

Here, Wen et al. review the current knowledge and progress in understanding the impact of Zika virus exposure on mammalian brain development and discuss potential underlying mechanisms.
Keywords: brain development, microcephaly, Zika virus
Abstract
The re-emergence of Zika virus (ZIKV), a mosquito-borne and sexually transmitted flavivirus circulating in >70 countries and territories, poses a significant global threat to public health due to its ability to cause severe developmental defects in the human brain, such as microcephaly. Since the World Health Organization declared the ZIKV outbreak a Public Health Emergency of International Concern, remarkable progress has been made to gain insight into cellular targets, pathogenesis, and underlying biological mechanisms of ZIKV infection. Here we review the current knowledge and progress in understanding the impact of ZIKV exposure on the mammalian brain development and discuss potential underlying mechanisms.
Zika virus (ZIKV) is a mosquito-borne flavivirus first isolated in 1947 from a sentinel rhesus macaque in the Ziika Forest region of Uganda (Dick et al. 1952). It had remained in relative obscurity for decades until outbreaks in the Pacific islands in 2007 and then French Polynesia in 2013 (Duffy et al. 2009; Cao-Lormeau et al. 2014; Musso 2015). Since the large ZIKV outbreak coincided with a rise of birth defects in Brazil in 2015, it has become a growing public health concern (Heymann et al. 2016). So far, active transmission has been reported in >70 countries and territories globally (http://www.cdc.gov/zika/geo/index.html). Most human infections of ZIKV are typically transmitted by the Aedes mosquito, but the virus can also spread directly through sexual contact and blood transfusion (Foy et al. 2011; Musso et al. 2015; D'Ortenzio et al. 2016; McCarthy 2016). Most notably, ZIKV can be vertically transmitted from infected mothers to their fetuses (Brasil et al. 2016; Driggers et al. 2016; Petersen et al. 2016). About 20% of ZIKV-infected individuals develop mild symptoms, which mostly resemble those caused by other arboviruses, such as dengue viruses (DENV) or chikungunya viruses. Distinct from these viruses, however, ZIKV can cause congenital malformations in the fetus, such as microcephaly (Mlakar et al. 2016; Rasmussen et al. 2016), and Guillain-Barré syndrome, meningo-encephalitis, myelitis, and ophthalmologic abnormalities in infected adults (Araujo et al. 2016; Cao-Lormeau et al. 2016; Fontes et al. 2016; Petersen et al. 2016; Rasmussen et al. 2016).
Microcephaly is a neurodevelopmental disorder, which is characterized by a marked reduction in brain size and intellectual disability caused by impaired cell proliferation and the death of cortical progenitor cells and their neuronal progeny (Barbelanne and Tsang 2014). Through January 26 of 2016, 10,441 suspected and 2366 confirmed ZIKV-associated microcephaly cases have been reported in Brazil (Zika-Epidemiological Report by the Pan-American Health Organization [PAHO] and World Health Organization [WHO]), the country that has experienced the highest ZIKV infection rates worldwide. The increase of microcephaly cases and the coincidental ZIKV outbreak led the WHO to declare a Public Health Emergency of International Concern in early 2016 (Heymann et al. 2016). This ignited tremendous interest and immediate efforts by scientists to understand the impact of ZIKV on human brain development and the mechanistic link between ZIKV infection and microcephaly. In the past year, rapid and stunning progress has been made toward developing stem cell-based cellular models, primary human tissues, and animal models (Table 1) to understand its pathogenesis, investigate underlying mechanisms, and develop therapeutics (Abbink et al. 2016; Cugola et al. 2016; Dudley et al. 2016; Garcez et al. 2016; Lazear et al. 2016; Li et al. 2016a,b; Miner et al. 2016a; Ming et al. 2016; Qian et al. 2016, 2017; Rossi et al. 2016; Tang et al. 2016; Wu et al. 2016; Xu et al. 2016; Hirsch et al. 2017). Findings from these model systems have helped scientists from the US Centers for Disease Control and Prevention (CDC) to conclude that ZIKV causes microcephaly (Rasmussen et al. 2016). Here, we focus our review on the recent progress in understanding how ZIKV exposure leads to mammalian brain development deficits.
Table 1.
In vitro two-dimensional (2D) and three-dimensional (3D) culture models and in vivo mouse and nonhuman primate models to study ZIKV infection and transmission
ZIKV infection mechanisms
Compared with other flaviviruses, one striking feature of the current ZIKV epidemic is the association of viral infection with a marked increase of risk for congenital microcephaly and serious neurologic complications (Petersen et al. 2016), which triggered widespread efforts to understand the molecular basis of infection. Similar to its close relatives in the Flaviviridae family, such as DENV, yellow fever, Japanese encephalitis, and West Nile viruses (WNV), ZIKV has an icosahedral outer envelope and a dense inner core containing one single-strand positive sense RNA genome between 10,000 and 11,000 base pairs in length (Chambers et al. 1990; Kuno and Chang 2007). The genome of ZIKV encodes a single polyprotein that is post-translationally cleaved by host and viral proteases into three structural proteins (capsid [C], premembrane [prM], and envelope [E]) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (Fig. 1A).
Figure 1.

ZIKV genome structure and signaling transductions underlying ZIKV infection. (A) A diagram of ZIKV genomic RNA. (Red arrows) Mutations in the Brazilian strain of ZIKV compared with the French Polynesian strain; (blue arrows) mutations in the Asian strain compared with the African strain; (purple arrow) a mutation of the Brazilian strain in the E protein Asn154. (B) A diagram showing the viral replication pathway and pathogenesis pathways activated upon ZIKV infection.
Why did ZIKV only recently start to cause microcephaly even though it has been around for >50 years? One hypothesis is that nonsynonymous nucleotide mutations found in ZIKV strains from the current Brazilian outbreak may contribute to the increased incidence of microcephaly. For example, compared with the French Polynesian strain, the Brazilian strain contains mutations in three of the nonstructural proteins, including three in NS1 (K940E, T1027A, and M1143V), which is implicated in immune evasion; one in NS4B (T2509I), which is involved in the inhibition of type I interferon signaling; and one in NS5, which is known to mask the viral RNAs from host recognition (M2634V) (Mlakar et al. 2016). Given the roles of nonstructural proteins in viral RNA synthesis and replication, codon usage by the pandemic strain may be adapted to the human host by changes in these nonstructural proteins. For example, ZIKV NS5 binds to the interferon (IFN)-regulated transcriptional activator STAT2, which leads to the proteasomal degradation of STAT2, resulting in the silencing of type I and type III interferon-mediated signaling (Fig. 1B; Grant et al. 2016; Kumar et al. 2016). Direct comparison of African and Asian strains further yielded several mutations in NS5 and a shared mutation at the same position of NS5 in sequences from Colombia, Mexico, Panama, and Martinique (Adiga 2016), suggesting the possibility that mutation of amino acids at NS5 may facilitate viral replication in human cells. In addition to the adaptation of nonstructural proteins, the current pathology may also be consequent to modifications or mutations in the structural proteins of the Brazilian ZIKV strains, such as mutations in the prM protein and around the Asn-154 glycosylation site in the E protein. These modifications have been speculated to contribute to viral adhesion to host cells and facilitate unique aspects of ZIKV transmission (Sirohi et al. 2016). Furthermore, ZIKV also produces noncoding subgenomic flaviviral RNAs (sfRNAs) in their 3′ untranslated regions that accumulate during infection and resist degradation by host exonucleases in infected cells, which are directly linked to pathologic effects (Akiyama et al. 2016). All of these genetic and epigenetic hypotheses remain to be fully tested experimentally, which may provide critical insight into the viral–host interaction and pathogenesis of ZIKV infection.
