

Abstract
A method for the physical attachment of folded RNA libraries to their encoding DNA is presented as a way to circumvent the reverse transcription step during systematic evolution of RNA ligands by exponential enrichment (RNA-SELEX). A DNA library is modified with one isodC base to stall T7 polymerase and a 5′ “capture strand” which anneals to the nascent RNA transcript. This method is validated in a selection of RNA aptamers against human α-thrombin with dissociation constants in the low nanomolar range. This method will be useful in the discovery of RNA aptamers and ribozymes containing base modifications that make them resistant to accurate reverse transcription.
Graphical abstract
First characterized in 1990 independently by the laboratories of Szostak and Gold, oligonucleotide aptamers are DNA or RNA with high affinity for proteins or small molecules.1–4 Exhibiting advantages such as affinities comparable to those of antibodies, simplicity of synthesis, and general lack of immunogenicity, aptamers have found a place in the pharmaceutical market. The first aptamer drug approved by the FDA, Pegaptanib, targets age-related macular degeneration by binding to vascular endothelial growth factor (VEGF).5–10 Several other aptamers are in late-stage clinical trials, and an unknown number are in laboratory development.5–9 Aptamers are typically discovered using standard molecular biology techniques through an in vitro selection process termed SELEX (Systematic Evolution of Ligands by EXponential enrichment).4 DNA libraries with random regions flanked by constant regions are synthesized using standard phosphoramidite chemistry and can be used directly in the selection of DNA aptamers. RNA libraries are generated by transcription of a T7 promoter-containing random DNA library using T7 RNA polymerase. Libraries are mixed with a target small molecule or protein and the bound fraction is recovered using a variety of isolation techniques. The recovered library is amplified with the polymerase chain reaction (PCR) for DNA aptamers or reverse transcription-PCR for RNA aptamers, the single-stranded DNA or RNA is regenerated from the PCR product and the selection cycle is repeated until the library is sufficiently enriched with binders.
Limitations to standard aptamer libraries (nuclease sensitivity, limited chemical diversity) have been overcome by the use of non-natural nucleotide analogs.11–17 For instance, the incorporation of 2′-fluoro pyrimidines into RNA by T7 RNA polymerase variants results in significant serum nuclease resistance, a major requirement for drug development. To improve chemical diversity, base-modified DNA aptamers have been readily utilized, largely owing to the discovery that family B DNA polymerases (including Vent, KOD, and Pfu) can accommodate C5-substituted thymidine base analogs.3, 11–15, 18–22 In particular, SOMAmer technology, which incorporates short hydrophobic groups at the C5 position of uridine, improved the success rate of obtaining high-affinity aptamers from 30% to 80% for hundreds of targets.20 Recently, our group reported a method termed SELection of Modified Aptamers, or SELMA, allowing for the incorporation of large modifications into DNA libraries, which was successfully used to obtain multivalent glycoclusters that mimic a conserved epitope on the HIV envelope protein gp120.23–26
However, compared to DNA, successes with base-modified RNA aptamer libraries have not been as wide-spread, despite the superior serum nuclease resistance of 2′-F-modified RNA and greater folding space sampling of RNA libraries. An obstacle to development of base-modified RNA-SELEX is that it would require that two different types of enzymes (RNA polymerase and reverse transcriptase) tolerate the modified bases. Here we present the successful generation and selection of RNA libraries in which the folded RNA is physically attached to the dsDNA that encodes it. The concept of displaying a difficult-to-amplify oligonucleotide on analogous dsDNA was introduced by Szostak and coworkers,27 and has been put into practice by several groups.23, 28, 29 The present method circumvents the need for reverse-transcription in the amplification of RNA libraries and could be applied to base modifications for which reverse transcription is inefficient. We propose that this method could be useful in the future for introduction of alkyne-modified RNA analogues and post-transcriptional click modification in an RNA-version of SELMA.
