

Abstract
Acute intoxication with organophosphates (OPs) can trigger seizures that progress to status epilepticus, and survivors often exhibit chronic neuropathology, cognitive impairment, affective disorders, and/or electroencephalographic abnormalities. Understanding how acute injury transitions to persistent neurological sequelae is critical to developing medical countermeasures for mitigating damage following OP-induced seizures. Here, we used in vivo magnetic resonance imaging (MRI) to monitor the spatiotemporal patterns of neuropathology for 1 month after acute intoxication with diisopropylfluorophosphate (DFP). Adult male Sprague Dawley rats administered pyridostigmine bromide (0.1 mg/kg, im) 30 min prior to successive administration of DFP (4 mg/kg, sc), atropine sulfate (2 mg/kg, im), and 2-pralidoxime (25 mg/kg, im) exhibited moderate-to-severe seizure behavior. T2-weighted and diffusion-weighted MR imaging prior to DFP exposure and at 3, 7, 14, 21, or 28 days postexposure revealed prominent lesions, tissue atrophy, and ventricular enlargement in discrete brain regions. Lesions varied in intensity and/or extent over time, with the overall magnitude of injury strongly influenced by seizure severity. Importantly, lesions detected by MRI correlated spatially and temporally with histological evidence of brain pathology. Analysis of histogram parameters extracted from frequency distributions of regional apparent diffusion coefficient (ADC) values identified the standard deviation and 90th percentile of the ADC as robust metrics for quantifying persistent and progressive neuropathological changes. The interanimal and interregional variations observed in lesion severity and progression, coupled with potential reinjury following spontaneous recurrent seizures, underscore the advantages of using in vivo imaging to longitudinally monitor neuropathology and, ultimately, therapeutic response, following acute OP intoxication.
Keywords: diffusion-weighted MRI, in vivo imaging, organophosphate, T2-weighted MRI
Acute intoxication with organophosphate (OP) cholinesterase inhibitors, which include pesticides and nerve agents, is estimated to cause 300 000 deaths and 1 million life-threatening intoxications each year (Gunnell et al., 2007; Pereira et al., 2014). Acute OP poisoning inhibits acetylcholinesterase (AChE), leading to significantly increased levels of acetylcholine in peripheral and central cholinergic synapses. Overstimulation of central cholinergic synapses can trigger convulsions that progress to status epilepticus (SE) (Chen, 2012; de Araujo Furtado et al., 2012), defined clinically as continuous seizure activity lasting longer than five minutes (Billington et al., 2016). Individuals who survive OP-induced SE often experience significant morbidity in the form of chronic neuropathology, cognitive impairment, affective disorders, and recurrent seizures (Chen, 2012; Collombet, 2011; Eddleston and Phillips, 2004; Jett, 2007; Pereira et al., 2014; Yamasue et al., 2007; Yanagisawa et al., 2006).
The pathogenic mechanisms underlying the persistent neurological sequelae of acute OP intoxication remain speculative. The majority of research focused on this question has employed preclinical models of acute intoxication with OP nerve agents (Chen, 2012; Collombet, 2011; de Araujo Furtado et al., 2012; Pereira et al., 2014). However, due to increasing concern of the public health threat posed by the more readily available OP pesticides, preclinical models of acute intoxication with diisopropylfluorophosphate (DFP), an OP pesticide, are emerging as important alternative models of acute OP intoxication. DFP is structurally similar to the OP nerve agent soman, and, like soman, it rapidly produces electrographic SE (Deshpande et al., 2010; Kadar et al., 1992; Kadriu et al., 2009; Rojas et al., 2015). Preclinical studies have demonstrated that DFP-induced SE elicits progressive neuronal cell loss (Flannery et al., 2016; Kim et al., 1999; Li et al., 2011; Rojas et al., 2015), persistent neuroinflammation (Damodaran and Abou-Donia, 2000; Flannery et al., 2016; Liu et al., 2012; Rojas et al., 2015), and delayed cognitive impairment (Brewer et al., 2013; Flannery et al., 2016; Rojas et al., 2016; Wright et al., 2010). What is not known is whether DFP-induced neuropathology resolves over time, whether adverse outcomes are caused by seizure activity and/or the toxic effects of the chemical or whether these endpoints are causally linked. Addressing these questions will require quantifiable metrics for assessing the spatiotemporal progression and resolution of brain injury across multiple brain regions, which is challenging using standard histopathological approaches.
In vivo magnetic resonance imaging (MRI) can overcome these challenges by enabling prospective evaluation of the spatiotemporal progression of neuropathology following acute OP intoxication. In particular, T2-weighted MRI and diffusion-weighted MRI, the clinical standards for anatomic imaging and the detection of ischemic injury, respectively, have proven highly effective for visualizing seizure-induced neuropathology in clinical case studies and preclinical models of seizures induced by triggers other than OPs (Gomes and Shinnar, 2011; Liachenko et al., 2015; Mendes and Sampaio, 2016; Milligan et al., 2009). These non-invasive, non-destructive techniques enable longitudinal, high-resolution anatomic and physiologic imaging within individual animals, thereby reducing the number of animals needed, while simultaneously increasing statistical power. However, there is a critical need to identify metrics for quantifying OP-induced neuropathology in preclinical MRI experiments in order to rigorously evaluate correlations among endpoints, and assess the efficacy of candidate therapeutics in drug trials.
There are several recent reports of using MRI to characterize the development of brain lesions in preclinical models of acute intoxication with the nerve agent soman and the pesticide paraoxon (Bhagat et al., 2001; Gullapalli et al., 2010; Rosman et al., 2012; Shrot et al., 2012; Testylier et al., 2007). However, despite histological evidence of neuropathology that persists for months postexposure (Collombet, 2011; de Araujo Furtado et al., 2012; Pereira et al., 2014), with one exception (Rosman et al., 2012), these studies have focused on neuropathological changes during the first week postexposure (Bhagat et al., 2001; Bhagat et al., 2005; Carpentier et al., 2008; Gullapalli et al., 2010; Rosman et al., 2012; Shrot et al., 2012; Testylier et al., 2007). There is, therefore, a paucity of in vivo imaging data describing how OP-induced lesions evolve over longer postexposure time scales during which persistent neurological sequelae in preclinical models have been documented. The purpose of the present study was to use in vivo MRI to monitor the spatiotemporal patterns of neuropathology over the first month postexposure in a rat model of acute DFP intoxication, and to identify imaging metrics for quantifying neuropathological changes thought to be associated with clinically relevant behavioral outcomes.
MATERIALS AND METHODS
Animals and DFP Exposures
Animals were maintained in facilities fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and all studies were performed with regard to the alleviation of pain and suffering under protocols approved by the University of California, Davis Institutional Animal Care and Use Committee. Adult male Sprague Dawley rats (250–280 g; Charles River Laboratories, Hollister, California) were housed individually in standard plastic cages under controlled environmental conditions (22 ± 2 °C, 40%–50% humidity) with a normal 12 h light/dark cycle. Food and water were provided ad libitum.
As illustrated in a schematic of the acute DFP intoxication model (Figure 1), unanesthetized rats were administered DFP (Sigma Chemical Company, St Louis, MO) at 4 mg/kg in a total volume of 300 µl via sc injection. DFP was diluted with sterile ice cold phosphate buffered saline (PBS) within 5 min of administration. To increase survival, animals were administered pyridostigmine bromide (TCI America, Portland, OR) in saline at 0.1 mg/kg, im, 30 min prior to DFP, and, immediately following DFP exposure, atropine sulfate (Sigma Chemical Company) at 2.0 mg/kg, im, and pralidoxime (2-PAM, Sigma Chemical Company) at 25 mg/kg in saline, im. These drugs significantly reduce mortality by blocking the peripheral parasympathomimetic symptoms associated with acute OP intoxication (Kim et al., 1999). Vehicle (VEH) control animals were injected with 300 µl PBS sc in place of DFP, but were similarly pretreated with pyridostigmine bromide and posttreated with atropine and 2-PAM. At 6 h postexposure, animals were injected sc with 10 ml of 5% dextrose in saline to replace lost fluids and to prevent hypoglycemia. Once returned to their home cages, rats were weighed daily and provided rat chow softened in water for 3–5 days until they were able to locate and consume standard chow and water.