Study of ZIKV infection in multiple cell types indicated that ZIKV gains entry into host cells through the interaction of the virus envelope glycoprotein with cell surface receptors DC-SIGN, AXL, TYRO3, and TIM-1 and that AXL, a receptor tyrosine kinase mediating inflammatory processes, appears to play a major role (Fig. 1B; Hamel et al. 2015). The TAM ligand Gas6 acts as a cofactor to recruit the virus to the AXL receptor, resulting in the internalization of ZIKV through clathrin-mediated endocytosis (Meertens et al. 2017). ZIKV-containing vesicles then translocate to Rab5+ endosomes and induce the transcription of Toll-like receptor 3 (TLR3), DDX58, and IFIH1 as well as several interferon-stimulated genes, leading to suppression of the innate immune response and subsequent productive infection (Hamel et al. 2015; Meertens et al. 2017). Blocking AXL by an engineered AXL decoy receptor, MYD1; the AXL kinase inhibitor R428; or a nonactivating antibody specific for the extracellular domain of AXL inhibits ZIKV infection in glial cells, confirming the importance of this receptor for ZIKV entry into host cells (Retallack et al. 2016; Meertens et al. 2017).
Whether the AXL-mediated ZIKV entry is cell type-specific and the extent to which other receptors and factors are involved remain elusive. Axl knockout mice exhibit similar levels of ZIKV RNA loading compared with wild-type mice (Miner et al. 2016b). Furthermore, although AXL is highly expressed in neural progenitor cells (NPCs) (Nowakowski et al. 2016; Onorati et al. 2016), the anti-AXL antibody fails to reduce ZIKV infection of NPCs (Meertens et al. 2017), and genetic ablation of AXL is not able to block ZIKV entry or ZIKV-mediated cell death in human induced pluripotent stem cell (hiPSC)-derived NPCs or cerebral organoids (Wells et al. 2016), suggesting that AXL is not required for ZIKV infection in NPCs. Future work is needed to elucidate mechanisms underlying ZIKV entry in NPCs and shed light onto the potential therapeutic strategy of blocking ZIKV entry.
Vertical transmission and transplacental infection route of ZIKV
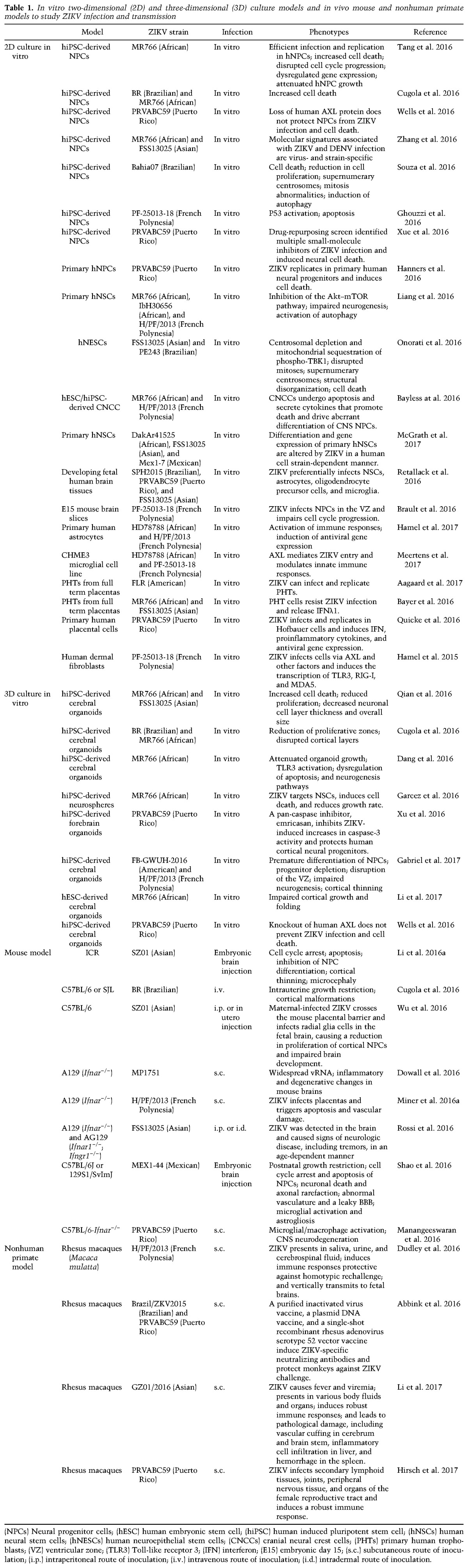
ZIKV antigen and RNA were present in amniotic fluid and placental tissues from ZIKV-infected women as well as fetal and newborn brain tissues diagnosed with fetal microcephaly (Calvet et al. 2016; Driggers et al. 2016; Martines et al. 2016b; Noronha et al. 2016; Bhatnagar et al. 2017), suggesting that ZIKV is a classic TORCH (toxoplasma, other, rubella, cytomegalovirus, and herpes) pathogen and gains access to the fetal brain during pregnancy by crossing the placental barrier (Fig. 2).
Figure 2.
Vertical transmission of ZIKV and its impact on human fetal development. ZIKV could be transmitted to a pregnant woman via the bite of an infected Aedes mosquito, which could be further vertically transmitted from the infected mother to the fetus by infecting placental trophoblasts and macrophages (Hofbauer cells) and crossing the placental barrier.