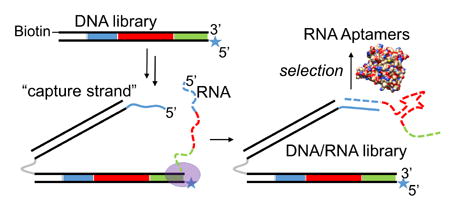
We hypothesized that a DNA library could be designed such that each DNA duplex could capture its corresponding RNA transcript on a “capture arm” (Figure 1, F–H). Generation of this library starts with singly biotinylated, double stranded DNA (Figure 1, A), from which the coding strand is removed using streptavidin magnetic beads. A capture oligonucleotide is annealed to the non-coding strand (B, C) and extended using Bst 2.0 DNA polymerase (DNAP) to regenerate the double stranded DNA (dsDNA) with an 18 atom hexaethyleneglycol (HEG) spacer (gray) and 124-base single-stranded DNA (ssDNA) extension 5′ of the coding strand (D). The 5′ end of the capture strand contains 21 bases (5′ GCT CGT TCT CCT TCC CTC TCC, shown in blue) that complement the 5′ end of the RNA strand encoded in the DNA library. An 82-base oligonucleotide is annealed to the capture strand, rigidifying it while maintaining ssDNA at the 5′ end (E). T7 RNA polymerase (RNAP) and nucleotide triphosphates are then added to initiate transcription of the DNA library. The 5′ end of the non-coding strand contains an unnatural base, isodC, which causes T7 polymerase to stall30 at the end of the template (F, G), allowing enough time for the 5′ ends of the capture strand and RNA transcript to anneal “intrastructurally” (G). The T7 RNAP dissociates and the DNA/RNA library (H) is amenable to selection. Amplification of the selected library with a biotinylated forward primer and 5′ isodC reverse primer regenerates the dsDNA library in its original form (A), completing a single selection cycle. The rigidifying oligonucleotide serves to increase the likelihood that the RNA and capture strand will come into contact and anneal. Within the capture strand, the HEG spacer is critical for maintaining the ssDNA nature of the 5′ capture sequence; without this spacer, the 3′ end of non-coding strand would be extended in step (C→D), creating a full-length complement of the capture arm that would block its ability to anneal to RNA.
Figure 1.
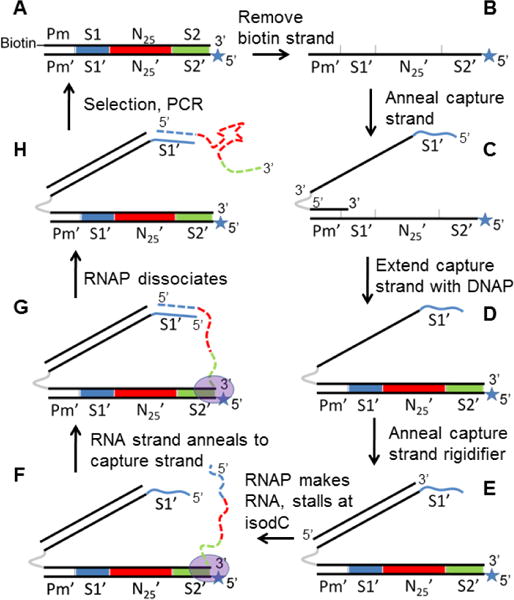
Generation and selection of a DNA-displayed RNA library. Black solid lines = DNA; dotted colored lines = RNA; grey = hexaethyleneglycol spacer; blue line = 5′ terminal capture sequence complementary to S1; Pm = T7 promoter sequence; S1′ = capture strand sequence; S2′ = reverse primer; N25 = random region; star = isodeoxycytidine; oval = T7 polymerase.
A considerable amount of effort went into the design of the sequences of the capture arm of the DNA display construct. Notably, predicted secondary structure was minimized by trial-and-error with the use of mFOLD prediction.31 Minimization of secondary structure allowed for annealing of the 82-base rigidifying oligonucleotide at low temperature (50° C) despite its high predicted melting temperature (Tm~75° C at 1 nM concentration). This is a crucial requirement because denaturation of the double stranded DNA library at higher temperatures will destroy the integrity of the library. The capture sequence itself (GCTCGTTCTCCTTCCCTCTCC) was designed to have minimal secondary structure as well as a high Tm with RNA. A key feature for high Tm is the lack of A-U base pairs, which are known to destabilize RNA-DNA hybrids considerably.32 As a result, we were able to design a capture sequence with Tm of ~70 °C (at 1 nM concentration), a strong non-covalent linkage between the RNA and encoding DNA, capable of forming at the 37 °C transcription temperature.
