–15]. ILK contributes to anoikis resistance by regulating key integrin-mediated processes including cell adhesion and fibronectin-ECM assembly [13,16,17]. This study explored the action of the parent quinazoline doxazosin and the lead derivative DZ-50 against human renal cancer. The anoikis-driven antitumor action of quinazolines significantly impairs renal cancer metastasis.
2. Materials and methods
2.1. Cell lines and transfections
Human renal cancer cell lines 786-0 and Caki were obtained from American Type Culture Collection (Rockville, MD, USA). Cells were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum and antibiotics (penicillin G/streptomycin). Human microvascular brain-endothelial cells (HBMEC) were cultured in endothelial-cell basal medium (EGM-2) (Cambrex, East Rutherford, NJ, USA).
2.2. Cell viability assay
Cell viability was assessed using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium) assay. Subconfluent cell cultures were exposed to increasing doses of doxazosin (0–25 μM) or DZ-50 (0–10 μM). Doxazosin mesylate was obtained from Sigma (St. Louis, MO, USA) and the lead compound DZ-50 was synthesized in our laboratory [10].
2.3. Cell adhesion assay
After treatment, cells were seeded in 6-well plates (5 × 104 cells per well) coated with fibronectin (BD Biosciences Discovery Labware, Bedford, MA, USA). Following a 10-min attachment period, attached cells were fixed with methanol (100% v/v) and counted in three ×100 fields per well.
2.4. Migration assay
Confluent cell monolayers were wounded and exposed to either doxazosin (25 μM) or DZ-50 (10 μM). After 9 and 24 h, cells migrating to the wounded areas were counted under a microscope and cell migration potential was calculated as the average number of cells observed in five random fields per well.
2.5. Transendothelial migration assay
Sterile cover slips were coated with Matrigel (Becton Dickson, Franklin Lakes, NJ, USA) and HBMEC cells (6.25 × 104) were seeded to form a monolayer. Cells were allowed to adhere on the Matrigel for 24 h. RCC 786-0 were labeled with 10 mg/ml octadecyl indocarbocyanines (DiI) (Molecular Probes, Invitrogen, Carlsbad, CA, USA), resuspended in EGM-2MV medium (Cambrex, East Rutherford, NJ, USA) and added to the HBMEC monolayer (8 × 103 cells per cover slip). Cells were fixed in 2% (v/v) paraformaldehyde in phosphate buffer saline and stained with DAPI nucleic acid stain (Molecular Probes, Invitrogen, Carlsbad, CA, USA) to detect nuclear presence under confocal microscopy.
2.6. Western blot analysis
Antibodies against focal adhesion kinase (FAK), AKT, phospho-Akt (Ser473), p-GSK3β, total GSK3β, caspase 8, and caspase 3 were from Cell Signaling Technology (Beverly, MA, USA). The phospho-FAK (Y397) antibody was obtained from Sigma (St. Louis, MO, USA). Protein levels were normalized to α-actin (Oncogene Research Products, La Jolla, CA, USA). Cell lysates (30-μg protein) in radio-immunoprecipitation assay buffer (150 mM NaCl, 50 mM Tris [pH 8.0], 0.5% deoxycholic acid, 1% (v/v) NP-40 with 1 mM phenyl methyl-sulfonyl fluoride) were subjected to sodium-dodecyl-sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and Hybond-C blotting (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Membranes were incubated with the primary antibody followed by species-specific horseradish peroxidase–labeled secondary antibodies. Signal detection was achieved with SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific Inc, Rockford, IL, USA).
2.7. Apoptosis evaluation
RCC 786-0 were treated with either DZ-50 or doxazosin for 24 h, fixed in 4% (v/v) formaldehyde, and incubated with Annexin-V antibody (R&D Systems, Minneapolis, MN, USA), followed by exposure to fluorescein isothiocyanate (FITC)–conjugated goat antimouse secondary antibodies. Analysis for Annexin-V staining was conducted using Partec FloMax (Partec, Munster, Germany).
2.8. Caspase-8 activity assay
Caspase-8 activation was evaluated in 786-0 cells after exposure to various agents for 24 h: DZ-50 (10 μM), doxazosin (25 μM), TRAIL (100 ng/ml), and Velcade (100 nM). Caspase-8-specific activity was measured by luminescence using Caspase-Glo 8 Assay Systems (Promega, Madison, WI, USA).
2.9. Cell-cycle analysis
Cell-cycle analysis was performed via fluorescence-activated cell sorting (FACS). Cells were fixed in ethanol 70% (v/v) and stained with propidium iodide (50 μg/ml) containing 20 μg/ml RNase. Flow cytometry-based cell-cycle analysis was conducted using Partec FloMax (Partec, Munster, Germany).
2.10. Anoikis evaluation
To determine the ability of the quinazoline compounds to induce anoikis, cells were plated on dishes coated with either fibronectin or the nonadhesive polyhydroxyethylmethacrylate (poly-HEMA) and incubated in culture medium for 24 h. Apoptotic cells (under adherent and nonadherent conditions) were detected and quantitated using the Annexin V-based flow cytometric assay.
2.11. In vivo experimental metastasis assay
Human RCC 786-0 were inoculated (2 × 106 cells per 80 μl) in the tail vein of male nude mice (age: 4–6 wk). After 2 wk, treatment was initiated: Doxazosin (100 mg/kg) or DZ-50 (100 mg/kg) was administered three times per week for 3 wk via oral gavage as previously described [9]. Treated and vehicle-control mice were sacrificed, and lungs, kidneys, and prostates were excised and examined for metastatic lesions.