Recent studies suggest that the placenta is the key mediator for vertical transmission of ZIKV from infected mothers to fetal brains (Bayer et al. 2016; Quicke et al. 2016). Due to its unique structural, cellular, and immunological properties, the placenta acts as a barrier against bacterial and viral infections. Several lines of evidence suggest that ZIKV passes through the placenta to reach the fetus by directly infecting placental cells and disrupting the placental barrier (Fig. 2). Placental damage from ZIKV infection, such as chronic placentitis, has been observed in human placental tissues from infected mothers (Noronha et al. 2016), in which the placental macrophages (known as Hofbauer cells) and trophoblasts are the main target cells of ZIKV infection (Bayer et al. 2016; Quicke et al. 2016). This transplacental infection route of ZIKV has also been demonstrated in vivo in pregnant immuno-incompetentors, a specific SJL strain of mouse models (Table 1; Fig. 3), including maternal inoculation via a subcutaneous route, resulting in placental damage and fetal demise (Miner et al. 2016a); infection of the pregnant mother through an intravenous route, leading to intrauterine growth restriction (Cugola et al. 2016); and intraperitoneal injection of the virus, leading to infection of the fetal brain in developing mice (Wu et al. 2016). RNA and infectious ZIKV particles, but not DENV, were found in maternal trophoblasts and fetal endothelial cells in the placenta (Miner et al. 2016a) as well as the dorsal ventricular zone (VZ) of the fetal brain (Wu et al. 2016). These findings suggest that ZIKV might have greater tropism for placental cells than other flaviviruses and further confirm the tropism for placental cells lining the maternal–fetal interface. In order to replicate in host cells, ZIKV overcomes interferon-mediated host immune responses by inducing the degradation of the IFN-regulated transcriptional activator STAT2 (Grant et al. 2016; Kumar et al. 2016). Under immuno-competent conditions, ZIKV infection in Hofbauer cells and trophoblasts in the placenta induces the production of type I or type III IFN and proinflammatory cytokines, resulting in antiviral gene expression (Bayer et al. 2016; Quicke et al. 2016). Consistent with this notion, blocking IFN–receptor signaling by an anti-Ifnar antibody in wild-type pregnant mice enhanced transplacental infection of ZIKV. Functionally, infection of ZIKV in placentas can induce apoptosis of trophoblasts and vascular damage, which leads to disruption of the placental barrier and thus compromises protection against viral infection and conveys ZIKV to the fetus (Miner et al. 2016a).
Figure 3.
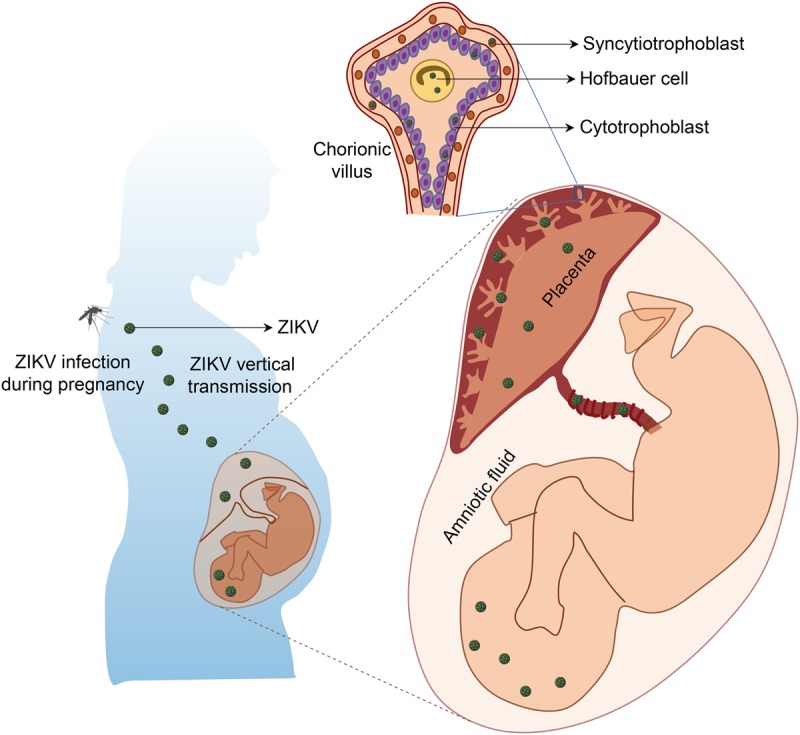
Investigation of neurotropism of ZIKV infection in vitro and in vivo. The effects of ZIKV on brain development and the underlying mechanisms could be explored with in vitro models, such as NPCs, astrocytes, and three-dimensional (3D) brain organoids derived from either hiPSCs or primary human brain tissue, or in vivo animal models, such as mouse models at multiple developmental stages (i.e., prenatal and postnatal stages) by infection of ZIKV through subcutaneous inoculation, intravenous injection, or intraperitoneal injection in pregnant immune-competent mice or direct injection of ZIKV into developing fetal mouse brains.
Placental insufficiency and inflammatory responses could lead to fetal development deficits (Fig. 4). The findings of vascular injury, the reduced number and irregular shape of fetal capillaries, and apoptotic trophoblasts are clinically relevant, as placental insufficiency is associated with intrauterine growth restriction. Moreover, increased levels of IFN-γ-inducible protein 10 (IP-10), interleukin-6 (IL-6), IL-8, vascular endothelial growth factor (VEGF), monocyte chemoattractive protein 1 (MCP-1), and granulocyte colony-stimulating factor (G-CSF) have been found in the amniotic fluid of ZIKV-infected pregnant women with neonatal microcephaly (Ornelas et al. 2017). Exposure of these cytokines to the fetal brain not only causes cell death but also dysregulates the differentiation and proliferation of NPCs, therefore providing an interacting set of mechanisms underlying ZIKV-induced microcephaly.
Figure 4.

Mechanisms underlying impaired brain development upon ZIKV infection. ZIKV directly targets NPCs in the developing brain and activates innate immune response, which could lead to dysregulation of genes involved in cell cycle, neurogenesis, and apoptosis, resulting in increased cell death, disrupted cell cycle progression, reduced proliferation, and premature differentiation. On the other hand, infection of ZIKV in placenta and glial cells, including astrocytes and microglia, could lead to placental insufficiency and activation of immune response (inflammation), which may elicit non-cell-autonomous effects on NPCs, neurons, and vasculature, resulting in impaired neurogenesis and microcephaly.
Targets of ZIKV infection in the developing mammalian brain
To understand the impact and mechanisms of ZIKV infection on human brain development and the link of ZIKV to microcephaly, a key step is to identify cell types that are particularly vulnerable to viral infection in the developing brain after ZIKV breaches the placental barrier. Following clinical observations that ZIKV could be found in brains of fetuses from infected pregnant women (Driggers et al. 2016; Mlakar et al. 2016; Bhatnagar et al. 2017), a hiPSC-based cellular model provided the first evidence that ZIKV efficiently targets human cortical NPCs, whereas immature neurons are less susceptible to ZIKV infection (Fig. 3; Tang et al. 2016). Compared with ZIKV, WNV has been demonstrated to have a very different tropism of infection with a bias toward neurons (Brault et al. 2016). Several groups have further shown that ZIKV infection of brain organoids, three-dimensional (3D) cellular models of human brain development, leads to reduced thickness of both NPC and neuronal layers and an overall reduction in organoid size and surface folding, resembling microcephaly (Table 1; Fig. 3; Cugola et al. 2016; Dang et al. 2016; Garcez et al. 2016; Qian et al. 2016; Li et al. 2017). Strong colocalization of ZIKV E protein in NESTIN+SOX2+ NPCs, rather than TBR2+ intermediate neural progenitors or CTIP2+ neurons in 3D brain organoids, again indicates relatively specific tropism of ZIKV toward NPCs (Cugola et al. 2016; Dang et al. 2016; Garcez et al. 2016; Qian et al. 2016). These results have also been recapitulated in mouse models, showing efficient ZIKV infection and replication in NPCs located in the VZ and subventricular zones (SVZs) of the fetal mouse cortex, leading to cortical thinning and microcephaly (Fig. 3; Cugola et al. 2016; Li et al. 2016a; Onorati et al. 2016; Shao et al. 2016; Wu et al. 2016). Collectively, these studies provide strong evidence that NPCs in the developing brain are particularly vulnerable to ZIKV infection.
