

Abstract
Upon detachment from the extracellular matrix, tumor epithelial cells and tumor-associated endothelial cells are capable of overcoming anoikis, gain survival benefits, and hence contribute to the process of metastasis. The focal-adhesion complex formation recruits the association of key adaptor proteins such as FAK (focal-adhesion kinase). Vimentin, paxillin, and talin are responsible for mediating the interaction between the actin cytoskeleton and integrins. Talin is an early-recruited focal-adhesion player that is of structural and functional significance in mediating interactions with integrin cytoplasmic tails leading to destabilization of the transmembrane complex and resulting in rearrangements in the extracellular integrin compartments that mediate integrin activation. Talin-mediated integrin activation plays a definitive role in integrin-mediated signaling and induction of downstream survival pathways leading to protection from anoikis and consequently resulting in cancer progression to metastasis. We recently reported that talin expression is significantly increased in prostate cancer compared with benign and normal prostate tissue and that this overexpression correlates with progression to metastatic disease implicating a prognostic value for talin during tumor progression. At the molecular level, talin is functionally associated with enhanced survival and proliferation pathways and confers anoikis resistance and metastatic spread of primary tumor cells via activation of the Akt survival pathway. In this review, we discuss the growing evidence surrounding the value of talin as a prognostic marker of cancer progression to metastasis and as therapeutic target in advanced prostate cancer, as well as the current understanding of mechanisms regulating its signaling activity in cancer.
Keywords: Talin, Integrins, Anoikis, Metastasis, Migration, Invasion, Focal adhesions
1. Introduction
Cancer metastasis is a multistep and complex process involving the spread of malignant cells from a primary tumor to distant sites. It specifically includes epithelial to mesenchymal transition (EMT) and penetration of the basement membrane, degradation of the extracellular matrix (ECM) and invasion of surrounding tissues, cell migration, anchorage-independent growth, apoptosis evasion, intravasation into existing and newly formed blood and lymph vessels, transportation through the vessels and extravasation, establishment of surviving cancer cells at distant sites, and outgrowth of secondary tumors (Chambers et al., 2002; Gupta and Massague, 2006; Sakamoto and Kyprianou, 2010). Cancer metastasis imposes the biggest problem to treatment and prognosis of the disease and is the main cause of death of cancer patients. Approximately 90% of all cancer deaths arise from the metastatic spread of tumors (Christofori, 2006). The tumor microenvironment has been addressed as a critical regulator of cancer progression and metastasis (Aguirre-Ghiso, 2007; Barkan et al., 2010; Chambers et al., 2002). It consists of the tumor stroma and surrounding tissue, composed of endothelial cells, pericytes adjacent to the endothelial cells, invading inflammatory cells and leucocytes, fibroblasts, and extensive ECM structures (Hanna et al., 2009). ECM represents a major component of the microenvironment as it is in immediate contact with the tumor cells and functions as a source of growth factors and cytokines which are critical for different aspects of tumor biology and progression. In this review, we aim to summarize the most recent evidence on the functional contribution of the ECM to cancer metastasis with particular focusing on talin, a key player in focal-adhesion activity regulating cancer cell survival throughout migration.
2. ECM in Control of Microenvironment
Alterations in the expression of ECM-related genes have been highlighted in gene expression profiling of cancers that are related to poor prognosis and metastasis (Barkan et al., 2010). Further, alterations in the gene expression signature of tumors can result in extensive remodeling of the ECM, an occurrence associated with poor outcomes (Chang et al., 2005a,b). Changes in the ECM components such as increased production and organization of fibronectin have been implicated in inducing metastatic growth. Except remodeling, the ECM can undergo degradation by matrix metalloproteinases (MMP) that are prominently secreted by stroma or tumor cells (Jodele et al., 2006; Vlodavsky et al., 2002). MMPs, by degrading the ECM, may contribute to the establishment of a microenvironment that supports tumor dormancy or its switch to metastatic ability (Barkan et al., 2010). Many growth factors like fibroblast growth factors (FGFs) and vascular endothelial growth factors (VEGFs) bind to heparin and to heparan sulfate, a component of many ECM proteoglycans (Hynes, 2009). Heparanase, an important protein ubiquitously expressed in human tumors, is an endoglycosidase that cleaves heparan sulfate chains of proteoglycans and is associated with the cell surface and the ECM in different tissues (Vlodavsky et al., 2002). Degradation and remodeling of the ECM by heparanase releases angiogenic factors and stimulates the angiogenic switch required in cancer metastasis (Roy and Marchetti, 2009; Vlodavsky et al., 2002).
Tumor-associated MMPs can also stimulate processes associated with EMT, thus contributing in a further way to metastasis progression (Orlichenko and Radisky, 2008; Przybylo and Radisky, 2007; Radisky and Radisky, 2010). During EMT, cells progressively downregulate their apical and basolateral epithelial-specific tight and adherens junction proteins, and reexpress mesenchymal molecules (Christofori, 2006; Huber et al., 2005; Thiery, 2002). Nonmotile, polarized epithelial cells, by dissolving their cell–cell junctions, convert into individual nonpolarized, motile, and invasive mesenchymal cells. Functional expression of the epithelial cell–cell adhesion molecule E-cadherin is lost, whereas the expression of the mesenchymal cell–cell adhesion molecule N-cadherin is induced, a process also known as the cadherin switch (Yilmaz and Christofori, 2009). Loss of E-cadherin gene expression is frequently found during tumor progression in most epithelial cancers; thus, this protein has been established as a clinical indicator for poor prognosis and metastasis (Bissell and Radisky, 2001; Cavallaro and Christofori, 2004). Forced downregulation of E-cadherin activity promotes tumor invasion and metastasis in vivo (Perl et al., 1998). As E-cadherin plays a key role in epithelial structural integration and homeostasis, its expression is under strict control. In the tumor microenvironment, a number of growth factors can induce EMT including, transforming growth factor β (TGF-β), epidermal growth factor (EGF), insulin-like growth factor (IGF), and FGF. E-cadherin is selectively downregulated by EGF receptor (EGFR), c-Met, insulin-like growth factor receptor I (IGF-RI), FGF receptors (FGFRs), while the non-RTK c-Src induces phosphorylation of E-cadherin and catenins, resulting in their ubiquitylation by the E3 ligase Hakai, and subsequent endocytosis and degradation (Fujita et al., 2002).
Expression of cell adhesion molecules on cancer epithelial and endothelial cells is a dynamic and highly regulated process under the presence of growth factors, cytokines, and chemokines, and highly dependent on the composition of the ECM (Cooper et al., 2002; Khatib et al., 1999; Stewart et al., 2004). The extracellular binding activity of integrins is regulated from the inside of the cell (inside-out signaling), while the binding of the ECM elicits signals that are transmitted into the cell (outside-in signaling) (Clark and Brugge, 1995; Shattil et al., 2010). Integrins can shift between high- and low-affinity conformations for ligand binding, and this shift from low- to high-affinity state results in integrin activation (Legate et al., 2009). Failure to activate integrin results in aberrant development and pathological conditions such as bleeding disorders, skin blistering, and leukocyte-adhesion deficiencies (Giancotti and Ruoslahti, 1999).
Cell responses to extracellular stimuli, such as regulating cell–cell and cell–substrate attachment, and increasing cell motility are accompanied by changes in the expression and function of adhesion receptors including the integrin family (Albelda, 1993; Stewart et al., 2004). Integrins are composed of two subunits α and β, and each α–β combination has its own binding specificity and signaling properties, while most integrins recognize several ECM proteins (Hynes, 1987; Shattil et al., 2010). In mammals, 18 α and 8 β subunits combine to form 24 specific dimmers in a noncovalent bound manner, which exhibit different ligand-binding properties (Moser et al., 2009). Integrin subunits have large extracellular domains (~800 amino acids) that are responsible for ligand binding, small single transmembrane domains of 20 amino acids, and short cytoplasmic tails ranging from 13 to 70 amino acids, except that of β4 (Moser et al., 2009). Integrin clustering occurs by inside-out signals resulting in formation of hetero-oligomers that stimulate the recruitment of protein complexes to integrin cytoplasmic domains (Critchley and Gingras, 2008). Clustering is important for inducing integrin recycling, outside-in signaling, and transduction by adhesion-based intracellular structures (Puklin-Faucher and Sheetz, 2009). Binding of ligands to integrin extracellular domains by the homodimerization of integrin transmembrane domains (α-to-α or β-to-β) (Li et al., 2003a,b), or by the release of integrins from cytoskeletal complexes, leads to the free diffusion of integrins in the plane of the membrane resulting in integrin clustering (Kucik, 2002). Interactions of integrin cytoplasmic domains with each other or cytoplasmic proteins lead to rearrangements of the integrins that induce its activation (O’Toole et al., 1991, 1994). Integrin β chains interact with actin-binding proteins (e.g., talin and filamin), which form mechanical links to the cytoskeleton, and other proteins like focal-adhesion kinase (FAK), integrin-linked kinase (ILK), and novel proteins that link integrins to signaling mechanisms and, in some cases, mediate integrin-dependent gene regulation (e.g., JAB1; Liu et al., 2000). Conformational changes induced by external ligands can be propagated across the plasma membrane, leading to alteration of the α- and β-integrin tails (Kim et al., 2003). As integrins bind to ECM, they associate with the cytoskeleton and promote the assembly of actin filaments (the α6β4 integrin associates with keratin filaments through the uniquely large β4 cytoplasmic domain; Giancotti and Ruoslahti, 1999). Further, organization of actin filaments into larger fibers supported by integrin clustering in turn promotes more integrin clustering, thus enhancing the matrix binding and organization by integrins in a positive feedback system (Giancotti and Ruoslahti, 1999). Consequently, ECM proteins, integrins, and cytoskeletal proteins assemble into aggregates forming focal adhesions (Burridge and Chrzanowska-Wodnicka, 1996). The cell type and composition of the surrounding matrix, along with tissue origin, determine which set of integrins are critical in transducing downstream survival signals (Chiarugi and Giannoni, 2008; Giancotti, 2000). Integrins play a pivotal role in normal homeostasis as well as oncogenic transformation (Chiarugi and Giannoni, 2008; Giancotti, 2000; Guo and Giancotti, 2004; Ramsay et al., 2007).
Ample evidence indicates abnormal integrin expression as prostate cancer progresses to an advanced stage with most α and β subunits shown to be downregulated (Fornaro et al., 2001; Goel et al., 2008; Knudsen and Miranti, 2006). Integrins directly interfere with the ECM and are connected to the actin cytoskeleton through focal adhesions thus regulating structural rearrangements and signaling pathways essential in cell movement (Juliano et al., 2004; Webb et al., 2002). Members of the Rho family of GTPases contribute in the regulation of the actin cytoskeleton, with RhoA, Rac, and CDC42 influencing stress fibers, lamellipodia, and filopodia (Juliano et al., 2004). Other signaling components of the focal-adhesion complex also influence the actin–myosin machinery, and integrins can closely modulate the functional interaction and signaling potential of those focal-adhesion proteins (Juliano et al., 2004). Moreover, MAP kinases as the principal downstream effectors of Ras signaling are regulated by integrins; thus, integrins partially take part in the regulation of cell proliferation and differentiation (Pearson et al., 2001). In breast, prostate, melanoma, and fibrosarcoma cell lines, a high ERK/p38 activity ratio favors tumor growth and activation of α5β1 integrin is proposed as a determinant for the in vivo ERK/p38 mediated growth promoting activity (Aguirre-Ghiso et al., 2003). Integrins may also promote cancer progression by regulating survival, invasion, and angiogenesis, leading to metastasis (Avraamides et al., 2008; Finger and Giaccia, 2010; Jin and Varner, 2004; Moschos et al., 2007; Tantivejkul et al., 2004).