2.12. Statistical analysis
One-way analysis of variance was performed using the StatView statistical program to determine the statistical significance between values (p < 0.05).
3. Results
3.1. Doxazosin and DZ-50 induce renal-cancer cell death
Treatment of human RCC 786-0 and Caki with either doxazosin or DZ-50 resulted in a dose-dependent loss of cell viability (Fig. 1A and 1B). DZ-50 induced cell death at a lower dose (5 μM) than doxazosin (25 μM). Time-course analysis revealed that DZ-50 (10 μM) significantly induced cell death within 12–24 h compared with doxazosin’s effect detected at 48 h post treatment (25 μM) (Fig. 1C–1D).
Fig. 1.
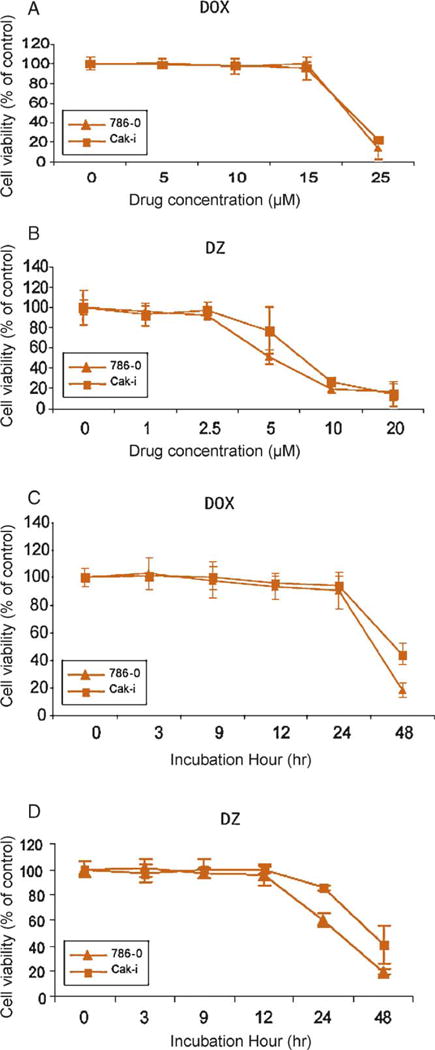
Effect of doxazosin and DZ-50 on cell death of 786-0 and Caki renal carcinoma cells. Human renal cell carcinoma 786-0 and Caki cells were treated with increasing concentrations of (A) doxazosin (0–25 μM) or (B) DZ-50 (0–20 μM) for 48 h, and cell death was determined using the MTT assay. Time-course assessment of loss of cell viability is shown in response to (C) doxazosin (25 μM) and (D) DZ-50 (10 μM). Numerical values represent the average of three experiments performed in triplicate (plus or minus standard error of the mean).
DOX = doxazosin; DZ = DZ-50.
3.2. Inhibition of renal cancer cell adhesion and invasion by quinazolines
The adhesion, migration, and invasive properties of 786-0 cells were subsequently evaluated. Exposure to either doxazosin (25 μM) or DZ-50 (10 μM) resulted in significant suppression of RCC adhesion potential to laminin- and fibronectin-coated surfaces in a time-dependent manner (Fig. 2A and 2B). Both quinazoline drugs significantly suppressed the migration and invasion ability of RCC (Fig. 2C and 2D).
Fig. 2.
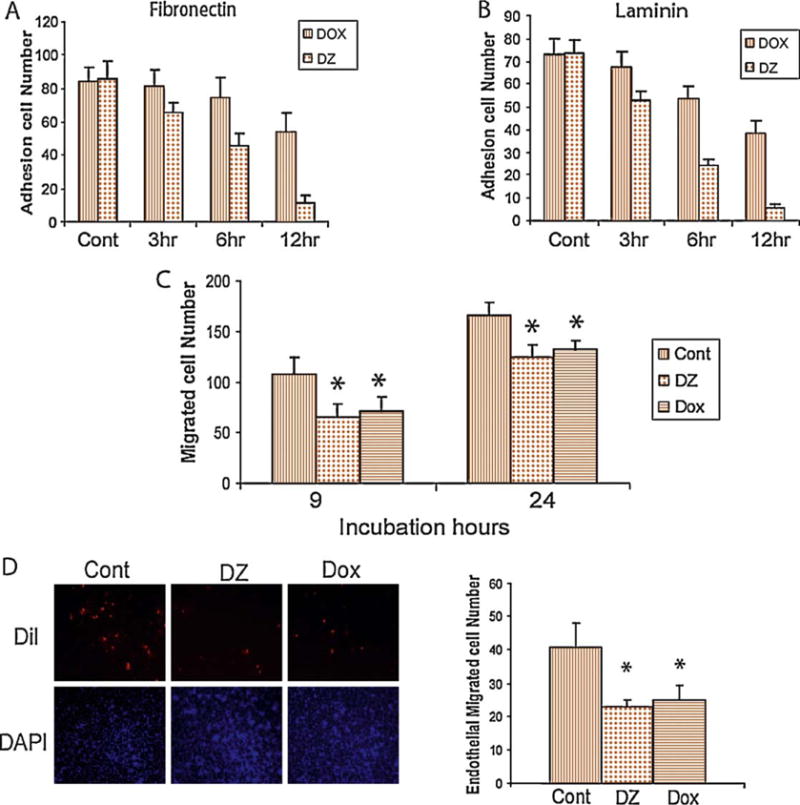
Effect of doxazosin and DZ-50 on migration, adhesion, and invasion in 786-0 and Caki renal carcinoma cells. The number of adherent cells on (A) laminin-coated and (B) fibronectin-coated plates was determined after 3–12 h of exposure to either drug as indicated. (C) Cell migration potential was determined on the basis of a wounding assay. The number of migrating cells was assessed after 9 and 24 h of doxazosin (25 μM) and DZ-50 (10 μM) treatment. (D) Results from the transendothelial migration invasion assay. Quantitative analysis of the data is shown on the left.