FIG. 1.

A, Schematic illustrating the diisopropylfluorophosphate (DFP) exposure paradigm and experimental design. Amongst DFP-exposed rats, 72% survived to 12 h post exposure; of those, 82% survived until euthanized. Imaging and histopathological examinations were performed on 28 DFP-exposed and 7 vehicle control rats at varying days post exposure (DPE) based on their injection group. The number of DFP-injected animals (DFP) and vehicle controls (VEH) imaged per injection group is listed at the end of each row. Note that subsets of animals were imaged at 1–7 days prior to DFP exposure (baseline). B, Scale used to score seizure severity. SLUD = salivation, lacrimation, urination, defecation, which are classic symptoms of cholinergic crisis.
Scoring Seizure Behavior
Seizure severity was quantified using a scale (Figure 1) previously used to characterize seizures in a preclinical model of acute DFP intoxication (Deshpande et al., 2010). Seizure behavior was scored at 5 min intervals from 0 to 120 min post-DFP, and at 20 min intervals from 120 to 240 min post-DFP (≥16 observations per animal). Seizure severity was initially evaluated as the maximal seizure severity score (SSmax) over the 4 h postexposure period. However, because the temporal profile of seizure behavior varied significantly across animals with equivalent SSmax, seizure severity was also assessed as the average of the seizure scores over the 4 h post-DFP (SSAVR).
In vivo Magnetic Resonance Imaging and Analysis
MRI scans were performed at the Center for Molecular and Genomic Imaging using a Bruker Biospec 70/30 (7T) preclinical MR scanner (Bruker BioSpin MRI, Ettlingen Germany) equipped with a 116 mm internal diameter B-GA12S gradient (450 mT/m, 4500 T/m/s), a 72 mm internal diameter linear transmit coil, and a four-channel, rat-brain phased array in cross-coil configuration for signal reception. Images were acquired and reconstructed, and parametric maps were generated, using Paravision 5.1 (Bruker BioSpin, MRI). Animals were imaged at one or two time points, ranging from 3 to 28 days postexposure, with a subset of animals additionally imaged 1–7 days prior to injection for baseline comparisons (Figure 1). Immediately prior to imaging, animals were anesthetized with isoflurane/O2 (Piramal Healthcare, Bethlehem PA) using 2.0%–3.0% vol/vol to induce and 1.0%–2.0% vol/vol to maintain anesthesia. Once anesthetized, animals were stereotactically restrained, and imaged for up to 2 h. During this time, body temperature was maintained at 37 °C using warm air, and anesthesia was adjusted to maintain a respiration rate of 50–70 breaths per min. Both temperature and respiration rate were monitored using SAII (Small Animal Instruments, Inc, Stony Brook, New York) small-animal monitoring equipment. Multislice, T2-weighted, Rapid Acquisition with Repeated Echoes (RARE) transaxial images were collected over 10 min using the following parameters: repetition time (TR) = 6100 ms; effective echo time (TE) = 60 ms; RARE factor = 8; averages = 4; field of view (FOV) = 35 × 25 mm2, with an in-plane data matrix of 280 × 200, resulting in a data set resolution of 0.125 × 0.125 mm2; 44 slices with a 0.5 mm thickness spanning approximately 7.5 to −13.5 bregma. Diffusion tensor imaging (DTI) data were collected using a multislice, multishot, 2D gradient echo planar imaging sequence with the following parameters: TR = 5500 ms, TE = 30 ms, FOV = 35 × 25 mm2 (280 × 200 data matrix), 22 slices with a 0.5 mm thickness, 30 diffusion weighted directions with b = 1600 s/mm2, 8 segments, 4 averages, and a total acquisition time of 103 min.
Image segmentation and extraction of voxel-wise apparent diffusion coefficient (ADC) data were performed in PMOD v3.5 (PMOD Technologies, Zurich, CHE). Delineations of bilateral brain region volumes of interest (VOIs) were guided by Paxinos and Watson’s The Rat Brain in Stereotaxic Coordinates (Paxinos and Watson, 2007) and manually traced on sequential A0 (no diffusion weighting) images generated from the DTI scan. The axial extent of brain-region VOIs, listed as “distance from bregma”/ “extent in mm”, were: hippocampus, −2.5/3.5; piriform cortex, 2.0/5.0; thalamus, −1.5/2.5. ADC histogram metrics (mean, 10th percentile, 90th percentile, standard deviation, skewness, kurtosis) were calculated from extracted ADC voxel data in Graph Pad Prism v5.01 (GraphPad Software Inc, La Jolla, California). Lateral ventricular volumes were calculated in a multistep process in AMIRA v6.0 (FEI, Hillsboro, Oregon). First, preliminary VOIs were manually traced on ADC parametric maps around lateral ventricles including approximately 1.0 mm of surrounding tissue as a buffer. Final VOIs for analysis were generated by thresholding of preliminary VOI’s to ADC values of 1.1 µm2/ms based on visual inspection of the first 10 animals and correction of any artifacts. Hippocampal and ventricular volumes were calculated by multiplication of voxel counts across segmented slices by the voxel volume.
Histology
Brains were harvested at 3, 7, 14, 21, or 28 days post exposure (DPE) from animals deeply anesthetized with 4% isoflurane in oxygen and subsequently perfused transcardially with cold PBS followed by cold 4% paraformaldehyde. After postfixation in 4% paraformaldehyde for a minimum of one week, brains were serially trimmed and placed into four to six paraffin-embedded blocks. Five-micron-thick sections were mounted on positive charged adhesion slides, air-dried overnight at 37 °C and subsequently deparaffinized and rehydrated. A subset of sections from each brain was stained with hematoxylin-eosin (H&E). Adjacent sections were immunostained for NeuN (A60, MAB#377, 1:400, Chemicon International, Temecula, California), GFAP (Z0334, 1:600, Dako North America, Carpinteria, California), and IBA1 (19-19741, 1:600, Wako, Richmond, Virginia) to assess neuronal cell loss, astrogliosis and microglial activation, respectively. Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in methanol, and sections subsequently rehydrated through serial dilution of the alcohol with water. Antigen retrieval procedures included either a step of heat-induced epitope retrieval using Dako Target Retrieval Solution (S1699) or, where appropriate, an enzymatic digestion with Dako Proteinase K (S3020), following the manufacturer’s instructions. The antibody and blocking solutions, and all rinses, were PBS-Tween 20 (0.02%). Sections were blocked in 10% normal horse serum for 20 min. All antigen: antibody complexes were visualized via a 30 min incubation with Dako Envision+ System-horseradish peroxidase (HRP)-conjugated secondary antibodies (mouse K4001 or rabbit K4003) followed by reaction with Vector NovaRed for peroxidase (SK-4800). Sections were counterstained with Mayer’s hematoxylin. Negative controls were incubated with diluent supplemented with normal serum, rather than primary antibody. Standardized, positive control tissue was included for each antibody.
Neuropathology was scored by a veterinary neuropathologist blinded to treatment using a scale ranging from 0 to 3, according to the following criteria: (a) acidophilic neuronal necrosis (NN) in the prosencephalon: 0, absent; 1, focal and a few neurons; 2, multifocal and more than one layer of neurons; 3, segmental and more than one layer of neurons; (b) IBA1 immunoreactivity in the prosencephalon: 0, absent; 1, focal; 2, multifocal; 3, segmental; (c) GFAP immunoreactivity in the prosencephalon: 0, absent; 1, focal; 2, multifocal; 3, segmental; (d) Mineralization in thalamic nuclei: 0, absent; 1, multifocal to diffuse; 2, diffuse with neuronal ferrugination (dystrophic mineralization in association with dead neurons); 3, diffuse with ferrugination and spongy change.