In addition to NPCs, studies of ZIKV infection in organotypic cultures from primary human brain tissue or necropsy brain tissues from fetuses and newborns found that astrocytes, microglia, and oligodendrocyte precursor cells located throughout the developing cortex can be productively infected by ZIKV, while the infection rate of neurons is low (Table 1; Noronha et al. 2016; Retallack et al. 2016). ZIKV infection of astrocytes and microglia has further been demonstrated in primary cell cultures, iPSC-derived astrocytes, and 3D forebrain organoids (Fig. 3; Lindqvist et al. 2016; Qian et al. 2016; Retallack et al. 2016; Xu et al. 2016; Meertens et al. 2017). Glial cells, including astrocytes and microglia, are known to be essential for normal brain development and function and play an important role in neurodevelopmental disorders by activating the neuro–immune interaction (Ma et al. 2005; Molofsky et al. 2012; Reemst et al. 2016). Upon ZIKV infection of primary human astrocytes, an increase in the production of viral particles over time has been observed, indicating active viral replication in these cells (Hamel et al. 2017; Tricarico et al. 2017). Other developmentally relevant stem cell populations are also highly susceptible to ZIKV infection, such as neocortical and spinal cord neuroepithelial stem cells (NESCs) (Table 1), which are the earliest population of resident neural stem cells (NSCs) present during neurodevelopment (Onorati et al. 2016), and cranial neural crest cells, which give rise to most cranial bones and exert paracrine effects on the developing brain (Bayless et al. 2016).
Impact of ZIKA infection on neural progenitors
How does ZIKV infection lead to microcephaly? There are both cell-autonomous and non-cell-autonomous effects (Fig. 4). A number of recent studies revealed dysregulated cell cycle progression in NPCs, which leads to reduced NPC proliferation and impaired neurogenesis (Ming et al. 2016). A dramatic reduction in cell density and a significant decrease of EdU-labeled or Ki67-expressing proliferating cells upon ZIKV infection have been demonstrated in monolayer human NPC cultures and 3D brain organoids (Cugola et al. 2016; Qian et al. 2016; Souza et al. 2016; Tang et al. 2016). Consistent with these results from human cell models, NPC proliferation is also suppressed in the ZIKV-infected developing mouse brain, which contains significantly fewer mitotic cells positive for phosphorylated H3 or Ki67 in the VZ/SVZ and intermediate zone, accompanied by more centrosomes at the ventricular surface facing away from the nuclei and a marked decrease in the intensity and band thickness of cortical layers (Li et al. 2016a; Wu et al. 2016). The reduction of NPC proliferation appears to be caused by perturbation of cell cycle progression. Analysis of DNA content by flow cytometry suggested that ZIKV infection leads to S-phase arrest in human NPCs (Tang et al. 2016). Similar results have been shown in the ZIKV-infected mouse fetal brain in vivo (Nguyen et al. 2016). Most of the infected cells in the VZ are arrested in S, G1, or G2 phase, and NPCs in M phase are dramatically reduced (Li et al. 2016a; Shao et al. 2016). Interestingly, impaired cell cycle progression appears to be specific for ZIKV infection but not for WNV infection (Brault et al. 2016). Perturbation of cell cycle progression may result from impaired dynamic gene expression during neurogenesis and disrupted mitosis (Dang et al. 2016; Souza et al. 2016). Indeed, mitotic dysfunction has been observed in ZIKV-infected NPC cultures, in part due to mitochondrial sequestration of centrosomal phospho-TBK1 (TANK-binding kinase 1) and redirection of TBK1 activity away from mitosis to innate immune signaling (Onorati et al. 2016). Disrupted mitosis further leads to accumulation of chromosomal abnormalities and cell division defects, resulting in not only impaired NPC proliferation but also cell death (Onorati et al. 2016; Souza et al. 2016). Global transcriptome analyses (RNA sequencing) revealed that many genes involved in cell proliferation, differentiation, and migration and organ development are down-regulated, with a particular enrichment of down-regulated genes in cell cycle-related and DNA replication/repair pathways upon ZIKV infection of human and mouse NPCs (Fig. 4; Li et al. 2016a; Tang et al. 2016; Zhang et al. 2016). Intriguingly, multiple significantly down-regulated genes are microcephaly-associated genes (Li et al. 2016a; Tang et al. 2016; Zhang et al. 2016), many of which encode proteins localized at the centrosome and play an important role in cell cycle progression (Gilmore and Walsh 2013), suggesting a direct mechanistic link of ZIKV infection to microcephaly at the molecular level.
Cell death also contributes to ZIKV-induced microcephaly, which has been detected extensively in human fetal brains infected with ZIKV (Fig. 4; Driggers et al. 2016; Mlakar et al. 2016). Studies in hiPSC-derived NPCs and 3D brain organoids also revealed that ZIKV infection leads to significant caspase-3 activation and increased cell death, resulting in diminished cortical layers (Cugola et al. 2016; Garcez et al. 2016; Qian et al. 2016; Souza et al. 2016; Tang et al. 2016; Li et al. 2017). In contrast, DENV infection does not trigger apoptosis or viral replication in brain organoids (Li et al. 2017). In one recently reported clinical case (Driggers et al. 2016), post-mortem analysis revealed diffuse cerebral cortex thinning in a fetal brain infected by ZIKV. Consistent with cell model-based research findings (Cugola et al. 2016; Qian et al. 2016; Li et al. 2017), excessive apoptosis was present in the neocortex, while well-differentiated neurons and primitive cells in the germinal matrix appeared to be unaffected. Investigations of ZIKV infection in mouse models further confirmed that ZIKV leads to cellular damage accompanied by caspase-3 activation and DNA fragmentation in NPCs, resulting in a reduction of the cortical NPC pool and smaller brains with significant damage to brain structure (Li et al. 2016a; Miner et al. 2016a; Shao et al. 2016; Wu et al. 2016). Notably, enrichment of genes related to apoptosis-related pathways are up-regulated upon ZIKV infection in human NPCs (Tang et al. 2016; Zhang et al. 2016). The apoptosis-induced cell death could be mediated by activation of tumor suppressor protein p53 (TP53), as the ZIKV infection significantly up-regulated TP53 gene and protein expression levels and Ser15 phosphorylation, which are correlated with genotoxic stress and apoptosis induction (Ghouzzi et al. 2016; Zhang et al. 2016). Both p53 and caspase inhibitors can efficiently prevent ZIKV-induced NPC death (Souza et al. 2016; Xu et al. 2016; Zhang et al. 2016).