DOX = doxazosin; DZ = DZ-50; Cont = control; DiI = octadecyl indocarbocyanines; DAPI = DAPI nucleic acid stain.
*p < 0.05.
3.3. Anoikis induction by DZ-50 and apoptosis by doxazosin
Treatment with either drug (24–48 h) resulted in activation of caspase 8 and sequential activation of caspase 3, as indicated by the cleaved enzyme fragment detected by Western blotting. As shown in Fig. 3A, DZ-50 exposure led to a more profound caspase-8 cleavage compared with that of doxazosin, even at lower concentrations (10 μM). Specific caspase-8 activity was also determined in response to DZ-50 and doxazosin, as well as two agents known to induce apoptosis via the death receptor pathway: Trail and Velcade (Fig. 3B). A significant increase in caspase-8 activity was detected after 12 h of treatment. Annexin-V staining revealed that both drugs induced apoptosis within 24 h (Fig. 3C–3D).
Fig. 3.
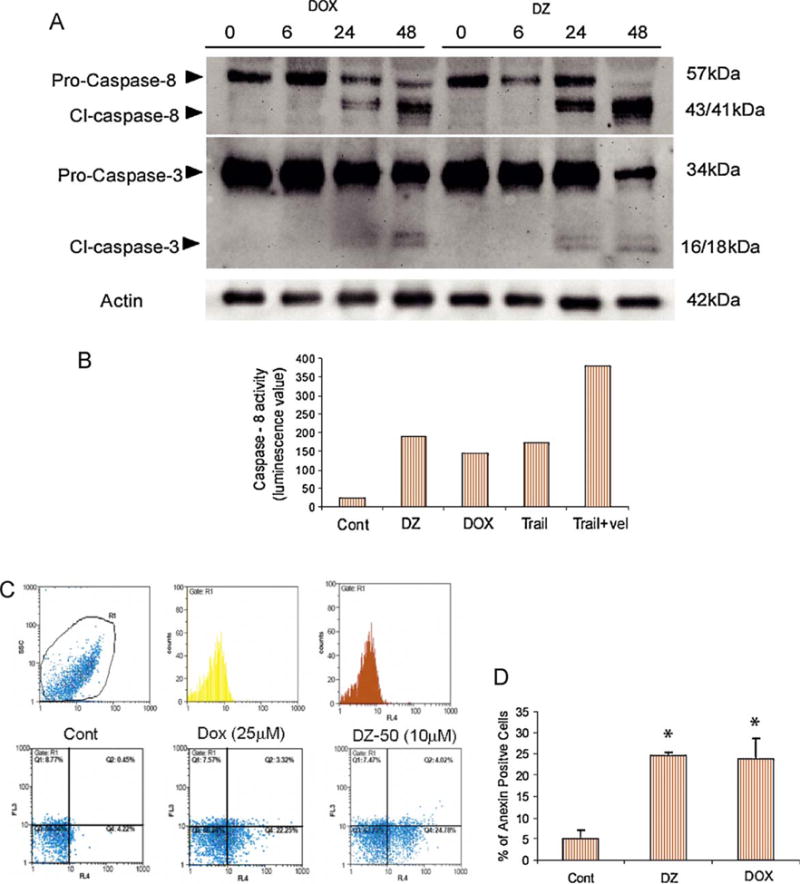
Apoptotic response of renal cancer cells to quinazolines. Human renal cancer 786-0 cells were treated with doxazosin or DZ-50 for increasing periods of time (0–48 h) and (A) cleavage of caspase 8 and caspase 3 was determined by Western blotting using the respective antibodies. (B) Data from evaluation of caspase-8-specific activity in 786-0 cells after treatment with DZ-50 (DZ; 10 μM), doxazosin (DOX; 25 μM), Trail (100 ng/ml), and Velcade (Vel; 100 nM) for 24 h. Average values of caspase-8-specific activity are shown from duplicate experiments. (D) Induction of apoptosis was determined by Annexin-V staining (flow cytometry analysis) following treatment with either DZ-50 or doxazosin for 24 h (right panel). Left panel indicates the average percentage of Annexin-V positive cells.
Cont = control.
*p < 0.01.
The pan-caspase inhibitor ZVAD-FMK significantly inhibited the morphologic changes associated with apoptosis in response to doxazosin in a dose-dependent manner (10–30 μM), while in DZ-50-treated cells, the caspase inhibitor failed to exert a significant effect on apoptosis or detachment (anoikis) (Fig. 4A–4C). The significance of the death-receptor pathway was assessed using stable clones of 786-0 human RCC expressing C-Flip, an endogenous inhibitor of FADD-mediated activation. In response to doxazosin, C-Flip expressing 786-0 cells exhibited a significant decrease in cell viability, while DZ-50 failed to trigger a similar effect (Fig. 4D and 4E).