Statistics
All statistical analyses were performed in SAS (version 9.4, SAS Institute, Inc, Cary North Carolina) and GraphPad Prism (version 5.01, GraphPad Software, Inc, San Diego, California). Results with P values <0.05 were considered significant. Analysis of variance was used to compare mean ventricular and hippocampal volumes on the day of euthanasia among groups of animals defined by the combination of treatment (DFP-intoxicated vs VEH) and scheduled day of euthanasia, followed by posthoc tests to identify specific differences using Tukey’s studentized range test (HSD) to adjust for multiple comparisons. Ventricular volumes were transformed using the natural log prior to analysis to meet assumptions of the model. Multiple images over time, per animal, were included in linear mixed-effects regression models with DPE as a continuous predictor and a random effect for animal to evaluate rates of ventricular expansion. Differences in MRI metrics derived from histogram analysis obtained from the scan taken immediately prior to euthanasia were determined between DFP-exposed animals (28/28) at different days postexposure and vehicle animals (7/7). These metrics were evaluated across regions using linear, mixed-effects regression models, including region as a within-animal repeated measure and a random effect for animal. The MRI metrics exhibiting the most consistent differences between DFP-exposed and vehicle animals were correlated to histopathological scoring, seizure severity, and time post-DFP-exposure using Spearman correlations. Correlations utilized all DFP-exposed animals (28/28) and vehicle animals (7/7). MRI metrics and histopathological findings from vehicle control animals were uniform across the 28-days study and, therefore, vehicle data were pooled during analysis.
RESULTS
Seizures and Survival following Acute DFP Intoxication
Acute intoxication with DFP (4 mg/kg, sc) produced severe seizure behavior, defined as a seizure severity score (SS) of ≥ 3, within minutes in 96% (46/48) of animals injected with DFP (Figure 1B). The majority (approx. 70%) of DFP animals that exhibited seizure behavior survived until euthanized between 3 and 28 days postexposure (DPE). Of the surviving animals, 28 were used for imaging studies. Beginning at 14 DPE, four DFP-intoxicated animals displayed spontaneous recurrent seizures during handling, characterized by forelimb clonus, rearing and falling, and loss of the righting reflex.
Acute and Chronic Neuropathology Detected by MRI in DFP-Intoxicated Animals
Acute DFP intoxication produced striking bilateral lesions visualized in T2-weighted (T2w) and diffusion-weighted (DW) images (Figure 2). Lesions at 3 and 7 DPE were found within the hippocampus, amygdala, olfactory and piriform cortex, dorsolateral and medial thalamus, and cerebral cortex in both T2w and DW images. No lesions were observed in the cerebellar or hypothalamic brain regions. Lesion morphology ranged from distinct, hyperintense striations to regions of diffuse enhancement. However, lesions were confined to the anatomical borders of limbic brain regions. At 3 DPE, distinct bands of hyperintensity were observed in the hippocampus, with a discrete band of hyperintensity along the ventral hippocampal midline, consistent with the lancunosum moleculare layer, and a more diffuse band of hyperintensity in the septal region of the CA1. Hyperintensity in the cerebral cortex varied amongst animals, and, when present, was localized to the dorsal and lateral outer-tissue layers and was faint in comparison to that observed in limbic structures. Overall, T2w enhancement diminished and became more diffuse from 3 to 21 DPE, and was replaced by small, discrete areas of hypointensity. By 28 DPE, regions of T2w hyperintensity had resolved and been replaced by hypointensity along the septal hippocampal midline (Figure 2), and perivascular regions of the dorsolateral and medial thalamus.
FIG. 2.

Acute DFP intoxication-induced neuropathology, detected by anatomic MRI. The temporal progression of acute and chronic lesions induced by acute DFP intoxication is depicted in representative transaxial, T2-weighted and diffusion-weighted images acquired at baseline, 3, and 7 days post-DFP from the same animal and at 28 days post-DFP from a second animal with comparable seizure severity. Across time points, images were selected to match bregma to ≤ 250 µm. Red arrows indicate hippocampal lesions; green arrows indicate ventriculomegaly; blue arrows indicate the appearance and resolution of acute thalamic lesions.
The ADC for water within tissue, a standard quantitative metric derived from diffusion-weighted imaging (DWI) experiments, is sensitive to tissue microstructure. Increased ADC reflects reductions in the biological barriers impeding the movement of water, and is associated with increased extracellular water (vasogenic edema) or decreased cellular and stromal density. In contrast, decreased ADC, or restricted diffusion, is associated with increased intracellular water (cytotoxic edema) or increased cellular density. Parametric maps of ADC generated from DW images of DFP animals at early time points (3 and 7 DPE) postexposure indicated regions of either decreased or increased diffusion values that overlapped spatially with regions of T2w hyperintensity. Thalamic lesions were predominantly diffusion restricted (reduced ADC), specifically within the ventral reuniens, ventromedial, mediodorsal, and dorsal lateral thalamic regions. By comparison, lesions displayed a striated pattern of elevated and restricted diffusion localized along the three layers of the piriform cortex, and the CA and DG regions of the hippocampus, suggesting differential effects of DFP intoxication depending on the brain region and neuronal sublayer (Figures 3 and 4). The cerebral cortex appeared to be minimally affected. At later time points (14 through 28 DPE), lesions apparent on ADC maps decreased in size but maintained intralesional heterogeneity as characterized by areas of increased diffusion comingled with areas of decreased diffusion. These lesions coincided with the development of substantial ventriculomegaly (Figures 2 and 4A). Persistent lesions were localized within areas of damage observed in three DPE images, and most often included pockets of restricted diffusion and T2w hypointensity in the dorsolateral and ventromedial thalamus, and bands of elevated diffusion alongside restricted diffusion paralleling the midline of the hippocampus (Figures 3A and 4A). MRI-detected lesions in the piriform cortex were diminished or absent from 21 DPE and beyond, and, when apparent, were diffusion restricted. By comparison, no lesions were visualized in the cerebral cortex after seven DPE, and no new lesions arose in previously unaffected brain areas.
FIG. 3.

Acute DFP intoxication results in delayed ventriculomegaly and hippocampal atrophy. A, Representative, parametric maps of ADC at the level of the hippocampus (top) and caudate putamen (bottom) displaying ventricular enlargement (green arrows). B, Trajectory plots illustrating the effect of day postexposure and initial seizure severity on ventricular volume. Data points connected by line segments indicate repeated scans within a single animal. Vehicle (VEH) controls (green triangles) show no change in ventricular volume over time (slope = 0.07; P = 0.26). DFP animals with average seizure severity (SS) < 3 (black boxes) or ≥ 3 (red inverted triangles) exhibit significant growth of ventricular volume compared with VEH animals (P < 0.015); however, DFP animals with SS ≥ 3 experience growth at a faster rate than their lower seizure severity peers (P = 0.03). C, Hippocampal volume is significantly reduced following DFP exposure (P < 0.05 after Tukey HSD), although this effect is not apparent until three weeks post exposure. *P < 0.05; **P < 0.01; ns = no significant difference.
FIG. 4.

Quantification of brain damage using apparent diffusion coefficient (ADC). A, Representative, parametric maps of ADC at the level of the thalamus, piriform cortex, and hippocampus at baseline and at varying days postexposure (DPE). Colored outlines indicate slices through volumes of interest (VOIs), delineated along anatomic structures, whose pixel data contributed to the frequency distributions illustrated below. B, Frequency distributions of pixel-wise ADC values extracted from representative VOIs of the thalamus (left), piriform cortex (middle), and hippocampus (right) at baseline and 3, 7, and 28 DPE demonstrate both pronounced enhancement and restriction of diffusion within affected regions, resulting in a general broadening of the ADC distribution following acute DFP intoxication.
