

SUMMARY
Targeting the tumor vasculature with antibody-drug conjugates (ADCs) is a promising anti-cancer strategy that, in order to be realized, must overcome several obstacles, including identification of suitable targets and optimal warheads. Here, we demonstrate that the cell surface protein CD276/B7-H3 is broadly overexpressed by multiple tumor types on both cancer cells and tumor-infiltrating blood vessels, making it a potentially ideal dual-compartment therapeutic target. In preclinical studies CD276-ADCs armed with a conventional MMAE warhead destroyed CD276-positive cancer cells, but were ineffective against tumor vasculature. In contrast, pyrrolobenzodiazepine-conjugated CD276-ADCs killed both cancer cells and tumor vasculature, eradicating large established tumors and metastases, and improving long-term overall survival. CD276 targeted dual-compartment ablation could aid in development of highly selective broad-acting anti-cancer therapies.
Keywords: cancer, vasculature, angiogenesis, endothelium, TEM, ADC, MMAE, PBD, CD276, Abcb1
Graphical abstract
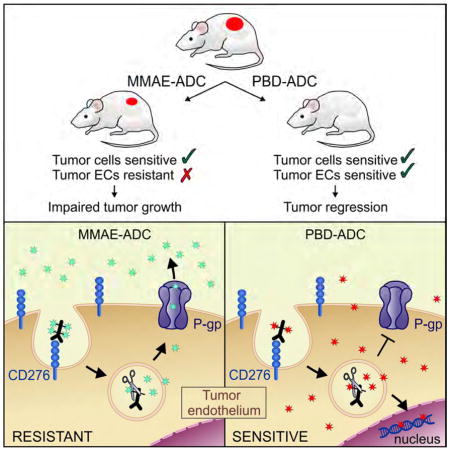
Seaman et al. show that CD276 is broadly overexpressed in cancer cells and tumor vascular cells and demonstrate anti-CD276-drug conjugates as promising anti-cancer reagents. The selection of conjugated drugs is important because tumor vascular cells can be resistant to a drug to which tumor cells are sensitive.
INTRODUCTION
Monoclonal antibodies (mAbs) have revolutionized cancer therapy. When harnessed appropriately their exquisite specificity enables them to selectively accumulate in tumors thereby reducing off-target toxicities. With over 15 mAbs clinically approved for cancer therapy, they have become a standard part of oncologic treatment regimens, targeting various antigens elevated on tumor cells such as HER2 or EGFR (Weiner, 2015). Despite these successes, in many cases the efficacy of naked antibodies has been limited, spurring the development of more potent immunotoxins and antibody-drug conjugates (ADCs). Unlike function blocking antibodies, ADC can be effective even if the target itself plays no role in tumor growth or is functionally redundant, provided that target is selectively overexpressed on the surface of tumor or tumor stromal cells. Major obstacles limiting the clinical deployment of immunotoxins include inherent immunogenicity of the xenotoxin, which limits repeated dosing, and the serious problem of vascular leak syndrome (VLS). mAbs bearing small molecule drugs, or ADCs, represent a viable alternative, as they are essentially invisible to the immune system and do not evoke VLS. ADCs also require much lower drug doses than naked antibodies which could eventually help in reducing manufacturing costs.
More than 40 ADCs are in clinical development and two are currently used clinically to treat cancer - brentuximab vedotin, an anti-CD30 monomethyl auristatin E (MMAE)-linked ADC, for treatment of Hodgkin lymphoma and trastuzumab emtansine (T-DM1), the HER2 binding mAb trastuzumab linked to the cytotoxic agent DM1, for treatment of HER2 positive breast cancer (Kim and Kim, 2015). The success of trastuzumab emtansine demonstrates that ADCs can effectively target not only hematological cancers but also solid tumors.
Clinically approved ADCs and most, if not all, ADCs in clinical development primarily target the tumor cells, and are therefore limited by tumor cell heterogeneity and genomic instability, which can lead to the emergence of drug resistant variants following chronic drug exposure. Resistance can potentially arise from a multitude of mechanisms, such as decreased target expression, poor internalization, drug recycling to the cell membrane, upregulation of detoxification enzymes, and increased drug efflux. One strategy to improve anti-tumor activity involves combining tumor cell targeting drugs with agents that can destroy stromal cells of the tumor microenvironment that facilitate tumor growth and spread. The tumor vasculature represents a particularly appealing therapeutic target as many tumors cannot grow beyond 1 to 2 mm in size without evoking new blood vessel growth, and tumor vessels provide a key escape route for metastatic spread (Folkman, 1971; Kerbel, 2008). The endothelial cells (ECs) lining the vasculature are also more accessible to mAbs and other high molecular weight drugs that have difficulty penetrating tumors. Furthermore, tumor vessels are genetically more stable than tumor cells and are therefore less likely to develop resistance through mutation and selection (Abdollahi and Folkman, 2010). Although anti-angiogenic agents that block the VEGF/VEGFR axis have helped prolong overall survival, their tumoricidal activity is almost always incomplete, and additional approaches to target tumor vasculature are needed in order to combat VEGF-independent neovascularization and tumor cell co-option of pre-existing vessels.
While the theoretical potential of targeting the tumor vasculature using ADCs is widely recognized (Gerber et al., 2009), so far limited data exists to support this concept. For example, even though prostate specific membrane antigen (PSMA) overexpression has been detected in tumor vessels of many human cancers and two PSMA-targeting ADCs have undergone extensive clinical testing for prostate cancer (Olson and Israel, 2014), these agents have only been shown to target PSMA-positive prostate tumor cells in vivo. There are several possible explanations for the paucity of validated ADCs targeting tumor vasculature. In the case of PSMA, the target is not overexpressed on rodent tumor versus normal vessels, unlike in humans, precluding targeting validation studies using PSMA-deficient mice (Huang et al., 2004). Optimal warheads for vascular targeted ADCs are also unclear as the endothelium, which has been exposed to blood-borne toxins throughout human evolution, may harbor intrinsically high levels of resistance to many drugs commonly used to arm ADCs. Another major hurdle involves the identification of the most suitable targets with the specificity needed to selectively target tumor vasculature while sparing normal vessels. Fortunately, advances in genomics and proteomics have led to the identification of cell surface tumor endothelial markers, some of which may have the desired specificity to build an effective tumor vascular-targeted ADC (Oh et al., 2004; Seaman et al., 2007; St Croix et al., 2000).
Comprehensive analysis of the vascular transcriptome identified CD276 as a cell surface tumor endothelial marker that could distinguish pathological and physiological angiogenesis in mice and humans (Seaman et al., 2007). CD276, also known as B7 homologue 3 (B7-H3), shares up to 30% amino acid identity with other B7 family members. While the physiological functions of CD276 remain unclear, CD276 protein is highly expressed in tumor vessels of human lung, breast, colon, endometrial, renal and ovarian cancer, but not in the angiogenic vessels of normal ovary. Thus, CD276 directed therapeutic mAbs may have a higher degree of specificity for tumor vessels than current anti-angiogenic agents that cannot distinguish physiological and pathological angiogenesis. Moreover, CD276 protein is also frequently overexpressed on tumor cells (Brunner et al., 2012; Qin et al., 2013; Zang et al., 2010). CD276 overexpression by tumor cells and Tumor ECs (TECs) makes it an appealing target for the development of therapeutic agents to simultaneously destroy both cell types.
Recently, an Fc-enhanced humanized anti-CD276 antibody, enoblituzumab, was shown to delay the growth of a variety of primary tumor types in preclinical studies and has advanced to Phase I clinical trials (Loo et al., 2012). Another humanized antibody, 8H9, originally identified based on its selective reactivity with human tumors cells, was later found to recognize CD276 and is also in Phase I clinical trials (Ahmed et al., 2015; Modak et al., 2001). However, these mAbs recognize human but not rodent CD276 precluding an assessment of the contribution of CD276 positive tumor-associated stromal cells in therapeutic targeting, and potential toxicities caused by inappropriate targeting of CD276 positive normal cells in preclinical rodent models. It is also unclear if the potency of current CD276 mAbs can be enhanced by blocking CD276 function or through conversion to an ADC. Finally, the role of CD276 mAbs in targeting late-stage metastatic disease has not yet been explored. Here, we set out to assess the value of CD276 as a target for primary and metastatic disease and determine if rationally designed CD276 ADCs could be used to simultaneously ablate tumor cells and tumor associated stroma.
RESULTS
CD276 is Broadly Expressed by Tumor Cells and Tumor Vasculature but is Dispensable for Tumor Growth
To evaluate the suitability of CD276 as a therapeutic target, we performed immunohistochemical (IHC) staining on formalin-fixed paraffin-embedded (FFPE) human tissues, including 1342 tumor and 245 normal samples. We screened several commercial antibodies for those that recognized both mouse and human CD276 and could be directly evaluated for specificity using FFPE tumor sections derived from Cd276 wildtype (WT) or Cd276 knockout (KO) mice, generated as described below. We identified a goat anti-human CD276 polyclonal antibody (pAb) that reacted specifically with murine tumor vessels from Cd276 WT but not KO mice (Figure S1A). Using this goat pAb, we verified the overexpression of CD276 in many human malignancies (Figures 1A, S1B and S1C). CD276 was strongly expressed (+++ score) by tumor vasculature in 51% of all tumors examined while the tumor cells stained strongly in 30% of all tumors (Figures 1B and 1C, and Table 1). Many tumors expressed CD276 in both tumor cells and tumor vessels, but in certain cancer types, such as colorectal and pancreatic cancer, the stromal compartment was often more strongly positive than the tumor cell compartment. Prostate tumors showed the opposite pattern, with tumor cells overexpressing CD276 more often than stromal cells. Although ECs were the most prominent staining stromal cell type, in some cancers additional perivascular stromal cells, most likely pericytes and fibroblasts, also stained positive (see Figure 1C, bottom panel). Importantly, CD276 was found to be strongly expressed in the stroma (54%) and tumor cells (31%) of triple-negative breast cancers, providing a potential target for this difficult-to-treat tumor type (Table 1). Although staining was undetectable in most normal tissues, the pAb displayed low reactivity with sinusoidal ECs of liver and the epithelial cells of skin, bladder, uterus and esophagus (Table 1 and Figures S1D–S1E). However, for at least some of these tissues low level staining was likely caused by non-specific cross-reactivity of the pAb as a similar pattern was observed in corresponding tissues of both Cd276 WT and KO mice (Figure S1F).
Figure 1. CD276 is Overexpressed in Various Human Tumors.

(A) IHC of FFPE tissues was used to evaluate CD276 expression in various human tumors or corresponding normal organs.
(B) Tumor cell staining was ranked as low (+), moderate (++) or strong (+++), as shown in this renal cell cancer example.
(C) Stromal cell staining was ranked as low (+), moderate (++) or strong (+++), as shown in this colon cancer example.
Scale bar in (A) to (C): 100 μm. See also Figure S1.
Table 1.
IHC staining of human normal and tumor tissues.