Fig. 4.
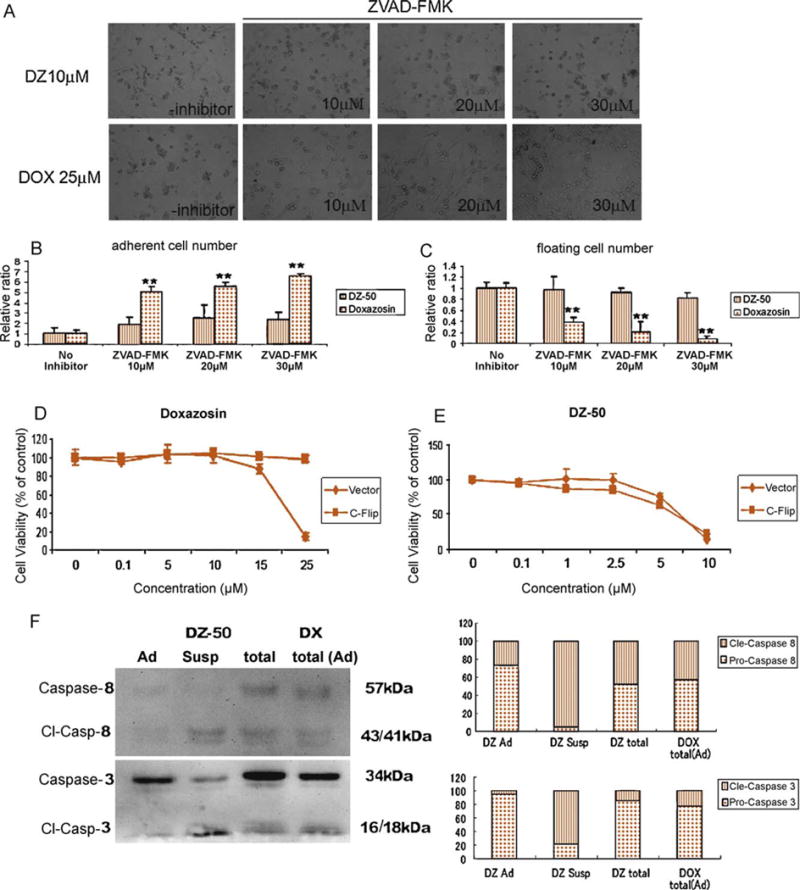
Induction of apoptosis by doxazosin and anoikis by DZ-50 in renal cancer cells 0-786, which were treated with either doxazosin or DZ-50 for 48 h in the presence or absence of the pan–caspase inhibitor ZVAD-FMK (10–30 μM). (A) Cell morphology was examined under light microscopy; (B) the ratio of adherent cells relative to the control cells (no inhibitor) was quantified; (C) the ratio of floating (suspension) cells relative to controls was also determined. Response of C-Flip and vector stably expressing 786-0 renal cancer cells to (D) doxazosin and (E) DZ-50, respectively (dose response). (F) Comparison of caspase 8 and caspase 3 cleavage in adherent and detached cells are shown together with total cells (adherent plus suspension) after DZ-50 treatment (10 μM) for 24 h. Doxazosin treatment failed to induce detachment within 24 h.
DOX = doxazosin; DZ = DZ-50; Cont = control; susp = suspension; ad = adherent.
To determine the detachment dynamics in response to DZ-50, activation of caspase 8 and caspase 3 was assessed at 24 h post treatment. Both compounds led to activation of caspase 8 and caspase 3 (Fig. 3A). Most cleaved forms of caspase 8 and caspase 3, however, were observed in detached cells (suspension), while there was low caspase activity under adherent conditions (Fig. 4F). Caspase activation was consequential to DZ-50-triggered detachment compared with doxazosin’s effect under adherent conditions (Fig. 4F).
3.4. Targeting focal adhesion complex interactions by DZ-50
To study the mechanism driving DZ-50-mediated anoikis, the integrin-focal adhesion signaling repertoire was investigated. As shown on Fig. 5A, exposure of RCC 786-0 to DZ-50 inhibited phosphorylation of focal adhesion-dependent FAK (Y397) within the first 3 h, and by 12 h there was a significant decrease in phosphorylated FAK levels. This was paralleled by a decrease in phosphorylated AKT (Ser473) levels in response to DZ-50. Since GSK-3β, a downstream target of ILK-1, contributes to anoikis resistance in other cell systems [15,18], its phosphorylation pattern was assessed in response to quinazolines. As shown in Fig. 5A, a marked reduction in phosphorylated GSK-3β was detected after 12 h of treatment with DZ-50. In contrast, no significant differences in either the expression levels or phosphorylation status of focal adhesion signaling effectors were elicited by doxazosin (Fig. 5A). To dissect the anoikis effect of DZ-50 in RCC, we subsequently examined whether formation of the focal adhesion complexes mediated by integrin β1, a cell-adhesion regulator [19], is targeted by DZ-50. Fig. 5B reveals that DZ-50 disrupted ILK-1, FAK, and paxillin binding with integrin β1, while doxazosin had only a modest effect. Confocal microscopy confirmed reduced FAK phosphorylation and revealed disruption of focal adhesion complexes by DZ-50 (Fig. 5C, arrows).