Figure 2 shows the various library species, as visualized in native (A) and denaturing (B) PAGE gels. For lanes 5–7, comparison of the two gels shows clearly that bands in the native gel corresponding to Figure 1 species C, D, and E each separate into two or three strands of the appropriate size in the denaturing gel. The lane 8 band highlighted with a box in the native gel was hypothesized to be the DNA-displayed RNA species. To confirm this assignment, the band was excised from the native gel, and the material extracted from the gel slice was rerun on a denaturing gel (lane 10), giving rise to four bands matching the separate component strands. Extra bands in lane 8 show that library generation results in additional species, which we hypothesized to be excess RNA transcripts and other species resulting from transcription that do not anneal to the library. This was confirmed (lanes 9) by a control transcription of the DNA library lacking a capture arm (Figure 1, form A), which produced most of the bands present in Lane 8, but not the one attributed to the self-annealed RNA-DNA library, species G (boxed band).
Figure 2.
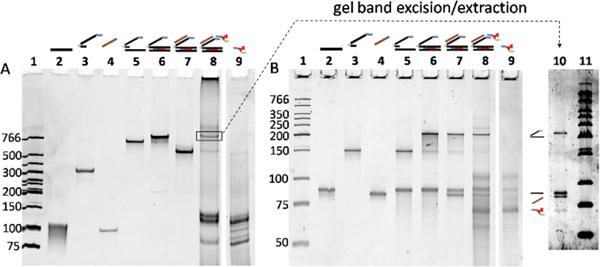
Non-denaturing (A) and denaturing (B) gels of library generation. In both gels, lanes 2–4 are controls that show migration of the individual purchased oligos; lanes 5–8 show the migration of species produced when these components are combined and used to generate the library; lane 9 is a control showing that RNA transcription of an ordinary DNA duplex produces several additional bands observed in lane 8. Lane contents: 1) LMW ladder (NEB); 2) non-coding library strand (10 pmol); 3) capture arm (10 pmol); 4) rigidifier (10 pmol); 5) library annealed to capture arm (0.25 pmol); 6) extension product, 0.25 pmol; 7) extension product annealed to rigidifier (0.25 pmol); 8) transcribed library (0.25 pmol) 9) transcribed library without capture arm. The box indicates the final RNA-displayed library, which was excised from the gel, eluted from the gel slice and rerun on a denaturing gel (lane 10). Gel conditions: A) 10% acrylamide, 29:1 acrylamide:bis-acrylamide, stained with ethidium bromide. B) 8 M urea, 12.5% acrylamide, 29:1 acrylamide:bis-acrylamide, stained with Sybr Gold; lanes 1–9 contain the same samples in both gels.
To verify that the RNA transcript was being captured “intrastructurally” on its own DNA, with sufficient fidelity for selections, we performed a proof-of-principle experiment to show that a sequence with known binding activity could be enriched from a spiked library. To this end, we generated a random RNA-displayed DNA library spiked with 1:1000 of an NheI restriction site sequence. After incubation of the library with a biotinylated DNA oligo complementary to the NheI sequence, the bound fraction was isolated with streptavidin magnetic beads and binders were amplified and regenerated. After two rounds of selection, a significant proportion of the recovered DNA contained the selected sequence (as evidenced by its NheI restriction susceptibility), indicating that the RNA was correctly captured by the DNA encoding it (Figure S1). To control for the possibility that the NheI-cleavable PCR product resulted from traces of the bait sequence contaminating the sample, a parallel selection was performed with the transcription step omitted. In this negative control, more PCR cycles were required to amplify selected library, and the resulting library was not susceptible to NheI cleavage.