| Tissue | n | Cell typea | Positive (%) | |||
|---|---|---|---|---|---|---|
|
| ||||||
| − | + | ++ | +++ | |||
| Normal | ||||||
|
| ||||||
| Brain | 18 | N/A | 100 | 0 | 0 | 0 |
| Bladder | 20 | N/A | 80 | 20 | 0 | 0 |
| Breast | 7 | N/A | 57 | 43 | 0 | 0 |
| Cervix | 9 | N/A | 56 | 44 | 0 | 0 |
| Colon | 78 | N/A | 43 | 56 | 1 | 0 |
| Esophagus | 8 | N/A | 75 | 25 | 0 | 0 |
| Heart | 13 | N/A | 100 | 0 | 0 | 0 |
| Ovary | 11 | N/A | 45 | 55 | 0 | 0 |
| Kidney | 8 | N/A | 100 | 0 | 0 | 0 |
| Lung | 22 | N/A | 95 | 5 | 0 | 0 |
| Liver | 20 | N/A | 35 | 55 | 10 | 0 |
| Muscle | 3 | N/A | 100 | 0 | 0 | 0 |
| Pancreas | 23 | N/A | 78 | 22 | 0 | 0 |
| Prostate | 5 | N/A | 60 | 40 | 0 | 0 |
|
| ||||||
| Total normal tissue: | 245 | 65 | 33 | 1 | 0 | |
|
| ||||||
| Tumor | ||||||
|
| ||||||
| Bladder | 59 | TC | 17 | 20 | 26 | 37 |
| SC | 3 | 10 | 43 | 44 | ||
|
| ||||||
| Breast: Unselected | 208 | TC | 26 | 13 | 27 | 34 |
| SC | 2 | 8 | 32 | 58 | ||
|
| ||||||
| PR, ER, or HER2+ | 34 | TC | 12 | 9 | 38 | 41 |
| SC | 0 | 6 | 32 | 62 | ||
|
| ||||||
| Triple negative | 55 | TC | 35 | 16 | 18 | 31 |
| SC | 4 | 9 | 33 | 54 | ||
|
| ||||||
| Cervical | 70 | TC | 38 | 26 | 20 | 16 |
| SC | 2 | 17 | 51 | 30 | ||
|
| ||||||
| Colorectal | 104 | TC | 30 | 28 | 28 | 14 |
| SC | 0 | 1 | 18 | 81 | ||
|
| ||||||
| Esophageal: SCC | 109 | TC | 13 | 8 | 23 | 56 |
| SC | 6 | 7 | 19 | 68 | ||
|
| ||||||
| Glioma | 41 | TC | 17 | 17 | 17 | 49 |
| SC | 5 | 5 | 24 | 66 | ||
|
| ||||||
| Kidney: RCC and TCC | 35 | TC | 40 | 17 | 29 | 14 |
| SC | 3 | 11 | 46 | 40 | ||
|
| ||||||
| Liver: HCC | 60 | TC | 25 | 13 | 32 | 30 |
| SC | 0 | 3 | 30 | 67 | ||
|
| ||||||
| Lung: SCC | 210 | TC | 27 | 16 | 26 | 31 |
| SC | 3 | 12 | 45 | 40 | ||
|
| ||||||
| Mesothelioma | 30 | TC | 33 | 20 | 27 | 20 |
| SC | 20 | 23 | 33 | 24 | ||
|
| ||||||
| Melanoma | 79 | TC | 22 | 6 | 40 | 32 |
| SC | 16 | 3 | 29 | 52 | ||
|
| ||||||
| Ovarian | 68 | TC | 26 | 34 | 22 | 18 |
| SC | 4 | 25 | 43 | 28 | ||
|
| ||||||
| Pancreatic | 116 | TC | 59 | 17 | 17 | 7 |
| SC | 0 | 7 | 35 | 58 | ||
|
| ||||||
| Prostate | 64 | TC | 3 | 8 | 34 | 55 |
| SC | 63 | 13 | 19 | 5 | ||
|
| ||||||
| Total tumor tissue: | 1342 | TC | 27 | 16 | 26 | 30 |
| SC | 6 | 9 | 33 | 51 | ||
TC: Tumor cell staining, SC: Stromal cell staining
To assess CD276 expression patterns further, we generated a rabbit mAb that specifically reacted with mouse, rat and human CD276 proteins in several assays including immunoprecipitation, immunoblotting, flow cytometry and immunofluorescence (IF) staining of frozen sections (Figures 2A–2E and S2A). IF staining with the rabbit mAb revealed CD276 expression in both tumor cells and tumor vessels, while CD276 protein was undetectable in all normal tissues tested. Upregulation of CD276 in tumors compared to adjacent normal tissues was most striking at the normal/tumor margins, for example in MDA-MB-231 breast cancer lung metastases where both the human tumor cells and murine tumor vessels were CD276 positive (Figure 2C). In the case of MC38 liver metastases, CD276 was only detected in CD276 WT mice on tumor-infiltrating host vessels, but not normal adjacent liver, and tumor vessel staining was undetectable in Cd276 KO mice confirming antibody specificity (Figure 2D). IF staining also revealed upregulation of CD276 in clinical samples of human colon cancer liver metastases whereas vWF, a pan endothelial marker, was found in both tumor vessels and normal adjacent sinusoidal liver endothelium (Figure 2E). Importantly, high CD276 expression was detected in co-opted vasculature and perivascular stromal cells of human colon cancer liver and lung metastases (Figures 2F and 2G). Taken together, these studies demonstrate that CD276 protein expression patterns are highly conserved.
Figure 2. CD276 is Overexpressed in Tumors but has Minimal Impact on Tumor Growth.

(A) CD276 proteins were immunoprecipitated (IP) from MC38 parent cells or MC38 cells overexpressing full-length mouse (mCD276), rat (rCD276) or human (hCD276) CD276 and detected by immunoblotting.
(B) Detection of human CD276 overexpressed in MC38 cells using flow cytometry.
(C) CD276 IF staining (green) of an MDA-MB-231 breast cancer lung metastasis (yellow arrows indicate tumor margins). CD31 positive vessels are counterstained blue.
(D) CD276 IF staining (green) of MC38 colon liver metastasis from Cd276 WT or KO mice.
(E) vWF (top panel) or CD276 (bottom panel) IF staining of human colon cancer liver metastases. The two panels were taken from serial sections.
Normal (N) / tumor (T) margins in (D) and (E) are indicated by a white dash. Scale bar in (C) to (E): 50μm.
(F) CD276 IHC staining of human colon cancer lung metastases with co-opted vasculature. Tumor cells (T) have expanded inside the alveolar air space and are separated from stromal cells (S) by a thin layer of cytokeratin 7 (CK7) positive alveolar type I cells. Top and bottom panels represent serial sections, and arrows (inset) indicate the location of a large co-opted CD276+ blood-filled vessel. Staining is representative of 5 independent cases with vessel co-option.
(G) CD276 IF staining of human colon cancer liver metastases with co-opted vasculature. A co-opted portal triad is found near the center of the section (inset), and is identified by large CD31+ vessels (red) next to bile ducts (arrows) that stain strongly for cytokeratin 19 (CK19; yellow). Surrounding colon tumor cells also express low levels of CK19. Arrowheads indicate the CD276 positive co-opted vessels. Staining is representative of 5 independent cases with vessel co-option. Scale bar in (F) to (H): 100μm.
(H) CD276 IF staining of MC38 tumor vessels from Cd276 WT and Cd276 KO mice. Note that CD276 staining (green) was undetectable in desmin-positive vascular pericytes (red) of MC38 tumors (yellow arrowheads). CD31 staining (blue) was used to highlight the location of the endothelium in the KO. Scale bar: 10 μm.
(I) Tumor cells were inoculated subcutaneously into athymic nude (DLD-1, HCT-116, and UACC melanoma) or C57BL/6 (MC38 colon) Cd276 WT or KO mice and tumor growth monitored. n = 10/ group (MC38), n = 12/group (DLD-1), n = 8 (KO) or 11 (WT) per group (HCT-116) and n = 9/group UACC.
See also Figure S2.
To determine if host CD276 was functionally important for tumor growth, we generated Cd276 knockout (KO) mice on both an immunocompetent (C57BL/6) and immunodeficient (athymic nude) background (Figure S2B–S2D). The knockout vector was designed to remove exon 2 containing the start codon and signal peptide, creating a null allele. Cd276 KO mice were born at expected Mendelian frequencies, showed no obvious phenotype and were fertile, consistent with previous studies and supporting the idea that CD276 function may be largely redundant or dispensable under normal unchallenged conditions (Suh et al., 2003). To verify the loss of CD276 in tumor stroma in Cd276 KO mice in the absence of any confounding tumor cell expression, MC38 colon cancer cells that express very low endogenous CD276 were rendered Cd276 null (MC38-276−/−) using CRISPR-Cas9. Cd276 null cells were then injected into Cd276 WT or KO mice. CD276 protein expression in MC38-276−/− tumors was high in tumor endothelium of Cd276 WT but not Cd276 KO mice (Figure 2H and S2E). Cd276 WT and KO mice were then challenged with various murine and human tumors and tumor growth rates compared (Figures 2I and S2F). These studies revealed a modest but statistically significant tumor growth delay of 34%, 48% and 36% in Cd276 KO mice bearing DLD-1, HCT-116 and KM12SM colon tumors, respectively. However, in MC38 colon, SW620 colon and UACC melanoma no difference in tumor growth was observed between Cd276 WT and KO mice. Based on this, we conclude that stromal derived CD276, depending on the particular tumor, plays either a minor or no obvious role in promoting tumor growth.
To determine if CD276 expression in tumor cells plays an important role in regulating tumor growth, next we used CRISPR-Cas9 to disrupt Cd276 in B16 melanoma cells, which express high CD276 levels endogenously (Figure S2G). Comparison of B16-276−/− and parent B16 tumor growth in C57BL6 mice revealed an indistinguishable growth rate (Figure S2H). Thus, endogenous CD276 expression in either tumor cells or tumor associated stromal cells had little or no obvious functional consequence on tumor growth.
MMAE-linked Anti-CD276 ADC Kills CD276+ Tumor Cells
The overexpression of CD276 in carcinomas of diverse origin suggests CD276 represents a promising target for mAbs that deliver toxic payloads or elicit antibody-dependent cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC). To test this idea, we created fully-human anti-CD276 mAbs that recognize both mouse and human CD276 proteins. To avoid problems associated with in vivo tolerance we performed antibody selection in vitro using a diverse human single chain Fv (scFv) yeast display library. Following sequential rounds of selection against the purified native human and mouse CD276 proteins we identified an IgG1 antibody, m276, that recognized native mouse, monkey and human CD276 with similar affinity (24 to 33 nM) and rat CD276 with 10-fold higher affinity (3 nM) (Figure 3A). Although m276 did not perform adequately when used as a primary antibody to stain frozen or FFPE tumor sections, following FITC labelling and i.v. injection into tumor bearing mice the antibody was readily detected in tumor vasculature but not normal tissues by IF staining of frozen tissue (Figure S3A).
Figure 3. m276-MMAE Elicits Potent Anti-Tumor Activity Against Various Human Tumor Xenografts Without Evidence of Toxicities.

(A) The affinity of the monovalent m276 Fab for human, mouse, rat and monkey (cynomolgus) CD276 was determined using Biacore analysis.
(B, C) Cell viability assays were used to measure the activity of m276-MMAE against HT29 (B) and OVCAR3 (C) cells. To verify specificity, competition with unlabeled m276 was performed. Error bars in (B) and (C) = SD.
(D–H) Subcutaneous growth of colon HCT-116 (D), KM12 (E), HT29 (F), ovarian OVCAR3 (G), or orthotopic breast MDA-MB-231 (H) tumors. Treatments with vehicle, MMAE, m276, or m276-MMAE were initiated when tumors reached an average size of ~100mm3 and were administered on the days shown (green arrows). n = 10/group (DH). Error bars in (D) to (H) = SEM.
(I) Six OVCAR3 tumors that had relapsed following treatment with 3 mg/kg of m276-MMAE (biwk x 3.5) were retreated with 3 mg/kg (biwk x 3) starting on day 55 and then 10 mg/kg (biwk x 5) starting on day 76. Individual tumor measurements are shown.
See also Figures S3–S5.
IgG1 is the human isotype most effective at stimulating ADCC or CDC in humans and mice, and other anti-CD276 IgG1s have been shown to elicit ADCC in preclinical studies (Ahmed et al., 2015; Loo et al., 2012) suggesting that m276 could also have therapeutic activity on its own. However, preliminary studies in mice indicated that m276 had little, if any, anti-tumor activity. To determine if m276 could be exploited as an ADC, we verified that m276 was rapidly internalized upon binding CD276 on cells (Figure S3B). The initial anti-CD276 ADC we generated used the tubulin binding anti-mitotic drug MMAE, attached to m276 through a cathepsin B cleavable valine-citrulline linker (Figure S3C). We chose vcMMAE because the same drug-linker technology is used in the US FDA-approved brentuximab vedotin and several other ADCs in clinical development, and previous reports suggest that tumor vessels may be particularly sensitive to auristatin-like compounds (Visintin et al., 2015). Upon target binding and internalization the dipeptide linker can be cleaved by lysosomal proteases releasing cell-permeable MMAE which can then freely diffuse into nearby cells in a target-independent manner resulting in substantial bystander killing (Li et al., 2016). m276 was labelled with a previously optimized average 4:1 MMAE drug to antibody ratio (DAR). Uptake studies using immunofluorescently labelled m276-MMAE antibody confirmed that the ADC was rapidly taken up into cells with kinetics that were indistinguishable from m276 parent antibody (data not shown). Next, we checked the potency of m276-MMAE against 293 cells that express CD276 endogenously (Seaman et al., 2007). m276-MMAE specifically killed CD276+ 293 cells, while parent m276 and a nontargeting ADC (m825-MMAE) had no activity against 293 cells (Figures S3D and S3E). m276-MMAE displayed potent cytotoxic activity against CD276+ HT29 colon and OVCAR3 ovarian tumor cell lines, which could be competed with parent m276 (Figures 3B and 3C).
Next, m276-MMAE was tested for cross-reactivity with hematopoietic cells as previous studies found low CD276 expression in dendritic cells, monocytes, and T-cells (Chapoval et al., 2001; Suh et al., 2003). Flow cytometry on Cd276 WT and KO bone marrow and spleen cells failed to identify specific m276 reactivity with these and other hematopoietic cells (Figures S4A–S4D), and m276-MMAE treatment did not alter hematopoietic cell populations in Cd276 WT versus KO mice (Figure S4E and S4F). We further found m276-MMAE to be highly stable in human and monkey serum, although some instability was seen in mouse serum (Figure S5A). MMAE-vc-PABC linker instability in mouse serum was recently shown to result from carboxylesterase 1C enzymatic cleavage in mouse but not primate or human serum (Dorywalska et al., 2016).
Encouraged by these results, m276-MMAE was next tested for efficacy in vivo. m276-MMAE displayed dose-dependent anti-tumor activity against CD276 positive HCT-116, KM12, and HT29 colon, OVCAR3 ovarian, and MDA-MB-231 breast tumor xenografts, inducing complete regression of many tumors at doses as low as 3 mg/kg (Figures 3D–3H). Importantly, treatment with 10 mg/kg of the parent m276 antibody (Figures 3D–3H) or 0.2 mg/kg of free MMAE (Figure 3G), equivalent to the free drug load on m276-MMAE at 10 mg/kg, had no significant impact on tumor growth. Furthermore, m276-MMAE had no discernable impact on the mouse bodyweight or wound closure rates compared to treatment with vehicle (Figure S5B and S5C).