Integrin antagonists, including function-blocking antibodies and peptide molecules with high affinity and specificity, are currently under investigation (Avraamides et al., 2008; Fig. 4.1). MEDI-522, a humanized ανβ3 antibody, was the first anti-integrin agent to be tested in clinical trials for advanced malignancies showing promising results in tumor perfusion and inhibition of angiogenesis (McNeel et al., 2005; Zhang et al., 2007). CNTO 95, a human αvβ3/αvβ5 antibody, reduced angiogenesis and tumor growth in human melanoma xenografts in nude mice and rats and was further tested in Phase I/II clinical trials for advanced solid tumors (Mullamitha et al., 2007; Trikha et al., 2004). Further, cilengitide, a cyclic peptide antagonist of integrins αvβ3/αvβ5, was developed and evaluated in Phase I/II clinical trials for recurrent malignant glioma, where significantly enhanced progression-free survival was observed (Nabors et al., 2007). In addition, cilengitide was further applied in Phase II trials for non-small-cell lung cancer, melanoma, and pancreatic cancer in combination with other chemotherapeutic agents (Beekman et al., 2006; Friess et al., 2006). Another drug in clinical trials, ATN-161, is a peptide inhibitor of integrin α5β1 that reduced metastases and improved survival when combined with chemotherapy in colon cancer animal studies and was further processed for Phase II clinical trials for patients with solid tumors resulting in prolonged stable disease (Cianfrocca et al., 2006; Stoeltzing et al., 2003).
Figure 4.1.
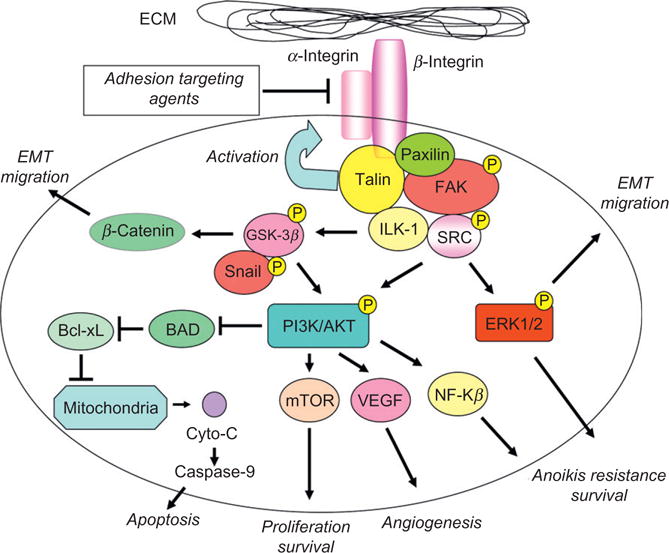
ECM–cell communication is dictated by focal-adhesion players. Activation of integrins by talin induces multiple downstream signaling cascades via formation and functional activation of the focal-adhesion complex. The integrity of focal adhesions is constitutively maintained by active interaction between the focal-adhesion complex players (such as FAK, paxilin, and Src) downstream that regulate phosphorylation and activation of the PI3K/AKT survival pathway resulting in anoikis resistance, increased angiogenesis, and survival after cell detachment from the ECM. ERK1/2 and GSK-3β are concurrently activated conferring not only a survival advantage for the tumor cells but also promoting the migratory and invasive properties via induction of EMT.
3. Epithelial to Mesenchymal Transition
EMT is temporary and reversible phenomenon characterized by the acquisition of mesenchymal phenotype by cancer cells resulting in loss of cell–cell adhesion, loss of cell polarity, and the acquisition of migratory and invasive properties leading to cancer progression and metastatic spread (Thiery et al., 2009; Yilmaz and Christofori, 2009). TGF-β is a prominent regulator of EMT. In response to TGF-β, Smad2 and 3 are activated and form trimers with Smad4, which upon nuclear translocation, regulate transcription of target genes by facilitating the interaction with other DNA binding transcription factors. EMT in response to TGF-β is achieved through a well-characterized transcription program that involves three families of transcription factors, the Snail, ZEB, and helix-loop-helix bHLH families (Xu et al., 2009). Their expression is induced either through a Smad-dependent mechanism or indirectly through activation of other transcription factors or relief of repression (Xu et al., 2009). Upon activation, these three families of transcription factors suppress expression of epithelial markers and in turn activate mesenchymal marker gene expression. Orchestrated signaling between TGF-β and Ras/Raf/MEK/MAPK is required for maintenance of EMT in various epithelial cell types (Zavadil and Bottinger, 2005). Other proteins which are engaged by TGF-β-induced EMT are the small GTPases RhoA and Rac1 (Bakin et al., 2002; Bhowmick et al., 2001), phosphoinositol-3 kinase (PI3K; Bakin et al., 2000), ILK (Lee et al., 2004), and the Jagged1/Notch signaling pathway (Zavadil et al., 2004).
EGF increases N-cadherin expression as a consequence of increased expression of TWIST, SLUG, and Snail in cancer cells, while expression of E-cadherin is significantly repressed (Lo et al., 2007). Chromatin immunoprecipitation studies reveal that EGF induces binding of nuclear signal transducer and activator of transcription 3 (STAT3) to the TWIST promoter; moreover, immunohistochemical analysis of primary breast carcinomas indicates positive correlations between nonnuclear EGFR and TWIST, and between activated STAT3 and TWIST (Lo et al., 2007). Although induction of ZEB1 by TGF-β promotes EMT, IGF-I is also found to play a role in the regulation of ZEB1. IGF-I upregulates ZEB1 expression in epithelial prostate cancer cells toward EMT ultimately promoting prostate tumor cell migration in vitro (Graham et al., 2008). In prostate cancer cells displaying a mesenchymal phenotype, ZEB1 inhibition reverses the EMT phenotype by increased expression of E-cadherin and downregulation of N-cadherin and fibronectin (Graham et al., 2008). FGFR1 is another receptor commonly overexpressed in advanced prostate cancer. In an inducible FGFR1 prostate mouse model, it was shown that activation of FGFR1 led stepwise progression to adenocarcinoma that was linked to EMT (Acevedo et al., 2007). After induction with FGF, mesenchymal-like cells expressed lower and more diffuse levels of E-cadherin, and a high percentage of these cells lost cytokeratin expression associated with loss of epithelial differentiation; at the same time, expression of vimentin was increased in those cells in comparison to adjacent glandular epithelial cells (Acevedo et al., 2007). Changes in the composition of the ECM are also able to induce EMT, as shown for collagen I that induces EMT in non-small-cell lung cancer cells (Shintani et al., 2008). Moreover, hyaluronan promotes anchorage-independent growth and invasiveness, induces gelatinase production, and stimulates PI3K/Akt pathway activity in phenotypically normal kidney and mammary epithelial cells (Zoltan-Jones et al., 2003).
The intermediate filament protein vimentin is mainly expressed in mesenchymal cells and has been annotated a marker for EMT. Correlation between vimentin and inhibition of E-cadherin mediated cell adhesion has been shown along with increased vimentin protein levels and loss of E-cadherin expression in invasive breast or lung cancer cells (Polette et al., 1998; Sommers et al., 1994). In addition, expression of vimentin in combination with cytokeratins has been strongly associated with highly aggressive and metastatic breast cancer. Estrogen receptor (ER)-negative cells exhibiting aggressive phenotypes express vimentin, whereas noninvasive ER-positive cells do not (Sommers et al., 1989). Vimentin downregulation leads to a significant decrease in tumor cell invasion or migration, evidence indicating a functional contribution of vimentin invasion and metastasis (Gilles et al., 1999; Hendrix et al., 1997). In prostate cancer, expression of vimentin is very modest, in well-differentiated or in moderately differentiated tumors, while poorly differentiated tumors and bone metastases showed high vimentin expression that correlates with high invasive ability (Zhao et al., 2008). Moreover, silibinin, the major active constituent of silymarin isolated from milk thistle (Silybum marianum), not only exerts growth inhibitory effects by induction of apoptosis together with cell cycle arrest but also reduces the invasive and migrating abilities in bone metastatic prostate cancer cells by inhibiting vimentin and MMP-2 (Wu et al., 2009).
Paxillin is a 68-kDa focal-adhesion protein, with four tandem LIM domains at the C-terminus, involved in growth factor receptor, integrin, and oncogenic signaling such as Src (Salgia et al., 1999). As part of the focal-adhesion complex, paxillin is tyrosine phosphorylated in response to growth factors such as PDGF and EGF, IL-3, and neuropeptides like bombesin (Charlesworth et al., 1996; Rankin et al., 1996; Salgia et al., 1995). Multiple kinases, including FAK and ILK, directly interact with paxillin, while activated β1 and β2 integrins also lead its activation via protein phosphorylation (Charlesworth et al., 1996). Indeed, a mutant paxillin protein lacking both FAK-binding sites characteristically exhibits reduced tyrosine phosphorylation (Thomas et al., 1999). Activation of FAK by anchoring to the cell membrane and recruitment of paxillin sufficiently stimulates ERK and c-Jun NH (2)-terminal kinases (JNKs) in a PI3K/Akt-independent manner (Igishi et al., 1999). Paxillin overexpression results in stimulation of squamous cell carcinoma lines to migrate on type IV collagen and through reconstituted basement membrane which is dependent on ERK activity (Crowe and Ohannessian, 2004).
Gene silencing of mutant paxillin led to reduction of cell viability, while A127T Paxillin showed an increase in tumor growth, cell proliferation, and invasion in vivo (Jagadeeswaran et al., 2008). Fibronectin promotes tyrosine (Tyr118) phosphorylation of paxillin, via activation of the focal-adhesion complex, resulting in cell invasiveness in gastric tumor cells (Li et al., 2009). In bladder tumors, IGF-IR activation does not significantly affect their growth, but it notably promotes migration and stimulates in vitro wound healing and invasion (Metalli et al., 2010). These IGF-IR mediated effects are apparently dependent upon the activation of Akt and MAPK pathways, as well as IGF-I-induced Akt- and MAPK-dependent phosphorylation of paxillin (Metalli et al., 2010). In prostate cancer cells, Paxilllin is shown to regulate Erk signaling and cell proliferation after induction of EGFR by dihydrotestosterone (DHT) via androgen receptor (AR) binding and MMP-mediated release of EGFR ligands (Sen et al., 2010).
4. Anoikis Resistance in Metastasis
Anoikis, a Greek word meaning homelessness, is a unique mode of apoptosis induced after loss of cell adhesion to ECM (Sakamoto and Kyprianou, 2010). The role of ECM as a suppressor of apoptosis has been well established, and anoikis, following loss of cell anchorage, is of physiological relevance for development, tissue homeostasis, and disease progression including cancer metastasis (Rennebeck et al., 2005). The ability of cancer cells to survive in the absence of adhesion to the ECM (anoikis resistance) enables them to develop anchorage independence, disseminate from the primary tumor, invade a distant site, and establish a metastatic lesion (Chiarugi and Giannoni, 2008). Cell detachment from the ECM induces downregulation of the antiapoptotic Bcl2 family member Bcl-xL along with significant increase of Fas ligand (FasL) which activates the death receptor pathway (Rosen et al., 2000, 2002). Alterations in cell–cell adhesion molecules, protein kinases and phosphatases, integrin-associated signaling molecules, or apoptosis regulators can confer resistance to anoikis and promote progression to metastasis (Liotta and Kohn, 2004). Focal-adhesion complexes and downstream survival pathways play a definitive role in integrin-mediated signal transduction leading to protection from anoikis (Chiarugi and Giannoni, 2008; Sakamoto and Kyprianou, 2010; Sakamoto et al., 2010; Fig. 4.1).