With this evidence that our RNA library was effectively displayed on its encoding DNA, we attempted the more rigorous test of our system by using it to select RNA sequences that could bind to a protein. As our target, we chose human α-thrombin, a commonly used target in proof-of-principle studies.33 The library described above was prepared without the added NheI cleavage sequence and was then panned for binding to biotinylated thrombin over 10 rounds of selection with capture on streptavidin beads. Enrichment for binders was evident after the 7th round as indicated by the decreased number of PCR cycles required to recover the library. Stringency was then increased by lowering the thrombin concentration from 10 to 1 nM and shortening incubation time from 1 hour to 5 minutes; after the 10th round of selection the library was cloned and 10 members sequenced and analyzed for thrombin binding. As Table 1 indicates, all but two of the isolated sequences showed similarity in both sequence and predicted secondary structure.
Table 1.
Sequences and thrombin binding of library clones after round 10. Dissociation constants are derived from triplicate measurement in nitrocellulose filter binding assays, and the error reported is the standard error of the curve fit. Each oligo also contains 5′ and 3′ sequences complementary to the capture strand and reverse primer, respectively, which are presented together with binding curves for each clone in the Supplementary Information.
| Clone ID | Sequence | Kd (nM) | Fbmax |
|---|---|---|---|
| 1 | UGUUACUCAC A UAGCGAAG CU | 44.7 ± 4.6 | 95.6 ± 4.4 |
| 2 | CCGGCGUCAC G UAGACAAA CU | 3.5 ± 0.3 | 80.6 ± 1.6 |
| 3 | CGGGACUCAC G UAGACAAU CU | 2.4 ± 0.3 | 65.3 ± 1.9 |
| 4 | UGCGGAUAAC G UAGCAAAG CU | 10.0 ± 0.9 | 84.5 ± 2.3 |
| 5 | CUACCGU GAGAACGAGC GACUC | NB | ND |
| 6 | CGGGACACU GGAACAUAAA GUU | 13.8 ± 1.9 | 74.5 ± 3.2 |
| 7 | CCGAAGCUCGGAGAAGCACAGAAGC | NB | ND |
| 8 | GGGAUUGCAC G UAGCGUAG CU | 1.6 ± 0.4 | 63.5 ± 3.3 |
| 9 | GGGUGUUCAC G UAGAGUAG CU | 1.4 + 0.2 | 92.3 ± 3.1 |
| 10 | GCUUUGACAC G UACAAUAU GU | 36.4 ± 2.8 | 52.9 ± 1.7 |
Thrombin binding affinity was measured for the corresponding RNAs of the 10 clones in nitrocellulose filter binding assays. For the eight related clones, dissociation constants (Table 1) ranged from 1.4 to 44.7 nM. Interestingly, the predicted RNA folds for these sequences, as determined by mFOLD, were strikingly similar to each other and to a previously isolated thrombin RNA aptamer for which a co-crystal structure (PDB ID 3DD2) has been obtained.34, 35 A prominent feature of the aptamer fold in the crystal structure is adenosine stacking, where the aptamer contains a stem motif followed by the bulge sequence AACA opposite a single adenosine, followed by another 3-base pair stem, 8 base loop structure. Two adenosines (ACA) in the bulge sequence (red) sandwich an adenosine (red) from the loop CUGAAGUA. The fourth adenosine also stacks against Arg233 of α-thrombin. In the aptamers obtained in the present study, a similar motif is found, where there is a bulge containing an AAXA opposite a lone adenosine (with the exception of clone 6, which contains AAXG opposite an adenosine), followed by a 3-base-pair stem, 6 base loop with a terminal adenosine in the otherwise variable loop. To confirm that our aptamers bind thrombin through their similar motif, we synthesized a truncated variant of clone 9, containing only the portion highlighted in the red box (Figure S4, ESI). Nitrocellulose filter binding and biolayer interferometry (BLI) measurements confirmed tight binding to thrombin (2 and 0.4 nM, respectively). Together, these data suggest that our aptamers adopt a conformation similar to those previously reported, and bind to the same region of thrombin, exosite II.