Ventriculomegaly was variably apparent in DFP-exposed animals at three DPE, increased rapidly thereafter, and was characterized by substantial enlargement of the lateral, third, and cerebral aqueduct components of the ventricular system (Figure 3). Ventricular volumes were significantly larger in DFP-intoxicated animals at late time points (pooled scans at 21 and 28 DPE) compared with early time points (pooled scans at 3 and 7 DPE), and both pooled groups had larger ventricular volumes than vehicle controls (P < 0.05 after Tukey HSD test). Although there were no significant differences in hippocampal volumes between vehicle controls and DFP animals at 7 DPE, by 21 and 28 DPE, hippocampal volumes were significantly reduced in DFP-exposed animals relative to vehicle controls (P < 0.05 after Tukey HSD, Figure 3C).
Quantification of DFP-Induced Brain Damage Using ADC Histogram Characteristics
Methodologies for capturing lesion heterogeneity, including voxel-based analyses and statistical mapping (Liachenko et al., 2015) were considered; however, because of significant changes in brain morphology following DFP intoxication (tissue atrophy and ventriculomegaly), automated brain registration and/or segmentation was not attempted. Therefore, higher-order histogram parameters were extracted from frequency distributions of regional ADC values (Just, 2014; Kang et al., 2011). Distributions of ADC values within the hippocampus, piriform cortex, and thalamus were dramatically altered following acute DFP intoxication (Figure 4). Six parameters characterizing ADC distributions were evaluated as potential quantitative metrics of DFP-induced neuropathology: mean, 10th percentile, 90th percentile, standard deviation, kurtosis, and skewness. The spatiotemporal changes in these ADC metrics associated with acute DFP intoxication are summarized in Table 1. Several parameters were able to distinguish DFP-exposed animals from vehicle controls; however, the 90th percentile (90thADC) and standard deviation (SDADC) of the ADC were the most consistent in differentiating DFP-exposed vs vehicle animals across brain regions and times postexposure (Table 1). Briefly, SDADC increased significantly across brain regions at 3 and 7 DPE, but was variably altered in the hippocampus and piriform cortex at later time points. Conversely, the 90thADC was elevated in DFP exposed animals at 14, 21, and 28 DPE, but was unchanged in the thalamus at earlier time points. Mean and 10thADC values, when significantly different than vehicle, were decreased at three DPE but increased at later time points (Table 1). The kurtosis and skewness of ADC distributions showed the least differences between DFP-exposed and vehicle animals (data not shown). Although these two metrics were significantly altered in DFP animals under some circumstances, no consistent trends were observed across regions and time points. In all brain regions, there was a significant, although weak, correlation between mean ADC and DPE (piriform cortex, rs = 0.54, 95% CI = 0.20–0.76; hippocampus, rs = 0.57, 95% CI = 0.23–0.77; thalamus, rs = 0.50, 95% CI = 0.15–0.73; P < 0.01). Overall, in the hippocampus and piriform cortex, metrics that reflected the observed increase in ADC over time (mean and 90thADC) showed more consistent differences between DFP-exposed vs vehicle control animals at 14 through 28 DPE, whereas the standard deviation of ADC distributions, a measure of regional heterogeneity, was most robust at distinguishing DFP animals from vehicle controls at earlier time points (3 and 7 DPE).
TABLE 1.
Spatiotemporal Effects of DFP Intoxication on ADC Distribution Characteristics
| Brain Region | DPE | Mean | 10th percentile | 90th percentile | SD |
|---|---|---|---|---|---|
| 3 | NS | NS | ↑*** | ↑*** | |
| 7 | ↑* | NS | ↑** | ↑* | |
| Hippocampus | 14 | ↑** | NS | ↑*** | ↑* |
| 21 | ↑*** | ↑*** | ↑** | NS | |
| 28 | ↑*** | ↑*** | ↑*** | ↑*** | |
| 3 | NS | ↓* | ↑** | ↑*** | |
| 7 | NS | NS | ↑* | ↑* | |
| Piriform Cortex | 14 | ↑** | ↑* | ↑*** | NS |
| 21 | ↑** | ↑* | ↑** | ↑** | |
| 28 | ↑* | NS | ↑*** | NS | |
| 3 | ↓*** | ↓*** | NS | ↑*** | |
| 7 | NS | NS | NS | ↑** | |
| Thalamus | 14 | NS | NS | ↑*** | ↑* |
| 21 | ↑*** | ↑** | ↑*** | ↑*** | |
| 28 | NS | NS | ↑*** | ↑*** |
Abbreviations: NS, not significant; ADC, apparent diffusion coefficient; SD, Standard Deviation.
Statistical significance was calculated using a repeated measures regression model comparing DFP-intoxicated animals to vehicle controls: ↑, DFP group > vehicle group; ↓, DFP group < vehicle group.
P < 0.05
P < 0.01
P < 0.001
ADC Histogram Characteristics Correlate With Neuronal Necrosis and Neuroinflammation
To validate and further characterize the MRI data, subsets of animals at each time point were euthanized to collect brain tissue for correlative histopathological assessment. A detailed description of this histopathological assessment is provided in a companion paper (Siso et al., 2016, Tox Sci, this issue), and neuronal necrosis scores of individual animals as a function of brain region and DPE are available in supplementary material (Supplementary Table 1). Lesions detected by T2w and DW MR imaging corresponded spatially to regions of neuronal necrosis and inflammation as identified by histology (Figure 5). Furthermore, ADC heterogeneity within brain lesions mapped to different neuropathological events: regions of restricted diffusion colocalized with regions of intense IBA1 immunoreactivity (indicative of microglial/macrophage infiltration), and regions of enhanced diffusion colocalized with areas of increased GFAP immunoreactivity (indicative of astrogliosis) and pan neuronal necrosis on H&E stained sections (Figure 5). At later time points, hypointense thalamic lesions on T2w images that display restricted diffusion corresponded to areas of profound tissue mineralization as determined in histological analyses.
FIG. 5.

Spatial registration of lesions detected by T2 weighted images (T2w) and parametric maps of the apparent diffusion coefficient (ADC) with areas of severe neuronal necrosis, microgliosis, and astrocytic activation, as assessed by H&E staining, IBA1 immunoreactivity, and GFAP immunoreactivity, respectively. Arrows denote lesions in the dorsolateral thalamus (top), ventromedial thalamus (middle) and piriform cortex (bottom). Micrograph magnifications: hemisphere, ×0.8; piriform cortex inset, ×2.
Regional neuronal necrosis, microgliosis, and astrogliosis were scored by a veterinary neuropathologist blinded to treatment, using a scale of 0–3, with a score of 0 referring to no pathology and a score of 3 corresponding to severe pathology. Histopathology scores positively correlated with mean ADC, SDADC, and 90thADC within the same brains at the same time points (Table 2). SDADC was significantly correlated with neuronal necrosis and both inflammatory metrics (IBA1 and GFAP) across all brain regions. The 90thADC was significantly correlated with neuropathological endpoints in the hippocampus and piriform cortex, but not in the thalamus. Neuronal necrosis was best predicted by SDADC followed by the 90thADC. In regions other than the thalamus, the 90thADC and SDADC varied, though not significantly, in their correlations to neuroinflammatory metrics, with the 90thADC being marginally superior in its correlation to GFAP. In contrast, mean ADC values correlated with astrogliosis in the piriform cortex but were weakly correlated with histopathological assessment in the hippocampus, and did not correlate with neuropathology in the thalamus.
TABLE 2.