NPC apoptosis upon ZIKV infection can also be caused by activation of the immune response (Fig. 4). Infection by the Asian strain of ZIKV, which is more closely related to current epidemic strains, leads to significant up-regulation of viral response genes involved in IFN response and the type II IFN signaling, TLR signaling, and TNFα signaling pathways in human NPCs (Zhang et al. 2016). On the other hand, the prototypical African ZIKV strain does not elicit such a virus response in human NPCs, suggesting that different ZIKV strains have specific neurotropisms and unique molecular signatures. Activation of an immune response has also been observed in mouse fetal brains infected with another Asian ZIKV strain, SZ01 (Li et al. 2016a), which was isolated from a patient infected in Samoa (Deng et al. 2016), and a Mexican ZIKV strain, MEX1-44, which also belongs to the Asian lineage (Shao et al. 2016). Notably, many genes involved in cytokine production and response, such as IL1B, TNF, CXCL10, IFNB1, TLR3, and OAS2, are dramatically up-regulated upon SZ01 or MEX1-44 ZIKV infection (Li et al. 2016a; Shao et al. 2016), suggesting that cytokines may play a key role in the pathogenesis of ZIKV infection. Activation of an immune response, such as activation of the innate immune receptor TLR3, may mediate the ZIKV-induced apoptosis and impaired neurogenesis (Fig. 1). Indeed, inhibition of TLR signaling by a TLR3 competitive inhibitor largely rescued an African strain of ZIKV MR766-induced apoptosis and organoid shrinkage, while a TLR3 agonist, poly(I:C), mimicked the phenotypes induced by ZIKV infection in brain organoids (Dang et al. 2016). Importantly, ZIKV infection and TLR3 activation share common molecular signatures, including dysregulation of gene expressions involved in neurogenesis and apoptosis (Dang et al. 2016), suggesting that ZIKV infection could activate TLR3 signaling, which in turn perturbs neurogenesis and apoptotic pathways. It remains to be addressed whether all strains of ZIKV could activate the TLR3 pathway and whether such activation is context-dependent and cell type-dependent.
Non-cell-autonomous effects of ZIKV-infected glial cells
NPC infection alone cannot explain some of the phenotypes observed in microcephaly and other congenital malformations caused by ZIKV infection, such as calcifications in the cortical plate, axonal damage, microglial nodules, and gliosis (Driggers et al. 2016; Martines et al. 2016a; Mlakar et al. 2016). Glial cells, including astrocytes and microglia, have been demonstrated to be two major targets of ZIKV (Noronha et al. 2016; Retallack et al. 2016; Meertens et al. 2017; Tricarico et al. 2017). Extensive microglial activation and astrogliosis, indicated by a significant increase in the number of IBA1+ microglia and GFAP+ astrocytes, have been detected in ZIKV-infected mouse fetal brains (Fig. 4; Shao et al. 2016). AXL, which is highly expressed in glial cells (Nowakowski et al. 2016; Meertens et al. 2017), may mediate the entry of ZIKV. Binding of ZIKV to AXL activates AXL kinase activity to induce oxidative stress and subsequently trigger inflammation and innate antiviral immune responses, including the up-regulation of TLRs, NOD-like receptors (NLRs), and RIG-I-like receptors (RLRs), which are involved in the activation of the inflammasome pathway (Hamel et al. 2017; Meertens et al. 2017; Tricarico et al. 2017). Intriguingly, compared with the African strain of ZIKV, the Asian strain induces antiviral responses in human astrocytes, including up-regulation of TLR and RLR downstream genes such as SPP1, TRAF3, and TBK1, with much faster kinetics and to a greater extent (Hamel et al. 2017). Notably, activation of inflammasome genes, such as NLR family Pyrin domain-containing 3 (NLRP3), can subsequently trigger release of mature cytokines such as IL-1β (Meertens et al. 2017; Tricarico et al. 2017).
Consequences of glia-mediated inflammation upon ZIKV infection are largely unknown. One possibility is neuronal death and dysfunction (Fig. 4), such as axonal damage. Although ZIKV shows limited infection in neurons (Brault et al. 2016; Cugola et al. 2016; Li et al. 2016a; Qian et al. 2016; Retallack et al. 2016; Tang et al. 2016; Wu et al. 2016), massive neuronal death and dysfunction upon ZIKV infection have been observed in human brains and organoids as well as mouse fetal brains (Cugola et al. 2016; Driggers et al. 2016; Li et al. 2016a; Mlakar et al. 2016; Qian et al. 2016; Shao et al. 2016; Wu et al. 2016). Glial cells are known to play a key role in flavivirus-mediated neurological disorders by activating the neuro–immune interaction. For example, microglia activation has been shown to drive neuronal and synaptic loss as well as memory dysfunction upon WNV infection via the complement pathway, a key component of innate immune pathogen defense during early postnatal development (Vasek et al. 2016). Considering the central role of inflammation in neurological disorders, future studies are needed to test the model that infection and activation of astrocytes and microglia upon ZIKV infection triggers a strong immune response, which in turn promotes neuronal death and dysfunction.
Another possible consequence of glia-mediated inflammation upon ZIKV infection is vascular abnormalities (Fig. 4). Previous studies suggest that inflammation is associated with abnormal vasculature, such as alterations in permeability (Friedl et al. 2002; Puhlmann et al. 2005). Consistently, ZIKV infection has been shown to alter vascular integrity, including vessel density and diameter, in the developing mouse brain and lead to a leaky blood–brain barrier (Shao et al. 2016). A direct causal link between glia-mediated inflammation and abnormal vasculature upon ZIKV infection remains to be established. Interestingly, blood–brain barrier leakage may further lead to neuronal calcification (Miklossy et al. 2005; Keller et al. 2013), a key feature identified in the ZIKV-infected microcephalic fetal brain (Driggers et al. 2016; Martines et al. 2016a; Mlakar et al. 2016). Future studies are also needed to examine the role of blood–brain barrier deficiency in brain calcification caused by ZIKV infection.