Mechanistically, anoikis resistance is controlled by constitutive activation of FAK (Frisch et al., 1996), EGFR-mediated Src activation that leads to MEK/Erk and PI3-K/Akt-1 signaling (Demers et al., 2009), increased cytosolic c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long (c-FLIP) that inhibits CD95-induced caspase-8 activation and apoptosis (Shain et al., 2002), overexpression of β-catenin that regulates the function of the LEF/TCF family of transcription factors, and protects cancer cells from suspension-induced anoikis (Orford et al., 1999). Increased Bcl-xL has been also implicated in resistance to anoikis during colorectal tumor progression (Coll et al., 2002). Talin1 enhances prostate cancer cell adhesion, migration, and invasion by activating survival signals in response to anoikis (Sakamoto et al., 2010). Src phosphorylation of Bif-1 suppresses the interaction between Bif-1 and Bax, impairing Bax activation during anoikis (Yamaguchi et al., 2008), while p38 mitogen-activated protein kinase (p38MAPK) is necessary in activation of Bax after its translocation to mitochondria and induction of mitochondrial outer membrane permeabilization that results in cytochrome c release and apoptosis (Owens et al., 2009).
Different death regulators can initially confer resistance to anoikis after detachment of tumor cells from the ECM by triggering survival pathways, while they subsequently serve as mediators of cell migration, invasion, and metastasis. For instance, galectins comprise an intriguing group of molecules, overexpression of which in malignant epithelial cells, as well as tumor-associated stroma cells, is directly correlated with acquisition of a metastatic phenotype (Lahm et al., 2004; Oka et al., 2004). In particular, Gal-3 translocation properties make this molecule taking a central role in facilitating malignant transformation and resistance to anoikis, depending on its subcellular localization. Cytoplasmic Gal-3 is antiapoptotic, whereas the nuclear presence of Gal-3 exerts proapoptotic properties (Rennebeck et al., 2005). Gal-3 regulates cell cycle progression by blocking cyclins A and E and by stimulating p27 and p21 (cell cycle-dependent kinase inhibitors), resulting in cell cycle arrest and inhibition of anoikis (Kim et al., 1999). In addition, this protein has in vitro angiogenic properties by inducing endothelial cell migration (Nangia-Makker et al., 2000). TrkB, a neurotrophic tyrosine kinase, upon binding to its ligand is activated and functions as a key suppressor of anoikis, conferring anoikis resistance and promoting metastasis (Douma et al., 2004). Moreover, caveolin (cav-1) is identified as a tumor promoter in bladder, esophageal, and prostate cancers (Fong et al., 2003; Rajjayabun et al., 2001; Satoh et al., 2003). Cav-1 is able to suppress anoikis by both activating Akt and blocking two inhibitors of Akt, PP1 and PP2A (Li et al., 2003a,b). Another potential anoikis inducer is peroxisome proliferator receptor-gamma (PPAR-γ). Inhibition of PPAR-γ results in downregulation of integrin α5 expression, thus impairing FAK signaling and ultimately pronounced anoikis manifestation in squamous and hepatocellular carcinomas (Sakamoto and Kyprianou, 2010). In prostate cancer epithelial cells, IGF interacts with integrin signaling pathways toward anoikis resistance via enhancing tumor cell proliferation and decreasing cell adhesion (Goel et al., 2004). Loss FLIP induces tumor cell death upon detachment from the ECM, while it has no effect on adherent cancer cells (Mawji et al., 2007a,b). Overexpression of the Bcl-2 protein itself can also confer resistance to anoikis ultimately leading to loss of therapeutic response to chemotherapy (Coates et al., 2010).
5. Focal-Adhesion Complex: Securing Cell–ECM Interactions
The focal-adhesion complex formation at the cytoplasmic face of the cell membrane includes the connection of adaptor proteins such as FAK, ILK, vimentin, talin, and paxillin to the cellular actin cytoskeleton as well as to integrins (Fig. 4.1). FAK is a cytoplasmic tyrosine kinase recognized as a key mediator of signaling by integrins in both normal and cancer cells. FAK is activated by integrins through disruption of an autoinhibitory intramolecular interaction between its kinase domain and the amino-terminal FERM domain (Zhao and Guan, 2009). FERM-mediated nuclear localization of FAK promotes enhanced cell survival through the inhibition of tumor suppressor p53 activation during development but also in cancer progression (Lim et al., 2008). Transient dimerization of FAK molecules leads to increased phosphorylation and activation of Tyr397 (Parsons, 2003). The phosphorylation on Tyr397 creates a high-affinity binding site for the SH2 domain of Src family kinases and leads to the recruitment and activation of Src through the formation of a kinase complex (Schaller et al., 1994). Tyr397-dependent activation of FAK and the recruitment of Src have been implicated in the efficient tyrosine phosphorylation of additional sites on FAK as well as the FAK-binding protein paxillin (Owen et al., 1999; Schaller et al., 1999). In addition, binding of the adaptor protein growth factor receptor bound 2/homologue of Drosophila melanogaster “son of sevenless” protein (GRB2/SOS) to the FAK Tyr925 plays a significant role in activating the prosurvival Ras/Raf/MEK/MAPK pathway (Schlaepfer et al., 2004).
ILK interacts within the focal-adhesion plaques with several adaptor and signaling components of the formed complex resulting in its activation and localization (Persad and Dedhar, 2003). The amino-terminal domain of ILK contains four ankyrin repeats, which are essential for localization of ILK to focal adhesions. LIM domains (first described in LIN-11, ISL1, and MEC-3) are protein–protein interaction domains with cysteine-rich zinc-finger structures that usually interact with tyrosine-containing motifs. ILK mediates the phosphorylation of diverse intracellular substrates including protein kinase B (PKB/Akt), glycogen synthase kinase-3 (GSK3), and myosin light chain (Persad and Dedhar, 2003). ILK can phosphorylate AKT on Ser473 and GSK3β on Ser9 in a PI3K-dependent manner (Delcommenne et al., 1998; Olski et al., 2001), while the PI3K-regulating tumor suppressor phosphatase and tensin homologue deleted on the chromosome 10 (PTEN) negatively regulates ILK kinase activity (Persad et al., 2000).
Src is the founding member of a family of comprising nine family members: Src, Fyn, Yes, Lck, Hck, Blk, Fgr, Lyn, and Yrk (Thomas and Brugge, 1997). Each member consists of an amino-terminal domain, an SH3 and SH2 domain, a tyrosine kinase domain, and a carboxy-terminal negative regulatory element (Thomas and Brugge, 1997). Increased Src kinase activity in cancer is correlated with tyrosine phosphatase-mediated dephosphorylation of the carboxy-terminal negative regulatory element, increased Src protein levels and/or altered protein stability, an increase in upstream receptor tyrosine kinase activity, or loss of key regulatory proteins (Playford and Schaller, 2004). Src family kinases can interact with tyrosine kinase receptors, such as EGFR and the VEGF receptor, affect cell proliferation via the Ras/ERK/MAPK pathway, and can also regulate gene expression via transcription factor control such as of STAT molecules (Kim et al., 2009). Src kinase activity is downregulated by the tyrosine carboxy-terminal Src kinase (Csk) that phosphorylates the negative tyrosine residue in the carboxy-terminal tail of Src, resulting in intramolecular interaction that induces an inactive conformation of Src (Nada et al., 1991). Tyrosine phosphorylation of FAK creates a high-affinity binding site for Src, thereby leading to the formation of a stable FAK–Src complex that promotes the phosphorylation of many FAK-associated Src substrates including CAS, paxillin, and p190RhoGAP which have a central role in the reorganization of the actin cytoskeleton and migration (Guarino, 2010; Playford and Schaller, 2004). Overexpression of Src in colon cancer cells enhances primary tumor growth without an increase in metastasis (Irby et al., 1997), while decreased Src protein suppresses cell proliferation in vitro and in vivo (Staley et al., 1997). The effect of elevated Src activity in tumor cells is certainly pleiotropic, as it significantly impacts multiple processes involved tumorogenesis, including cell–cell adhesion, apoptosis, angiogenesis, tumor cell growth, invasion, and EMT (Playford and Schaller, 2004).
Talins are about 270-kDa proteins in size consisting of an N-terminal 47-kDa head domain and a ~220-kDa C-terminal flexible rod domain (Moser et al., 2009). The talin head consists of a FERM (4.1, ezrin, radixin, moesin) domain composed of three subdomains (F1, F2, and F3) and an F0 subdomain with no homology to known domains. The F3 subdomain is responsible for binding to the integrin cytoplasmic tales (Calderwood, 2004; Garcia-Alvarez et al., 2003; Moser et al., 2009; Papagrigoriou et al., 2004). The talin rod domain is composed of a series of helical bundles that contain multiple binding sites for the F-actin-binding protein vinculin and a second integrin-binding site (Critchley and Gingras, 2008). Recent crystallography studies indicated that the structure of talin residues 1359–1659 which contains nine α-helices that are organized into a unique fold with two distinct domains that interact with vinculin and F-actin (Gingras et al., 2010). The C-terminus contains a THATCH (talin/HIP1R/Sla2p actin tethering C-terminal homology) domain that mediates dimerization and provides a direct linkage between talin and F-actin (Senetar et al., 2004; Smith and McCann, 2007).
5.1. Function of talin in integrin activation and focal adhesions
The integrin-binding site for the talin head was mapped to the membrane-proximal NPxY motif, a common binding motif for phosphotyrosine-binding domain-containing proteins (Calderwood et al., 2002; Moser et al., 2009; Uhlik et al., 2005), and it has been revealed that mutations within the NPxY motif of both β1 and β3 integrins, as well as mutations in the talin phosphotyrosine-binding domain, abolish talin binding and decrease integrin affinity (Bouaouina et al., 2008; Tadokoro et al., 2003; Wegener et al., 2007). Dok1, Numb, and tensin have not shown similar properties as talin in regulating integrin activation (Calderwood et al., 2002; McCleverty et al., 2007). That noticeable difference might occur as the talin head has an additional binding site on the β integrin tail, whereas Dok1 binds only to the region surrounding the NPxY motif (Oxley et al., 2008). The Talin F3 subdomain contains an extra loop of amino acids that binds to membrane-proximal sequences in the β3 integrin tail; thus, it is proposed that talin first encounters the β integrin tail by binding the NPxY motif through its phosphotyrosine-binding domain, and the loop sequence subsequently interacts with membrane-proximal sequences within the β tail to displace the α integrin tail (Moser et al., 2009). Since talin contains two β integrin-binding sites, one within the FERM and the other within the rod domain, talin homodimers have up to four integrin-binding sites, thus providing a biochemical base to talin’s ability to act as an integrin cross-linker in order to promote clustering (Moser et al., 2009).
Talin is a recruited component of the focal-adhesion complex to functionally interact with integrin cytoplasmic tails (Horwitz et al., 1986; Lewis and Schwartz, 1995; Sakamoto and Kyprianou, 2010; Fig. 4.1). The binding of cytoplasmic proteins to the integrin intracellular domains destabilizes the transmembrane complex and results in rearrangements in the extracellular integrin compartments that lead to integrin activation (Shattil et al., 2010). The role of talin in the regulation of integrin function was originally demonstrated in Chinese hamster ovary cells, where it was shown that talin induces a dramatic shift in the affinity of a normally inactive integrin (Calderwood et al., 1999, 2002). Talin interconnects β integrin cytoplasmic tails and actin filaments by directly binding both, and binding of talin head domain to integrin β tails is selectively abrogated by a single point mutation that disrupts integrin localization to talin-rich focal adhesions (Calderwood et al., 1999). Further, overexpression of a fragment of talin containing the head domain leads to activation of integrin αIIbβ3 that is dependent on the presence of both the talin head domain and the integrin β3 cytoplasmic tail (Calderwood et al., 1999). Knockout studies further revealed a role for talin as a key regulator of integrin affinity for its ligands, and diverse structural studies defined the mechanism via which talin accomplishes that (Moser et al., 2009). Talin orthologs have been identified in all multicellular eukaryotes studied; vertebrates encode two talin isoforms, talin1 and talin2, whereas lower eukaryotes encode only a single talin isoform corresponding to talin1 (Monkley et al., 2001; Senetar et al., 2007).