Spearman Correlation of ADC Distribution Characteristics and Histopathological Assessment
| ADC Mean | ADC 90th percentile |
ADC SD |
|||||
|---|---|---|---|---|---|---|---|
| rs (95%CI) | P value | rs (95%CI) | P value | rs (95%CI) | P value | ||
| Hippocampus | NN | 0.4 (0.07–0.64) | * | 0.68 (0.44–0.82) | *** | 0.80 (0.62–0.89) | *** |
| IBA1 | 0.45 (0.13–0.68) | ** | 0.71 (0.48–0.84) | *** | 0.71 (0.48–0.84) | *** | |
| GFAP | 0.61 (0.34–0.78) | *** | 0.81 (0.65–0.90) | *** | 0.68 (0.43–0.82) | *** | |
| Total | 0.48 (0.15–0.70) | ** | 0.74 (0.52–0.86) | *** | 0.74 (0.53–0.86) | *** | |
| PiriformCortex | NN | 0.22 (−0.13 to 0.51) | NS | 0.62 (0.35–0.78) | *** | 0.65 (0.39–0.80) | *** |
| IBA1 | 0.21 (−0.14 to 0.51) | NS | 0.54 (0.25–0.74) | *** | 0.56 (0.27–0.75) | *** | |
| GFAP | 0.5 (0.18–0.71) | ** | 0.67 (0.42–0.82) | *** | 0.56 (0.26–0.75) | *** | |
| Total | 0.38 (0.03–0.63) | * | 0.70 (0.46–0.84) | *** | 0.59 (0.31–0.77) | *** | |
| Thalamus | NN | −0.11 (−0.43 to 0.23) | NS | 0.25 (−0.1 to 0.53) | NS | 0.77 (0.58–0.88) | *** |
| IBA1 | −0.11 (−0.43 to 0.24) | NS | 0.21 (−0.14–0.51) | NS | 0.72 (0.50–0.85) | *** | |
| GFAP | 0.06 (−0.28 to 0.39) | NS | 0.32 (−0.02 to -0.59) | NS | 0.66 (0.40–0.81) | *** | |
| Total | −0.1 (−0.43-0.25) | NS | 0.18 (−0.17 to 0.49) | NS | 0.71 (0.48–0.84) | *** | |
Abbreviations: ADC, apparent diffusion coefficient; DPE, day post exposure; SD, Standard Deviation; NN, neuronal necrosis assessed by H&E; IBA1, microgliosis assessed by ionized calcium-binding adapter molecule 1 immunoreactivity; GFAP, astrogliosis assessed by glial fibrillary acidic protein immunoreactivity; Total, sum of NN, IBA1, and GFAP scores; rs, Spearman correlation coefficient; 95%CI, 95 percent confidence interval; NS, not significant.
Confidence interval
P < 0.05
P < 0.01
P < 0.0001
Initial Seizure Behavior Predicts Lesion Severity and Brain Atrophy
Initial analyses (Fig 3B) suggested that seizure severity influences the extent of neuropathology. Specifically, DFP-exposed animals that displayed the most severe seizure behavior (SSAVR > 3.0) had a significantly higher rate of ventricular expansion than those with milder seizure behavior (SSAVR < 3.0, P = 0.03) who, in turn, had a higher rate of expansion than the vehicle controls (P = 0.01). Animals whose seizure scores were below the threshold associated with SE (SSMAX < 3.0) during the first 4 h after DFP administration lacked detectable abnormalities on T2w images, and had histopathological assessments (Siso et al., 2016, Tox Sci, this issue) similar to vehicle controls (Figure 6). Furthermore, animals that reached a SSMAX ≥ 3.0, suggesting induction of status epilepticus, but whose SSAVR was relatively lower than their peers, had less severe lesions and brain abnormalities when assessed by MRI and histopathology (Figure 6B). Average seizure severity amongst DFP-exposed animals over the first 4 h post DFP administration (SSAVR) was positively correlated with the 90thADC and SDADC in the hippocampus (SDADC, rs = 0.67, 95% CI = 0.42–0.82; 90thADC, rs = 0.55, 95% CI = 0.26–0.75; P < 0.001 for both), thalamus (SDADC, rs = 0.74, 95% CI = 0.54–0.86, P < 0.001; 90thADC, rs = 0.13, 95% CI = −0.22 to 0.44 NS), and piriform cortex (SDADC, rs = 0.55, 95%CI = 0.26–0.74, P < 0.001; 90thADC, rs = 0.40, 95% CI = 0.07–0.64, P = 0.02).
FIG. 6.
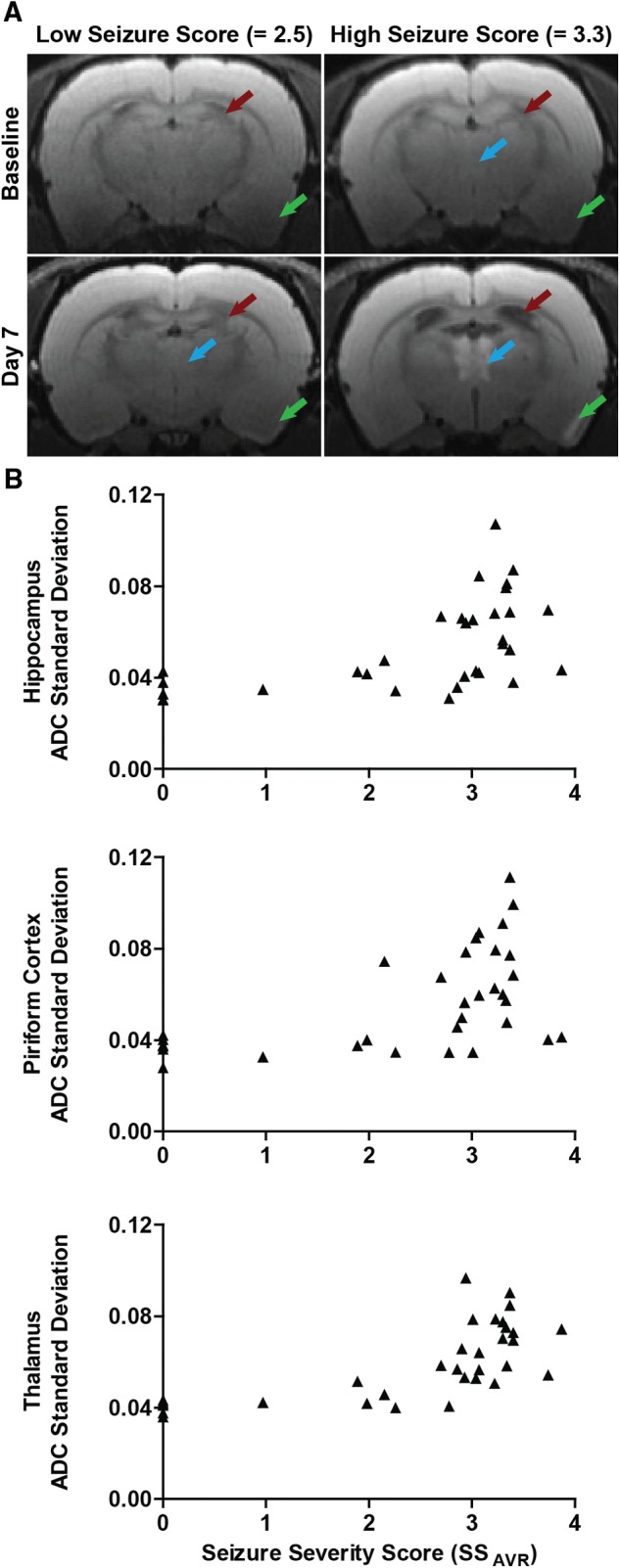
Brain damage assessed by MRI is positively correlated with seizure severity. A, Diffusion-weighted MR images of DFP-intoxicated animals with differing average seizure severity scores (SSAVR). Purple arrows identify hyperintensity in the thalamus; green arrows, hyperintensity in the piriform cortex; red arrows, expansion of the lateral ventricles. The lack of significant brain lesions in the animal with the low seizure score was confirmed by histopathological examination. B, Seizure severity, defined as the average seizure score over the first 4 h post exposure, is significantly correlated with tissue injury in the hippocampus (rs = 0.67; P < 0.001), piriform cortex (rs = 0.55; P < 0.001) and thalamus (rs = 0.74; P < 0.001), as assessed by the standard deviation of the apparent diffusion coefficient (ADC).