Although a single 3 mg/kg dose of ADC was able to elicit up to 71% tumor growth delay (Figure S5D), the most striking tumor regressions were observed with a repeated dosing schedule, i.e. twice per week for a total of 6 or 7 treatments (Figure 3H). However, most tumors eventually recurred 3 to 4 months post therapy, even following doses up to 30 mg/kg. To determine if tumors that initially regressed and then recurred following therapy were still sensitive to ADC, we re-treated 6 relapsed OVCAR3 tumors ranging from 500 to 800 mm3 with a second cycle of 3 mg/kg, the same dose used at the outset, and found that tumors were still highly responsive (Figure 3I). However, following the 5th treatment of cycle 2 tumors again relapsed, so the dose was raised to 10 mg/kg and mice were treated twice per week until study end. Tumors initially responded to the higher dose but then relapsed while on therapy. IF staining of the relapsed tumors revealed CD276 levels comparable to that in untreated OVCAR3 control tumors (Figure S5E), indicating that target antigen loss was not responsible for the reduced anti-tumor activity. Long-term treatment was well tolerated with no significant changes in body weights (data not shown).
Although KM12 colon tumor xenografts showed an 86% tumor growth delay following 10 mg/kg ADC treatments (Figure 3E), this response was less potent than in the other tumor models where substantial tumor regressions were observed (Figures 3D, 3F–3H). To understand why KM12 tumors were intrinsically more resistant we assessed CD276 expression in tumor cells by flow cytometry and found KM12 was heterogeneous for CD276, with only 11% of the tumor cells positive, while the other tumor cell lines were uniformly positive (Figure S5F). CD276 IF staining of KM12 tumors revealed focal patches of CD276 positive tumor cells, while most tumor cells failed to stain (Figure S5G). In contrast, tumor vessels were uniformly positive in each of the tumor types analyzed (data not shown). Taken together, these data suggest tumor cell CD276 expression may be a primary determinant of early tumor growth responses to m276-MMAE ADC in vivo.
Tumor Endothelium is Intrinsically Resistant to the m276-MMAE ADC
To determine which cells were responding to the m276-MMAE ADC in vivo, we performed co-IF staining for CD276 and apoptosis. In the KM12 model, 24 hours after m276-MMAE exposure apoptotic tumor cells were readily detected in CD276 positive tumor cell patches but, surprisingly, no apoptosis was detected in CD276 positive tumor vasculature (Figure S6A) suggesting that tumor vasculature may be relatively resistant to m276-MMAE ADC. To directly assess the stromal cells’ contribution to the anti-tumor response, we evaluated m276-MMAE efficacy in Cd276 WT and KO mice. Although parental MC38 cells have an extremely low level of endogenous CD276 that is undetectable in most assays, to prevent the possibility of any direct tumor-cell targeting by the ADC we injected mice with Cd276 null MC38-276−/− colon cancer cells. Surprisingly, MC38-276−/− tumors in vivo did not show any discernable growth delay in response to 3 mg/kg of the m276-MMAE (Figure S6B), despite clear expression of CD276 in tumor endothelium of Cd276 WT mice (Figure 2H). Notably, no decrease in vessel density was seen in MC38 tumors following m276-MMAE treatment (Figures 4A and 4B). Similar results were found in Pan02 pancreatic tumors (Figure S6C) that also express high CD276 levels in tumor vasculature but low levels in tumor cells (Figure S3A). These results suggest that the tumor vasculature may be resistant to MMAE, a warhead commonly used on current ADCs.
Figure 4. CD276 ADC Activity against CD276+ Tumor Endothelium.

(A) CD276 (green) and CD31 (red) IF staining of MC38 tumors 48 hr post treatment with vehicle (control), m276-MMAE (3 mg/kg) or m276-PBD (1 mg/kg). Scale bar: 100μm.
(B) Mean vessel area (% of field) in MC38 tumors 48 hr post treatment with vehicle (control), m276-MMAE or m276-PBD. N.S.; non-significant. n = 25 fields/group, *p<0.001.
(C) Flow cytometry was used to measure CD276 or CD31 levels in bEnd.3 Normal ECs (B-NECs) or MC38 Tumor ECs (M-TECs).
(D) Cell viability of B-NECs or M-TECs following treatment with m276-MMAE and m276-MMAE. Error bars in (B) and (D) = SD.
See also Figure S6.
To better understand the MMAE resistance of tumor endothelium, we sought to culture TECs in vitro for drug screening as TECs largely retain their in vivo gene expression patterns for several passages in vitro (Xiao et al., 2014). We established conditional (temperature dependent) immortalized MC38-derived colon TECs (M-TECs), by isolating CD31+CD276+ TECs from MC38 tumors (Figure 4C). We also assessed drug sensitivity of CD31+CD276− bEnd.3 Normal ECs (B-NECs). B-NECs and M-TECs were both highly resistant to m276-MMAE ADC in vitro (Figure 4D), suggesting an alternative payload is needed to effectively target CD276+ TECs.
MMAE resistance in tumor cells can be caused by expression of ATP-Binding Cassette (ABC) transporter P-glycoprotein (P-gp, ABCB1, MDR1)(Chen et al., 2015; Yu et al., 2015) and P-gp expression has been found in tumor endothelium (Akiyama et al., 2012). Interestingly, we found P-gp expression in tumor endothelium both in vitro and in vivo (Figures 5A–5C), suggesting MMAE resistance in TECs may be similar to tumor cells. To directly evaluate the role of P-gp, we measured MMAE free drug activity against P-gp negative HT29 parent cells and HT29/P-gp cells engineered to express P-gp ectopically at levels similar to that found in M-TECs. HT29/P-gp cells (IC50: 3.4 nM) were 19-fold more resistant to MMAE than HT29 cells (IC50: 0.18 nM), and drug sensitivity was completely restored by treatment with the P-gp inhibitors zosuquidar (Figures 5D), PSC-833 or verapamil (data not shown). Zosuquidar increased M-TEC sensitivity to free MMAE 22-fold (Figure 5E) and similar results were obtained with PSC-833 or verapamil (data not shown), confirming that P-gp contributes to MMAE resistance of tumor endothelium.
Figure 5. m276-PBD helps Circumvent P-gp Mediated Resistance.

(A) Co-immunofluorescence staining using anti-P-gp (green) and anti-CD31 (red) antibodies was performed on DMS-273 tumors. The yellow arrowheads mark a P-gp positive vessel. Non-specific rabbit IgG was used as a specificity control. Scale bar: 50 μm.
(B) RT-PCR was used to evaluate ABCB1 mRNA expression in HT29, HT29/P-gp and M-TECs. The β-actin (ACTB) mRNA was used as a loading control.
(C) Immunoblotting was used to evaluate P-gp protein expression in HT29, HT29/P-gp and M-TECs. GAPDH was used as a loading control.
(D) Cell viability assays were used to measure the activity of the cell permeable MMAE free drug against HT29 and HT29/P-gp cells. P-gp activity was inhibited by treating cells with 1 μM zosuquidar (zos).
(E) Cell viability assays were used to measure the activity of the cell permeable MMAE and PBD free drugs against M-TECs. P-gp activity was inhibited by treating cells with 1 μM zosuquidar.
(F) Cell viability assays were used to measure the activity of the cell permeable PBD free drug against HT29 and HT29/P-gp cells. P-gp activity was inhibited by treating cells with 1 μM zosuquidar (zos). Error bars in (D) to (F) = SD.
m276-PBD Kills CD276+ Tumor Cells and Tumor ECs
Due to resistance of M-TECs, we tested alternative free drugs against M-TECs and found they were 142-fold more sensitive to pyrrolobenzodiazepine (PBD) dimers than free MMAE (MMAE IC50: 9.8 nM vs. PBD IC50: 0.069 nM, Figure 5E). PBD dimers are DNA damaging agents that covalently bind to guanine residues in the minor groove of DNA, disrupting transcription and cell division and resulting in the induction of apoptosis (Hartley, 2011). Importantly, previous studies suggest that the PBD dimers used in our study may be a poor substrate for P-gp drug efflux (Jeffrey et al., 2013; Kung Sutherland et al., 2013). Consistent with this, P-gp did not significantly impact the resistance of HT29/P-gp cells to free PBD, and only a modest 1.9-fold increase in sensitivity of M-TECs was observed following treatment with zosuquidar (Figures 5E and 5F). Similar results were obtained with PSC-833 or verapamil (data not shown).
PBD-linked ADCs targeting multiple cell surface receptors, including CD19, CD25, CD33, CD70, CD123, and DLL3 are currently in clinical development, including one in Phase III clinical trials (NCT02785900) (Kung Sutherland et al., 2013). PBD-ADCs in clinical development use an optimal DAR of 2:1 and a cathepsin B cleavable linker that, like vcMMAE, enables release of cell permeable free drug from targeted cells resulting in substantial bystander activity (Kung Sutherland et al., 2013; Li et al., 2016). To generate m276-PBD, we introduced 2 PBD dimer molecules per antibody, one on each GlcNAc sugar residue in the CH2 domain using a cathepsin B cleavable linker (Figure S7A). In vitro viability assays revealed that the m276-PBD ADC killed CD276 positive M-TEC’s with an IC50 of 7.2 nM but showed no killing of B-NECs (Figure 4D).
To directly assess the impact of CD276 cell surface protein levels on cytotoxicity, we compared m276-PBD cytotoxicity in MC38-276−/− cells that lack CD276 with MC38 parent cells, which expressed an average of 1711 antibody binding sites (ABS) per cell and an MC38 clone that expressed ~20,000 ABS (Figure S7B). We also compared clones derived from CD276-transfected MC38 cells that harbored up to 200,000 ABS. Cytotoxicity corresponded to CD276 levels, with the highest expresser displaying the most sensitivity (IC50 of 0.15 nM) (Figure S7B). Although some killing was observed at the highest drug doses tested in cell lines with under 20,000 ABS, this was deemed non-specific as it could not be competed with a 20-fold excess of unlabeled m276 competitor antibody and similar cytotoxicity was observed in Cd276 null cells. These studies demonstrate that selective killing of MC38 cells with the m276-PBD in vitro requires more than 20,000 ABS per cell.
To determine if m276-PBD targeted the tumor stroma, MC38 tumors were established in Cd276 WT and KO mice and tumor growth was measured following treatment with m276-PBD. In contrast to the MMAE-linked ADC, which had no detectable activity in this model (Figure S6B), m276-PBD elicited 75% tumor growth inhibition in Cd276 WT mice at only 1 mg/kg (Figure 6A). Although modest (up to 30%) inhibition of tumor growth was also observed in m276-PBD treated Cd276 KO mice, this was likely caused by linker instability at the amide bond (Figure S7A) which can result in premature drug release in preclinical mouse models (Dorywalska et al., 2016). IF staining revealed apoptotic tumor endothelium 8 hours post m276-PBD treatment (Figure S7C) followed by a striking loss of endothelium by 24 hours, with low levels of CD276 on the few remaining CD31 positive vessels identified (Figures 4A and 4B).
Figure 6. m276-PBD Eradicates Common Tumor Types and is Well Tolerated.

(A) Subcutaneous MC38 tumor growth in Cd276 WT or KO mice. Treatments with vehicle (control) or 1 mg/kg m276-PBD (red arrows) were initiated once tumors reached an average size of 60mm3. TGI; Tumor growth inhibition, n = 15/group, *p<0.0001, **p=0.02
(B) Growth of subcutaneous MC38 or MC38/CD276-c3 (M/CD276) tumors in Cd276 WT or KO mice. Treatments with vehicle (control), free PBD, or 1mg/kg m276-PBD (red arrows) began when tumors reached an average size of 100mm3. n = 10 to 14 mice/group.
(C) CD276 (green) and CD31 (red) IF staining of MC38/CD276-c3 tumors from Cd276 WT mice. Note both tumor cells and tumor vasculature are CD276 positive. Scale bar: 50μm.
(D) Growth of subcutaneous human DMS-273 lung tumors in athymic nude mice treated with PBS (vehicle), m276-MMAE (green arrows), or m276-PBD (red arrows) starting when tumors reached an average size of 140mm3.
(E) Body weights of the DMS-273 tumor-bearing mice from (D).
(F) Growth of subcutaneous human HCT-116 colon tumors in athymic nude mice treated with PBS (vehicle), or m276-PBD (red arrows) once tumors reached an average size of ~140mm3.
(G) Growth of Py230 breast tumors inoculated orthotopically into syngeneic C57BL6 mice and treated with PBS (vehicle) or 1mg/kg of m276-PBD (biwk x 2 - red arrows) starting when tumors reached an average size of ~100 mm3. Error bars in (A), (B), (D) (F) and (G) = SEM.
(H) Kaplan-Meier survival analysis from the Py230 study shown in (G). Log-rank analysis: p<0.0001 m276-PBD vs. vehicle. n=15/group
(I) Body weights of the Py230 tumor-bearing mice from the study in (G). Error bars in (E) and (I) = SD. See also Figure S7.
