

Abstract
Activation of the hepatocyte growth factor receptor Met induces a morphogenic response and stimulates the formation of branching tubules by Madin-Darby canine kidney (MDCK) epithelial cells in three-dimensional cultures. A constitutively activated ErbB2/Neu receptor, NeuNT, promotes a similar invasive morphogenic program in MDCK cells. Because both receptors are expressed in breast epithelia, are associated with poor prognosis, and hepatocyte growth factor (HGF) is expressed in stroma, we examined the consequence of cooperation between these signals. We show that HGF disrupts NeuNT-induced epithelial morphogenesis, stimulating the breakdown of cell-cell junctions, dispersal, and invasion of single cells. This correlates with a decrease in junctional proteins claudin-1 and E-cadherin, in addition to the internalization of the tight junction protein ZO-1. HGF-induced invasion of NT-expressing cells is abrogated by pretreatment with a pharmacological inhibitor of the mitogen-activated protein kinase kinase (MEK) pathway, which restores E-cadherin and ZO-1 at cell-cell junctions, establishing the involvement of MEK-dependent pathways in this process. These results demonstrate that physiological signals downstream from the HGF/Met receptor synergize with ErbB2/Neu to enhance the malignant phenotype, promoting the breakdown of cell-cell junctions and enhanced cell invasion. This is particularly important for cancers where ErbB2/Neu is overexpressed and HGF is a physiological growth factor found in the stroma.
INTRODUCTION
Normal epithelia are highly organized tissues composed of a single sheet of polarized cells connected by cellular junctions and attached to the extracellular matrix (ECM) via heteromeric protein complexes (Balda and Matter, 2003). This highly organized architecture suggests that dynamic and reciprocal interactions take place between epithelial cells and the surrounding ECM (Somasiri and Roskelley, 1999; Bissell et al., 2002). Tight regulation of these interactions dictates cell behavior and contributes to tissue homeostasis. Specific cellular processes during embryogenesis, such as neural crest cell migration, require a transient phenotypic modulation where epithelial cells lose their cell-cell junctions and acquire a motile mesenchymal phenotype (Savagner, 2001). This process, called epithelial-mesenchymal transition (EMT), can be initiated by different growth factors (hepatocyte growth factor [HGF], epidermal growth factor [EGF], transforming growth factor-β [TGFβ]) or extracellular matrix components, such as collagen. EMT also occurs in tumor dissemination and metastasis, where the phenotypic changes are more potent (Thiery, 2002).
The development of three-dimensional cell culture models that mimic the complexity of epithelial architecture, and the use of animal models, have allowed the identification of important modifiers of epithelial integrity, although for many, their mechanisms of action remain poorly understood (Schmeichel and Bissell, 2003; Kuperwasser et al., 2004; Wrobel et al., 2004). A role for the HGF/scatter factor (SF) receptor Met and HER2/ErbB2/Neu, a member of the EGFR family, in epithelial remodeling has been proposed from genetic ablation studies (Lee et al., 1995; Schmidt et al., 1995; Uehara et al., 1995; Ebens et al., 1996; Andrechek et al., 2002; Chan et al., 2002; Leu et al., 2003). Whole organ cultures and cell culture models suggest that Met and ErbB2/Neu are associated with the morphogenic and functional differentiation of normal breast epithelium (Yang et al., 1995; Niemann et al., 1998; Jones and Stern, 1999). Using a nontransformed kidney epithelial cell model (MDCK), we have previously shown that ligand-induced activation of Met and constitutive activation of ErbB2/Neu each promote the inherent morphogenic program of MDCK cells, that is the conversion of cysts of polarized epithelial cells into branching tubules in three-dimensional collagen gels (Maroun et al., 1999; Khoury et al., 2001).