DISCUSSION
In the present study, in vivo MRI of rats acutely intoxicated with DFP revealed prominent lesions, tissue atrophy, and ventricular enlargement that varied in intensity and/or extent over time, with the overall magnitude of injury positively correlated to seizure severity. Importantly, lesions detected by MRI correlated spatially and temporally with histological evidence of neuropathology (Siso et al., 2016, Tox Sci, this issue).
Changes in T2 values have been suggested as a robust biomarker of early injury following OP poisoning (Bhagat et al., 2005; Gullapalli et al., 2010). We observed prominent T2 hyperintense lesions in DFP-intoxicated animals at 3 and 7 DPE in brain regions exhibiting histological evidence of significant neuronal necrosis and neuroinflammation. However, in stark contrast to histological analyses indicating sustained neuropathology for up to one month postexposure (Flannery et al., 2016), T2w hyperintensity markedly decreased in extent and intensity with increasing time postexposure. This observation suggests that T2w hyperintensity per se may not adequately reflect progressive chronic neuropathology. However, it is possible that more quantitative metrics of T2 imaging, including lesion volume measures and T2 value maps, would better correlate with neuropathology at these later time points (Liachenko et al., 2015). Alternatively, T2w hypointensity may be useful as a marker of chronic damage because both the hippocampus and thalamus of DFP animals exhibited T2w hypointensity at later time points corresponding to areas of mineralization.
Diffusion-weighted imaging of DFP-intoxicated animals revealed lesions characterized by heterogeneous diffusion. Specifically, we observed areas of increased diffusion comingled with areas of decreased diffusion within the same lesion. At early time points, ADC heterogeneity colocalized with T2w hyperintensity, suggesting that the heterogeneity was the result of vasogenic and cytotoxic edema, which have opposing effects on diffusion, occurring simultaneously in the same brain region. Beyond the first week postexposure, when T2w hyperintensity was significantly decreased, ADC heterogeneity likely reflected multiple types of pathological response within the same brain region, which often localized to specific neuronal cell layers, with areas of healthy tissue immediately adjacent to areas of damaged tissue. This marked intralesional heterogeneity observed in DW images and parametric ADC maps within a given brain region likely explains why mean ADC values within regions of interest did not consistently correspond to robust lesions observed in T2w and DW images.
Evaluation of higher-order histogram parameters (Just, 2014; Kang et al., 2011) revealed SDADC and 90thADC as more robust metrics for identifying significant DFP-induced pathology by MRI, particularly at time points when the mean ADC values indicated no difference between DFP animals and vehicle controls. These metrics also consistently correlated with histological assessments of neuronal necrosis and neuroinflammation in the hippocampus and piriform cortex. The effectiveness of SDADC and 90thADC as markers of DFP-induced pathology likely stems from the fact that, as metrics of heterogeneity, they are better suited for characterizing the heterogeneity of DFP-induced pathology. The particular effectiveness of the 90thADC at the late time points is consistent with histological analyses, indicating predominantly astrogliosis and reduced neuronal cell density, which are associated with increased ADC. Unevenly distributed increases in ADC would shift voxels in the frequency distribution to the right. These findings are consistent with the histological analyses of DFP-induced neuropathology, suggesting that the 90thADC and mean ADC may be useful in tracking persistent tissue remodeling in these brain regions following acute DFP intoxication. Notably, the SDADC was the only metric that correlated with neuropathology in the thalamus. Likely, this is because the thalamus exhibited not only gliosis and reduced neuronal cell density at late time points, but also extensive and marked mineralization, thereby producing opposing effects on tissue diffusion. Overall, these metrics indicate that lesions detected early after DFP intoxication do not resolve after one week but rather continue to evolve for at least one month postexposure, as evidenced by changing ADC heterogeneity, increasing mean ADC values and a shift from T2w hyperintensity to T2w hypointensity with time postexposure.
Previous T2-weighted MRI studies of acute OP intoxication, which did not extend beyond the first week postexposure, have described brain lesions with similar regional distribution, but there is a lack of consensus regarding the nature and temporal progression of these lesions. Studies of soman in guinea pigs (Gullapalli et al., 2010) or paraoxon in rats (Rosman et al., 2012; Shrot et al., 2012; Shrot et al., 2015) similarly reported T2w hyperintensity and/or increased T2 values relative to vehicle controls early; however, by 7 DPE, these lesions persisted in the soman model (Gullapalli et al., 2010), but resolved in the paraoxon model (Rosman et al., 2012; Shrot et al., 2012; Shrot et al., 2015). In contrast, in soman studies in the rat (Bhagat et al., 2001; Bhagat et al., 2005), T2 values decreased across brain regions during the first 24 h, and then normalized by one week. In our rat DFP model, we observed T2w hyperintensity during the first week postexposure, consistent with studies of the guinea pig model acute soman intoxication (Gullapalli et al., 2010). However, in contrast to the soman study, T2w hyperintensity in the DFP model diminished substantially over time, which more closely resembles findings in the paraoxon model (Shrot et al., 2012; Shrot et al., 2015). Diffusion-weighted MRI studies of soman (Bhagat et al., 2001; Bhagat et al., 2005; Carpentier et al., 2008; Testylier et al., 2007) or paraoxon (Rosman et al., 2012) reported reduced ADC in cholinergic brain regions at 24 h postexposure. Only two studies looked beyond 24 h, and these reported decreased mean ADC values at 7 DPE (Bhagat et al., 2001; Bhagat et al., 2005). In contrast, in our DFP model we observed significantly increased mean ADC values at 7 DPE. The reasons for the apparent discrepancies in MR metrics across models are unknown, but likely include the use of different OPs and/or species. Discrepancies may also result from differences in seizure severity and/or duration, but since this outcome is often not reported, it is difficult to assess its contribution to interstudy variability.
A major question is the role of seizure activity in mediating the neurological sequelae of acute OP intoxication. Several lines of evidence support a predominant role for seizure activity. First, our results demonstrated a strong correlation between MRI metrics of brain injury and initial seizure, and a similar relationship was reported in a guinea pig model of soman exposure (Gullapalli et al., 2010). These findings are consistent with previous correlations of seizure severity and histological assessments in experimental models of acute intoxication with OP nerve agents (McDonough and Shih, 1997). Second, the types of damage documented in our DFP model (edema, ventricular enlargement, hippocampal atrophy, thalamic mineralization) are similar to those documented in clinical epilepsy (Mendes and Sampaio, 2016; Milligan et al., 2009) and preclinical models of seizures triggered by mechanistically diverse chemicals such as kainic acid (Gayoso et al., 2003; Wolf et al., 2002) and pilocarpine (Fabene and Sbarbati, 2004; Lafreniere et al., 1992). However, evidence of differences in the lesions detected by T2w and DW imaging across models of acute OP intoxication suggest chemical influences independent of seizure activity. This is consistent with previous evidence suggesting that despite having the same primary mechanism of action (AChE inhibition), OPs elicit unique toxicity profiles, likely via non-cholinergic mechanisms (Pope, 1999).
Interestingly, the temporal progression of ADC changes reported in preclinical models of acute OP intoxication is strikingly similar to that observed in preclinical and clinical stroke. As in acute OP intoxication (Bhagat et al., 2001), diffusion in ischemic-reperfusion injury is acutely restricted and associated with neuronal cell death (Farr and Wegener, 2010). Over subsequent weeks, ADC increases above baseline within infarct lesions, and is characterized by a significant inflammatory response, as is the case following acute OP intoxication (Kanekar et al., 2012; van der Zijden et al., 2008). Furthermore, the thalamic lesions detected by MRI and corresponding mineralization observed in our DFP-intoxication animals are also reported in clinical (Ansari et al., 1990) and preclinical (Makinen et al., 2008; Watanabe et al., 1998) studies of hypoxic-ischemic injury. These similarities suggest that the MRI data reflect similar pathogenic mechanisms, such as persistent calcium dysregulation (Deshpande et al., 2010; Makinen et al., 2008) and neuroinflammation (de Araujo Furtado et al., 2012; Flannery et al., 2016; Liu and Chopp, 2016; Weinstein et al., 2010) that may contribute to secondary injury in both models.