Because m276-PBD can indeed target tumor vasculature in vivo, next we used genetically engineered models to determine if CD276 positive tumor cells were also a direct target for the m276-PBD. We compared the activity of the PBD-ADC (1 mg/kg biwk x 2) on subcutaneous tumors derived from MC38-276−/− cells or CD276 positive MC38/CD276-c3 (MC38/CD276) tumors grown in Cd276 KO mice. Exogenous levels of CD276 in MC38/CD276 tumor cells are similar to endogenous levels found in CD276 positive human tumor cell lines (compare Figure S7B with S5F). As an additional control, mice were treated 4 times with 0.02 mg/kg of free PBD, equivalent to the free drug load on m276-PBD at 1 mg/kg. As shown in Figure 6B, m276-PBD was able to substantially delay the growth of MC38/CD276 tumors but not MC38 in the Cd276 KO, while free drug had no detectable activity. These results demonstrate that CD276 positive tumor cells are effectively targeted by m276-PBD and that this high molecular weight full-size (IgG1) antibody (160 kDa), despite the known drug diffusion barriers of tumors, can reach tumor cells in sufficient amounts to achieve substantial anti-tumor activity. To determine if simultaneous targeting of the tumor and tumor vasculature would further improve anti-tumor efficacy, we tested m276-PBD against MC38/CD276 tumors grown subcutaneously in Cd276 WT mice where both tumor cells and tumor vessels were CD276 positive (Figure 6C). We observed more potent anti-tumor activity than when only CD276 positive vessels, or tumor cells alone were targeted. Strikingly, most tumors in this group regressed and 60% of the mice were tumor-free 100 days post treatment (Figure 6B).
m276-PBD Displays Broad Tumoricidal and Anti-Metastatic Activity In Vivo
Next we tested m276-PBD against established 100–200 mm3 subcutaneous tumors where endogenous CD276 was expressed by both the tumor cells and tumor vasculature. First, DMS-273 human lung tumor xenografts were treated with either 4 doses of m276-PBD (0.5 mg/kg) or 6 doses of the m276-MMAE (3 mg/kg). Only the PBD conjugate caused sustained regression in 100% of tumors (Figure 6D), while body weights were unaffected by either ADC (Figure 6E). Remarkably, no tumors were found in the m276-PBD treated group 6 months after therapy when mice were sacrificed for post-mortem examination. HCT-116 human colon tumor xenografts responded similarly to m276-PBD therapy, with no tumors detected in the ADC treatment arm six months post-therapy (Figure 6F).
Next, m276-PBD was tested against Py230 murine breast tumors grown orthotopically in the mammary fat pad of C57BL6 mice. This model is both triple-negative and CD276 positive (Figure S5F). Treatments with m276-PBD twice per week for two weeks (biwk x 2) were initiated once tumors reached an average size of 100mm3. m276-PBD blocked the growth of all the tumors in the study (Figure 6G). Although 2 of the 15 mice in this study eventually relapsed, overall survival was significantly extended (Figure 6H). Importantly, the PBD-ADC was well tolerated as mice showed no changes in grooming activity and both body weights and wound closure rates were unaltered by treatment (Figures 6I and S5C). T-cells, dendritic cells, monocytes and other hematopoietic cell populations were also unaffected by m276-PBD treatment (Figures S7D and S7E). Taken together, these studies demonstrate that the CD276 PBD-ADC is highly potent against both mouse and human tumors representative of three of the most prevalent tumor types, i.e. lung, colon and breast cancer.
Based on the widespread overexpression of CD276 in tumors derived from various anatomical locations and the strong tumoricidal activity of the PBD-ADC, we hypothesized that the m276-PBD may also display activity against established late-stage metastases. We first created colon cancer liver metastases via intrasplenic injection of luciferase-tagged human HCT-116-luc tumor cells. Using bioluminescence imaging (BLI) to monitor tumor growth, 8 days post inoculation (dpi) mice were sorted into groups of equal average tumor burden and treatments initiated (biwk x 2). m276-PBD treatment significantly reduced tumor burden and prolonged overall survival with no tumors detectable in 88% of the treatment arm at 5 months (Figures 7A–7C). In the second model, experimental breast cancer lung metastases were generated through i.v. injection of luciferase-tagged 4T1-luc breast tumor cells into syngeneic Balb/c mice. 10 days later, once lung metastases were well established mice were sorted into 3 groups with equal average tumor burden and were either untreated or treated with m276 or m276-PBD (biwk x 2). As we saw previously, overall survival was significantly extended (Figure 7D). IF staining of 4T1 lung metastases revealed strong CD276 expression in both tumor cells and tumor vasculature, and CD31+ vessel density in tumors was significantly reduced 48h post treatment with m276-PBD (Figures 7E–7G). These data suggest that CD276 may be a useful ADC target for treatment of late-stage metastatic disease.
Figure 7. m276-PBD Blocks Metastasis and Extends Overall Survival.

(A) BLI of liver metastases following intrasplenic injection of HCT-116-luc colon cancer cells. 8 days post-inoculation (dpi) mice were sorted into two groups of equal average tumor burden and treatments initiated with PBS (vehicle) or m276-PBD (1 mg/kg) twice per week for two weeks (biwk x 2). Note the decrease in bioluminescence in the m276- PBD treated group at 35 dpi.
(B) Quantification of tumor burden from the HCT-116 liver metastasis study shown in (A). Error bars = SEM.
(C) Kaplan-Meier survival analysis of HCT-116 liver metastasis study shown in (A). Logrank analysis: p<0.0001 m276-PBD vs. vehicle. n=15–17/group.
(D) Kaplan-Meier survival analysis of vehicle (PBS), m276 and m276-PBD in an experimental 4T1 breast cancer lung metastasis model. Treatments began 10 days after i.v. tumor cell inoculation and were administered twice per week (red arrows). Logrank analysis: p=0.0002 m276-PBD vs. untreated, p<0.0001 m276-PBD vs. m276. Nonsignificant: untreated vs. m276. n = 5 to 8/group.
(E) CD276 IF staining in 4T1 breast cancer lung metastasis. Note the reduced number of CD31+ vessels within tumor nodules 48 hr post treatment with 1 mg/kg m276-PBD. Scale bar: 100μm.
(F) Higher magnification of insets from (E). Arrowheads highlight some CD31+ tumor vessels that are also CD276+. Scale bar: 20μm.
(G) Tumor vessel density in 4T1 lung metastases 48 hr post treatment with 1 mg/kg m276-PBD. n=30 metastases/group. Error bars = SD.
DISCUSSION
We previously identified CD276 as the most differentially expressed cell-surface tumor endothelial marker that could distinguish physiological and pathological angiogenesis (Seaman et al., 2007). Expanding on this, here we found that CD276 is expressed not only on angiogenic tumor vasculature but also on established vasculature that has been co-opted by the tumor. Because vessel co-option may contribute to resistance to current anti-angiogenic therapies (Bridgeman et al., 2016; Kuczynski et al., 2016), CD276 mAbs could aid in the effective targeting of both angiogenic and non-angiogenic tumor vasculature. In addition to tumor vessels, tumor cells also frequently overexpress CD276 (Brunner et al., 2012; Qin et al., 2013; Zang et al., 2010). We further validated CD276 expression on human tumors. Remarkably, each solid tumor type examined displayed moderate to strong CD276 staining in at least 50% of the samples analyzed, suggesting that m276-ADCs could potentially benefit a substantial fraction of cancer patients. Following ADC binding, internalization and cleavage, released MMAE and PBD are both membrane permeable and can elicit strong bystander activity (Li et al., 2016), suggesting that these ADCs could also have activity against tumors with heterogeneous CD276 expression. However, human clinical studies correlating receptor levels with tumor response data will be required to determine if CD276 staining can be used as a biomarker to identify patients most likely to benefit from CD276 therapies.
From an ADC standpoint it is important to understand the functional consequences of CD276 overexpression in both tumor cells and tumor associated stroma, because targeting a protein that normally functions in tumor suppression could potentially promote the selection of a more aggressive tumor cell population thereby offsetting initial anti-tumor responses. Most studies investigating the impact of CD276 overexpression on patient outcome indicate that high CD276 levels correlate with poor prognosis (Arigami et al., 2010; Loos et al., 2010). Our studies using Cd276 KO models supports the idea that CD276 overexpression has modest or no detectable effect on tumor growth. Our results also indicate that the current commercially available CD276 antibodies are not optimal for detecting CD276 in FFPE tumor sections and that further improvements in antibody sensitivity could increase the fraction of receptor positive samples identified, as previously shown for other antibodies (Cheang et al., 2006). Thus, further work to develop even more robust CD276 antibodies for IHC of FFPE tissues is warranted.
To test the hypothesis that CD276-directed therapeutic agents could simultaneously target tumor cells and tumor vasculature (Seaman et al., 2007), we created a fully-human mAb called m276. While m276 had no activity on its own, the Fc-enhanced anti-CD276 mAb enoblituzumab did elicit tumor growth delays in preclinical studies, but was unable to elicit long-term sustained responses (Loo et al., 2012). To generate a more potent CD276-targeted drug, we first conjugated m276 to MMAE. In preclinical studies m276-MMAE showed potent tumoricidal activity but, surprisingly, no obvious cytotoxic activity towards CD276 positive tumor endothelium at 3 mg/kg, a drug dose that displayed potent activity against tumor cells.
By exploring alternative payloads we found that TECs were intrinsically more sensitive to PBD dimers than MMAE, and that a PBD-linked m276 ADC could ablate tumor vasculature. By comparing MC38 (CD276 negative) tumor responses in Cd276 WT versus Cd276 KO mice, we demonstrate that CD276 positive stroma is important for m276-PBD killing. The increased sensitivity of tumor endothelium to m276-PBD compared to m276-MMAE was partly due to the fact that these cells express P-gp and that our PBD dimers, unlike MMAE, were hardly impacted by P-gp mediated efflux. As expected based on its dual targeting ability, m276-PBD consistently displayed more potent anti-tumor activity than m276-MMAE in vivo, eliciting long-standing tumor regressions without notable toxicities.
Based on its dual-compartment targeting ability, m276-PBD appears to be a more promising ADC than m276-MMAE for most tumors. However, m276-MMAE may also have clinical utility, for example, in prostate adenocarcinoma where tumor cells are more frequently CD276 positive than the tumor vasculature (Table 1). Also, based on its strong preference for rapidly dividing cells, m276-MMAE could potentially display a more favorable therapeutic window when targeting tumors with a high proliferation rate. Also, because MMAE and PBD bind different targets (microtubules vs. DNA) and have distinct mechanisms of action it follows that acquired resistance, should it develop, is likely to arise through different mechanisms that could also vary according to cancer type. Therefore, if tumor cells develop resistance to one m276-ADC, they may still be sensitive to the other. Accordingly, it is possible that the most useful regimen will involve a combined or alternating treatment schedule, allowing for dose reductions of individual ADCs and reducing toxicities while minimizing potential for development of resistance.
PBDs can bind DNA minor groves with minimal distortion of DNA structure (Hartley, 2011). This unique feature of PBDs helps prevent detection by DNA repair mechanisms, resulting in persistent DNA cross-links and minimizing the possibility of resistance. Although downregulation of CD276 is a potential mechanism of resistance, we have not found evidence for receptor loss in tumors that have recurred following m276-ADC treatment. The tumor vasculature is comprised of non-malignant host cells that are genetically more stable than the tumor cells. Therefore, if this target population is able to develop resistance to the m276-PBD then the mechanisms are likely to be different than in the tumor cells. Although the high potency and selectively of the CD276-ADCs may be sufficient to avoid resistance in many cases (as demonstrated by the large number of cures observed in our preclinical models) further studies are required to determine if tumors that have relapsed have intrinsic or acquired resistance, and the underlying mechanisms.
Although CD276 is overexpressed in tumors, the fact that CD276 can be detected in normal tissues raises concerns for toxicity (Loo et al., 2012; Modak et al., 2001; Seaman et al., 2007). We observed no weight loss with either ADC, no alterations in wound healing, no changes in hematopoietic cell populations in CD276 WT versus KO mice, and no other overt signs of toxicity. The relatively high number of antibody binding sites needed for cytotoxicity (>20,000 for MC38 cells) indicates that many normal cells may be protected from adverse effects because their CD276 levels fall below a threshold needed for cytotoxicity. MMAE is known to selectively target dividing cells, providing an additional safeguard that helps protect normal quiescent cells from inadvertent targeting. While PBD dimers may not have as stringent a requirement for proliferating cells as MMAE, the potent tumoricidal activity of the m276-PBD in vivo suggests that the stressful conditions of the tumor microenvironment may also predispose CD276 positive cancer or tumor-associated vascular cells to cell killing. Notwithstanding these considerations, toxicity studies in primates will be required to fully establish the safety profile of these ADCs in vivo.
Our studies help shed light on a recently failed Phase II clinical trial using an MMAE-linked ADC targeting PSMA (NCT01856933). The trial, which was initiated based on a high level of PSMA expression observed in the tumor neovasculature of human glioblastoma multiforme (GBM) (Nomura et al., 2014), found no evidence of PSMA-ADC efficacy (Elinzano et al., 2016). Because PSMA is not overexpressed in rodent tumor vessels (Huang et al., 2004), unlike in humans, preclinical studies with the PSMA ADC were limited to tumor cell models wherein only the tumor cells were PSMA-positive (Olson and Israel, 2014). Our work suggests that the GBM trials likely failed due to the selection of an inappropriate warhead, and suggest that PSMA may still be a valid neovascular drug target for GBM and other human tumor types through the re-engineering of a PBD-linked PSMA ADC.