Conclusions and perspectives
Since the WHO declared ZIKV infection a Public Health Emergency of International Concern on February 1 of 2016, tremendous progress has been made in understanding the impact of ZIKV infection on human brain development and its underlying biological mechanisms. Remarkable evidence across multiple model systems, including two-dimensional (2D) NSC cultures, 3D neurospheres and human brain organoids, acute human fetal tissue cultures, animal models, and clinical fetal samples, all consistently point to one main conclusion: Vertical transmission of ZIKV via a transplacental route targets NPCs and glial cells in the developing brain, which causes cell death and reduced NPC proliferation, resulting in impaired brain development. Despite this rapid progress, these initial studies merely scratch the surface, and many questions remain to be answered in the near future. First, is the neurotropism of ZIKV strain-specific or human population-specific? Systematic comparison of strain-specific effects on cells derived from different populations will help to determine molecular and cellular signatures of vulnerability to ZIKV infection. Ultimately, genetically engineered ZIKV or iPSC lines can provide a more definitive answer. Previous infection with other flaviviruses, such as DENV and yellow fever viruses, may also underpin the apparent geographical region-specific differences in ZIKV effects (Bardina et al. 2017). Second, how does each of the individual ZIKV viral proteins and ZIKV-derived small RNAs elicit cytopathic effects on specific cellular properties? Investigation of their functions in both viral infection and host response will provide key insights into the pathogenesis of ZIKV infection. Third, what host factors and pathways mediate the entry of ZIKV into NPCs, one of the cell types most vulnerable to ZIKV in the developing brain? NPCs are known to be in direct contact and exposed to cerebrospinal fluid at the ventricle. The possibility that this interaction make the NPCs direct target remains to be explored. Genome-wide screens using RNAi or CRISPR/Cas9 may facilitate the identification of these host factors and pathways. Fourth, what is the role of glia and microglia activation in ZIKV-mediated brain developmental deficits? Future studies are needed to address whether and how glia and microglia activation and inflammation affect neuronal and other cellular functions in the ZIKV-infected developing brain. Overall, collaborative efforts from the scientific community across many disciplines, including virology, neuroscience, stem cell biology, and developmental biology, may contribute knowledge and technology in a rapid response to this global health emergency.
Acknowledgments
We apologize for not citing many original papers due to space limitations. We thank Kimberly M. Christian for comments. Research in the authors’ laboratories was supported by grants from the National Institutes of Health (R01MH105128, R21MH110160, R35NS097370, and U19AI131130 to G.-L.M., and R37NS047344, U19MH106434, P01NS097206, and R21HD086820 to H.S.), the Maryland Stem Cell Research Fund (G.-L.M. and H.S.), and the Foundation for Prader-Willi Research (G.-L.M.) and start-up funds from Emory University (Z.W.).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.298216.117.
References
- Aagaard KM, Lahon A, Suter MA, Arya RP, Seferovic MD, Vogt MB, Hu M, Stossi F, Mancini MA, Harris RA, et al. 2017. Primary human placental trophoblasts are permissive for Zika virus (ZIKV) replication. Sci Rep 7: 41389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbink P, Larocca RA, De La Barrera RA, Bricault CA, Moseley ET, Boyd M, Kirilova M, Li Z, Ng'ang'a D, Nanayakkara O, et al. 2016. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science 353: 1129–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adiga R. 2016. Phylogenetic analysis of the NS5 gene of Zika virus. J Med Virol 88: 1821–1826. [DOI] [PubMed] [Google Scholar]
- Akiyama BM, Laurence HM, Massey AR, Costantino DA, Xie X, Yang Y, Shi PY, Nix JC, Beckham JD, Kieft JS. 2016. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 354: 1148–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo LM, Ferreira ML, Nascimento OJ. 2016. Guillain-Barre syndrome associated with the Zika virus outbreak in Brazil. Arq Neuropsiquiatr 74: 253–255. [DOI] [PubMed] [Google Scholar]
- Barbelanne M, Tsang WY. 2014. Molecular and cellular basis of autosomal recessive primary microcephaly. Biomed Res Int 2014: 547986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardina SV, Bunduc P, Tripathi S, Duehr J, Frere JJ, Brown JA, Nachbagauer R, Foster GA, Krysztof D, Tortorella D, et al. 2017. Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science 356: 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET Jr, Cherry S, Sadovsky Y, Coyne CB. 2016. Type III interferons produced by human placental trophoblasts confer protection against Zika virus infection. Cell Host Microbe 19: 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayless NL, Greenberg RS, Swigut T, Wysocka J, Blish CA. 2016. Zika virus infection induces cranial neural crest cells to produce cytokines at levels detrimental for neurogenesis. Cell Host Microbe 20: 423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar J, Rabeneck DB, Martines RB, Reagan-Steiner S, Ermias Y, Estetter LB, Suzuki T, Ritter J, Keating MK, Hale G, et al. 2017. Zika virus RNA replication and persistence in brain and placental tissue. Emerg Infect Dis 23: 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasil P, Pereira JP Jr, Raja Gabaglia C, Damasceno L, Wakimoto M, Ribeiro Nogueira RM, Carvalho de Sequeira P, Machado Siqueira A, Abreu de Carvalho LM, Cotrim da Cunha D, et al. 2016. Zika virus infection in pregnant women in Rio de Janeiro—preliminary report. N Engl J Med 375: 2321–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault JB, Khou C, Basset J, Coquand L, Fraisier V, Frenkiel MP, Goud B, Manuguerra JC, Pardigon N, Baffet AD. 2016. Comparative analysis between flaviviruses reveals specific neural stem cell tropism for Zika virus in the mouse developing neocortex. EBioMedicine 10: 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvet GA, Santos FB, Sequeira PC. 2016. Zika virus infection: epidemiology, clinical manifestations and diagnosis. Curr Opin Infect Dis 29: 459–466. [DOI] [PubMed] [Google Scholar]
- Cao-Lormeau VM, Roche C, Teissier A, Robin E, Berry AL, Mallet HP, Sall AA, Musso D. 2014. Zika virus, French polynesia, South pacific, 2013. Emerg Infect Dis 20: 1085–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao-Lormeau VM, Blake A, Mons S, Lastere S, Roche C, Vanhomwegen J, Dub T, Baudouin L, Teissier A, Larre P, et al. 2016. Guillain-Barre Syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet 387: 1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers TJ, Hahn CS, Galler R, Rice CM. 1990. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 44: 649–688. [DOI] [PubMed] [Google Scholar]
- Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JL, Guimaraes KP, Benazzato C, Almeida N, Pignatari GC, Romero S, et al. 2016. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534: 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang J, Tiwari SK, Lichinchi G, Qin Y, Patil VS, Eroshkin AM, Rana TM. 2016. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 19: 258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng YQ, Zhao H, Li XF, Zhang NN, Liu ZY, Jiang T, Gu DY, Shi L, He JA, Wang HJ, et al. 2016. Isolation, identification and genomic characterization of the Asian lineage Zika virus imported to China. Sci China Life Sci 59: 428–430. [DOI] [PubMed] [Google Scholar]