This study provides the first description of MRI data in the rat model of acute DFP intoxication, and establishes that brain injury quantified by in vivo MRI correlates both spatially and temporally with brain damage detected by histology. Imaging measures that reflect tissue heterogeneity, specifically, the standard deviation and 90th percentile of the ADC, are robust metrics for quantifying persistent and progressive neuropathological changes. The severity of brain lesions assessed by MRI was dependent upon the intensity of seizure behavior during the first 4 h after administration of DFP. Interanimal, and interregional variation in lesion severity and progression, coupled with potential reinjury following spontaneous recurrent seizures, underscore the advantages of using in vivo imaging to longitudinally monitor neuropathology and therapeutic response following acute OP intoxication. Further studies are needed to determine whether the metrics identified herein are effective at predicting the severity of clinically relevant behavioral deficits observed following acute OP intoxication.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Jennifer Fung and Charles Smith (UC Davis Center for Molecular and Genomic Imaging) for their assistance with animal care and imaging, Dr Simon Cherry (UC Davis) for advice on experimental design, and both Dr Heike Wulff (UC Davis) and Dr Daniel Tancredi (UC Davis) for providing helpful feedback on early drafts of the manuscript.
FUNDING
CounterACT Program, National Institutes of Health Office of the Director and the National Institute of Neurological Disorders and Stroke [U54 NS079202 to P.J.L.], the National Institute of General Medical Sciences [T32 GM099608 to B.A.H.], the David and Dana Loury Foundation (predoctoral fellowship to B.A.H.] and the ARCS Foundation (predoctoral fellowship to B.A.H.]. The sponsors were not involved in the study design, the collection, analysis and interpretation of data, in the writing of the paper or in the decision to submit the work for publication.
REFERENCES
- Ansari M. Q., Chincanchan C. A., Armstrong D. L. (1990). Brain calcification in hypoxic-ischemic lesions: an autopsy review. Pediatr. Neurol. 6, 94–101. [DOI] [PubMed] [Google Scholar]
- Bhagat Y. A., Obenaus A., Hamilton M. G., Kendall E. J. (2001). Magnetic resonance imaging predicts neuropathology from soman-mediated seizures in the rodent. Neuroreport 12, 1481–1487. [DOI] [PubMed] [Google Scholar]
- Bhagat Y. A., Obenaus A., Hamilton M. G., Mikler J., Kendall E. J. (2005). Neuroprotection from soman-induced seizures in the rodent: evaluation with diffusion- and T2-weighted magnetic resonance imaging. Neurotoxicology 26, 1001–1013. [DOI] [PubMed] [Google Scholar]
- Billington M., Kandalaft O. R., Aisiku I. P. (2016). Adult status epilepticus: a review of the prehospital and emergency department management. J. Clin. Med. 5, pii: E74. doi: 10.3390/jcm5090074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer K. L., Troendle M. M., Pekman L., Meggs W. J. (2013). Naltrexone prevents delayed encephalopathy in rats poisoned with the sarin analogue diisopropylflurophosphate. Am. J. Emerg. Med. 31, 676–679. [DOI] [PubMed] [Google Scholar]
- Carpentier P., Testylier G., Dorandeu F., Segebarth C., Montigon O., Foquin A., Lahrech H. (2008). Hyperosmolar treatment of soman-induced brain lesions in mice: evaluation of the effects through diffusion-weighted magnetic resonance imaging and through histology. Toxicology 253, 97–103. [DOI] [PubMed] [Google Scholar]
- Chen Y. (2012). Organophosphate-induced brain damage: mechanisms, neuropsychiatric and neurological consequences, and potential therapeutic strategies. Neurotoxicology 33, 391–400. [DOI] [PubMed] [Google Scholar]
- Collombet J. M. (2011). Nerve agent intoxication: recent neuropathophysiological findings and subsequent impact on medical management prospects. Toxicol. Appl. Pharmacol. 255, 229–241. [DOI] [PubMed] [Google Scholar]
- Damodaran T. V., Abou-Donia M. B. (2000). Alterations in levels of mRNAs coding for glial fibrillary acidic protein (GFAP) and vimentin genes in the central nervous system of hens treated with diisopropyl phosphorofluoridate (DFP). Neurochem. Res. 25, 809–816. [DOI] [PubMed] [Google Scholar]
- de Araujo Furtado M., Rossetti F., Chanda S., Yourick D. (2012). Exposure to nerve agents: from status epilepticus to neuroinflammation, brain damage, neurogenesis and epilepsy. Neurotoxicology 33, 1476–1490. [DOI] [PubMed] [Google Scholar]
- Deshpande L. S., Carter D. S., Blair R. E., DeLorenzo R. J. (2010). Development of a prolonged calcium plateau in hippocampal neurons in rats surviving status epilepticus induced by the organophosphate diisopropylfluorophosphate. Toxicol. Sci. 116, 623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleston M., Phillips M. R. (2004). Self poisoning with pesticides. BMJ 328, 42–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabene P. F., Sbarbati A. (2004). In vivo MRI in different models of experimental epilepsy. Curr. Drug Targets 5, 629–636. [DOI] [PubMed] [Google Scholar]
- Farr T. D., Wegener S. (2010). Use of magnetic resonance imaging to predict outcome after stroke: a review of experimental and clinical evidence. J. Cereb. Blood Flow Metab. 30, 703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery B. M., Bruun D. A., Rowland D. J., Banks C. N., Austin A. T., Kukis D. L., Li Y., Ford B. D., Tancredi D. J., Silverman J. L., et al. (2016). Persistent neuroinflammation and cognitive impairment in a rat model of acute diisopropylfluorophosphate intoxication. J. Neuroinflammation 13, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayoso M. J., Al-Majdalawi A., Garrosa M., Calvo B., Diaz-Flores L. (2003). Selective calcification of rat brain lesions caused by systemic administration of kainic acid. Histol. Histopathol. 18, 855–869. [DOI] [PubMed] [Google Scholar]
- Gomes W. A., Shinnar S. (2011). Prospects for imaging-related biomarkers of human epileptogenesis: a critical review. Biomark. Med. 5, 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullapalli R. P., Aracava Y., Zhuo J., Helal Neto E., Wang J., Makris G., Merchenthaler I., Pereira E. F., Albuquerque E. X. (2010). Magnetic resonance imaging reveals that galantamine prevents structural brain damage induced by an acute exposure of guinea pigs to soman. Neurotoxicology 31, 67–76. [DOI] [PubMed] [Google Scholar]
- Gunnell D., Eddleston M., Phillips M. R., Konradsen F. (2007). The global distribution of fatal pesticide self-poisoning: systematic review. BMC Public Health 7, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jett D. A. (2007). Neurological aspects of chemical terrorism. Ann. Neurol. 61, 9–13. [DOI] [PubMed] [Google Scholar]
- Just N. (2014). Improving tumour heterogeneity MRI assessment with histograms. Br. J. Cancer 111, 2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadar T., Cohen G., Sahar R., Alkalai D., Shapira S. (1992). Long-term study of brain lesions following soman, in comparison to DFP and metrazol poisoning. Hum. Exp. Toxicol. 11, 517–523. [DOI] [PubMed] [Google Scholar]