Little or no binding was detected for the remaining two sequences, clones 5 and 7. The predicted fold of clones 5 and 7 (Figure 3) is such that the 5′ capture strand complement region (green) engages in extensive base pairing, which would be predicted to inhibit the capture of the RNA by the DNA and therefore render the DNA display ineffective for these clones. Because library generation produces excess free RNA transcripts, abundant thrombin-binding free RNA present in later rounds may anneal to DNA capture strands belonging to sequences such as 5 and 7 that are unable to self-capture, thereby rescuing them. Alternative explanations for the presence of non-binding sequences are that they are simply particularly efficient PCR substrates, or that they might bind to thrombin in a manner that is dependent on the presence of their DNA tag.
Figure 3.
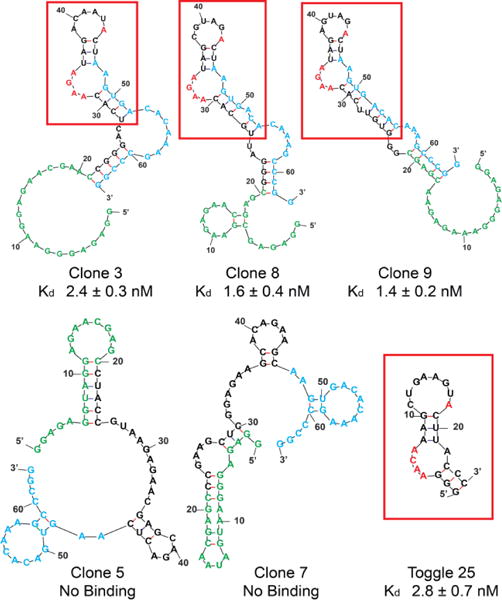
mFold predictions of most stable secondary structures for selected clones. The red boxes and letters highlight the stem loop structure and sequence common to most binding clones, and to the “Toggle 25” aptamer previously reported by Sullenger and Long (Refs. 30, 31). Green = capture strand complement. Blue = complement of reverse primer.
In summary, we have described and verified a method for DNA display of RNA. In the future, the benefits of using RNA in selections (structural diversity, amenability to 2′ modifications for nuclease resistance) can be coupled with substantial post-transcriptional modification in a SELMA-type experiment.23 Applications other than SELMA may also benefit from DNA display of RNA. For instance, it will be useful in RNA-SELEX using unnatural NTPs that are accepted by T7 RNA polymerase, but then render the RNA a poor template for reverse transcriptase. It should be noted that the display method described herein uses commercially available enzymes and DNA oligonucleotide synthesis, and an entire library generation and selection cycle can be performed in a day. Further applications of this method for in vitro selections will be reported in due course.
Supplementary Material
Acknowledgments
The generous support of the National Institutes of Health (NIH/NIAID R01AI090745) is gratefully acknowledged.
Footnotes
Electronic Supplementary Information (ESI) available: supplementary figure, experimental procedures, oligo sequences and filter binding data. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 2.Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 3.Gold L, Janjic N, Jarvis T, Schneider D, Walker JJ, Wilcox SK, Zichi D. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a003582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoltenburg R, Reinemann C, Strehlitz B. Biomol Eng. 2007;24:381–403. doi: 10.1016/j.bioeng.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Kanwar JR, Roy K, Maremanda NG, Subramanian K, Veedu RN, Bawa R, Kanwar RK. Curr Med Chem. 2015;22:2539–2557. doi: 10.2174/0929867322666150227144909. [DOI] [PubMed] [Google Scholar]
- 6.Keefe AD, Pai S, Ellington A. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sundaram P, Kurniawan H, Byrne ME, Wower J. Eur J Pharm Sci. 2013;48:259–271. doi: 10.1016/j.ejps.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 8.Sun H, Zhu X, Lu PY, Rosato RR, Tan W, Zu Y. Mol Ther Nucleic Acids. 2014;3:e182. doi: 10.1038/mtna.2014.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santosh B, Yadava PK. Biomed Res Int. 2014;2014:540451. doi: 10.1155/2014/540451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruckman J, et al. J Biol Chem. 1998;273:20556–20567. doi: 10.1074/jbc.273.32.20556. [DOI] [PubMed] [Google Scholar]