Structurally, the talin head can effectively compete with the αIIb tail for binding to the β3 tail in platelets (Vinogradova et al., 2000). Similarly, fluorescence energy transfer (FRET) experiments showed that the talin head domain binding to integrin causes a spatial separation of the integrin tails, which is associated with increased basal integrin ligand binding (Kim et al., 2003). Further, knockdown experiments in Caenorhabditis elegans revealed the importance of talin in integrin activation. Integrins are essential for embryonic development, muscle cell adhesion and contraction, and migration of nerve cell axons and gonadal distal tip cells in C. elegans, and downregulation of talin showed that this protein is required not only for cell adhesion and cytoskeletal organization but also for the dynamic regulation of integrin signals during cell migration (Cram et al., 2003). Similarly, elegant genetic studies on D. melanogaster established that talin is essential for integrin function, acting to stably linking ECM associated integrin clusters to the cytoskeleton (Brown et al., 2002). Platelets from mice lacking talin1 are unable to activate integrins in response to all known major platelet agonists (Petrich et al., 2007), and talin-deficient platelets display a severe hemostatic defect and are completely resistant to arterial thrombosis (Nieswandt et al., 2007). The autoinhibitory interaction between the talin C-terminus and the phosphotyrosine-binding domain that blocks the integrin-binding pocket implies a tight regulation of integrin activation by talin Goksoy et al., 2008). The large C-terminal rod domain of talin interacts with the talin head domain and allosterically holds back talin in a closed conformation, and in addition, the talin rod domain specifically masks a region of the phosphotyrosine-binding domain–integrin-binding site (Goksoy et al., 2008). The lipid second messenger phosphotidylinositol-4,5-bisphosphate [PtdIns(4,5)P2] is found to be an activator of talin when it binds to it, as it induces a conformational change that disrupts the auto-inhibitory interaction between talin domains, enhancing its binding to integrins (Goksoy et al., 2008; Martel et al., 2001).
The Ras GTPases is a critical group of cytoplasmic signaling proteins that mediate integrin–talin interactions (Kinbara et al., 2003). In addition to GTPases that are encoded by the H-ras, K-ras, and N-ras, the Ras family also includes R-ras (R-ras, R-ras2/TC21, and R-ras3/M-ras), Ral (RalA and RalB), RAP (RAP1A, 1B, 2A, and 2B), and Rheb (Rheb1 and 2; Kinbara et al., 2003). RAP1 directly stimulates integrin activation (Enserink et al., 2002; Katagiri et al., 2000, 2002), while RAP1 GTPases interact with RAP1–GTP-interacting adaptor molecule (RIAM), a member of the MRL (Mig-10/RIAM/Lamellipodin) protein family, to promote talin-dependent integrin activation (Lee et al., 2009). The association of RAP1 and RIAM is sufficient to recruit talin1 to integrins, resulting in integrin activation (Lee et al., 2009). In lymphoid cells, overexpression of RIAM induced cell spreading, lamellipodia formation, and the active conformation of β1 and β2 integrins along with cell adhesion, while RIAM knockdown displaced RAP1–GTP from the plasma membrane and abrogated RAP1-induced adhesion (Lafuente et al., 2004). In a dynamic functional exchange, RAP1 activation induces association of talin with RAP1 and RIAM, thus signaling αIIbβ3 integrin–talin1 interactions (Shattil et al., 2010). Interestingly, RIAM overexpression stimulates, while RIAM loss blocks talin recruitment by αIIbβ3 integrin (Watanabe et al., 2008). The tyrosine residues that are located within the β1 and β3 integrin NPxY motif can be phosphorylated by kinases of the Src family resulting in integrin-dependent migration of v-src transformed fibroblasts, but when these tyrosine residues are mutated, the migration ability of the cells is reversed (Law et al., 1996; Wennerberg et al., 2000).
Additional insights into integrin β1-dependent cell spreading revealed that this process was delayed in GD25 fibroblasts expressing the β1A–Y783F/Y795F double mutation compared to wild-type GD25-β 1A. FAK tyrosine phosphorylation and activation were severely blocked by β1-dependent adhesion in GD25-β1A–Y783F/Y795F mutated cells when compared to that in wild-type GD25-β1A or mutants in which only a single tyrosine (β1A–Y783F or β1A–Y795F) was altered (Wennerberg et al., 2000). Since talin is also known to form a homodimer, the integrin-binding sites of talin could be effectively masked (Goldmann et al., 1994; Ratnikov et al., 2005). The talin FERM domain which interacts with integrins is masked in the intact molecule, and after calpain-mediated cleavage of talin, the generated head and rod domain fragments facilitate talin binding to the β3 cytoplasmic tail (Calderwood et al., 2002; Yan et al., 2001). Talin N-terminal head and isolated phosphotyrosine-binding domains bind to β3 cytoplasmic tail with sixfold higher affinity than full-length talin (Calderwood et al., 2002; Yan et al., 2001). Thus, conformational changes mediated by Calpain result in the exposure of the integrin-binding site.
Phosphatidylinositol-4,5-bisphosphase (PIP2) induces talin binding to integrin β1 tail by potentially triggering conformational changes in talin, mediating the exposure of the integrin-binding site (Martel et al., 2001). Further evidence established that phosphatidylinositol phosphate kinase type Ic-90 (PIPKIc-90) is activated after PIP2 binding to talin, an interaction that induces PIPKIc-90 mediated PIP2 synthesis and talin binding to integrin β cytoplasmic tails in a positive feedback regulation (Di Paolo et al., 2002; Ling et al., 2002). Recruitment of PIPKIc-90 to focal adhesions and its interaction with talin occurs after its phosphorylation by Src kinase, leading to a 20-fold increase in PIPKIc-90 affinity for talin (Ling et al., 2003). This interaction implicates a positive feedback loop; activated Src promotes PIP2 synthesis and talin binding to integrin, resulting in further adhesion and Src activation (Ratnikov et al., 2005). Talin is phosphorylated at serine and threonine residues (Beckerle, 1990; Turner et al., 1989), a process that regulates its uniform redistribution to submembranous locations (Beckerle et al., 1989).
The functional involvement of talin in regulating cell adhesion to the ECM is critical for cell migration (e.g., during embryonic development, immune responses, cardiovascular function, angiogenesis), tumor invasion, and metastasis (Frame and Norman, 2008). Significantly enough, talin is capable of providing the critical mechanical linkage between activated integrins and the actin cytoskeleton, an essential catalytic factor for focal-adhesion signaling pathways (Zhang et al., 2008). This concept gains support from the recent findings on the activation of FAK–Src complex and AKT survival signaling in talin overexpressing prostate cancer cells leading to enhanced metastasis (Sakamoto et al., 2010).
Kindlins emerge as functional partners assisting talin in the activation of integrins. Kindlins belong to a family of evolutionarily conserved proteins that contain a FERM domain and they take their name after the gene mutated in Kindler syndrome. There are three kindlin family members in mammals: kindlin-1 (Unc-112 related protein 1, URP1), kindlin-2 (Mig2), and kindlin-3 (URP2; Siegel et al., 2003). Kindlin-1 is mainly expressed in epithelial cells in skin, intestine, and kidney, kindlin-2 is expressed in most tissues with skeletal and smooth muscle cells, while kindlin-3 expression only occurs in hematopoietic cells (Siegel et al., 2003; Ussar et al., 2006). The FERM domain near the C-terminus in kindlins is similar with the FERM domain of talin (Moser et al., 2009), while F3 subdomain mediates interaction of kindlins with β-integrin cytoplasmic tails (Shattil et al., 2010). Most integrin β tails, except those containing the NPxY motif that is recognized by phosphotyrosine-binding domains, also possess a membrane distal NxxY motif that functions as a binding site for multiple integrin-binding proteins including kindlins (Moser et al., 2009).
Besides β1-integrin and β3-integrin, other proteins that bind kindlins are ILK (Mackinnon et al., 2002; Montanez et al., 2008) and migfilin (Wu 2005). Kindlin-2 is required for actin polarization, cell spreading, and ILK localization into focal adhesions and enhances talin-mediated integrin activation (Montanez et al., 2008), while assists migfilin in linking focal adhesions to filamin and the actin cytoskeleton and functions in the orchestration of actin assembly and cell shape modulation (Tu et al., 2003). UNC-112 (the C. elegans ortholog of kindlin-1) colocalizes with integrins while loss of its expression results in a similar muscle detachment phenotype as that seen in α or β integrin mutants (Rogalski et al., 2000). Kindlin-1, kindlin-2, and kindlin-3 are capable of regulating the activation of specific integrins, but only when talin is concurrently interacting with the integrin β tails (Larjava et al., 2008). Ligand binding to and activation of αIIbβ3 or α5β1 integrins in CHO cells is enhanced by the talin head domain, and however, neither kindlin-1 nor kindlin-2 can mediate integrin activation in the absence of the talin head domain (Ma et al., 2008; Montanez et al., 2008). Cotransfection of kindlin-2 and talin head domain results in a synergistic enhancement of integrin αIIbβ3 activation (Ma et al., 2008). A role for kindlins in cell–ECM adhesion is further supported from studies on kindlin-2 colocalization to cell–ECM adhesion sites and direct interaction with the focal-adhesion proteins ILK and migfilin, whereas depletion of kindlin-2 impairs cell spreading (Tu et al., 2003).
5.2. Talin as a metastasis marker and therapeutic target
Upon detachment from the ECM, tumor epithelial cells and tumor-associated endothelial cells are capable of overcoming anoikis, gain survival benefits, and hence contribute to the process of metastasis. The cytoskeletal rearrangements and molecular changes that tumor cells experience during EMT, invasion, and metastasis, in the context of the tumor microenvironment, determine the plasticity of the tumor cells and their sensitivity to anoikis. The focal-adhesion complex formation recruits the functional integrity of talin as a key adaptor protein mediating the interaction between the actin cytoskeleton and integrins. Integrins function to anchor epithelial as well as endothelial cells to the ECM and activate intracellular signaling pathways that lead to tumor growth and vascular growth, respectively. The concept of angiogenesis and its significance in cancer progression, pioneered by Folkman several decades ago (Folkman, 1990), gradually led to the therapeutic targeting of the signaling pathways regulating the process, such as VEGF-A and its receptor VEGFR2, angiopoietins and Tie2 receptor, PDGF-β and its receptor PDGFRβ, and the Dll4-Notch1 pathway (Roodink and Leenders, 2010).
Indeed several antiangiogenic agents are approved by the Food and Drug Administration (FDA) for the treatment of human cancer; Bevacizumab (humanized antibody) targeting all isoforms of VEGF-A represents first-line treatment for patients with metastatic colorectal cancer, non-small-cell lung cancer, and metastatic breast cancer; Sorafenib (small compound multitargeted receptor tyrosine kinase inhibitor) inhibiting VEGFR2 and PDGFRβ has been approved for the treatment of metastatic renal cell cancer and advanced hepatocellular carcinoma. Sunitinib (small compound kinase inhibitor) directed like Sorafenib against VEGFR2 and PDGFRβ is approved for advanced renal cell cancer and gastrointestinal stromal tumors (in patients who failed imatinib treatment). Last, IFNα induces apoptosis in endothelial cells and interferes with endothelial cell adhesion by downregulating expression of VEGF and basic FGF (Roodink and Leenders, 2010). Specific targeting of the existing tumor endothelium is achieved with the L19 single chain antibody which is directed against the ED-B fragment of fibronectin, a splice variant serving as an angiogenesis marker and found only in the ECM of newly formed vessels in actively growing tumors (Santimaria et al., 2003). Plexin D1 has also emerged as a potential target for inhibition of established vasculature, as it is specifically expressed on activated tumor vasculature and tumor cells in solid tumors of different origin (Roodink et al., 2009). Several novel vascular disrupting agents (VDAs) have emerged as anticancer drugs that selectively impair tumor vasculature by targeting established blood vessels but not neovascularization (Kanthou and Tozer, 2009; Tozer et al., 2005).