Fig. 5.
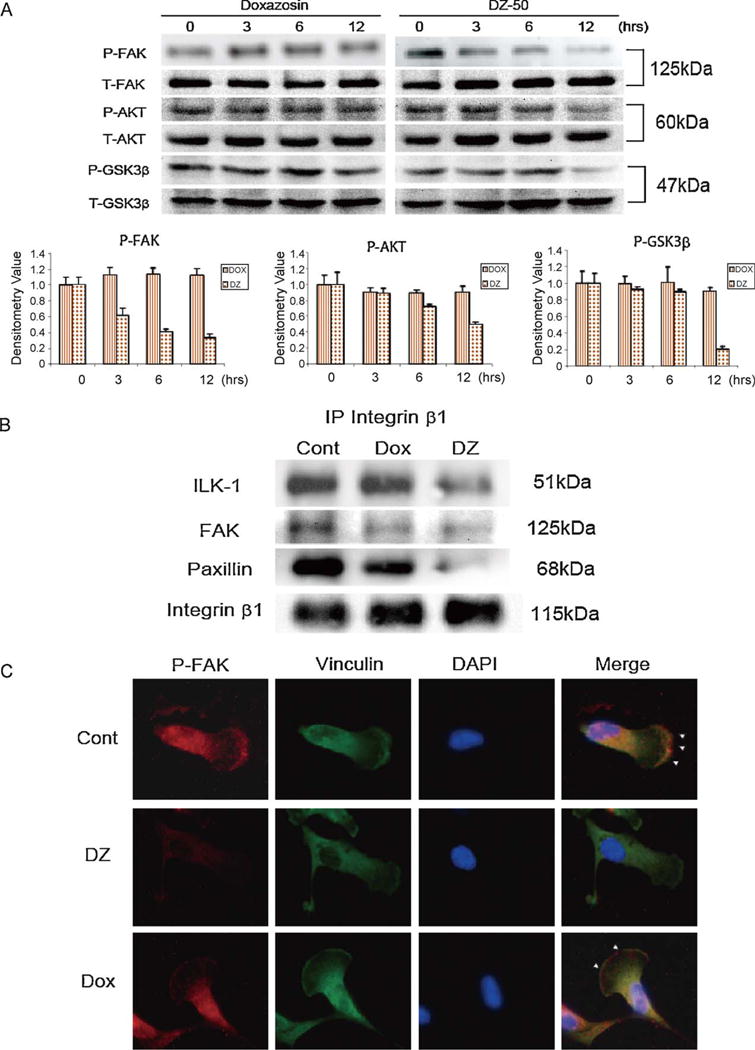
DZ-50 focal adhesion complex targeted by DZ-50 in renal cancer cells. (A) The kinetics of phosphorylation of anoikis-related focal adhesion kinase (FAK) (Y367), AKT (Ser473), and GSK3b signaling players were determined after incubation with DZ-50 (10 μM) and doxazosin (25 μM) for increasing time periods. Relative expression of specific phosphorylated proteins relative to total protein levels (FAK, AKT, and GSK3β) was determined by densitometry. (B) The consequences of quinazolines DZ-50 and doxazosin on renal cell carcinoma 0-786 on the interaction of integrin β1-mediated focal adhesion players was investigated after treatment with either drug for 12 h by immunoprecipitation analysis; the profiling of the association with integrin-linked kinase (ILK)-1, FAK, and paxillin was detected by Western blotting. (C) Phosphorylation status of FAK as examined using confocal microscopy. Vinculin was used as a focal adhesion marker. Colors used for nuclear staining: red = P-FAK; green = vinculin; blue = DAPI stain.
DOX = doxazosin; DZ = DZ-50; Cont = control.
3.5. Quinazoline-mediated inhibition of renal cell carcinoma metastasis in vivo
Fig. 6A indicates that treatment with either doxazosin or DZ-50 at pharmacologically relevant doses significantly decreased formation of RCC lung metastatic lesions compared with the control group of vehicle-treated mice (p = 0.0206 and p = 0.0357, respectively, for doxazosin and DZ-50).
Fig. 6.