- Kadriu B., Guidotti A., Costa E., Auta J. (2009). Imidazenil, a non-sedating anticonvulsant benzodiazepine, is more potent than diazepam in protecting against DFP-induced seizures and neuronal damage. Toxicology 256, 164–174. [DOI] [PubMed] [Google Scholar]
- Kanekar S. G., Zacharia T., Roller R. (2012). Imaging of stroke—Part 2: pathophysiology at the molecular and cellular levels and corresponding imaging changes. AJR Am. J. Roentgenol. 198, 63–74. [DOI] [PubMed] [Google Scholar]
- Kang Y., Choi S. H., Kim Y. J., Kim K. G., Sohn C. H., Kim J. H., Yun T. J., Chang K. H. (2011). Gliomas: histogram analysis of apparent diffusion coefficient maps with standard- or high-b-value diffusion-weighted MR imaging-correlation with tumor grade. Radiology 261, 882–890. [DOI] [PubMed] [Google Scholar]
- Kim Y. B., Hur G. H., Shin S., Sok D. E., Kang J. K., Lee Y. S. (1999). Organophosphate-induced brain injuries: delayed apoptosis mediated by nitric oxide. Environ. Toxicol. Pharmacol. 7, 147–152. [DOI] [PubMed] [Google Scholar]
- Lafreniere G. F., Peredery O., Persinger M. A. (1992). Progressive accumulation of large aggregates of calcium-containing polysaccharides and basophilic debris within specific thalamic nuclei after lithium/pilocarpine-induced seizures. Brain Res. Bull. 28, 825–830. [DOI] [PubMed] [Google Scholar]
- Li Y., Lein P. J., Liu C., Bruun D. A., Tewolde T., Ford G., Ford B. D. (2011). Spatiotemporal pattern of neuronal injury induced by DFP in rats: a model for delayed neuronal cell death following acute OP intoxication. Toxicol. Appl. Pharmacol. 253, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachenko S., Ramu J., Konak T., Paule M. G., Hanig J. (2015). Quantitative Assessment of MRI T2 response to kainic acid neurotoxicity in rats in vivo. Toxicol. Sci. 146, 183–191. [DOI] [PubMed] [Google Scholar]
- Liu C., Li Y., Lein P. J., Ford B. D. (2012). Spatiotemporal patterns of GFAP upregulation in rat brain following acute intoxication with diisopropylfluorophosphate (DFP). Curr. Neurobiol. 3, 90–97. [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Chopp M. (2016). Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol. 144, 103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinen S., van Groen T., Clarke J., Thornell A., Corbett D., Hiltunen M., Soininen H., Jolkkonen J. (2008). Coaccumulation of calcium and beta-amyloid in the thalamus after transient middle cerebral artery occlusion in rats. J. Cereb. Blood Flow Metab. 28, 263–268. [DOI] [PubMed] [Google Scholar]
- McDonough J. H. Jr, Shih T. M. (1997). Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci. Biobehav. Rev. 21, 559–579. [DOI] [PubMed] [Google Scholar]
- Mendes A., Sampaio L. (2016). Brain magnetic resonance in status epilepticus: a focused review. Seizure 38, 63–67. [DOI] [PubMed] [Google Scholar]
- Milligan T. A., Zamani A., Bromfield E. (2009). Frequency and patterns of MRI abnormalities due to status epilepticus. Seizure 18, 104–108. [DOI] [PubMed] [Google Scholar]
- Paxinos G., Watson C. (2007). The Rat Brain in Stereotaxic Coordinates, 6th ed Academic Press/Elsevier, Amsterdam; Boston. [Google Scholar]
- Pereira E. F., Aracava Y., DeTolla L. J. Jr, Beecham E. J., Basinger G. W. Jr, Wakayama E. J., Albuquerque E. X. (2014). Animal models that best reproduce the clinical manifestations of human intoxication with organophosphorus compounds. J. Pharmacol. Exp. Ther. 350, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope C. N. (1999). Organophosphorus pesticides: do they all have the same mechanism of toxicity?. J. Toxicol. Environ. Health B Crit. Rev. 2, 161–181. [DOI] [PubMed] [Google Scholar]
- Rojas A., Ganesh T., Lelutiu N., Gueorguieva P., Dingledine R. (2015). Inhibition of the prostaglandin EP2 receptor is neuroprotective and accelerates functional recovery in a rat model of organophosphorus induced status epilepticus. Neuropharmacology 93, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A., Ganesh T., Manji Z., O'Neill T., Dingledine R. (2016). Inhibition of the prostaglandin E2 receptor EP2 prevents status epilepticus-induced deficits in the novel object recognition task in rats. Neuropharmacology 110, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosman Y., Eisenkraft A., Krivoy A., Schein O., Makarovski I., Shrot S., Ramaty E., Shilderman E. B., Kapon J., Gilat E., et al. (2012). Using MRI for the assessment of paraoxon-induced brain damage and efficacy of antidotal treatment. J. Appl. Toxicol. 32, 409–416. [DOI] [PubMed] [Google Scholar]
- Shrot S., Anaby D., Krivoy A., Makarovsky I., Rosman Y., Bloch-Shilderman E., Lazar S., Bar-Shir A., Cohen Y. (2012). Early in vivo MR spectroscopy findings in organophosphate-induced brain damage-potential biomarkers for short-term survival. Magn. Reson. Med. 68, 1390–1398. [DOI] [PubMed] [Google Scholar]
- Shrot S., Tauber M., Shiyovich A., Milk N., Rosman Y., Eisenkraft A., Kadar T., Kassirer M., Cohen Y. (2015). Early brain magnetic resonance imaging can predict short and long-term outcomes after organophosphate poisoning in a rat model. Neurotoxicology 48, 206–216. [DOI] [PubMed] [Google Scholar]
- Testylier G., Lahrech H., Montigon O., Foquin A., Delacour C., Bernabe D., Segebarth C., Dorandeu F., Carpentier P. (2007). Cerebral edema induced in mice by a convulsive dose of soman. Evaluation through diffusion-weighted magnetic resonance imaging and histology. Toxicol. Appl. Pharmacol. 220, 125–137. [DOI] [PubMed] [Google Scholar]
- van der Zijden J. P., van der Toorn A., van der Marel K., Dijkhuizen R. M. (2008). Longitudinal in vivo MRI of alterations in perilesional tissue after transient ischemic stroke in rats. Exp. Neurol. 212, 207–212. [DOI] [PubMed] [Google Scholar]
- Watanabe H., Kumon Y., Ohta S., Sakaki S., Matsuda S., Sakanaka M. (1998). Changes in protein synthesis and calcium homeostasis in the thalamus of spontaneously hypertensive rats with focal cerebral ischemia. J. Cereb. Blood Flow Metab. 18, 686–696. [DOI] [PubMed] [Google Scholar]
- Weinstein J. R., Koerner I. P., Moller T. (2010). Microglia in ischemic brain injury. Future Neurol. 5, 227–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf O. T., Dyakin V., Vadasz C., de Leon M. J., McEwen B. S., Bulloch K. (2002). Volumetric measurement of the hippocampus, the anterior cingulate cortex, and the retrosplenial granular cortex of the rat using structural MRI. Brain Res. Brain Res. Protoc. 10, 41–46. [DOI] [PubMed] [Google Scholar]
- Wright L. K., Liu J., Nallapaneni A., Pope C. N. (2010). Behavioral sequelae following acute diisopropylfluorophosphate intoxication in rats: comparative effects of atropine and cannabinomimetics. Neurotoxicol. Teratol. 32, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasue H., Abe O., Kasai K., Suga M., Iwanami A., Yamada H., Tochigi M., Ohtani T., Rogers M. A., Sasaki T., et al. (2007). Human brain structural change related to acute single exposure to sarin. Ann. Neurol. 61, 37–46. [DOI] [PubMed] [Google Scholar]
- Yanagisawa N., Morita H., Nakajima T. (2006). Sarin experiences in Japan: acute toxicity and long-term effects. J. Neurol. Sci. 249, 76–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.