The recent identification of talin1 as a significant promoter of invasion and anoikis resistance of human prostate cancer cells, and its loss leading to a diminished in vivo metastatic ability (Sakamoto et al., 2010), implicated an appealing therapeutic targeting value for this anoikis player in the context of the microenvironment and specifically the tumor vascularity. Mechanistically, talin1 upregulation leads to activation of FAK/AKT signaling and anoikis resistance, in accordance with independent reports linking the AKT survival signaling to anoikis resistance (Chang et al., 2005a,b; Fig. 4.1). The clinical significance of talin’s contribution to the metastatic cascade comes from recent findings showing significant talin1 upregulation in primary tumors and metastatic prostate cancer compared to the normal prostate gland (Sakamoto et al., 2010). Talin1 expression was significantly higher in poorly differentiated prostate tumors (Gleason >8) compared to moderately differentiated (tumors Gleason 6 and 7). In the same study, Sakamoto and colleagues also documented a striking inverse correlation between talin1 and E-cadherin in human prostate tumors and metastatic lesions. An earlier proteomics-based analysis reported that highly metastatic cells expressed significantly high levels of talin1 (>16-fold, out of 440 proteins screened) compared to cells with low metastatic potential (Everley et al., 2004). Further analysis of talin1 protein expression in the transgenic mouse model of prostate cancer (TRAMP) revealed a greater than twofold increase in talin1 expression in advanced disease compared to early stage tumors (Sakamoto et al., 2010).
Doxazosin and related quinazoline-based α1-adrenoceptor antagonists trigger apoptosis in benign epithelial, smooth muscle, and endothelial cells, in addition to malignant epithelial cells (Arencibia et al., 2005; Kyprianou et al., 2009), through an α1-adrenoreceptor-independent mechanism (Kyprianou, 2003; Shaw et al., 2004). This apoptotic action proceeds via the death receptor pathway, engaging caspase-8 and FADD mechanism, activation of TGF-β1 signaling, and targeting the AKT survival pathway (Benning and Kyprianou, 2002; Garrison and Kyprianou, 2006; Partin et al., 2003). The quinazolines can also synergize with ionizing radiation to exert a potent antitumor effect against castration resistant prostate cancer (CRPC; Cuellar et al., 2002). Moreover, epidemiological studies documented a significantly decreased risk ratio for prostate cancer in patients treated with quinazoline-based adrenoceptor antagonists, indicating that the apoptotic and antiangiogenic effects of these drugs, at the cellular level, translate into a chemoprevention action for prostate cancer initiation (Harris et al., 2007). Finally, our drug optimization efforts led to the generation of novel quinazoline-based compounds with more potent antiangiogenic effects against CRPC. DZ-50, the lead derivative, impairs prostate tumor vascularity by inducing anoikis of endothelial and tumor epithelial cells (Garrison et al., 2007). Current efforts focus on interrogation of the molecular mechanisms driving the apoptotic and antiangiogenic effects exerted by quinazolines with talin being the prime candidate as a molecular therapeutic target toward increased efficacy and limited toxicity.
6. Concluding Remarks and Future Directions
We have reviewed the evidence and current understanding of the role of talin as a prognostic marker of cancer progression and as therapeutic target in advanced metastatic disease, as well as the molecular mechanisms regulating its signaling activity in the context of the tumor microenvironment. Talin is an early-recruited focal-adhesion protein that binds to critical adhesion molecules, including the integrins, FAK, and ILK, resulting in integrin activation and signaling (Fig. 4.1). Upon activation, integrins increase the functional communication between cells and the ECM, serving as unique bidirectional transducers of both extracellular and intracellular signals. Since in talin-deficient mice, integrins fail to aggregate into clusters and connect to the cytoskeleton (similar to integrin-deficient phenotype), integrin function is clearly considered as talin dependent. Integrin activation and signaling via the focal-adhesion complex is associated with downregulation of E-cadherin and EMT induction via activation of transcriptional repressors (Snail, Slug, Twist, or ZEB1/2; Bolos et al., 2010). Cell adhesion molecules participate in the inflammatory response following radiotherapy and integrin-mediated adhesion to ECM induces survival and antiapoptotic signals that confer resistance to radiation therapy (Cordes, 2006; Cordes et al., 2006; Hallahan et al., 1996). Reversing anoikis resistance or facilitating anoikis and inhibiting angiogenesis are recognized as attractive avenues to be exploited therapeutically (Kyprianou et al., 2009).
Acknowledgments
Our work described was supported by grants from the National Institute of Health, RO1CA10757506, and the Department of Defense, W81XWH-08-1-0431.
Abbreviations
- AR
androgen receptor
- BPH
benign prostate hyperplasia
- cav-1
caveolin
- c-FLIP
c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long
- CRPC
castration resistant prostate cancer
- Csk
carboxy-terminal Src kinase
- DHT
dihydrotestosterone
- ECM
extracellular matrix
- EGF
epidermal growth factor
- EGFR
EGF receptor
- EMT
epithelial to mesenchymal transition
- ER
estrogen receptor
- FAK
focal-adhesion kinase
- FasL
Fas ligand
- FDA
Food and Drug Administration
- FGF
fibroblast growth factor
- FGFR1
FGF receptor-1
- FRET
fluorescence energy transfer
- GRB2/SOS
growth factor receptor bound 2/homologue of Drosophila melanogaster “son of sevenless” protein
- GSK3
glycogen synthase kinase-3
- IGF
insulin-like growth factor
- IGF-RI
insulin-like growth factor receptor I
- ILK
integrin-linked kinase
- JNK
c-Jun NH (2)-terminal kinases
- MMP
matrix metalloproteinases
- p38MAPK
p38 mitogen-activated protein kinase
- PDGF
platelet derived growth factor
- PDGFRβ
platelet derived growth factor receptor β
- PI3K
phosphoinositol-3 kinase
- PIP2
phosphatidylinositol-4,5-bisphosphase
- PIPKIc-90
phosphatidylinositol phosphate kinase type Ic-90
- PKB/Akt
protein kinase B
- PPAR-γ
peroxisome proliferator receptor-gamma
- PtdIns(4,5)P2
phosphotidylinositol-4,5-bisphosphate
- PTEN
phosphatase and tensin homologue deleted on the chromosome 10
- RIAM
RAP1–GTP-interacting adaptor molecule
- RTK
receptor tyrosine kinase
- STAT3
signal transducer and activator of transcription 3
- TGF-β
transforming growth factor β
- TRAMP
transgenic mouse model of prostate cancer
- URP1
Unc-112 related protein 1
- VDA
vascular disrupting agent
- VEGF
vascular endothelial growth factor
- VEGFR2
VEGF receptor 2
References
- Acevedo VD, Gangula RD, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12:559–571. doi: 10.1016/j.ccr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Estrada Y, et al. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Res. 2003;63:1684–1695. [PubMed] [Google Scholar]
- Albelda SM. Role of integrins and other cell adhesion molecules in tumor progression and metastasis. Lab Invest. 1993;68:4–17. [PubMed] [Google Scholar]
- Arencibia JM, Del Rio M, et al. Doxazosin induces apoptosis in LNCaP prostate cancer cell line through DNA binding and DNA-dependent protein kinase down-regulation. Int J Oncol. 2005;27:1617–1623. [PubMed] [Google Scholar]
- Avraamides CJ, Garmy-Susini B, et al. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604–617. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakin AV, Tomlinson AK, et al. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Rinehart C, et al. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci. 2002;115:3193–3206. doi: 10.1242/jcs.115.15.3193. [DOI] [PubMed] [Google Scholar]
- Barkan D, Green JE, et al. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer. 2010;46:1181–1188. doi: 10.1016/j.ejca.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerle MC. The adhesion plaque protein, talin, is phosphorylated in vivo in chicken embryo fibroblasts exposed to a tumor-promoting phorbol ester. Cell Regul. 1990;1:227–236. doi: 10.1091/mbc.1.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerle MC, Miller DE, et al. Activation-dependent redistribution of the adhesion plaque protein, talin, in intact human platelets. J Cell Biol. 1989;109:3333–3346. doi: 10.1083/jcb.109.6.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekman KW, Colevas AD, et al. Phase II evaluations of cilengitide in asymptomatic patients with androgen-independent prostate cancer: scientific rationale and study design. Clin Genitourin Cancer. 2006;4:299–302. doi: 10.3816/CGC.2006.n.012. [DOI] [PubMed] [Google Scholar]
- Benning CM, Kyprianou N. Quinazoline-derived alpha1-adrenoceptor antagonists induce prostate cancer cell apoptosis via an alpha1-adrenoceptor-independent action. Cancer Res. 2002;62:597–602. [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassi M, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolos V, Gasent JM, et al. The dual kinase complex FAK-Src as a promising therapeutic target in cancer. Onco Targets Ther. 2010;3:83–97. doi: 10.2147/ott.s6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaouina M, Lad Y, et al. The N-terminal domains of talin cooperate with the phosphotyrosine binding-like domain to activate beta1 and beta3 integrins. J Biol Chem. 2008;283:6118–6125. doi: 10.1074/jbc.M709527200. [DOI] [PubMed] [Google Scholar]
- Brown NH, Gregory SL, et al. Talin is essential for integrin function in Drosophila. Dev Cell. 2002;3:569–579. doi: 10.1016/s1534-5807(02)00290-3. [DOI] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Calderwood DA. Talin controls integrin activation. Biochem Soc Trans. 2004;32:434–437. doi: 10.1042/BST0320434. [DOI] [PubMed] [Google Scholar]
- Calderwood DA, Zent R, et al. The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J Biol Chem. 1999;274:28071–28074. doi: 10.1074/jbc.274.40.28071. [DOI] [PubMed] [Google Scholar]
- Calderwood DA, Yan B, et al. The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem. 2002;277:21749–21758. doi: 10.1074/jbc.M111996200. [DOI] [PubMed] [Google Scholar]
- Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- Chambers AF, Groom AC, et al. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- Chang HY, Nuyten DS, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci USA. 2005a;102:3738–3743. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LC, Huang CH, et al. Differential effect of the focal adhesion kinase Y397F mutant on v-Src-stimulated cell invasion and tumor growth. J Biomed Sci. 2005b;12:571–585. doi: 10.1007/s11373-005-7212-5. [DOI] [PubMed] [Google Scholar]
- Charlesworth A, Broad S, et al. The bombesin/GRP receptor transfected into Rat-1 fibroblasts couples to phospholipase C activation, tyrosine phosphorylation of p125FAK and paxillin and cell proliferation. Oncogene. 1996;12:1337–1345. [PubMed] [Google Scholar]
- Chiarugi P, Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol. 2008;76:1352–1364. doi: 10.1016/j.bcp.2008.07.023. [DOI] [PubMed] [Google Scholar]
- Christofori G. New signals from the invasive front. Nature. 2006;441:444–450. doi: 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]
- Cianfrocca ME, Kimmel KA, et al. Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH(2)), a beta integrin antagonist, in patients with solid tumours. Br J Cancer. 2006;94:1621–1626. doi: 10.1038/sj.bjc.6603171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Coates JM, Galante JM, et al. Cancer therapy beyond apoptosis: autophagy and anoikis as mechanisms of cell death. J Surg Res. 2010;164:301–308. doi: 10.1016/j.jss.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Coll ML, Rosen K, et al. Increased Bcl-xL expression mediates v-Src-induced resistance to anoikis in intestinal epithelial cells. Oncogene. 2002;21:2908–2913. doi: 10.1038/sj.onc.1205388. [DOI] [PubMed] [Google Scholar]