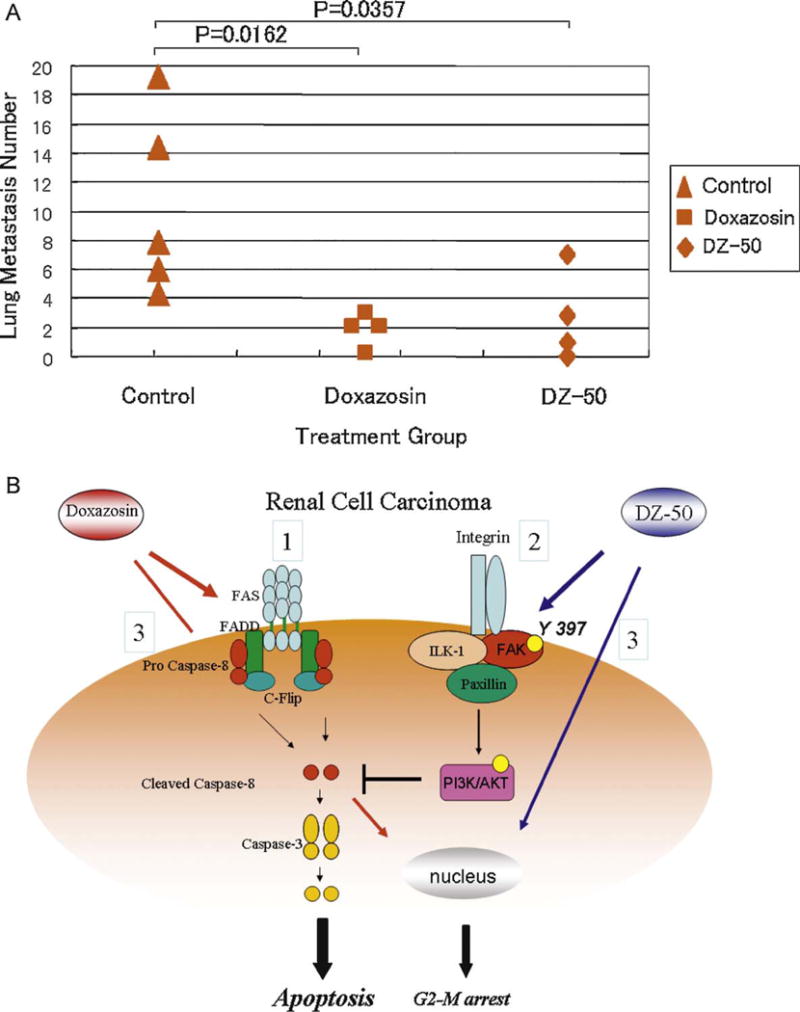
(A) DZ-50 and doxazosin suppressed renal cancer cell metastasis in vivo. Severe combined immunodeficient mice were injected with renal cell carcinoma (RCC) 786-0 in the tail vein. Two weeks after inoculation, mice were treated with doxazosin (100 mg/kg) (n = 4) and DZ-50 (100 mg/kg) (n = 4) via oral gavage three times a week. After 3 wk of treatment, doxazosin and DZ-50 significantly decreased the number of lung metastatic lesions compared to the untreated control mice (p = 0.0162 and p = 0.0357, respectively). (B) Proposed model of quinazoline cell-death action in RCC. Scenario 1: doxazosin-mediated death receptor triggers caspase-8 cleavage, leading to caspase-3 activation and “classic” apoptosis induction. Scenario 2: DZ-50 disrupts integrin-focal adhesion kinase (FAK)-mediated cell-survival pathways, which regulate cellular susceptibility to anoikis signaling stimuli. Scenario 3: quinazoline directly affects G2-M cell-cycle arrest of RCC.
3.6. Effect of doxazosin and DZ-50 on cell-cycle progression of renal cancer cells
Considering the reported effect of quinazolines on cell-cycle arrest in other cell systems [20,21], we examined the effect quinazolines on RCC cell-cycle progression. Within 12–24 h of treatment, both drugs blocked progression through the G2 checkpoint towards mitosis (G2-M arrest) that was associated with a marked decrease in CDC25c and CDK1 expression [22] (Fig. 7).
Fig. 7.
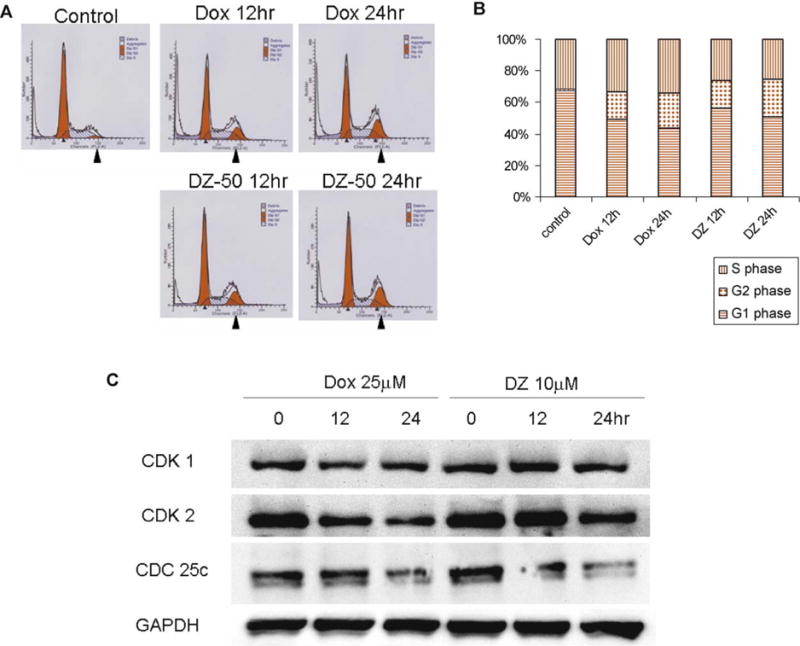
DZ-50 and doxazosin mediating G2-M arrest. (A) After treatment with either doxazosin (Dox) or DZ-50 for 12 and 24 h, cell-cycle analysis was evaluated using propidium iodide staining. (B) The ratio of each cycle phase is shown in barographs. (C) Expression of G2-M arrest-related cell cycle regulatory proteins was analyzed by Western blotting using specific antibodies (CDK-1, CDK-2, and CDC25c). Glyceraldehyde 3-phosphate dehydrogenase was used as a loading control.