- Dick GW, Kitchen SF, Haddow AJ. 1952. Zika virus. I. Isolations and serological specificity. Trans R Soc Trop Med Hyg 46: 509–520. [DOI] [PubMed] [Google Scholar]
- D'Ortenzio E, Matheron S, Yazdanpanah Y, de Lamballerie X, Hubert B, Piorkowski G, Maquart M, Descamps D, Damond F, Leparc-Goffart I. 2016. Evidence of sexual transmission of Zika virus. N Engl J Med 374: 2195–2198. [DOI] [PubMed] [Google Scholar]
- Dowall SD, Graham VA, Rayner E, Atkinson B, Hall G, Watson RJ, Bosworth A, Bonney LC, Kitchen S, Hewson R. 2016. A susceptible mouse model for Zika virus infection. PLoS Negl Trop Dis 10: e0004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driggers RW, Ho CY, Korhonen EM, Kuivanen S, Jaaskelainen AJ, Smura T, Rosenberg A, Hill DA, DeBiasi RL, Vezina G, et al. 2016. Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N Engl J Med 374: 2142–2151. [DOI] [PubMed] [Google Scholar]
- Dudley DM, Aliota MT, Mohr EL, Weiler AM, Lehrer-Brey G, Weisgrau KL, Mohns MS, Breitbach ME, Rasheed MN, Newman CM, et al. 2016. A rhesus macaque model of Asian-lineage Zika virus infection. Nat Commun 7: 12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, Pretrick M, Marfel M, Holzbauer S, Dubray C, et al. 2009. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med 360: 2536–2543. [DOI] [PubMed] [Google Scholar]
- Fontes CA, Dos Santos AA, Marchiori E. 2016. Magnetic resonance imaging findings in Guillain-Barre syndrome caused by Zika virus infection. Neuroradiology 58: 837–838. [DOI] [PubMed] [Google Scholar]
- Foy BD, Kobylinski KC, Chilson Foy JL, Blitvich BJ, Travassos da Rosa A, Haddow AD, Lanciotti RS, Tesh RB. 2011. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg Infect Dis 17: 880–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl J, Puhlmann M, Bartlett DL, Libutti SK, Turner EN, Gnant MF, Alexander HR. 2002. Induction of permeability across endothelial cell monolayers by tumor necrosis factor (TNF) occurs via a tissue factor-dependent mechanism: relationship between the procoagulant and permeability effects of TNF. Blood 100: 1334–1339. [PubMed] [Google Scholar]
- Gabriel E, Ramani A, Karow U, Gottardo M, Natarajan K, Gooi LM, Goranci-Buzhala G, Krut O, Peters F, Nikolic M, et al. 2017. Recent Zika virus isolates induce premature differentiation of neural progenitors in human brain organoids. Cell Stem Cell 20: 397–406.e5. [DOI] [PubMed] [Google Scholar]
- Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, Nascimento JM, Brindeiro R, Tanuri A, Rehen SK. 2016. Zika virus impairs growth in human neurospheres and brain organoids. Science 352: 816–818. [DOI] [PubMed] [Google Scholar]
- Ghouzzi VE, Bianchi FT, Molineris I, Mounce BC, Berto GE, Rak M, Lebon S, Aubry L, Tocco C, Gai M, et al. 2016. ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly and p53. Cell Death Dis 7: e2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore EC, Walsh CA. 2013. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip Rev Dev Biol 2: 461–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sanchez-Seco MP, Evans MJ, Best SM, et al. 2016. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19: 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, et al. 2015. Biology of Zika virus infection in human skin cells. J Virol 89: 8880–8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel R, Ferraris P, Wichit S, Diop F, Talignani L, Pompon J, Garcia D, Liegeois F, Sall AA, Yssel H, et al. 2017. African and Asian Zika virus strains differentially induce early antiviral responses in primary human astrocytes. Infect Genet Evol 49: 134–137. [DOI] [PubMed] [Google Scholar]
- Hanners NW, Eitson JL, Usui N, Richardson RB, Wexler EM, Konopka G, Schoggins JW. 2016. Western Zika virus in human fetal neural progenitors persists long term with partial cytopathic and limited immunogenic effects. Cell Rep 15: 2315–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann DL, Hodgson A, Sall AA, Freedman DO, Staples JE, Althabe F, Baruah K, Mahmud G, Kandun N, Vasconcelos PF, et al. 2016. Zika virus and microcephaly: why is this situation a PHEIC? Lancet 387: 719–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch AJ, Smith JL, Haese NN, Broeckel RM, Parkins CJ, Kreklywich C, DeFilippis VR, Denton M, Smith PP, Messer WB, et al. 2017. Zika Virus infection of rhesus macaques leads to viral persistence in multiple tissues. PLoS Pathog 13: e1006219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Westenberger A, Sobrido MJ, Garcia-Murias M, Domingo A, Sears RL, Lemos RR, Ordonez-Ugalde A, Nicolas G, da Cunha JE, et al. 2013. Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat Genet 45: 1077–1082. [DOI] [PubMed] [Google Scholar]
- Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, Power C, Hobman TC. 2016. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep 17: 1766–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno G, Chang GJ. 2007. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch Virol 152: 687–696. [DOI] [PubMed] [Google Scholar]
- Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS. 2016. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19: 720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, Zhang N, Shi L, Qin CF, Xu Z. 2016a. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19: 120–126. [DOI] [PubMed] [Google Scholar]
- Li XF, Dong HL, Huang XY, Qiu YF, Wang HJ, Deng YQ, Zhang NN, Ye Q, Zhao H, Liu ZY, et al. 2016b. Characterization of a 2016 clinical isolate of Zika virus in non-human primates. EBioMedicine 12: 170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Muffat J, Omer A, Bosch I, Lancaster MA, Sur M, Gehrke L, Knoblich JA, Jaenisch R. 2017. Induction of expansion and folding in human cerebral organoids. Cell Stem Cell 20: 385–396.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Luo Z, Zeng J, Chen W, Foo SS, Lee SA, Ge J, Wang S, Goldman SA, Zlokovic BV, et al. 2016. Zika virus NS4A and NS4B proteins deregulate Akt–mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell 19: 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist R, Mundt F, Gilthorpe JD, Wolfel S, Gekara NO, Kroger A, Overby AK. 2016. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J Neuroinflammation 13: 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma DK, Ming GL, Song H. 2005. Glial influences on neural stem cell development: cellular niches for adult neurogenesis. Curr Opin Neurobiol 15: 514–520. [DOI] [PubMed] [Google Scholar]
- Manangeeswaran M, Ireland DD, Verthelyi D. 2016. Zika (PRVABC59) infection is associated with T cell infiltration and neurodegeneration in CNS of immunocompetent neonatal C57Bl/6 mice. PLoS Pathog 12: e1006004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martines RB, Bhatnagar J, de Oliveira Ramos AM, Davi HP, Iglezias SD, Kanamura CT, Keating MK, Hale G, Silva-Flannery L, Muehlenbachs A, et al. 2016a. Pathology of congenital Zika syndrome in Brazil: a case series. Lancet 388: 898–904. [DOI] [PubMed] [Google Scholar]
- Martines RB, Bhatnagar J, Keating MK, Silva-Flannery L, Muehlenbachs A, Gary J, Goldsmith C, Hale G, Ritter J, Rollin D, et al. 2016b. Notes from the field: evidence of Zika virus infection in brain and placental tissues from two congenitally infected newborns and two fetal losses—Brazil, 2015. MMWR Morb Mortal Wkly Rep 65: 159–160. [DOI] [PubMed] [Google Scholar]