In summary, we describe the development of a CD276 targeting ADC that can simultaneously target both cancer cells and tumor associated vasculature. CD276 overexpression is associated with most solid tumors types, including many that are in urgent need of better targeted therapies such as triple-negative breast cancer, pancreatic cancer and glioblastomas. The widespread overexpression of CD276 in cancers from disparate anatomical sites suggests that CD276-ADCs may be a particularly valuable tool for the treatment of late-stage metastatic disease.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| m276 | This paper | N/A |
| m276-MMAE | This paper | N/A |
| m276-PBD | This paper | N/A |
| m825-MMAE | This paper | N/A |
| Rabbit Anti-CD276 (clone: EPNCIR122 | Abcam | Cat# ab134161 |
| Rabbit Anti-P-Glycoprotein (clone: EPR10364-57) | Abcam | Cat# ab170904 |
| Rabbit IgG, Monoclonal Isotype Control (clone: EPR25A) | Abcam | Cat# ab172730 |
| Rabbit Anti-Human von Willebrand Factor | Agilent Technologies | Cat# A0082 |
| Mouse Anti-Cytokeratin19 Alexa 647 (clone: A53-B/A2) | Biolegend | Cat# 628506 |
| Rat Anti-Mouse CD31, biotin (clone MEC13.3) | BD Biosciences | Cat # 553371 |
| Rat Anti-Mouse CD16/CD32, Purified (clone: 93) | eBioscience | Cat# 14-0161-81 |
| Rat Anti-Mouse IgM PE (clone: II/41) | eBioscience | Cat# 12-5790-81 |
| Rat Anti-Mouse Ly-6G (Gr-1) PE (clone: RB6-8C5) | eBioscience | Cat# 12-5931-81 |
| Rat Anti-Human/Mouse CD45R PE (clone: RA3-6B2) | eBioscience | Cat# 12-0452-81 |
| Rat Anti-Mouse CD4 PE (clone: GK1.5) | eBioscience | Cat# 12-0041-81 |
| Rat Anti-Mouse Ly-6C PE (clone: HK1.4) | eBioscience | Cat# 12-5932-80 |
| Rat Anti-Mouse Ly-6G (Gr-1) APC (clone: 1A8-Ly6g) | eBioscience | Cat# 17-9668-80 |
| Rat Anti-Mouse CD19 APC (clone: eBio1D3 (1D3) | eBioscience | Cat# 17-0193-80 |
| Hamster Anti-Mouse CD11c APC (clone: N418) | eBioscience | Cat# 17-0114-81 |
| Rat Anti-Mouse CD11b APC-eFluor 780 (clone: M1/70) | eBioscience | Cat# 47-0112-80 |
| Rat Anti-Mouse CD8a Biotin (clone: 53–6.7) | eBioscience | Cat# 13-0081-82 |
| Goat Anti-Human/Mouse Desmin | R&D Systems | Cat# AF3844 |
| Goat Anti-Human B7-H3 | R&D Systems | Cat# AF1027 |
| Rat Anti-Mouse PECAM-1 (clone: MEC 13.3) | Santa Cruz Biotechnology | Cat# sc-18916 |
| Mouse Anti-Cytokeratin 7 (clone: RCK105) | Santa Cruz Biotechnology | Cat# sc-23876 |
| Rat Anti-Mouse pan ECA (clone: MECA-32) | Santa Cruz Biotechnology | Cat# sc-19603 |
| Human IgG1 Lambda-UNLB | Southern Biotech | Cat# 0151L-01 |
| Goat Anti-FITC/Oregon Green, Alexa Fluor 488 | ThermoFisher | Cat# A-11096 |
| Donkey Anti-Goat IgG (H+L), Alexa Fluor 488 | ThermoFisher | Cat# A11055 |
| Mouse Anti-CD31/PECAM-1 (Clone: JC/70A) | ThermoFisher | Cat# MA5-13188 |
| Rat anti-CD31 (clone: 2H8) | ThermoFisher | Cat# MA3105 |
| Mouse Anti-P-glycoprotein (clone: C219) | ThermoFisher | Cat# NB6001036 |
| Texas Red Streptavidin | Vector Labs | Cat# SA-5006 |
| HRP Rabbit Anti-Goat IgG, F(ab′)2 Fragment Specific | Jackson ImmunoResearch | Cat# 305-035-047 |
| HRP Goat Anti-Mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 115-035-146 |
| FITC Goat Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat# 111-095-144 |
| FITC Goat Anti-Human IgG (H+L) | Jackson ImmunoResearch | Cat# 109-095-088 |
| Biotin-SP F(ab′)2 Fragment Donkey Anti-Rat IgG (H+L) | Jackson ImmunoResearch | Cat# 712-066-150 |
| DyLight 594 Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 715-515-150 |
| DyLight 405 Streptavidin | Jackson ImmunoResearch | Cat# 016-470-084 |
| Bacterial and Virus Strains | ||
| Electromax DH10B competent cells | ThermoFisher | Cat# 18290015 |
| Biological Samples | ||
| Tissue Microarrays | US Biomax | N/A |
| Human paraffin blocks | CHTN | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PSC833; MDR1/P-gp Inhibitor | Sigma Aldrich | Cat# SBPSC |
| Zosuqidar; MDR1/P-gp Inhibitor | MedChem Express | Cat# HY-15255 |
| Verapamil hydrochloride; MDR1/P-gp Inhibitor | MedChem Express | Cat# HY-A0064 |
| Monomethyl auristatin E (MMAE) | Levena Biopharma | Cat# T1004 |
| Pyrrolobenzodiazepine (PBD) dimer | Levena Biopharma | Custom synthesis |
| EGM-2 BulletKit | Lonza | Cat# CC-3162 |
| 20% paraformaldehyde | Electron Microscopy Sciences | Cat# 15713 |
| Triton X-100 | Sigma Aldrich | Cat# T9284 |
| Imidazole | Sigma Aldrich | Cat# 15513 |
| TO-PRO®-3 Iodide (642/661) | ThermoFisher | Cat# T3605 |
| Hoechst 33258, Pentahydrate (bis-Benzimide) | ThermoFisher | Cat# H3569 |
| Lipofectamine 2000 Transfection Reagent | ThermoFisher | Cat# 11668019 |
| PolyFect Transfection Reagent | Qiagen | Cat# 3101105 |
| VECTASHIELD HardSet Mounting Medium with DAPI | Vector Labs | Cat# H-1500 |
| Blocking Reagent | Roche | Cat# 11096176001 |
| EcoRI-HF | New England Biolabs | Cat # R3101S |
| NotI-HF | New England Biolabs | Cat # R3189S |
| XhoI | New England Biolabs | Cat# R0146S |
| NheI-HF | New England Biolabs | Cat# R3131S |
| HindIII-HF | New England Biolabs | Cat# R3104S |
| humanCD276(ED)-AP | This paper | N/A |
| mouseCD276(ED)-AP | This paper | N/A |
| ratCD276(ED)-AP | This paper | N/A |
| monkeyCD276(ED)-AP | This paper | N/A |
| Critical Commercial Assays | ||
| Dual Endogenous Enzyme-Blocking Reagent | Dako | Cat# S200380-2 |
| Biotin Blocking System | Dako | Cat# X059030-2 |
| ApopTag Red In Situ Apoptosis Detection Kit | EMD Millipore | Cat# S7165 |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| Ni-NTA agarose Beads | Qiagen | Cat# 30210 |
| Fluoreporter FITC protein labeling kit | ThermoFisher | Cat# F6434 |
| Pierce Fab Preparation Kit | ThermoFisher | Cat# 44985 |
| Quantum Simply Cellular anti-Human IgG | Polysciences Inc | Cat# BLI816A-1 |
| Vectastain Elite ABC Kit (Goat IgG) | Vector Labs | Cat# PK-6105 |
| Vectastain Elite ABC Kit (rabbit IgG) | Vector Labs | Cat# PK-6101 |
| ECL 2 Western Blotting Substrate | ThermoFisher | Cat# 80196X3 |
| SuperSignal West Dura Extended Duration Substrate | ThermoFisher | Cat# 34075 |
| Pierce Fab Micro Preparation Kit | ThermoFisher | Cat# 44685 |
| AlamarBlue Cell Viability Reagent | ThermoFisher | Cat# DAL1100 |
| SuperScript III First-Strand Synthesis System | ThermoFisher | Cat# 18080051 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | CRL-1573 |
| CHO | ATCC | CCL-61 |
| DLD-1 | ATCC | CCL-221 |
| SW620 | ATCC | CCL-227 |
| bEnd.3 | ATCC | CRL-2299 |
| Py230 | ATCC | CRL-3279 |
| L929 | ATCC | CCL-1 |
| B16 | DCTD | N/A |
| DMS-273 | DCTD | N/A |
| HCT-116 | DCTD | N/A |
| HCT-116-luc | (Xu et al., 2014) | N/A |
| HT-29 | DCTD | N/A |
| KM-12 | DCTD | N/A |
| MDA-MB-231 | DCTD | N/A |
| OVCAR3 | DCTD | N/A |
| Pan 02 | DCTD | N/A |
| UACC-62 | DCTD | N/A |
| MC38 | Jeffrey Schlom | N/A |
| KM12SM | Isaiah Fidler | N/A |
| 4T1-luc | (Xu et al., 2014) | N/A |
| CHO/huCD276 | This paper | N/A |
| L929/msCD276 | This paper | N/A |
| L929/huCD276 | This paper | N/A |
| MC38-276 −/− (CRISPR Cd276 KO) | This paper | N/A |
| MC38/CD276-c3 | This paper | N/A |
| M-TEC (MC38 Tumor ECs) | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Cd276−/− mouse | This paper | N/A |
| C57BL6-NCr | Charles River | Strain code: 027 |
| BALB/cAnNCrl | Charles River | Strain code: 028 |
| Crl:NU(NCr)-Foxn1nu (Athymic nude) | Charles River | Strain code: 490,491 |
| CB17/Icr-Prkdcscid/IcrCr (CB17 SCID) | Charles River | Strain code: 561 |
| Immortomouse; CBA;B10-Tg(H2Kb-tsA58)6Kio/Crl | Charles Rivers | Strain code: 238 HE |
| Oligonucleotides | ||
| Genotyping Primer: CD276-F GGACAGCCAGGGAAGTGG | This paper | Custom synthesis IDT |
| Genotyping Primer: CD276-R1 ACCTCTGATGGAGCAAATGC | This paper | Custom synthesis IDT |
| Genotyping Primer: CD276-R2 TGAATACAGCACCCATATAAGGC | This paper | Custom synthesis IDT |
| QPCR Primer: β-Actin-For GGCACCAGGGCGTGATG | This paper | Custom synthesis IDT |
| QPCR Primer: β-Actin-Rev AGGTCTCAAACATGATCTGGGTC | This paper | Custom synthesis IDT |
| QPCR Primer: Abcb1-For ATGGATGAGATTGAGAAAGCTGTC | This paper | Custom synthesis IDT |
| QPCR Primer: Abcb1-Rev TGACAAGTTTGAAGTAAATGCC | This paper | Custom synthesis IDT |
| CRISPR/Cas9 CD276 −/− TGGCACAGCTCAACCTCATC | This paper | Custom synthesis Origene |
| CRISPR/Cas9 CD276 −/− CTCGAAGCCCAGCATGACCC | This paper | Custom synthesis Origene |
| CRISPR/Cas9 CD276 −/− CAGCATGACCCTGGAGCCCA | This paper | Custom synthesis Origene |
| Recombinant DNA | ||
| humanCD276/pcDNA3.1(+) (full length) | This paper | N/A |
| RatCD276/pcDNA3.1(+) (full length) | This paper | N/A |
| mouseCD276/pcDNA3.1(+) (full length) | This paper | N/A |
| HuCD276(ED)/pAPtag-5 (extracellular domain) | This paper | N/A |
| MsCD276(ED)/pAPtag-5 (extracellular domain) | This paper | N/A |
| RatCD276(ED)/pAPtag-5 (extracellular domain) | This paper | N/A |
| CynoCD276(ED)/pAPtag-5 (extracellular domain) | This paper | N/A |
| Hu-MDR1/pcDNA3.1 | Michael Gottesman | N/A |
| pCas-Guide-EF1a-GFP | Origene | Cat# GE100018 |
| pcDNA3.1(+) Mammalian Expression Vector | ThermoFisher | Cat# V79020 |
| APTag5 Kit B | GenHunter | Cat# Q202 |
| Software and Algorithms | ||
| FlowJo 10.0.8r1 | FlowJo LLC | www.flowjo.com |
| BIAevaluation | Biacore | |
| GraphPad Prism Version 6.05 for Windows | GraphPad Software, La Jolla California USA | www.graphpad.com |
| FIJI | ImageJ | http://imagej.net/Fiji |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author Brad St. Croix (stcroixb@mail.nih.gov)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Clinical Samples
Anonymized human FFPE normal or cancer tissue samples were obtained from the Cooperative Human Tissue Network (CHTN) or the Laboratory of Pathology Tissue Resource Committee (TRC) at NIH with approval from the NIH Office of Human Subject Research. All clinical protocols were approved by institution-specific investigational review boards, with appropriate patient informed consent.