- 11.Gold L, et al. PLoS One. 2010;5:e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta S, et al. J Biol Chem. 2014;289:8706–8719. doi: 10.1074/jbc.M113.532580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keefe AD, Cload ST. Curr Opin Chem Biol. 2008;12:448–456. doi: 10.1016/j.cbpa.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 14.Lapa SA, Chudinov AV, Timofeev EN. Mol Biotechnol. 2016;58:79–92. doi: 10.1007/s12033-015-9907-9. [DOI] [PubMed] [Google Scholar]
- 15.Ohsawa K, Kasamatsu T, Nagashima J, Hanawa K, Kuwahara M, Ozaki H, Sawai H. Anal Sci. 2008;24:167–172. doi: 10.2116/analsci.24.167. [DOI] [PubMed] [Google Scholar]
- 16.Shoji A, Kuwahara M, Ozaki H, Sawai H. J Am Chem Soc. 2007;129:1456–1464. doi: 10.1021/ja067098n. [DOI] [PubMed] [Google Scholar]
- 17.Wang RE, Wu H, Niu Y, Cai J. Curr Med Chem. 2011;18:4126–4138. doi: 10.2174/092986711797189565. [DOI] [PubMed] [Google Scholar]
- 18.Kuwahara M, Ohsawa K, Kasamatsu T, Shoji A, Sawai H, Ozaki H. Nucleic Acids Symp Ser (Oxf) 2005:81–82. doi: 10.1093/nass/49.1.81. [DOI] [PubMed] [Google Scholar]
- 19.Kuwahara M, Sugimoto N. Molecules. 2010;15:5423–5444. doi: 10.3390/molecules15085423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohloff JC, Gelinas AD, Jarvis TC, Ochsner UA, Schneider DJ, Gold L, Janjic N. Mol Ther Nucleic Acids. 2014;3:e201. doi: 10.1038/mtna.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuwahara M, et al. Nucleic Acids Res. 2006;34:5383–5394. doi: 10.1093/nar/gkl637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuwahara M, et al. Molecules. 2010;15:8229. doi: 10.3390/molecules15118229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacPherson IS, Temme JS, Habeshian S, Felczak K, Pankiewicz K, Hedstrom L, Krauss IJ. Angew Chem Int Ed Engl. 2011;50:11238–11242. doi: 10.1002/anie.201105555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Temme JS, Drzyzga MG, MacPherson IS, Krauss IJ. Chemistry. 2013;19:17291–17295. doi: 10.1002/chem.201303848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Temme JS, Krauss IJ. Curr Protoc Chem Biol. 2015;7:73–92. doi: 10.1002/9780470559277.ch140233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Temme JS, MacPherson IS, DeCourcey JF, Krauss IJ. J Am Chem Soc. 2014;136:1726–1729. doi: 10.1021/ja411212q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ichida JK, Zou K, Horhota A, Yu B, McLaughlin LW, Szostak JW. J Am Chem Soc. 2005;127:2802–2803. doi: 10.1021/ja045364w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu H, Zhang S, Chaput JC. Nat Chem. 2012;4:183–187. doi: 10.1038/nchem.1241. [DOI] [PubMed] [Google Scholar]
- 29.Renders M, Miller E, Hollenstein M, Perrin D. Chemical Communications. 2015;51:1360–1362. doi: 10.1039/c4cc07588a. [DOI] [PubMed] [Google Scholar]
- 30.Tanasova M, Goeldi S, Meyer F, Hanawalt PC, Spivak G, Sturla SJ. ChemBioChem. 2015;16:1212–1218. doi: 10.1002/cbic.201500077. [DOI] [PubMed] [Google Scholar]
- 31.Zuker M. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin FH, Tinoco I., Jr Nucleic Acids Res. 1980;8:2295–2299. doi: 10.1093/nar/8.10.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. Nature. 1992;355:564–566. doi: 10.1038/355564a0. [DOI] [PubMed] [Google Scholar]
- 34.Long SB, Long MB, White RR, Sullenger BA. RNA. 2008;14:2504–2512. doi: 10.1261/rna.1239308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White R, Rusconi C, Scardino E, Wolberg A, Lawson J, Hoffman M, Sullenger B. Mol Ther. 2001;4:567–573. doi: 10.1006/mthe.2001.0495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.