- Cooper CR, Bhatia JK, et al. The regulation of prostate cancer cell adhesion to human bone marrow endothelial cell monolayers by androgen dihydrotestosterone and cytokines. Clin Exp Metastasis. 2002;19:25–33. doi: 10.1023/a:1013849123736. [DOI] [PubMed] [Google Scholar]
- Cordes N. Integrin-mediated cell-matrix interactions for prosurvival and antiapoptotic signaling after genotoxic injury. Cancer Lett. 2006;242:11–19. doi: 10.1016/j.canlet.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Cordes N, Seidler J, et al. beta1-integrin-mediated signaling essentially contributes to cell survival after radiation-induced genotoxic injury. Oncogene. 2006;25:1378–1390. doi: 10.1038/sj.onc.1209164. [DOI] [PubMed] [Google Scholar]
- Cram EJ, Clark SG, et al. Talin loss-of-function uncovers roles in cell contractility and migration in C. elegans. J Cell Sci. 2003;116:3871–3878. doi: 10.1242/jcs.00705. [DOI] [PubMed] [Google Scholar]
- Critchley DR, Gingras AR. Talin at a glance. J Cell Sci. 2008;121:1345–1347. doi: 10.1242/jcs.018085. [DOI] [PubMed] [Google Scholar]
- Crowe DL, Ohannessian A. Recruitment of focal adhesion kinase and paxillin to beta1 integrin promotes cancer cell migration via mitogen activated protein kinase activation. BMC Cancer. 2004;4:18. doi: 10.1186/1471-2407-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuellar DC, Rhee J, et al. Alpha1-adrenoceptor antagonists radiosensitize prostate cancer cells via apoptosis induction. Anticancer Res. 2002;22:1673–1679. [PubMed] [Google Scholar]
- Delcommenne M, Tan C, et al. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers MJ, Thibodeau S, et al. Intestinal epithelial cancer cell anoikis resistance: EGFR-mediated sustained activation of Src overrides Fak-dependent signaling to MEK/Erk and/or PI3-K/Akt-1. J Cell Biochem. 2009;107:639–654. doi: 10.1002/jcb.22131. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Pellegrini L, et al. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 2002;420:85–89. doi: 10.1038/nature01147. [DOI] [PubMed] [Google Scholar]
- Douma S, Van Laar T, et al. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430:1034–1039. doi: 10.1038/nature02765. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, et al. A novel Epac-specific cAMP analogue demonstrates independent regulation of RAP1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- Everley PA, Krijgsveld J, et al. Quantitative cancer proteomics: stable isotope labeling with amino acids in cell culture (SILAC) as a tool for prostate cancer research. Mol Cell Proteomics. 2004;3:729–735. doi: 10.1074/mcp.M400021-MCP200. [DOI] [PubMed] [Google Scholar]
- Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010;29:285–293. doi: 10.1007/s10555-010-9224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- Fong A, Garcia E, et al. Expression of caveolin-1 and caveolin-2 in urothelial carcinoma of the urinary bladder correlates with tumor grade and squamous differentiation. Am J Clin Pathol. 2003;120:93–100. doi: 10.1309/292N-HAYN-WAVR-EJ37. [DOI] [PubMed] [Google Scholar]
- Fornaro M, Manes T, et al. Integrins and prostate cancer metastases. Cancer Metastasis Rev. 2001;20:321–331. doi: 10.1023/a:1015547830323. [DOI] [PubMed] [Google Scholar]
- Frame M, Norman J. A tal(in) of cell spreading. Nat Cell Biol. 2008;10:1017–1019. doi: 10.1038/ncb0908-1017. [DOI] [PubMed] [Google Scholar]
- Friess H, Langrehr JM, et al. A randomized multi-center phase II trial of the angiogenesis inhibitor Cilengitide (EMD 121974) and gemcitabine compared with gemcitabine alone in advanced unresectable pancreatic cancer. BMC Cancer. 2006;6:285. doi: 10.1186/1471-2407-6-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, et al. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Krause G, et al. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 2002;4:222–231. doi: 10.1038/ncb758. [DOI] [PubMed] [Google Scholar]
- Garcia-Alvarez B, de Pereda JM, et al. Structural determinants of integrin recognition by talin. Mol Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- Garrison JB, Kyprianou N. Doxazosin induces apoptosis of benign and malignant prostate cells via a death receptor-mediated pathway. Cancer Res. 2006;66:464–472. doi: 10.1158/0008-5472.CAN-05-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison JB, Shaw YJ, et al. Novel quinazoline-based compounds impair prostate tumorigenesis by targeting tumor vascularity. Cancer Res. 2007;67:11344–11352. doi: 10.1158/0008-5472.CAN-07-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti FG. Complexity and specificity of integrin signalling. Nat Cell Biol. 2000;2:E13–E14. doi: 10.1038/71397. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Gilles C, Polette M, et al. Vimentin contributes to human mammary epithelial cell migration. J Cell Sci. 1999;112:4615–4625. doi: 10.1242/jcs.112.24.4615. [DOI] [PubMed] [Google Scholar]
- Gingras AR, Bate N, et al. The central region of talin has a unique fold that binds vinculin and actin. J Biol Chem. 2010;285:29577–29587. doi: 10.1074/jbc.M109.095455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel HL, Fornaro M, et al. Selective modulation of type 1 insulin-like growth factor receptor signaling and functions by beta1 integrins. J Cell Biol. 2004;166:407–418. doi: 10.1083/jcb.200403003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel HL, Li J, et al. Integrins in prostate cancer progression. Endocr Relat Cancer. 2008;15:657–664. doi: 10.1677/ERC-08-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goksoy E, Ma YQ, et al. Structural basis for the autoinhibition of talin in regulating integrin activation. Mol Cell. 2008;31:124–133. doi: 10.1016/j.molcel.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann WH, Bremer A, et al. Native talin is a dumbbell-shaped homodimer when it interacts with actin. J Struct Biol. 1994;112:3–10. doi: 10.1006/jsbi.1994.1002. [DOI] [PubMed] [Google Scholar]
- Graham TR, Zhau HE, et al. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008;68:2479–2488. doi: 10.1158/0008-5472.CAN-07-2559. [DOI] [PubMed] [Google Scholar]
- Guarino M. Src signaling in cancer invasion. J Cell Physiol. 2010;223:14–26. doi: 10.1002/jcp.22011. [DOI] [PubMed] [Google Scholar]
- Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Hallahan D, Kuchibhotla J, et al. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res. 1996;56:5150–5155. [PubMed] [Google Scholar]
- Hanna E, Quick J, et al. The tumour microenvironment: a novel target for cancer therapy. Oral Dis. 2009;15:8–17. doi: 10.1111/j.1601-0825.2008.01471.x. [DOI] [PubMed] [Google Scholar]
- Harris AM, Warner BW, et al. Effect of alpha1-adrenoceptor antagonist exposure on prostate cancer incidence: an observational cohort study. J Urol. 2007;178:2176–2180. doi: 10.1016/j.juro.2007.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix MJ, Seftor EA, et al. Experimental co-expression of vimentin and keratin intermediate filaments in human breast cancer cells results in phenotypic interconversion and increased invasive behavior. Am J Pathol. 1997;150:483–495. [PMC free article] [PubMed] [Google Scholar]
- Horwitz A, Duggan K, et al. Interaction of plasma membrane fibronectin receptor with talin—a transmembrane linkage. Nature. 1986;320:531–533. doi: 10.1038/320531a0. [DOI] [PubMed] [Google Scholar]
- Huber MA, Kraut N, et al. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: a family of cell surface receptors. Cell. 1987;48:549–554. doi: 10.1016/0092-8674(87)90233-9. [DOI] [PubMed] [Google Scholar]
- Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igishi T, Fukuhara S, et al. Divergent signaling pathways link focal adhesion kinase to mitogen-activated protein kinase cascades. Evidence for a role of paxillin in c-Jun NH (2)-terminal kinase activation. J Biol Chem. 1999;274:30738–30746. doi: 10.1074/jbc.274.43.30738. [DOI] [PubMed] [Google Scholar]
- Irby R, Mao W, et al. Overexpression of normal c-Src in poorly metastatic human colon cancer cells enhances primary tumor growth but not metastatic potential. Cell Growth Differ. 1997;8:1287–1295. [PubMed] [Google Scholar]
- Jagadeeswaran R, Surawska H, et al. Paxillin is a target for somatic mutations in lung cancer: implications for cell growth and invasion. Cancer Res. 2008;68:132–142. doi: 10.1158/0008-5472.CAN-07-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Varner J. Integrins: roles in cancer development and as treatment targets. Br J Cancer. 2004;90:561–565. doi: 10.1038/sj.bjc.6601576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jodele S, Blavier L, et al. Modifying the soil to affect the seed: role of stromal-derived matrix metalloproteinases in cancer progression. Cancer Metastasis Rev. 2006;25:35–43. doi: 10.1007/s10555-006-7887-8. [DOI] [PubMed] [Google Scholar]
- Juliano RL, Reddig P, et al. Integrin regulation of cell signalling and motility. Biochem Soc Trans. 2004;32:443–446. doi: 10.1042/BST0320443. [DOI] [PubMed] [Google Scholar]
- Kanthou C, Tozer GM. Microtubule depolymerizing vascular disrupting agents: novel therapeutic agents for oncology and other pathologies. Int J Exp Pathol. 2009;903:284–294. doi: 10.1111/j.1365-2613.2009.00651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri K, Hattori M, et al. RAP1 is a potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol Cell Biol. 2000;20:1956–1969. doi: 10.1128/mcb.20.6.1956-1969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri K, Hattori M, et al. RAP1 functions as a key regulator of T-cell and antigen-presenting cell interactions and modulates T-cell responses. Mol Cell Biol. 2002;22:1001–1015. doi: 10.1128/MCB.22.4.1001-1015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatib AM, Kontogiannea M, et al. Rapid induction of cytokine and E-selectin expression in the liver in response to metastatic tumor cells. Cancer Res. 1999;59:1356–1361. [PubMed] [Google Scholar]
- Kim HR, Lin HM, et al. Cell cycle arrest and inhibition of anoikis by galectin-3 in human breast epithelial cells. Cancer Res. 1999;59:4148–4154. [PubMed] [Google Scholar]
- Kim M, Carman CV, et al. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- Kim LC, Song L, et al. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6:587–595. doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- Kinbara K, Goldfinger LE, et al. Ras GTPases: integrins’ friends or foes? Nat Rev Mol Cell Biol. 2003;4:767–776. doi: 10.1038/nrm1229. [DOI] [PubMed] [Google Scholar]
- Knudsen BS, Miranti CK. The impact of cell adhesion changes on proliferation and survival during prostate cancer development and progression. J Cell Biochem. 2006;99:345–361. doi: 10.1002/jcb.20934. [DOI] [PubMed] [Google Scholar]
- Kucik DF. Rearrangement of integrins in avidity regulation by leukocytes. Immunol Res. 2002;26:199–206. doi: 10.1385/IR:26:1-3:199. [DOI] [PubMed] [Google Scholar]
- Kyprianou N. Doxazosin and terazosin suppress prostate growth by inducing apoptosis: clinical significance. J Urol. 2003;169:1520–1525. doi: 10.1097/01.ju.0000033280.29453.72. [DOI] [PubMed] [Google Scholar]
- Kyprianou N, Vaughan TB, et al. Apoptosis induction by doxazosin and other quinazoline alpha1-adrenoceptor antagonists: a new mechanism for cancer treatment? Naunyn Schmiedebergs Arch Pharmacol. 2009;380:473–477. doi: 10.1007/s00210-009-0462-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafuente EM, van Puijenbroek AA, et al. RIAM, an Ena/VASP and Profilin ligand, interacts with RAP1-GTP and mediates RAP1-induced adhesion. Dev Cell. 2004;7:585–595. doi: 10.1016/j.devcel.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Lahm H, Andre S, et al. Tumor galectinology: insights into the complex network of a family of endogenous lectins. Glycoconj J. 2004;20:227–238. doi: 10.1023/B:GLYC.0000025817.24297.17. [DOI] [PubMed] [Google Scholar]