HER2/ErbB2 and Met are independent prognostic markers for breast cancer (Yamashita et al., 1994; Ghoussoub et al., 1998). High levels of Met in human breast carcinomas and high levels of HGF in tumor stroma have been reported previously (Yamashita et al., 1994; Tuck et al., 1996; Kang et al., 2003). The amplification and overexpression of HER2/ErbB2 is associated with increased progression and metastasis in 25% of human breast carcinomas and is indicative of poor prognosis in breast, ovarian, and renal collecting duct carcinomas (Selli et al., 1997; Zhang et al., 1997; Pegram et al., 1998). Consistent with this, a constitutively activated ErbB2/Neu receptor, NeuNT, has been shown to induce mammary tumors in transgenic animals (Guy et al., 1992, 1996; Dankort et al., 1997). Transactivation of ErbB2/Neu by heregulin enhances cell invasion in some breast tumor cell lines (Meiners et al., 1998; Brandt et al., 1999), whereas in other cell lines, it promotes a remodeling of epithelial cell junctions consistent with a morphogenic response (Chausovsky et al., 1998). Synergy between an ErbB2/Neu signal and TGFβ is associated with cell invasion in culture and animal-based models (Siegel et al., 2003; Seton-Rogers et al., 2004; Ueda et al., 2004). These data suggest that the biological consequence of activation of ErbB2/Neu depends on how cells integrate an ErbB2/Neu signal in the context of other signals, and these may differ among different cancer cells. For example, activation of ErbB2/Neu may predispose epithelial cells to malignant transformation, where additional factors might be required to trigger the progression of invasive disease. Using stable MDCK cell lines, we show that the Met/HGF receptor converts an epithelial morphogenic program induced by activated ErbB2/Neu to cell invasion.
MATERIALS AND METHODS
Antibodies and Reagents
Antibodies used in this article are as follows: mouse anti-Neu antibodies antibody-3 and antibody-4 (Oncogene Science, Cambridge, MA), mouse anti-E-cadherin and β-catenin (BD Transduction Laboratories, Mississauga, Ontario, Canada), rabbit anti-ZO-1, occludin, claudin-1, and claudin-2 (Zymed Laboratories, South San Francisco, CA), and rabbit anti-phospho-Erk (New England Biolabs, Nepean, Ontario, Canada). The following antibodies were generously donated: mouse anti-gp135 from Dr. G. Ojakian (SUNY Health Science Center, Brooklyn, NY), mouse anti-p120catenin from Dr. A. Reynolds (Vanderbilt University School of Medicine, Nashville, TN), and rabbit anti-Erk from Dr. J. Blenis (Harvard University, Boston, MA). Secondary antibodies used were Alexa Fluor 488 and Alexa Fluor 555 donkey anti-mouse, as well as Alexa Fluor 488 and 555 donkey anti-rabbit (Molecular Probes, Eugene, OR). HGF was a generous gift from Dr. G. Van deWoude (Van Andel Research Institute, Grand Rapids, MI). EGF and heregulin βl were purchased from Roche Diagnostics (Mannheim, Germany) and Neomarkers (Fremont, CA), respectively. Inhibitors for MEK (UO126), phosphoinositide 3-kinase (PI3K) (LY294002), Src (PP2), and RhoA (Exoenzyme C3) were purchased as follows: UO126 was from Promega (Fisher Scientific, Nepean, Ontario, Canada), LY294002 was from BIOMOL Research Laboratories (Plymouth Meeting, PA), PP2 was from Calbiochem (San Diego, CA), and Exoenzyme C3 from Upstate Biotechnology (Cedarlane, Hornby, Ontario, Canada).
Cell Culture and Scatter Assay
MDCK cells were maintained in DMEM containing 10% fetal bovine serum (FBS) and 50 μg/ml Gentamicin (Invitrogen, Carlsbad, CA). Neu mutants (Dankort et al., 1997) were transfected into MDCK cells by the calcium phosphate method, and stable cell lines were selected in G418 (400 μg/ml) (Geneticin; Invitrogen). Cells (5 × 103) were seeded in 24-well dishes (Nalge Nunc International, Naperville, IL) for cell scatter studies. After 48 h, HGF was added to the medium at 5 U/ml for 24 h.
Migration Assay
Cells (5 × 105/well) were plated on six-well Transwell filters (Costar, Cambridge, MA) in the presence or absence of HGF (5 U/ml). Twenty-four hours later, filters were submerged in formalin phosphate buffer (Fisher Scientific) for 15 min, washed twice with distilled water, and stained with crystal violet for 15 min at room temperature, followed by several washes with water. Nonmigrating cells were scraped off the upper layer of the filter by using a cotton swab. Filters were then air-dried and photographed. To quantitate cell migration, filters were cut, solubilized in 10% acetic acid, and absorbance readings were taken at 596 nm. The results plotted are average numbers of four experiments for each cell line.
Matrigel Invasion Assay
Transwell filters (24-well; Costar) were coated with Matrigel (50-100 μg/cm2; BD Biosciences, Mississauga, Ontario, Canada). Cells (8 × 104/well) were plated and processed as described above. Where indicated, cells were grown in the presence of HGF for 7-15 d before plating them on Matrigel. Alternatively, EGF (50 ng/ml) was added to the assay for 24 h, or cells were grown in the presence of EGF for 7 d. For quantitation, filters were photographed and invading cells were counted using the Northern Eclipse image processing system (Empix Imaging, Toronto, Ontario, Canada).