Animals
The mice with Cd276 null or floxed alleles used in this study have been backcrossed to C57BL/6-NCrl (Charles Rivers) at least 10 generations. Athymic Cd276 KO mice on an immunodeficient background (Cd276−/−, nu/nu) were generated by crossing Cd276 KO mice on a C57BL/6 background with athymic NCr-nu/nu mice (Charles Rivers). Athymic nude Cd276+/+ or Cd276−/− littermates derived from Cd276+/− heterozygous intercrosses were randomly assigned to experimental groups. The immortomouse (Jat et al., 1991) (strain CBA;B10-Tg(H2Kb-tsA58)6Kio/Crl) was obtained from Charles River. All mice were bred and maintained in a pathogen-free facility certified by the Association for Assessment and Accreditation of Laboratory Animal Care International, and the study was carried out in accordance with protocols approved by the NCI Animal Care and Use Committee.
Cell Lines
The HEK293 (293), CHO, Py230, DLD-1, SW620 and bEnd.3 cell lines were obtained from the American Type Culture Collection (ATCC) and the B16, DMS-273, HCT116, HT29, KM-12, MDA-MB-231, OVCAR3, Pan02 and UACC-64 (UACC) cell lines were from the DCTD Tumor Repository at NCI (Frederick, MD). The MC38 cell line was a kind gift from Jeffrey Schlom and the KM12SM cell line was a kind gift from Isaiah Fidler. 4T1-luc and HCT-116-luc cells were previously described (Xu et al., 2014). CHO cells were maintained in Ham’s F12 medium supplemented with 10% fetal bovine serum (FBS). M-TECs and bEnd.3 were maintained in EGM-2 (Endothelial Cell Growth Medium) (Lonza). 293 cells were cultured in DMEM supplemented with 10% FBS and all other cells were cultured in RPMI 1640 supplemented with 10% FBS. Cells expressing CD276-AP, full-length CD276, or human P-gp were generated by lipofectamine 2000-mediated stable transfection.
METHOD DETAILS
Cd276 Gene Targeting and Animal Studies
The second exon of Cd276 was chosen for targeting because it contains the start codon and signal peptide. A LoxP site was introduced upstream and an Frt-loxP-Neo-Frt-loxP cassette was inserted downstream of exon 2 by sequential recombineering. The Cd276 conditional KO vector was electroporated into V6.4 129/SvJae X C57BL/6J hybrid F1 embryonic stem (ES) cells, and ES cell clones with one targeted allele were identified by Southern blot analysis by probing BamHI or SpeI digested DNA with PCR-generated probes. Targeted ES clones were microinjected into C57BL/6 blastocysts to generate chimeric mice and transfer the Cd276neo allele to the germline. Mice with a Cd276+/neo allele were identified by Southern blotting and crossed with either a transgenic β-actin-flp deleter strain to generate the conditional “floxed” allele or a transgenic β-actin-Cre deleter strain to generate the Cd276 null allele. Offspring from heterozygous intercrosses were born at the expected Mendelian frequency.
Genotyping
Mice were genotyped for Cd276 by mixing the following primers in a single PCR: F: 5′-GGACAGCCAGGGAAGTGG-3′, R1: 5′-ACCTCTGATGGAGCAAATGC-3′, R2: 5′-TGAATACAGCACCCATATAAGGC-3′. The expected amplicon sizes are 183 bp and 374 bp for the wildtype and null alleles, respectively.
Tumor Studies
Tumor studies were performed in female 7 to 10-week old C57BL6-NCr (B16, MC38, Py230), Balb/c-NCr (4T1), athymic NCr-nu/nu (DLD-1, HCT-116, KM12, KM12SM, MDA-MB-231, HT29, DMS-273 SW620, UACC) or CB17 SCID (OVCAR3) mice from Charles River Laboratories. For tumor studies involving Cd276 KO mice, only Cd276 WT and Cd276 KO littermates derived from Cd276 heterozygous intercrosses were used for comparison. 2.5 × 106 to 5 × 106 tumor cells were injected into the mammary fat pad (Py230 and MDA-MB-231) or subcutaneously into the flank. HCT-116, KM12 and DMS-273 tumors were co-injected with 50% Matrigel. Tumors were measured with a digital caliper, and tumor volumes were calculated using the formula LxW2x0.5 and presented as the mean ± SEM. For therapeutic studies, mice were sorted into groups containing the same average size tumors (usually ~100mm3) prior to initiation of therapy. To minimize tumor variation in these studies, at the time of sorting, mice were excluded from the analysis if their tumors were less than half the average tumor size for the group or more than double the average tumor size. Mice were treated with CD276 antibodies or free drug at the doses and schedules described in the individual figures. During initial studies the ADCs were administered intravenously (i.v.) through tail vein injection. However, during the course of this work we determined that i.v. and intraperitoneal (i.p.) dosing produced equivalent results. Because mouse tail vein injections are difficult to perform with 100% precision and consistency, in later studies ADCs were administered by i.p. injection.
Wound Healing Assay
C57BL6 mice were shaved and wounded on both flanks (2 wounds per mouse) with a 6 mm diameter uni-punch biopsy instrument (Premier). Wounds were measured daily with a digital caliper. Treatments with vehicle, 3 mg/kg m276-MMAE and 1 mg/kg m276-PBD were administered one day prior to wounding and on days 2 and 6 post wounding. Each treatment group contained 15 mice (30 wounds).
Cd276 Gene Targeting in Cells using CRISPR/Cas9
DNA constructs encoding the following three guide-RNAs in the pCas-Guide-GFP expression vector were purchased from Origene: TGGCACAGCTCAACCTCATC, CTCGAAGCCCAGCATGACCC, CAGCATGACCCTGGAGCCCA. A mixture of the three DNA constructs was transfected using Polyfect (Qiagen) into B16 parent cells or an MC38 clone that expressed a low but uniform level of CD276 cells. 48 hr after transfection, the transfected cell populations were sorted for GFP expression by flow cytometry, expanded in culture, then flow sorted and expanded again. The cells were then stained with m276 IgG1 followed by PE-labelled goat anti-human IgG1 (Invitrogen), and PE-negative single cells were sorted by flow cytometry, cloned and expanded. The phenotype and genotype of single clones were confirmed through FACS analysis and DNA sequencing respectively. Because the B16 Cd276 wildtype starting population is a heterogeneous pool, 8 validated knockout clones were combined to generate a single B16-276−/− knockout pool for comparison.
CD276 Expression Vectors
Full length human CD276 and rat CD276 expression vectors were made by excising the full-length CD276 sequences from EST IMAGE clone # 7472032 (human) or clone # 7104832 (rat) using the restriction enzymes EcoR1 and Not1, and cloning into the same sites of pcDNA3.1+. The full length mouse CD276 expression vector was made by excising the full-length Cd276 sequence from EST IMAGE clone #6397680 using the restriction enzymes EcoRI and XhoI and cloning into the same sites of pcDNA3.1+. cDNAs encoding the extracellular region of mouse (amino acids 1-248) and human (amino acids 1-465) CD276 were PCR amplified and directionally cloned into the alkaline phosphatase (AP)-Tag5 vector (Genhunter, Nashville, TN) by incorporating the restriction enzyme sites NheI and HindIII into the PCR forward and reverse primers respectively. cDNAs encoding the extracellular region of rat (amino acids 1-248) and monkey (Cynomolgus; amino acids 1-465) CD276 were synthesized and cloned between the NheI and HindIII sites of AP-Tag5. All vectors were amplified in E. coli. (DH10B) and sequence verified.
Purification of CD276(ED)-AP Fusion Proteins
Conditioned media from 293 stable transfectants expressing the extracellular domain (ED) of mouse, rat, human, or monkey (Cynomolgus) CD276 fused to alkaline phosphatase were collected, filtered, adjusted to pH 7.7 with NaH2PO4 and supplemented with 1% Triton-X 100, 150 mM NaCl and 3 mM Imidazole. His-tagged AP-fusion proteins were captured by Ni-NTA agarose beads (1:200 v/v; Qiagen), purified according to the manufactures protocol, and dialyzed into PBS. SDS-PAGE analysis followed by Coomassie blue staining revealed that all AP-fusion proteins were at least 95% pure.
Isolation of Conditionally Immortalized TECs
To generate MC38 tumor endothelial cells (M-TECs), MC38 tumors were injected subcutaneously into the immortomouse that contains a temperature sensitive SV40 TAg transgene (Jat et al., 1991). Tumors were excised and endothelial cells isolated using biotinylated anti-CD31 antibodies (BD Biosciences, clone MEC13.3) conjugated to streptavidin-linked magnetic beads as previously described (Seaman et al., 2007). To eliminate potential overgrowth by contaminating MC38 tumor cells, the isolated cells were expanded for one week at 33°C, CD31 bead-selected again, cloned by limiting cell dilution, evaluated for CD31 expression and positive clones pooled. Because the cultured TECs gradually lost CD276 expression with time, TECs were flow sorted to enrich for CD31+/CD276+ double positive cells, and a large frozen stock generated. Cells maintained strong CD276 positivity for several weeks and low passage cells were used for experiments. TECs were maintained at 33°C in EGM-2 media (Lonza) on collagen coated flasks and only shifted to 37°C (to silence the immortalizing SV40 TAg transgene) for experiments.
Development of Rabbit mAb Against CD276
A monoclonal antibody against CD276 was made in collaboration with Epitomics (now AbCam). Briefly, rabbits were injected sequentially with both recombinant huCD276(ED)-AP protein and CHO cells stably overexpressing full-length human CD276 on the cell surface (CHO/huCD276). Rabbits with the highest antibody titers were identified by comparing pre- and post-immunization bleeds for selective reactivity with L929 cells stably overexpressing either mouse or human CD276. Hybridoma supernatants that displayed specific reactivity with CD276-ED-AP by ELISA were screened for reactivity against CD276 on the cell surface by flow cytometry using L929, L929/msCD276 and L929/huCD276 cells. Clones that tested positive by flow cytometry were then tested for reactivity with CD276 in immunoprecipitation, immunoblotting, and immunofluorescence applications. One mAb that reacted with both mouse and human CD276 in each of the assays was selected for further cloning into a mammalian expression vector which was then used to generate stable 293-derived producer cells for antibody production and purification. The purified mAb antibody [called EPNCIR122] is available from Abcam (ab134161).
Immunoprecipitation and Immunoblotting
Supernatants or lysates were incubated overnight with rabbit anti-CD276 mAb (see above section: Development of Rabbit mAb Against CD276) or a nonspecific IgG control antibody (AbCam cat # ab172730). Precipitated proteins were eluted from protein A-agarose beads (Roche), separated by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore). Immunoblots were probed with a goat anti-CD276 polyclonal primary antibody (R&D), followed by HRP-conjugated anti-goat secondary antibody (Jackson Immunoresearch Laboratories), and visualized using Pierce enhanced chemiluminescence (ECL 2) Western Blotting Substrate (ThermoFisher Scientific) according to the supplier’s instructions. For P-glycoprotein detection, lysates separated by SDS-PAGE were transferred to PVDF and probed with mouse anti-P-glycoprotein antibody from ThermoFisher Scientific (clone C219), followed by HRP-conjugated anti-mouse secondary antibody (Jackson Immunoresearch). P-glycoprotein was visualized using the SuperSignal West Dura Extended Duration Substrate (ThermoFisher Scientific) according to the supplier’s instructions.
m276 Antibody Production and Purification
A large yeast display naïve single chain variable fragment (scFv) human antibody library was used to generate the anti-human CD276 antibodies described herein. The library was constructed using a collection of human antibody gene repertoires, including the genes used for the construction of a phage display Fab library (Zhu and Dimitrov, 2009) and those from more than 50 additional individuals. This library, being totally synthetic, is not subject to tolerance mechanisms found in normal immune responses and allowed the generation of antibodies against regions of the CD276-ED (extracellular domain) that are 100% conserved between mouse and human. In vitro selection of the yeast display library involved three rounds of sequential panning on biotinylated, purified recombinant CD276(ED)-AP (alkaline phosphatase) fusion proteins. For this, 10 μg of biotinylated hCD276(ED)-AP was incubated with approximately 5×1010 cells from the initial naïve antibody library in 50 ml PBSA (phosphate-buffered saline containing 0.1% bovine serum albumin) for 2 h, washed with PBSA and captured with streptavidin conjugated microbeads from Miltenyi Biotec using the AutoMACS system (Cologne, Germany). The sorted cells were amplified and the panning was repeated once with the human hCD276(ED)-AP, and once with the mouse mCD276(ED)-AP protein to enrich for cross-reactive binders. After characterizing several scFvs for binding specificity, cross-species reactivity and uptake, the lead antibody, designated m276, was converted into a full-size human IgG1 and a stable CHO producer cell line was generated. The expressed m276 IgG1 was collected from culture supernatants grown in serum free medium and purified by Protein A chromatography. Antibody preparations used for in vivo studies possessed <5% aggregates and endotoxin levels were below 1 EU/mg.