- Larjava H, Plow EF, et al. Kindlins: essential regulators of integrin signalling and cell-matrix adhesion. EMBO Rep. 2008;9:1203–1208. doi: 10.1038/embor.2008.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law DA, Nannizzi-Alaimo L, et al. Outside-in integrin signal transduction. Alpha IIb beta 3-(GP IIb IIIa) tyrosine phosphorylation induced by platelet aggregation. J Biol Chem. 1996;271:10811–10815. doi: 10.1074/jbc.271.18.10811. [DOI] [PubMed] [Google Scholar]
- Lee YI, Kwon YJ, et al. Integrin-linked kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition. Biochem Biophys Res Commun. 2004;316:997–1001. doi: 10.1016/j.bbrc.2004.02.150. [DOI] [PubMed] [Google Scholar]
- Lee HS, Lim CJ, et al. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J Biol Chem. 2009;284:5119–5127. doi: 10.1074/jbc.M807117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legate KR, Wickstrom SA, et al. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009;23:397–418. doi: 10.1101/gad.1758709. [DOI] [PubMed] [Google Scholar]
- Lewis JM, Schwartz MA. Mapping in vivo associations of cytoplasmic proteins with integrin beta 1 cytoplasmic domain mutants. Mol Biol Cell. 1995;6:151–160. doi: 10.1091/mbc.6.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Ren CH, et al. Caveolin-1 maintains activated Akt in prostate cancer cells through scaffolding domain binding site interactions with and inhibition of serine/threonine protein phosphatases PP1 and PP2A. Mol Cell Biol. 2003a;23:9389–9404. doi: 10.1128/MCB.23.24.9389-9404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Mitra N, et al. Activation of integrin alphaIIbbeta3 by modulation of transmembrane helix associations. Science. 2003b;300:795–798. doi: 10.1126/science.1079441. [DOI] [PubMed] [Google Scholar]
- Li D, Ding J, et al. Fibronectin promotes tyrosine phosphorylation of paxillin and cell invasiveness in the gastric cancer cell line AGS. Tumori. 2009;95:769–779. doi: 10.1177/030089160909500621. [DOI] [PubMed] [Google Scholar]
- Lim ST, Mikolon D, et al. FERM control of FAK function: implications for cancer therapy. Cell Cycle. 2008;7:2306–2314. doi: 10.4161/cc.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling K, Doughman RL, et al. Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature. 2002;420:89–93. doi: 10.1038/nature01082. [DOI] [PubMed] [Google Scholar]
- Ling K, Doughman RL, et al. Tyrosine phosphorylation of type I gamma phosphatidylinositol phosphate kinase by Src regulates an integrin-talin switch. J Cell Biol. 2003;163:1339–1349. doi: 10.1083/jcb.200310067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta LA, Kohn E. Anoikis: cancer and the homeless cell. Nature. 2004;430:973–974. doi: 10.1038/430973a. [DOI] [PubMed] [Google Scholar]
- Liu S, Calderwood DA, et al. Integrin cytoplasmic domain-binding proteins. J Cell Sci. 2000;113:3563–3571. doi: 10.1242/jcs.113.20.3563. [DOI] [PubMed] [Google Scholar]
- Lo HW, Hsu SC, et al. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67:9066–9076. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YQ, Qin J, et al. Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J Cell Biol. 2008;181:439–446. doi: 10.1083/jcb.200710196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon AC, Qadota H, et al. C. elegans PAT-4/ILK functions as an adaptor protein within integrin adhesion complexes. Curr Biol. 2002;12:787–797. doi: 10.1016/s0960-9822(02)00810-2. [DOI] [PubMed] [Google Scholar]
- Martel V, Racaud-Sultan C, et al. Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J Biol Chem. 2001;276:21217–21227. doi: 10.1074/jbc.M102373200. [DOI] [PubMed] [Google Scholar]
- Mawji IA, Simpson CD, et al. A chemical screen identifies anisomycin as an anoikis sensitizer that functions by decreasing FLIP protein synthesis. Cancer Res. 2007a;67:8307–8315. doi: 10.1158/0008-5472.CAN-07-1687. [DOI] [PubMed] [Google Scholar]
- Mawji IA, Simpson CD, et al. Critical role for Fas-associated death domain-like interleukin-1-converting enzyme-like inhibitory protein in anoikis resistance and distant tumor formation. J Natl Cancer Inst. 2007b;99:811–822. doi: 10.1093/jnci/djk182. [DOI] [PubMed] [Google Scholar]
- McCleverty CJ, Lin DC, et al. Structure of the PTB domain of tensin1 and a model for its recruitment to fibrillar adhesions. Protein Sci. 2007;16:1223–1229. doi: 10.1110/ps.072798707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeel DG, Eickhoff J, et al. Phase I trial of a monoclonal antibody specific for alphavbeta3 integrin (MEDI-522) in patients with advanced malignancies, including an assessment of effect on tumor perfusion. Clin Cancer Res. 2005;11:7851–7860. doi: 10.1158/1078-0432.CCR-05-0262. [DOI] [PubMed] [Google Scholar]
- Metalli D, Lovat F, et al. The insulin-like growth factor receptor I promotes motility and invasion of bladder cancer cells through Akt- and mitogen-activated protein kinase-dependent activation of paxillin. Am J Pathol. 2010;176:2997–3006. doi: 10.2353/ajpath.2010.090904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monkley SJ, Pritchard CA, et al. Analysis of the mammalian talin2 gene TLN2. Biochem Biophys Res Commun. 2001;286:880–885. doi: 10.1006/bbrc.2001.5497. [DOI] [PubMed] [Google Scholar]
- Montanez E, Ussar S, et al. Kindlin-2 controls bidirectional signaling of integrins. Genes Dev. 2008;22:1325–1330. doi: 10.1101/gad.469408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschos SJ, Drogowski LM, et al. Integrins and cancer. Oncology (Williston Park) 2007;21:13–20. [PubMed] [Google Scholar]
- Moser M, Legate KR, et al. The tail of integrins, talin, and kindlins. Science. 2009;324:895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- Mullamitha SA, Ton NC, et al. Phase I evaluation of a fully human anti-alphav integrin monoclonal antibody (CNTO 95) in patients with advanced solid tumors. Clin Cancer Res. 2007;13:2128–2135. doi: 10.1158/1078-0432.CCR-06-2779. [DOI] [PubMed] [Google Scholar]
- Nabors LB, Mikkelsen T, et al. Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J Clin Oncol. 2007;25:1651–1657. doi: 10.1200/JCO.2006.06.6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nada S, Okada M, et al. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. 1991;351:69–72. doi: 10.1038/351069a0. [DOI] [PubMed] [Google Scholar]
- Nangia-Makker P, Honjo Y, et al. Galectin-3 induces endothelial cell morphogenesis and angiogenesis. Am J Pathol. 2000;156:899–909. doi: 10.1016/S0002-9440(10)64959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieswandt B, Moser M, et al. Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J Exp Med. 2007;204:3113–3118. doi: 10.1084/jem.20071827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka N, Takenaka Y, et al. Galectins and urological cancer. J Cell Biochem. 2004;91:118–124. doi: 10.1002/jcb.10663. [DOI] [PubMed] [Google Scholar]
- Olski TM, Noegel AA, et al. Parvin, a 42 kDa focal adhesion protein, related to the alpha-actinin superfamily. J Cell Sci. 2001;114:525–538. doi: 10.1242/jcs.114.3.525. [DOI] [PubMed] [Google Scholar]
- Orford K, Orford CC, et al. Exogenous expression of beta-catenin regulates contact inhibition, anchorage-independent growth, anoikis, and radiation-induced cell cycle arrest. J Cell Biol. 1999;146:855–868. doi: 10.1083/jcb.146.4.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlichenko LS, Radisky DC. Matrix metalloproteinases stimulate epithelial-mesenchymal transition during tumor development. Clin Exp Metastasis. 2008;25:593–600. doi: 10.1007/s10585-008-9143-9. [DOI] [PubMed] [Google Scholar]
- O’Toole TE, Mandelman D, et al. Modulation of the affinity of integrin alpha IIb beta 3 (GPIIb-IIIa) by the cytoplasmic domain of alpha IIb. Science. 1991;254:845–847. doi: 10.1126/science.1948065. [DOI] [PubMed] [Google Scholar]
- O’Toole TE, Katagiri Y, et al. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen JD, Ruest PJ, et al. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19:4806–4818. doi: 10.1128/mcb.19.7.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens TW, Valentijn AJ, et al. Apoptosis commitment and activation of mitochondrial Bax during anoikis is regulated by p38MAPK. Cell Death Differ. 2009;16:1551–1562. doi: 10.1038/cdd.2009.102. [DOI] [PubMed] [Google Scholar]
- Oxley CL, Anthis NJ, et al. An integrin phosphorylation switch: the effect of beta3 integrin tail phosphorylation on Dok1 and talin binding. J Biol Chem. 2008;283:5420–5426. doi: 10.1074/jbc.M709435200. [DOI] [PubMed] [Google Scholar]
- Papagrigoriou E, Gingras AR, et al. Activation of a vinculin-binding site in the talin rod involves rearrangement of a five-helix bundle. EMBO J. 2004;23:2942–2951. doi: 10.1038/sj.emboj.7600285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Partin JV, Anglin IE, et al. Quinazoline-based alpha 1-adrenoceptor antagonists induce prostate cancer cell apoptosis via TGF-beta signalling and I kappa B alpha induction. Br J Cancer. 2003;88:1615–1621. doi: 10.1038/sj.bjc.6600961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson G, Robinson F, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Perl AK, Wilgenbus P, et al. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- Persad S, Dedhar S. The role of integrin-linked kinase (ILK) in cancer progression. Cancer Metastasis Rev. 2003;22:375–384. doi: 10.1023/a:1023777013659. [DOI] [PubMed] [Google Scholar]
- Persad S, Attwell S, et al. Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells. Proc Natl Acad Sci USA. 2000;97:3207–3212. doi: 10.1073/pnas.060579697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrich BG, Marchese P, et al. Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. J Exp Med. 2007;204:3103–3111. doi: 10.1084/jem.20071800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23:7928–7946. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- Polette M, Gilles C, et al. Association of fibroblastoid features with the invasive phenotype in human bronchial cancer cell lines. Clin Exp Metastasis. 1998;16:105–112. doi: 10.1023/a:1006572204497. [DOI] [PubMed] [Google Scholar]
- Przybylo JA, Radisky DC. Matrix metalloproteinase-induced epithelial-mesenchymal transition: tumor progression at Snail’s pace. Int J Biochem Cell Biol. 2007;39:1082–1088. doi: 10.1016/j.biocel.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Puklin-Faucher E, Sheetz MP. The mechanical integrin cycle. J Cell Sci. 2009;122:179–186. doi: 10.1242/jcs.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky ES, Radisky DC. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:201–212. doi: 10.1007/s10911-010-9177-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajjayabun PH, Garg S, et al. Caveolin-1 expression is associated with high-grade bladder cancer. Urology. 2001;58:811–814. doi: 10.1016/s0090-4295(01)01337-1. [DOI] [PubMed] [Google Scholar]
- Ramsay AG, Marshall JF, et al. Integrin trafficking and its role in cancer metastasis. Cancer Metastasis Rev. 2007;26:567–578. doi: 10.1007/s10555-007-9078-7. [DOI] [PubMed] [Google Scholar]
- Rankin S, Hooshmand-Rad R, et al. Requirement for phosphatidylinositol 3′-kinase activity in platelet-derived growth factor-stimulated tyrosine phosphorylation of p125 focal adhesion kinase and paxillin. J Biol Chem. 1996;271:7829–7834. doi: 10.1074/jbc.271.13.7829. [DOI] [PubMed] [Google Scholar]
- Ratnikov BI, Partridge AW, et al. Integrin activation by talin. J Thromb Haemost. 2005;3:1783–1790. doi: 10.1111/j.1538-7836.2005.01362.x. [DOI] [PubMed] [Google Scholar]