Tubulogenesis Assay
MDCK cells were suspended in a collagen matrix as described previously (Maroun et al., 1999). Briefly, 5 × 103 cells were resuspended in 500 μl of collagen solution (Vitrogen 100; Celtrix, Santa Clara, CA) prepared as specified by the manufacturer, and layered over 350 μl of the collagen solution in a 24-well plate. The cells were maintained in L-15 medium containing 5% FBS and allowed to form structures for 7 d. HGF (5 U/ml) and UO126 (5 and 20 μM) were then added to the medium either simultaneously or at a 2-d interval, as indicated. The medium was changed every 4 d. For the quantitation of the morphogenic response, 60 colonies in each of five independent cultures (wells) were scored for their ability to form branching tubules (structures whose length is 5 times their diameter), and the results were plotted as the average number of cysts able to undergo tubulogenesis per culture per 100.
Anoikis Assay
Anchorage-independent cell survival assay was performed as described previously (Frisch, 2000). Briefly, genomic DNA from normal (MDCK), wild-type Neu, and deregulated NeuNT-expressing cells maintained in suspension for 2 h was separated on a 1.5% agarose gel.
Immunoprecipitation and Western blotting
Subconfluent cells were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin and aprotinin, and 1 mM Na3VO4). Equal amounts of total protein (20-50 μg) were separated by electrophoresis on 8% SDS-polyacrylamide gels, transferred to a nitrocellulose membrane, and immunoblotted. Where indicated, cells were grown in the presence of HGF for 5 d. To quantitate Neu mutant expression levels, equal amounts of total protein (500 μg) were immunoprecipitated before gel separation.
Light Microscopy
Photographs of fixed collagen cultures and dispersed cells were taken using a digital image processing system (Northern Eclipse; Empix Imaging, Toronto, Ontario, Canada) at a magnification of 2.5× (bright field objective) and 10× (phase contrast).
Confocal Scanning Laser Microscopy
Collagen cultures were stained as described previously (Debnath et al., 2003) Briefly, slices from collagen cultures were fixed in 2% paraformaldehyde for 20 min at room temperature, rinsed three times with phosphate-buffered saline (PBS), and then permeabilized with PBS containing 0.2-0.5% Triton X-100 for 10 min. The slices were then washed three times in PBS/glycine (100 mM glycine) for 5 min each time at room temperature, and then incubated in blocking solution (PBS, 2% bovine serum albumin, 0.2% Triton X-100, and 0.05% Tween 20) for 30 min. Incubation with primary antibody was performed for 1 h (room temperature) to overnight (4°C), after which slices were washed three times (5 min per wash) and incubated with the secondary antibody for 40-50 min. Antibodies were diluted in blocking solution. Collagen slices were then washed twice with water, mounted in Immu-mount, and allowed to dry overnight at room temperature.
Live/dead staining was performed on live cultures by using calcein and ethidium homodimers from Molecular Probes (Eugene, OR) (Debnath et al., 2002). Images were photographed using Confocal LSM510 (Carl Zeiss, Thornwood, NY).
RESULTS
NeuNT Enhances Cell Migration
Stable expression of a constitutively activated ErbB2/Neu mutant receptor, NeuNT (NT-3, NT-6, and NT-9), but not a WT ErbB2/Neu receptor, Neu-1, induces a transient epithelial-mesenchymal (EM)-like transition in normal epithelial MDCK cells (Khoury et al., 2001). This transition is characterized by the loss of cell-cell junctions and epithelial cell dispersal as well as enhanced cell migration and is comparable with HGF-induced cell scatter (Figure 1B) (Khoury et al., 2001). To establish whether NeuNT-induced cell scatter is similar to that induced by HGF, we determined whether HGF synergizes with NeuNT to enhance cell migration by using a modified Boyden chamber. In the absence of HGF, MDCK cells overexpressing a WT Neu receptor (Neu-1) behave in a similar manner to vector-alone cells. In contrast, cells expressing an activated NeuNT (NT-6 and NT-3) show increased migration, where fivefold more cells have traversed the membrane 16 h postseeding (Figure 1, C and D). HGF treatment of NT-expressing cells enhances cell migration a further twofold, to a level that is comparable with, but not in excess of, HGF-treated control MDCK cells or cells expressing WT Neu (Figure 1, C and D). This demonstrates that although signals downstream from an activated ErbB2/Neu enhance cell migration, these signals are not as potent as HGF. Furthermore, signals downstream from NeuNT and HGF are not additive for cell migration.