- McCarthy M. 2016. Zika virus was transmitted by sexual contact in Texas, health officials report. BMJ 352: i720. [DOI] [PubMed] [Google Scholar]
- McGrath EL, Rossi SL, Gao J, Widen SG, Grant AC, Dunn TJ, Azar SR, Roundy CM, Xiong Y, Prusak DJ, et al. 2017. Differential responses of human fetal brain neural stem cells to Zika virus infection. Stem Cell Reports 8: 715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L, Labeau A, Dejarnac O, Cipriani S, Sinigaglia L, Bonnet-Madin L, Le Charpentier T, Hafirassou ML, Zamborlini A, Cao-Lormeau VM, et al. 2017. Axl mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep 18: 324–333. [DOI] [PubMed] [Google Scholar]
- Miklossy J, Mackenzie IR, Dorovini-Zis K, Calne DB, Wszolek ZK, Klegeris A, McGeer PL. 2005. Severe vascular disturbance in a case of familial brain calcinosis. Acta Neuropathol 109: 643–653. [DOI] [PubMed] [Google Scholar]
- Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, et al. 2016a. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 165: 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JJ, Sene A, Richner JM, Smith AM, Santeford A, Ban N, Weger-Lucarelli J, Manzella F, Ruckert C, Govero J, et al. 2016b. Zika virus infection in mice causes panuveitis with shedding of virus in tears. Cell Rep 16: 3208–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming GL, Tang H, Song H. 2016. Advances in Zika virus research: stem cell models, challenges, and opportunities. Cell Stem Cell 19: 690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlakar J, Korva M, Tul N, Popovic M, Poljsak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodusek V, et al. 2016. Zika virus associated with microcephaly. N Engl J Med 374: 951–958. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH. 2012. Astrocytes and disease: a neurodevelopmental perspective. Genes Dev 26: 891–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso D. 2015. Zika virus transmission from French Polynesia to Brazil. Emerg Infect Dis 21: 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso D, Roche C, Robin E, Nhan T, Teissier A, Cao-Lormeau VM. 2015. Potential sexual transmission of Zika virus. Emerg Infect Dis 21: 359–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HN, Qian X, Song H, Ming GL. 2016. Neural stem cells attacked by Zika virus. Cell Res 26: 753–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noronha L, Zanluca C, Azevedo ML, Luz KG, Santos CN. 2016. Zika virus damages the human placental barrier and presents marked fetal neurotropism. Mem Inst Oswaldo Cruz 111: 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski TJ, Pollen AA, Di Lullo E, Sandoval-Espinosa C, Bershteyn M, Kriegstein AR. 2016. Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell 18: 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onorati M, Li Z, Liu F, Sousa AM, Nakagawa N, Li M, Dell'Anno MT, Gulden FO, Pochareddy S, Tebbenkamp AT, et al. 2016. Zika virus disrupts phospho-TBK1 localization and mitosis in human neuroepithelial stem cells and radial glia. Cell Rep 16: 2576–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornelas AM, Pezzuto P, Silveira PP, Melo FO, Ferreira TA, Oliveira-Szejnfeld PS, Leal JI, Amorim MM, Hamilton S, Rawlinson WD, et al. 2017. Immune activation in amniotic fluid from Zika virus associated microcephaly. Ann Neurol 81: 152–156. [DOI] [PubMed] [Google Scholar]
- Petersen E, Wilson ME, Touch S, McCloskey B, Mwaba P, Bates M, Dar O, Mattes F, Kidd M, Ippolito G, et al. 2016. Rapid spread of Zika virus in the Americas—implications for public health preparedness for mass gatherings at the 2016 Brazil Olympic Games. Int J Infect Dis 44: 11–15. [DOI] [PubMed] [Google Scholar]
- Puhlmann M, Weinreich DM, Farma JM, Carroll NM, Turner EM, Alexander HR Jr. 2005. Interleukin-1β induced vascular permeability is dependent on induction of endothelial tissue factor (TF) activity. J Transl Med 3: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, et al. 2016. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165: 1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Jacob F, Song H, Ming GL. 2017. Using brain organoids to understand Zika virus-induced microcephaly. Development 144: 952–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O'Neal JT, Rajakumar A, Wrammert J, Rimawi BH, Pulendran B, et al. 2016. Zika virus infects human placental macrophages. Cell Host Microbe 20: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. 2016. Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med 374: 1981–1987. [DOI] [PubMed] [Google Scholar]
- Reemst K, Noctor SC, Lucassen PJ, Hol EM. 2016. The indispensable roles of microglia and astrocytes during brain development. Front Hum Neurosci 10: 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retallack H, Di Lullo E, Arias C, Knopp KA, Laurie MT, Sandoval-Espinosa C, Mancia Leon WR, Krencik R, Ullian EM, Spatazza J, et al. 2016. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc Natl Acad Sci 113: 14408–14413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi SL, Tesh RB, Azar SR, Muruato AE, Hanley KA, Auguste AJ, Langsjoen RM, Paessler S, Vasilakis N, Weaver SC. 2016. Characterization of a novel murine model to study Zika virus. Am J Trop Med Hyg 94: 1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Q, Herrlinger S, Yang SL, Lai F, Moore JM, Brindley MA, Chen JF. 2016. Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage. Development 143: 4127–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, Kuhn RJ. 2016. The 3.8 Å resolution cryo-EM structure of Zika virus. Science 352: 467–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza BS, Sampaio GL, Pereira CS, Campos GS, Sardi SI, Freitas LA, Figueira CP, Paredes BD, Nonaka CK, Azevedo CM, et al. 2016. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci Rep 6: 39775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM, et al. 2016. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 18: 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricarico PM, Caracciolo I, Crovella S, D'Agaro P. 2017. Zika virus induces inflammasome activation in the glial cell line U87-MG. Biochem Biophys Res Commun 10.1016/j.bbrc.2017.01.158. [DOI] [PubMed] [Google Scholar]
- Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez-Torres C, Frouin A, Wilton DK, et al. 2016. A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature 534: 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells MF, Salick MR, Wiskow O, Ho DJ, Worringer KA, Ihry RJ, Kommineni S, Bilican B, Klim JR, Hill EJ, et al. 2016. Genetic ablation of AXL does not protect human neural progenitor cells and cerebral organoids from Zika virus infection. Cell Stem Cell 19: 703–708. [DOI] [PubMed] [Google Scholar]
- Wu KY, Zuo GL, Li XF, Ye Q, Deng YQ, Huang XY, Cao WC, Qin CF, Luo ZG. 2016. Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res 26: 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Lee EM, Wen Z, Cheng Y, Huang WK, Qian X, Tcw J, Kouznetsova J, Ogden SC, Hammack C, et al. 2016. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat Med 22: 1101–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Hammack C, Ogden SC, Cheng Y, Lee EM, Wen Z, Qian X, Nguyen HN, Li Y, Yao B, et al. 2016. Molecular signatures associated with ZIKV exposure in human cortical neural progenitors. Nucleic Acids Res 44: 8610–8620. [DOI] [PMC free article] [PubMed] [Google Scholar]