- Rennebeck G, Martelli M, et al. Anoikis and survival connections in the tumor microenvironment: is there a role in prostate cancer metastasis? Cancer Res. 2005;65:11230–11235. doi: 10.1158/0008-5472.CAN-05-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski TM, Mullen GP, et al. The UNC-112 gene in Caenorhabditis elegans encodes a novel component of cell-matrix adhesion structures required for integrin localization in the muscle cell membrane. J Cell Biol. 2000;150:253–264. doi: 10.1083/jcb.150.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roodink I, Leenders WP. Targeted therapies of cancer: angiogenesis inhibition seems not enough. Cancer Lett. 2010;299:1–10. doi: 10.1016/j.canlet.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Roodink I, Verrijp K, et al. Plexin D1 is ubiquitously expressed on tumor vessels and tumor cells in solid malignancies. BMC Cancer. 2009;9:297. doi: 10.1186/1471-2407-9-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen K, Rak J, et al. Activated Ras prevents downregulation of Bcl-X(L) triggered by detachment from the extracellular matrix. A mechanism Ras induced resistance anoikis intestinal epithelial cells. J Cell Biol. 2000;149:447–456. doi: 10.1083/jcb.149.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen K, Shi W, et al. Cell detachment triggers p38 mitogen-activated protein kinase-dependent overexpression of Fas ligand. A novel mechanism Anoikis intestinal epithelial cells. J Biol Chem. 2002;277:46123–46130. doi: 10.1074/jbc.M207883200. [DOI] [PubMed] [Google Scholar]
- Roy M, Marchetti D. Cell surface heparan sulfate released by heparanase promotes melanoma cell migration and angiogenesis. J Cell Biochem. 2009;106:200–209. doi: 10.1002/jcb.22005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto S, Kyprianou N. Targeting anoikis resistance in prostate cancer metastasis. Mol Aspects Med. 2010;31:205–214. doi: 10.1016/j.mam.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto S, McCann RO, et al. Talin1 promotes tumor invasion and metastasis via focal adhesion signaling and anoikis resistance. Cancer Res. 2010;70:1885–1895. doi: 10.1158/0008-5472.CAN-09-2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgia R, Li JL, et al. Molecular cloning of human paxillin, a focal adhesion protein phosphorylated by P210BCR/ABL. J Biol Chem. 1995;270:5039–5047. doi: 10.1074/jbc.270.10.5039. [DOI] [PubMed] [Google Scholar]
- Salgia R, Li JL, et al. Expression of the focal adhesion protein paxillin in lung cancer and its relation to cell motility. Oncogene. 1999;18:67–77. doi: 10.1038/sj.onc.1202273. [DOI] [PubMed] [Google Scholar]
- Santimaria M, Moscatelli G, et al. Immunoscintigraphic detection of the ED-B domain of fibronectin, a marker of angiogenesis, in patients with cancer. Clin Cancer Res. 2003;9:571–579. [PubMed] [Google Scholar]
- Satoh T, Yang G, et al. Caveolin-1 expression is a predictor of recurrence-free survival in pT2N0 prostate carcinoma diagnosed in Japanese patients. Cancer. 2003;97:1225–1233. doi: 10.1002/cncr.11198. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, et al. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, et al. Complex formation with focal adhesion kinase: a mechanism to regulate activity and subcellular localization of Src kinases. Mol Biol Cell. 1999;10:3489–3505. doi: 10.1091/mbc.10.10.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Mitra SK, et al. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim Biophys Acta. 2004;1692:77–102. doi: 10.1016/j.bbamcr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Sen A, O’Malley K, et al. Paxillin regulates androgen- and epidermal growth factor- induced MAPK signaling and cell proliferation in prostate cancer cells. J Biol Chem. 2010;285:28787–28795. doi: 10.1074/jbc.M110.134064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senetar MA, Foster SJ, et al. Intrasteric inhibition mediates the interaction of the I/LWEQ module proteins Talin1, Talin2, Hip1, and Hip12 with actin. Biochemistry. 2004;43:15418–15428. doi: 10.1021/bi0487239. [DOI] [PubMed] [Google Scholar]
- Senetar MA, Moncman CL, et al. Talin2 is induced during striated muscle differentiation and is targeted to stable adhesion complexes in mature muscle. Cell Motil Cytoskeleton. 2007;64:157–173. doi: 10.1002/cm.20173. [DOI] [PubMed] [Google Scholar]
- Shain KH, Landowski TH, et al. Adhesion-mediated intracellular redistribution of c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long confers resistance to CD95-induced apoptosis in hematopoietic cancer cell lines. J Immunol. 2002;168:2544–2553. doi: 10.4049/jimmunol.168.5.2544. [DOI] [PubMed] [Google Scholar]
- Shattil SJ, Kim C, et al. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw YJ, Yang YT, et al. Pharmacological exploitation of the alpha1-adrenoreceptor antagonist doxazosin to develop a novel class of antitumor agents that block intracellular protein kinase B/Akt activation. J Med Chem. 2004;47:4453–4462. doi: 10.1021/jm049752k. [DOI] [PubMed] [Google Scholar]
- Shintani Y, Maeda M, et al. Collagen I promotes epithelial-to-mesenchymal transition in lung cancer cells via transforming growth factor-beta signaling. Am J Respir Cell Mol Biol. 2008;38:95–104. doi: 10.1165/rcmb.2007-0071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel DH, Ashton GH, et al. Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes Kindler syndrome. Am J Hum Genet. 2003;73:174–187. doi: 10.1086/376609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SJ, McCann RO. A C-terminal dimerization motif is required for focal adhesion targeting of Talin1 and the interaction of the Talin1 I/LWEQ module with F-actin. Biochemistry. 2007;46:10886–10898. doi: 10.1021/bi700637a. [DOI] [PubMed] [Google Scholar]
- Sommers CL, Walker-Jones D, et al. Vimentin rather than keratin expression in some hormone-independent breast cancer cell lines and in oncogene-transformed mammary epithelial cells. Cancer Res. 1989;49:4258–4263. [PubMed] [Google Scholar]
- Sommers CL, Byers SW, et al. Differentiation state and invasiveness of human breast cancer cell lines. Breast Cancer Res Treat. 1994;31:325–335. doi: 10.1007/BF00666165. [DOI] [PubMed] [Google Scholar]
- Staley CA, Parikh NU, et al. Decreased tumorigenicity of a human colon adenocarcinoma cell line by an antisense expression vector specific for c-Src. Cell Growth Differ. 1997;8:269–274. [PubMed] [Google Scholar]
- Stewart DA, Cooper CR, et al. Changes in extracellular matrix (ECM) and ECM-associated proteins in the metastatic progression of prostate cancer. Reprod Biol Endocrinol. 2004;2:2. doi: 10.1186/1477-7827-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeltzing O, Liu W, et al. Inhibition of integrin alpha5beta1 function with a small peptide (ATN-161) plus continuous 5-FU infusion reduces colorectal liver metastases and improves survival in mice. Int J Cancer. 2003;104:496–503. doi: 10.1002/ijc.10958. [DOI] [PubMed] [Google Scholar]
- Tadokoro S, Shattil SJ, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- Tantivejkul K, Kalikin LM, et al. Dynamic process of prostate cancer metastasis to bone. J Cell Biochem. 2004;91:706–717. doi: 10.1002/jcb.10664. [DOI] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, et al. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Thomas JW, Cooley MA, et al. The role of focal adhesion kinase binding in the regulation of tyrosine phosphorylation of paxillin. J Biol Chem. 1999;274:36684–36692. doi: 10.1074/jbc.274.51.36684. [DOI] [PubMed] [Google Scholar]
- Tozer GM, Kanthou C, et al. Disrupting tumour blood vessels. Nat Rev Cancer. 2005;5:423–435. doi: 10.1038/nrc1628. [DOI] [PubMed] [Google Scholar]
- Trikha M, Zhou Z, et al. CNTO 95, a fully human monoclonal antibody that inhibits alphav integrins, has antitumor and antiangiogenic activity in vivo. Int J Cancer. 2004;110:326–335. doi: 10.1002/ijc.20116. [DOI] [PubMed] [Google Scholar]
- Tu Y, Wu S, et al. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell. 2003;113:37–47. doi: 10.1016/s0092-8674(03)00163-6. [DOI] [PubMed] [Google Scholar]
- Turner CE, Pavalko FM, et al. The role of phosphorylation and limited proteolytic cleavage of talin and vinculin in the disruption of focal adhesion integrity. J Biol Chem. 1989;264:11938–11944. [PubMed] [Google Scholar]
- Uhlik MT, Temple B, et al. Structural and evolutionary division of phosphotyrosine binding (PTB) domains. J Mol Biol. 2005;345:1–20. doi: 10.1016/j.jmb.2004.10.038. [DOI] [PubMed] [Google Scholar]
- Ussar S, Wang HV, et al. The Kindlins: subcellular localization and expression during murine development. Exp Cell Res. 2006;312:3142–3151. doi: 10.1016/j.yexcr.2006.06.030. [DOI] [PubMed] [Google Scholar]
- Vinogradova O, Haas T, et al. A structural basis for integrin activation by the cytoplasmic tail of the alpha IIb-subunit. Proc Natl Acad Sci USA. 2000;97:1450–1455. doi: 10.1073/pnas.040548197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky I, Goldshmidt O, et al. Mammalian heparanase: involvement in cancer metastasis, angiogenesis and normal development. Semin Cancer Biol. 2002;12:121–129. doi: 10.1006/scbi.2001.0420. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Bodin L, et al. Mechanisms and consequences of agonist-induced talin recruitment to platelet integrin alphaIIbbeta3. J Cell Biol. 2008;181:1211–1222. doi: 10.1083/jcb.200803094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb DJ, Parsons JT, et al. Adhesion assembly, disassembly and turnover in migrating cells—over and over and over again. Nat Cell Biol. 2002;4:E97–E100. doi: 10.1038/ncb0402-e97. [DOI] [PubMed] [Google Scholar]
- Wegener KL, Partridge AW, et al. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Armulik A, et al. The cytoplasmic tyrosines of integrin subunit beta1 are involved in focal adhesion kinase activation. Mol Cell Biol. 2000;20:5758–5765. doi: 10.1128/mcb.20.15.5758-5765.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. Migfilin and its binding partners: from cell biology to human diseases. J Cell Sci. 2005;118:659–664. doi: 10.1242/jcs.01639. [DOI] [PubMed] [Google Scholar]
- Wu KJ, Zeng J, et al. Silibinin inhibits prostate cancer invasion, motility and migration by suppressing vimentin and MMP-2 expression. Acta Pharmacol Sin. 2009;30:1162–1168. doi: 10.1038/aps.2009.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lamouille S, et al. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Woods NT, et al. SRC directly phosphorylates Bif-1 and prevents its interaction with Bax and the initiation of anoikis. J Biol Chem. 2008;283:19112–19118. doi: 10.1074/jbc.M709882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B, Calderwood DA, et al. Calpain cleavage promotes talin binding to the beta 3 integrin cytoplasmic domain. J Biol Chem. 2001;276:28164–28170. doi: 10.1074/jbc.M104161200. [DOI] [PubMed] [Google Scholar]
- Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Cermak L, et al. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23:1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Pier T, et al. Effects of a monoclonal anti-alphavbeta3 integrin antibody on blood vessels—a pharmacodynamic study. Invest New Drugs. 2007;25:49–55. doi: 10.1007/s10637-006-9013-8. [DOI] [PubMed] [Google Scholar]
- Zhang X, Jiang G, et al. Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat Cell Biol. 2008;10:1062–1068. doi: 10.1038/ncb1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yan Q, et al. Vimentin affects the mobility and invasiveness of prostate cancer cells. Cell Biochem Funct. 2008;26:571–577. doi: 10.1002/cbf.1478. [DOI] [PubMed] [Google Scholar]
- Zoltan-Jones A, Huang L, et al. Elevated hyaluronan production induces mesenchymal and transformed properties in epithelial cells. J Biol Chem. 2003;278:45801–45810. doi: 10.1074/jbc.M308168200. [DOI] [PubMed] [Google Scholar]