Preparation of m276-MMAE ADC
The m276-MMAE ADC was made by conjugating the antibody to a maleimide activated drug linker containing monomethyl auristatin E (MMAE). The chemistry involved two reactions. In the first step, partial reduction of the antibody inter-chain disulfide bonds with tris (2-carboxyethyl)-phosphine hydrochloride (TCEP HCL) yielded a reduced antibody intermediate mixture containing free thiols. In the second step, the partially reduced antibody was reacted with excess maleimide activated drug linker to afford the crude ADC product with the desired average drug to antibody ratio (DAR) of approximately 4. Process related impurities were removed by ultrafiltration, and the final formulation (10 mg/mg of m276-MMAE in 20mM pH5.8 histidine buffer) was sterile filtered and packaged. Antibody preparations used for in vivo studies had a DAR of 4, possessed 5% aggregates and endotoxin levels were 0.275 EU/mg.
Preparation of m276-PBD ADC
The purified CHO cell expressed m276 IgG1 was directly used for glycan-based site-specific modification and conjugation as previously described (Zhu et al., 2014) with the following modifications. C2-Azide-Galacotose was used as substrate for the Fc-glycan modification. DBCO-PEG4-VA-PBD was used as payload for the conjugation following the click chemistry based approach (Baskin et al., 2007). To maintain solubility of the DBCO-linker-PBD, the azide-attached antibody itself was diluted with propylene glycol to a final concentration of 33%. The solution of DBCO-linker-PBD in propylene glycol was added to the antibody solution with a drug-to-antibody mole ratio of about 3:1. The final concentration of propylene glycol in the conjugation reaction was 50%. The reaction was allowed to proceed for at least 4 hr at room temperature. The finished reaction mixture was purified by size exclusion chromatography and dialyzed into PBS.
Antibody Affinity Measurements
m276 Fab was generated from full size IgG1 using the Pierce Fab Preparation Kit (ThermoFisher Scientific). Surface plasmon resonance was used to measure the binding affinity of the Fab to the CD276 extracellular domain on a BIAcore X100 instrument (GE Healthcare). Purified rat, mouse, monkey and human AP-CD276 fusion proteins were diluted in 10 mM sodium acetate buffer (pH 5.0) and immobilized on a CM5 biosensor chip using an amine coupling kit. The running buffer was HBS-EP (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% surfactant P20). The Fabs diluted with the running buffer were allowed to flow through the cells, at concentrations ranging from 0.05 nM to 500 nM. After 10 min of dissociation, the chip was regenerated with 10 mM acetate buffer, pH 4.0. The data were fitted with 1:1 binding model and the dissociation rate constant was estimated with BIAevaluation software.
m276-MMAE Serum Stability Test
Stability of m276-MMAE was performed by Seventh Wave Laboratories. Briefly, m276-MMAE was incubated with PBS or serum from mouse, rat, monkey (Cynomolgus), or humans for 0, 1, 2, 4, 7, 10, 14 or 21 days, flash frozen, then analyzed by solid phase extraction of free MMAE followed by LC-MS/MS analysis. Auristatin F was used as an internal control.
Flow Cytometry
Pilot studies revealed that CD276 on the surface of cultured cells is insensitive to brief trypsinization. Therefore, cells were trypsinized, rinsed in cold PBS/0.5%BSA (PBS/BSA), and labeled with m276 human anti-CD276 mAb or rabbit anti-CD276 mAb in PBS/BSA at 4°C. Cells were rinsed with PBS/BSA and incubated with FITC-conjugated goat anti-human IgG or goat anti-rabbit IgG secondary antibodies (Jackson Immunoresearch, West Grove, PA), rinsed again, and analyzed on a FACS Calibur flow cytometer (Becton Dickinson). The average number of m276 binding sites per cells was measured on a FACS Calibur using Quantum Simply Cellular microspheres (Bangs Laboratories, Inc.) according to the manufacturer’s instructions. For the hematopoietic lineage staining, bone marrow was harvested from the femurs and tibias of CD276 wildtype or knockout mice by flushing with cold 1xPBS (Invitrogen) / 0.1% bovine serum albumin (BSA, Sigma). Marrow was physically disrupted by pipetting, then filtered using a 30 μm mesh cell strainer (CellTrics - Sysmex Partec). Spleens were mechanically disrupted by passing through a 40 μm cell strainer (Thomas Scientific). Cells were washed twice with cold PBS/0.1%BSA prior to staining. Non-specific antibody binding to the FcR was blocked by pre-incubation with purified anti-CD16/CD32 antibodies (eBioscience, San Diego, CA). Mouse bone marrow and spleen cells were stained with antibodies to the following markers, or corresponding isotype matched controls: Gr-1-PE (RB6-8C5), IgM-PE (II/41), B220-PE (RA3-6B2), CD4-PE (GK1.5), Ly6C-PE-Cy7 (HK1.4), Ly6G-APC (1A8-Ly6G), CD19-APC (eBio1D3), CD11c-APC (N418), Mac1-APCeFluor780 (M1/70), or CD8-Biotin (53–6.7) followed by streptavidin-PE-Cy7 (eBioscience, San Diego, CA). Human m276 anti-CD276 or human IgG1 lambda control antibodies were labelled with FITC using the FluoReporter FITC Protein Labeling Kit from ThermoFisher according to the manufactures instructions. Staining was performed in cold 1x PBS / 0.1% BSA followed by fixation with 1% paraformaldehyde (Sigma) in PBS. Samples were analyzed using a BD Bioscience LSR II flow cytometer. All flow cytometry data were analyzed using FlowJo software (FlowJo, LLC).
Cell Viability Assays
Cell viability was measured using alamarBlue (ThermoFisher Scientific). Briefly, cells were plated into 96-wells, allowed to attach and grow for 24 hr, then triplicate wells were treated with ADCs, naked antibodies, free drugs, P-gp inhibitors or ADCs plus competitor antibodies. Three to five days later, when untreated control wells were 70 to 90% confluent, alamarBlue reagent was added to the plates according to the supplier’s instructions. Wells treated identically but without cells were used to subtract background. Fluorescence (ex: 570 nm, Em: 585 nm) was measured using a CLARIOstar microplate reader (BMG Labtech) and data analyzed using GraphPad Prism 6 software.
Immunohistochemistry
Formalin-fixed paraffin embedded (FFPE) sections were deparaffinized, treated with Dual Endogenous Enzyme-Blocking Reagent (Dako) followed by the Biotin blocking system (Dako) and blocked with 1% blocking reagent (Roche) in TBS (100 mM Tris (pH 7.5), 150 mM NaCl + 1% Triton X 100). Sections were incubated with goat anti-human CD276 (R&D catalogue # AF1027) or cytokeratin 7 (clone RCK105, Santa Cruz Biotechnology; catalogue #, sc-23876) for 2 hr at room temperature followed by signal amplification using a Vectastain ABC HRP Kit (Vector Laboratories). Human FFPE tissue arrays were obtained from US Biomax, and anonymized human FFPE normal or cancer tissue samples were obtained from the Cooperative Human Tissue Network (CHTN) or the Laboratory of Pathology Tissue Resource Committee (TRC) at NIH with approval from the NIH Office of Human Subject Research. The goat anti-human CD276 antibody from R&D used for IHC staining of human tissues was also found to react specifically with murine tumor vessels of CD276 WT but not KO mice, as shown in Figure S2. Although this antibody was the most suitable for CD276 IHC of FFPE sections, for some normal tissues such as prostate and liver a low level of staining was found in humans but not mouse tissues, precluding an evaluation of specificity through comparison of the CD276 WT and KO mice. Moreover, we did not observe similar CD276 staining when using our rabbit anti-CD276 mAb on frozen tissues. Therefore, at this time we cannot determine whether the low level staining observed with the goat pAb in certain normal tissues represents low level CD276 expression or cross-reactivity of the pAb with another protein.
Immunofluorescence Staining
For in vivo target identification, 75μg of m276 labeled with FITC (Fluoreporter FITC Protein kit, Invitrogen) was co-injected with 1mg non-specific human IgG intraperitoneally into Pan02 tumor-bearing Cd276 wildtype and Cd276 knockout mice. Animals were euthanized 3 hr later, and normal and tumor tissues were excised, frozen, cryosectioned, rinsed with PBS, and fixed with methanol:acetone. Sections were blocked in Tris-Buffered Saline (TBS) containing 1% blocking reagent (Roche) and signal was amplified using Alexa 488 goat anti-FITC antibodies (Invitrogen) followed by Alexa 488 donkey anti-goat antibodies (Jackson Immunoresearch Laboratories). For microvascular density studies and CD276 IF staining, frozen sections fixed with methanol:acetone were stained with rabbit anti-CD276 antibodies, and amplified with FITC goat anti-rabbit antibodies (Jackson Immunoresearch Laboratories) followed by Alexa 488 donkey anti-goat antibodies (Jackson Immunoresearch Laboratories). Endothelial cells were co-localized using rat anti-PV-1 (Meca-32) or rat anti-CD31 antibodies (clone MEC13.3, Santa Cruz) followed by biotin-labeled donkey anti-rat antibodies (Jackson) and Texas red-streptavidin (Vector laboratories). For vessel co-option staining, frozen sections of human colon liver metastasis (n=5) were stained with rabbit anti-CD276 and mouse anti-CD31 (clone: JC/70A; ThermoFisher Scientific), followed by FITC goat anti-rabbit antibodies and 488 donkey anti-goat antibodies or Alexa 594 donkey anti-mouse antibodies. After completing the CD276 and CD31 staining, sections were stained with Alexa 647 anti-Cytokeratin 19 [A53-B/A2]. Alexa 647 (CK19) was pseudocolored yellow. For pericyte staining a goat anti-Desmin (R&D Systems) antibody was used, and endothelium was stained with rabbit anti-CD276 mAb and hamster anti-CD31 antibodies (clone 2H8; ThermoFisher Scientific). The mean vessel area for primary tumors was determined using FIJI software by averaging the CD31-positive area taken from the areas with the highest vascular density (twenty-five microscopic fields from 5 different tumors), and presented as a percent of the total field area. The CD31-positive area within metastasis was determined by averaging the CD31-positive area taken from within the tumor nodule area (30 metastases from 5 different lungs), and presented as a percent of the total field area for each nodule. The tumor nodule area was identified by CD276 staining of tumor cells. To measure apoptosis following ADC treatment in vivo, CD276-positive tumor cells and vessels were labeled with rabbit anti-CD276 mAb and amplified with FITC goat anti-rabbit antibodies followed by Alexa 488 goat anti-FITC antibodies (ThermoFisher Scientific). Endothelial cells were labeled with rat anti-CD31 (clone MEC13.3) and rat anti-PV-1 (Meca-32) antibodies followed by biotin-labeled donkey anti-rat antibodies and Dylight™ 405 streptavidin (Jackson Immunoresearch Laboratories). Nuclei were labeled with TO-PRO-3 Iodide (ThermoFisher Scientific) and cells undergoing apoptosis were labeled with the ApopTag Red In Situ Apoptosis Detection Kit (Millipore, Cat#S7165). For Figure S5 (C) and (D), nuclei were pseudocolored blue, apoptotic cells white, and CD31/Meca-32 red. For all IF staining specificity was verified by substituting primary antibodies with isotype matched non-specific IgGs. All immunofluorescent images were captured using either a Carl Zeiss LSM 510 laser scanning confocal microscope.
m276 Cell Uptake Assay
CHO or CHO/CD276 cells grown on 8-well poly L lysine plates (Becton Dickinson) for 24 hr were rinsed with Hams F-12 culture medium containing 0.5% BSA (Hams/BSA) and stained with m276 on ice for 30 mins. Cells were rinsed with Hams/BSA and stained a further 30 min on ice with FITC-conjugated goat anti-human secondary antibodies (Jackson Immuno research), then transferred to a 37°C incubator for 90 mins, or else left on ice. After rinsing with PBS containing 0.5% BSA, cells were fixed with 2% paraformaldehyde in PBS, counterstained with Hoechst 33258 and visualized under a fluorescent microscope.
RT-PCR
RT-PCR was performed using total RNA isolated from cells using RNeasy mini kit (Qiagen) and cDNA was synthesized using Superscript III 1st strand synthesis system (Invitrogen). The primer pairs used were Abcb1-F: 5′-ATGGATGAGATTGAGAAAGCTGTC-3′, Abcb1-R: 5′-TGACAAGTTTGAAGTAAATGCC-3′; human β-actin-F: 5′-GGCACCAGGGCGTGATG-3′, mouse β-actin-F: 5′-GGCACCAGGGTGTGATGG-3′; hu/ms β-actin-R: 5′-AGGTCTCAAACATGATCTGGGTC-3′. Both Abcb1 primers and the β-actin (Actb) reverse primer were designed against sequences that are 100% conserved between mouse and human.
QUANTIFICATION AND STATISTICAL ANALYSIS
A Students t-test was used to calculate differences in tumor volumes between two groups. For comparisons between multiple groups, a one-way ANOVA was used with a Bonferroni post-test. For Kaplan Meier survival analysis, a Log-rank (Mantel-Cox) test was used to compare each of the arms. Differences between two groups were presented as the mean ± SEM or mean ± SD as noted in the Figure Legends. Experimental sample numbers (n) are indicated in the Figure Legends. All tests were two-sided and p values <0.05 were considered statistically significant. All statistical analysis was performed with GraphPad Prism 6.04.