Figure 1.
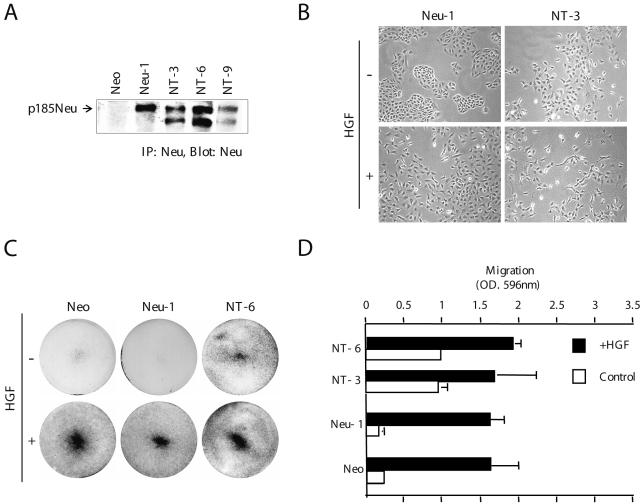
Additive effects of HGF and deregulated ErbB2/Neu enhance cell dispersal and motility. (A) Stable expression of ErbB2/Neu mutant receptors in MDCK cells. Neu protein was immunoprecipitated from RIPA cell lysates (500 μg of protein) with mouse antibody-4. Proteins were separated by SDS-PAGE (8%) and immunoblotted with antibody-3 (anti-Neu) antibody. (B) Stable MDCK cells expressing ErbB2/Neu mutants were grown in DMEM with 10% FBS in six-well plates, in the presence or absence of HGF. Representative clones were photographed using the Northern Eclipse image processing system (magnification, 10×). (C) Motility assays were performed using Transwell filters, as described in Materials and Methods. Representative photographs of crystal violet-stained filters were taken. (D) Filters were solubilized in 10% acetic acid, and the migration of Neu-expressing cells was determined by measuring the absorbance at a wavelength of 596 nm. Each value represents the average of four filters.
HGF and NeuNT Synergize to Promote Disruption of Organized Epithelia
We have shown previously that deregulated ErbB2/Neu (NT) induces an invasive morphogenic program in MDCK cells in a three-dimensional collagen matrix and promotes the formation of tubular structures (Khoury et al., 2001). This response is similar in morphology to that induced by HGF in MDCK cells. To establish whether HGF and NeuNT cooperate to enhance the morphogenic response in normal epithelia, we allowed cells expressing NeuNT to form branching tubules and stimulated these with HGF. HGF promotes a morphogenic response in cells expressing WT Neu, or a Neu mutant receptor with impaired ability to couple with signaling molecules (NYPD) (Figure 2A). In contrast, stimulation with HGF promotes the disruption of NeuNT-induced branching tubules into single cells, which invade the collagen matrix (Figure 2, A and B). To establish which NeuNT-dependent signals collaborate with HGF to promote the breakdown of organized epithelia-stable MDCK cell lines expressing Neu tyrosine add-back mutants that couple to selected signaling molecules (Dankort et al., 1997) were examined. Add-back mutants were generated using the Neu Tyrosine Phosphorylation-Deficient (NYPD) mutant by reverting individual phenylalanine substitutions to tyrosine residues (Dankort et al., 1997). The resulting mutants YB, YC, YD, and YE are tyrosine phosphorylated and catalytically active (Dankort et al., 1997). As shown previously (Khoury et al., 2001), the YD and YE add-back mutants stimulate epithelial remodeling of MDCK cells when seeded in collagen, whereas MDCK cells expressing the YA, YB, and YC receptor mutants form cysts in a similar manner to control MDCK cells (Figure 2C). To establish whether specific NeuNT add-back mutants cooperate with HGF for breakdown of organized epithelia, we allowed cells expressing each of the add-back mutants to form cysts (YA, YB, and YC) or morphogenic structures (YD and YE) for 7 d and stimulated these with HGF. HGF promotes a morphogenic response in cells expressing each of the add-back mutants (Figure 2C), demonstrating that single carboxy-terminal tyrosine residues are not sufficient for the breakdown of organized epithelial structures observed with cells expressing NeuNT.