4. Discussion
Acquisition of resistance to anoikis is the one of major hallmarks for cancer-cell metastatic potential. Thus, reversing such resistance provides a distinct therapeutic advantage in preventing metastasis. Disruption of cell-survival signals due to loss of integrin-ECM signaling is considered the first step in anoikis induction [10,11]. Integrin β1 recognizes the major adhesive ECM components, fibronectin and laminin, in its regulation of cell survival and mediates anoikis resistance [23,24] by activating intracellular signaling effectors such as FAK and phosphatidylinositol 3-kinase (PI 3-kinase)/AKT.
Our drug optimization efforts recently identified a quinazoline lead compound, DZ-50, which exerts potent antiangiogenic properties via anoikis induction, impairing tumor vascularity [9]. The present findings demonstrate that doxazosin induced apoptosis in two human RCC lines, 0-786 and Caki, and this cell-death action was significantly abrogated by C-Flip, an endogenous FADD-mediated apoptosis inhibitor, indicating the participation of the death-receptor apoptotic signaling pathway. These observations resonate with the mechanism of doxazosin induced in prostate cancer cells [8]. In contrast, DZ-50 induced RCC death via anoikis. DZ-50 inhibited FAK phosphorylation within 3 h and suppressed critical focal adhesion complex downstream survival signaling events, including activation of AKT and GSK-3β and disruption of integrin-mediated focal adhesion complexes, including FAK, ILK-1, and paxillin. Temporal analysis of cell death revealed that in response to DZ-50, anoikis occurs prior to apoptosis; moreover, DZ-50 reduces FAK phosphorylation as an early event via disruption of integrin β1-mediated focal adhesion complexes, thus sensitizing cells to anoikis. Ongoing studies are pursuing the focal adhesion partners critical for conferring anoikis resistance, (eg, talin) as potential targets of DZ-50-induced anoikis in metastatic tumors.
ILK regulates several integrin-mediated cellular processes, including cell adhesion, fibronectin-ECM assembly, and anchorage-dependent cell growth [13,16,17]. On cell adhesion, ILK is transiently activated and directly phosphorylates AKT Ser473 [25] and GSK3 [17]. In contrast, inhibiting ILK in cancer cells blocks AKT phosphorylation and cell survival. Therefore, ILK-1 emerges as a major regulator of anoikis resistance [18,26,27]. Our results establish that DZ-50 can disrupt the ILK-1/integrin β1 complex and reduce phosphorylation of GSK-3β, a downstream target of ILK-1 signaling, in RCC. In squamous cell carcinoma, the peroxisome proliferator-activated receptor-γ (PPARγ) antagonist inhibited cell adhesion by downregulation of integrin β5 and further inhibited integrin-dependent signals, including phosphorylation of FAK, ERK, and MAPK [28]. The present data, in accord with our previous findings, establish the ability of lead quinazoline compounds to impair cancer-cell invasion via an anoikis effect [9]. This study suggests a potential therapeutic value of quinazoline-mediated anoikis in targeting renal tumor vascularity and effectively impairing metastasis (Fig. 6B).
5. Conclusions
Quinazoline-based drugs trigger anoikis in renal cancer cells by targeting the focal adhesion survival signaling. This potent antitumor action against human RCC suggests a novel quinazoline-based therapy targeting renal cancer.
Acknowledgments
The authors thank Patrick Hensley for critically reading the manuscript and Lorie Howard for her assistance in the electronic submission of the manuscript.
Funding/Support and role of the sponsor: This work was supported by grants from the National Institutes of Health, R01 CA107575-6, and the Department of Defense, W81XWH-08-1-0431 (to N.K.). The sponsors were involved in design and conduct of the study; data collection, management, analysis, and interpretation; and preparation of the manuscript.
Footnotes
Author contributions: Natasha Kyprianou had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Kyprianou, Sakamoto.
Acquisition of data: Sakamoto.
Analysis and interpretation of data: Kyprianou, Sakamoto, Schwarze.
Drafting of the manuscript: Kyprianou, Sakamoto, Schwarze.
Critical revision of the manuscript for important intellectual content: Kyprianou.
Statistical analysis: Sakamoto.
Obtaining funding: Kyprianou, Sakamoto.
Administrative, technical, or material support: Kyprianou, Schwarze.
Supervision: Kyprianou.
Other (specify): None.
Financial disclosures: I certify that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: None.