Supplementary Material
Significance.
Tumors can evade current anti-angiogenic therapies, i.e. VEGF/VEGFR pathway inhibitors, by co-opting established non-angiogenic vessels and exploiting VEGF-independent pathways of neovascularization. VEGF therapies also block normal physiological angiogenesis and can result in on-target off-tumor toxicities, creating demand for more tumor-selective vascular targeting agents. CD276, a highly conserved cell surface protein overexpressed during pathological but not physiological angiogenesis, is an attractive target for the selective destruction of tumor vasculature. CD276 is also frequently overexpressed by tumor cells, providing a dual target for the development of ADCs that can simultaneously destroy both tumor cells and tumor vasculature. Here we describe the development of a dual-compartment targeted CD276 ADC with potential to aid in the treatment of multiple tumor types.
Highlights.
CD276/B7-H3 is broadly overexpressed on both cancer cells and tumor vasculature
Both angiogenic and non-angiogenic tumor vasculature express CD276
Anti-CD276-drug conjugates display potent anti-tumor and anti-metastatic activity
Pyrrolobenzodiazepine dimers are optimal warheads for targeting tumor vasculature
Acknowledgments
We thank Dr. Michael M. Gottesman for advice and P-gp plasmids and inhibitors and Drs. Bernard Violand, David Tung, Linping Zhang and Halle Zhang for helpful discussions. This work was supported by a METAvivor grant, a Cooperative Research and Development Agreement (CRADA) between BioMed Valley Discoveries and the National Cancer Institute (NCI), and CCR, part of NCI’s intramural research program, National Institutes of Health, Department of Health and Human Services (DHHS). S.S., Z.Z., S.S., X.M.Z, D.W., G.D., D.S.D. and BSC are inventors of intellectual property related to CD276 antibodies and ADCs and BSC and D.S.D. have received research support through a CRADA with BVD. The content of this publication does not necessarily reflect the views or policies of the DHHS nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, S.S.1, Z.Z., S.S.2, X.M.Z, D.S.D., and B.S.C.; Methodology, S.S.1, Z.Z., X.M.Z., M.Y.Y., L.T., D.W., G.D., A.C., E.Z., J.R.K., D.S.D., and B.S.C.; Investigation, S.S.1, Z.Z., M.Y.Y., M.B.H., K.M., C.S., H.M., J.S., Y.F., Y.W., J.L., A.C., E.Z., K.D.K., and B.S.C.; Writing – Original Draft, S.S.1 and B.S.C.; Writing – Review & Editing, all authors; Resources, S.S.2, S.W.S., S.D., D.B., N.J.; Supervision, Z.Z., X.M.Z., D.A.S., J.R.K., D.S.D., and B.S.C.
Steven Seaman,
Saurabh Saha
Supplemental Information includes 8 figures and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdollahi A, Folkman J. Evading tumor evasion: current concepts and perspectives of anti-angiogenic cancer therapy. Drug Resist Updat. 2010;13:16–28. doi: 10.1016/j.drup.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Ahmed M, Cheng M, Zhao Q, Goldgur Y, Cheal SM, Guo HF, Larson SM, Cheung NK. Humanized Affinity-matured Monoclonal Antibody 8H9 Has Potent Antitumor Activity and Binds to FG Loop of Tumor Antigen B7-H3. J Biol Chem. 2015;290:30018–30029. doi: 10.1074/jbc.M115.679852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama K, Ohga N, Hida Y, Kawamoto T, Sadamoto Y, Ishikawa S, Maishi N, Akino T, Kondoh M, Matsuda A, et al. Tumor endothelial cells acquire drug resistance by MDR1 up-regulation via VEGF signaling in tumor microenvironment. Am J Pathol. 2012;180:1283–1293. doi: 10.1016/j.ajpath.2011.11.029. [DOI] [PubMed] [Google Scholar]
- Arigami T, Narita N, Mizuno R, Nguyen L, Ye X, Chung A, Giuliano AE, Hoon DS. B7-h3 ligand expression by primary breast cancer and associated with regional nodal metastasis. Ann Surg. 2010;252:1044–1051. doi: 10.1097/SLA.0b013e3181f1939d. [DOI] [PubMed] [Google Scholar]
- Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgeman VL, Vermeulen PB, Foo S, Bilecz A, Daley F, Kostaras E, Nathan MR, Wan E, Frentzas S, Schweiger T, et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J Pathol. 2016 doi: 10.1002/path.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner A, Hinterholzer S, Riss P, Heinze G, Brustmann H. Immunoexpression of B7-H3 in endometrial cancer: relation to tumor T-cell infiltration and prognosis. Gynecol Oncol. 2012;124:105–111. doi: 10.1016/j.ygyno.2011.09.012. [DOI] [PubMed] [Google Scholar]
- Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, Dong H, Sica GL, Zhu G, Tamada K, Chen L. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. 2001;2:269–274. doi: 10.1038/85339. [DOI] [PubMed] [Google Scholar]
- Cheang MC, Treaba DO, Speers CH, Olivotto IA, Bajdik CD, Chia SK, Goldstein LC, Gelmon KA, Huntsman D, Gilks CB, et al. Immunohistochemical detection using the new rabbit monoclonal antibody SP1 of estrogen receptor in breast cancer is superior to mouse monoclonal antibody 1D5 in predicting survival. J Clin Oncol. 2006;24:5637–5644. doi: 10.1200/JCO.2005.05.4155. [DOI] [PubMed] [Google Scholar]
- Chen R, Hou J, Newman E, Kim Y, Donohue C, Liu X, Thomas SH, Forman SJ, Kane SE. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Mol Cancer Ther. 2015;14:1376–1384. doi: 10.1158/1535-7163.MCT-15-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorywalska M, Dushin R, Moine L, Farias SE, Zhou D, Navaratnam T, Lui V, Hasa-Moreno A, Casas MG, Tran TT, et al. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol Cancer Ther. 2016;15:958–970. doi: 10.1158/1535-7163.MCT-15-1004. [DOI] [PubMed] [Google Scholar]
- Elinzano H, Hebda N, Luppe D, Turchetti W, Rosati K, Sikov WM, Safran H. PSMA ADC for progressive glioblastoma: Phase II Brown University Oncology Research Group Study. J Clin Oncol. 2016;34 (suppl; abstr 2065) [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Senter PD, Grewal IS. Antibody drug-conjugates targeting the tumor vasculature: Current and future developments. MAbs. 2009;1:247–253. doi: 10.4161/mabs.1.3.8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley JA. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin Investig Drugs. 2011;20:733–744. doi: 10.1517/13543784.2011.573477. [DOI] [PubMed] [Google Scholar]
- Huang X, Bennett M, Thorpe PE. Anti-tumor effects and lack of side effects in mice of an immunotoxin directed against human and mouse prostate-specific membrane antigen. Prostate. 2004;61:1–11. doi: 10.1002/pros.20074. [DOI] [PubMed] [Google Scholar]
- Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, Kioussis D. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci U S A. 1991;88:5096–5100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey SC, Burke PJ, Lyon RP, Meyer DW, Sussman D, Anderson M, Hunter JH, Leiske CI, Miyamoto JB, Nicholas ND, et al. A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconjug Chem. 2013;24:1256–1263. doi: 10.1021/bc400217g. [DOI] [PubMed] [Google Scholar]
- Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EG, Kim KM. Strategies and Advancement in Antibody-Drug Conjugate Optimization for Targeted Cancer Therapeutics. Biomol Ther (Seoul) 2015;23:493–509. doi: 10.4062/biomolther.2015.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski EA, Yin M, Bar-Zion A, Lee CR, Butz H, Man S, Daley F, Vermeulen PB, Yousef GM, Foster FS, et al. Co-option of Liver Vessels and Not Sprouting Angiogenesis Drives Acquired Sorafenib Resistance in Hepatocellular Carcinoma. J Natl Cancer Inst. 2016:108. doi: 10.1093/jnci/djw030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, Stone I, Ryan MC, Sussman D, Lyon RP, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood. 2013;122:1455–1463. doi: 10.1182/blood-2013-03-491506. [DOI] [PubMed] [Google Scholar]
- Li F, Emmerton KK, Jonas M, Zhang X, Miyamoto JB, Setter JR, Nicholas ND, Okeley NM, Lyon RP, Benjamin DR, Law CL. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res. 2016;76:2710–2719. doi: 10.1158/0008-5472.CAN-15-1795. [DOI] [PubMed] [Google Scholar]
- Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, Burke S, Ciccarone V, Li H, Yang Y, et al. Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res. 2012;18:3834–3845. doi: 10.1158/1078-0432.CCR-12-0715. [DOI] [PubMed] [Google Scholar]
- Loos M, Hedderich DM, Friess H, Kleeff J. B7-h3 and its role in antitumor immunity. Clin Dev Immunol. 2010;2010:683875. doi: 10.1155/2010/683875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modak S, Kramer K, Gultekin SH, Guo HF, Cheung NK. Monoclonal antibody 8H9 targets a novel cell surface antigen expressed by a wide spectrum of human solid tumors. Cancer Res. 2001;61:4048–4054. [PubMed] [Google Scholar]
- Nomura N, Pastorino S, Jiang P, Lambert G, Crawford JR, Gymnopoulos M, Piccioni D, Juarez T, Pingle SC, Makale M, Kesari S. Prostate specific membrane antigen (PSMA) expression in primary gliomas and breast cancer brain metastases. Cancer Cell Int. 2014;14:26. doi: 10.1186/1475-2867-14-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh P, Li Y, Yu J, Durr E, Krasinska KM, Carver LA, Testa JE, Schnitzer JE. Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature. 2004;429:629–635. doi: 10.1038/nature02580. [DOI] [PubMed] [Google Scholar]
- Olson WC, Israel RJ. Antibody-drug conjugates targeting prostate-specific membrane antigen. Front Biosci (Landmark Ed) 2014;19:12–33. doi: 10.2741/4193. [DOI] [PubMed] [Google Scholar]
- Qin X, Zhang H, Ye D, Dai B, Zhu Y, Shi G. B7-H3 is a new cancer-specific endothelial marker in clear cell renal cell carcinoma. Onco Targets Ther. 2013;6:1667–1673. doi: 10.2147/OTT.S53565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C, St Croix B. Genes that distinguish physiological and pathological angiogenesis. Cancer Cell. 2007;11:539–554. doi: 10.1016/j.ccr.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, Lal A, Riggins GJ, Lengauer C, Vogelstein B, Kinzler KW. Genes expressed in human tumor endothelium. Science. 2000;289:1197–1202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- Suh WK, Gajewska BU, Okada H, Gronski MA, Bertram EM, Dawicki W, Duncan GS, Bukczynski J, Plyte S, Elia A, et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol. 2003;4:899–906. doi: 10.1038/ni967. [DOI] [PubMed] [Google Scholar]
- Visintin A, Knowlton K, Tyminski E, Lin CI, Zheng X, Marquette K, Jain S, Tchistiakova L, Li D, O’Donnell CJ, et al. Novel Anti-TM4SF1 Antibody-Drug Conjugates with Activity against Tumor Cells and Tumor Vasculature. Mol Cancer Ther. 2015;14:1868–1876. doi: 10.1158/1535-7163.MCT-15-0188. [DOI] [PubMed] [Google Scholar]
- Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. 2015;15:361–370. doi: 10.1038/nrc3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Harrell JC, Perou CM, Dudley AC. Identification of a stable molecular signature in mammary tumor endothelial cells that persists in vitro. Angiogenesis. 2014;17:511–518. doi: 10.1007/s10456-013-9409-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Stevens J, Hilton MB, Seaman S, Conrads TP, Veenstra TD, Logsdon D, Morris H, Swing DA, Patel NL, et al. COX-2 inhibition potentiates antiangiogenic cancer therapy and prevents metastasis in preclinical models. Sci Transl Med. 2014;6:242ra284. doi: 10.1126/scitranslmed.3008455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SF, Zheng B, Go M, Lau J, Spencer S, Raab H, Soriano R, Jhunjhunwala S, Cohen R, Caruso M, et al. A Novel Anti-CD22 Anthracycline-Based Antibody-Drug Conjugate (ADC) That Overcomes Resistance to Auristatin-Based ADCs. Clin Cancer Res. 2015;21:3298–3306. doi: 10.1158/1078-0432.CCR-14-2035. [DOI] [PubMed] [Google Scholar]
- Zang X, Sullivan PS, Soslow RA, Waitz R, Reuter VE, Wilton A, Thaler HT, Arul M, Slovin SF, Wei J, et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. Mod Pathol. 2010;23:1104–1112. doi: 10.1038/modpathol.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Dimitrov DS. Construction of a large naive human phage-displayed Fab library through one-step cloning. Methods Mol Biol. 2009;525:129–142. xv. doi: 10.1007/978-1-59745-554-1_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Ramakrishnan B, Li J, Wang Y, Feng Y, Prabakaran P, Colantonio S, Dyba MA, Qasba PK, Dimitrov DS. Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar. MAbs. 2014;6:1190–1200. doi: 10.4161/mabs.29889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.