References
- 1.Motzer RJ, Bander NH, Nanus DM. Renal-cell carcinoma. N Engl J Med. 1996;335:865–75. doi: 10.1056/NEJM199609193351207. [DOI] [PubMed] [Google Scholar]
- 2.Kirkali Z, Tuzel E, Mungan MU. Recent advances in kidney cancer and metastatic disease. BJU Int. 2001;88:818–24. doi: 10.1046/j.1464-4096.2001.02442.x. [DOI] [PubMed] [Google Scholar]
- 3.Schrader AJ, Varga Z, Pfoertner S, Goelden U, Buer J, Hofmann R. Treatment targeted at vascular endothelial growth factor: a promising approach to managing metastatic kidney cancer. BJU Int. 2006;97:461–5. doi: 10.1111/j.1464-410X.2006.05873.x. [DOI] [PubMed] [Google Scholar]
- 4.Gore ME, Szczylik C, Porta C, et al. Safety and efficacy of sunitinib for metastatic renal-cell carcinoma: an expanded-access trial. Lancet Oncol. 2009;10:757–63. doi: 10.1016/S1470-2045(09)70162-7. [DOI] [PubMed] [Google Scholar]
- 5.Kyprianou N. Doxazosin and terazosin suppress prostate growth by inducing apoptosis: clinical significance. J Urol. 2003;169:1520–5. doi: 10.1097/01.ju.0000033280.29453.72. [DOI] [PubMed] [Google Scholar]
- 6.Benning CM, Kyprianou N. Quinazoline-derived alpha1-adrenoceptor antagonists induce prostate cancer cell apoptosis via an alpha1-adrenoceptor-independent action. Cancer Res. 2002;62:597–602. [PubMed] [Google Scholar]
- 7.Partin JV, Anglin IE, Kyprianou N. Quinazoline-based alpha 1-adrenoceptor antagonists induce prostate cancer cell apoptosis via TGF-beta signalling and I kappa B alpha induction. Br J Cancer. 2003;88:1615–21. doi: 10.1038/sj.bjc.6600961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garrison JB, Kyprianou N. Doxazosin induces apoptosis of benign and malignant prostate cells via a death receptor-mediated pathway. Cancer Res. 2006;66:464–72. doi: 10.1158/0008-5472.CAN-05-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garrison JB, Shaw YJ, Chen CS, Kyprianou N. Novel quinazoline-based compounds impair prostate tumorigenesis by targeting tumor vascularity. Cancer Res. 2007;67:11344–52. doi: 10.1158/0008-5472.CAN-07-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–26. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13:555–62. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 12.Rennebeck G, Martelli M, Kyprianou N. Anoikis and survival connections in the tumor microenvironment is there a role in prostate cancer metastasis? Cancer Res. 2005;65:11230–5. doi: 10.1158/0008-5472.CAN-05-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, et al. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature. 1996;379:91–6. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- 14.Li F, Liu J, Mayne R, Wu C. Identification and characterization of a mouse protein kinase that is highly homologous to human integrin-linked kinase. Biochim Biophys Acta. 1997;1358:215–20. doi: 10.1016/s0167-4889(97)00089-x. [DOI] [PubMed] [Google Scholar]
- 15.Wu C, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol. 2001;155:505–10. doi: 10.1083/jcb.200108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radeva G, Petrocelli T, Behrend E, et al. Overexpression of the integrin-linked kinase promotes anchorage-independent cell cycle progression. J Biol Chem. 1997;272:13937–44. doi: 10.1074/jbc.272.21.13937. [DOI] [PubMed] [Google Scholar]
- 17.Cieslik K, Zembowicz A, Tang JL, Wu KK. Transcriptional regulation of endothelial nitric-oxide synthase by lysophosphatidylcholine. J Biol Chem. 1998;273:14885–90. doi: 10.1074/jbc.273.24.14885. [DOI] [PubMed] [Google Scholar]
- 18.Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nature Rev. 2005;5:51–63. doi: 10.1038/nrc1524. [DOI] [PubMed] [Google Scholar]
- 19.Crowe DL, Ohannessian A. Recruitment of focal adhesion kinase and paxillin to beta1 integrin promotes cancer cell migration via mitogen activated protein kinase activation. BMC Cancer. 2004;4:18. doi: 10.1186/1471-2407-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kintscher U, Wakino S, Kim S, et al. Doxazosin inhibits retinoblastoma protein phosphorylation and G(1)→S transition in human coronary smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:1216–24. doi: 10.1161/01.atv.20.5.1216. [DOI] [PubMed] [Google Scholar]
- 21.Arencibia JM, Del Rio M, Bonnin A, Lopes R, Lemoine NR, Lopez-Barahona M. Doxazosin induces apoptosis in LNCaP prostate cancer cell line through DNA binding and DNA-dependent protein kinase down-regulation. Int J Oncol. 2005;27:1617–23. [PubMed] [Google Scholar]
- 22.DiPaola RS. To arrest or not to G(2)-M cell-cycle arrest: Commentary re: A.K. Tyagi et al., Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth inhibition, G(2)-M arrest, and apoptosis. Clin Cancer Res. 2002;8:3311–4. [PubMed] [Google Scholar]
- 23.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–32. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 24.Goel HL, Languino LR. Integrin signaling in cancer. Cancer Treat Res. 2004;119:15–31. doi: 10.1007/1-4020-7847-1_2. [DOI] [PubMed] [Google Scholar]
- 25.Lynch DK, Ellis CA, Edwards PA, Hiles ID. Integrin-linked kinase regulates phosphorylation of serine 473 of protein kinase B by an indirect mechanism. Oncogene. 1999;18:8024–32. doi: 10.1038/sj.onc.1203258. [DOI] [PubMed] [Google Scholar]
- 26.Attwell S, Roskelley C, Dedhar S. The integrin-linked kinase (ILK) suppresses anoikis. Oncogene. 2000;19:3811–5. doi: 10.1038/sj.onc.1203711. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda T, Chen K, Shi X, Wu C. PINCH-1 is an obligate partner of integrin-linked kinase (ILK) functioning in cell shape modulation, motility, and survival. J Biol Chem. 2003;278:51324–33. doi: 10.1074/jbc.M309122200. [DOI] [PubMed] [Google Scholar]
- 28.Masuda T, Wada K, Nakajima A, et al. Critical role of peroxisome proliferator-activated receptor gamma on anoikis and invasion of squamous cell carcinoma. Clin Cancer Res. 2005;11:4012–21. doi: 10.1158/1078-0432.CCR-05-0087. [DOI] [PubMed] [Google Scholar]