Figure 2.

HGF promotes the disruption of organized epithelia in MDCK cells overexpressing deregulated ErbB2/Neu (NT). (A) Stable lines of MDCK cells expressing wild-type, mutant, and deregulated Neu receptors were grown in collagen I. Five days later, cultures were stimulated with HGF (5 U/ml) for 10 d, and then fixed and photographed. (B) Quantitation of the morphogenic response to HGF treatment in Neu-expressing MDCK cells. Data from representative clones are shown. Columns represent cysts (blank), tubules (solid), and various structures (striped). Results are plotted as the average number of structures per 100. Each value represents the mean average from at least four independent experiments. (C) Representative clones of stable MDCK cell lines expressing Neu add-back mutants (YA, YB, YC, YD, and YE) were grown in collagen I, stimulated with HGF (5U/ml) as in A, and then fixed and photographed. Tubules are structures whose length is 5 times their diameter. Structures represent mainly dispersed structures, but also include spikes, unbranched tubules, and protrusions.
In cells expressing NeuNT, the breakdown of organized epithelia and cell dispersal observed in response to HGF, occurs in a time-dependent manner, leading to the loss of organized epithelial structures within 10 d post-HGF treatment (Figure 3A), whereas HGF treatment of MDCK cells overexpressing WT Neu or any of the add-back mutants (YA, YB, YC, YD, and YE) promotes the formation of branching tubules over this time period, and no breakdown of epithelial structures is observed at later time periods (Figures 2C and 3A; our unpublished results). Moreover, heregulin and EGF, ligands for the EGFR family receptors, fail to induce the breakdown of organized epithelia observed with HGF in NeuNT-expressing cells, and the branched tubular structures are maintained (Figure 3B).
Figure 3.

HGF but not EGF promotes the disruption of organized epithelia in NeuNT-expressing MDCK cells in a time-dependent manner. (A) Cultures were fixed at different time points after HGF addition to follow the progression of epithelial disruption. Time points indicated refer to the time when HGF was added (i.e., day 4 corresponds to 4 days of HGF treatment) (magnification, 10×). (B) MDCK cells grown in collagen I were stimulated with EGF and heregulin β1 at 20 and 100 ng/ml, respectively (magnification, 10×).
When seeded in a matrix of collagen I, MDCK cells form three-dimensional cyst structures. These cysts consist of a ball of polarized cells surrounding a hollow lumen formed through apoptosis of centrally located cells (Fournier et al., 1996; Ojakian et al., 1997; O'Brien et al., 2002). Cell polarity and cell death can be monitored by immunofluorescence by using the apical marker gp135 and calcein/ethidium homodimer reagents that identify dead versus live cells (Debnath et al., 2002). Using this assay, we show that gp135 localizes to the luminal side in NeuNT-induced tubular structures, similar to the fully polarized MDCK cysts and HGF-induced tubules (Figure 4). In all conditions, the lumen is filled with apoptotic cells as revealed by ethidium homodimer red staining, surrounded by live cells (green staining) (Figure 4). The single cells that disperse from NeuNT-dependent tubules in response to HGF stain for calcein (green), demonstrating that they are alive and invading the collagen matrix (Figure 4). These data demonstrate that signals downstream from the Met receptor synergize with an activated NeuNT to promote the loss of epithelial integrity and cell invasion, whereas either receptor signal alone promotes a morphogenic program.
Figure 4.
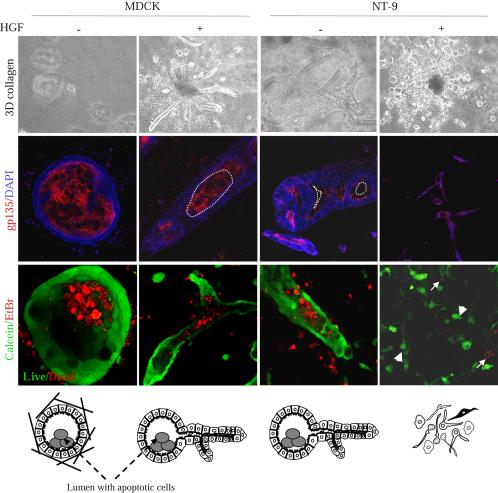
HGF and deregulated ErbB2/Neu synergize to promote collagen invasion. Live three-dimensional collagen cultures of MDCK cells and NeuNT-expressing cells were treated with HGF for 10 d and stained with calcein for live cells (green; arrowhead) and ethidium homodimers for dead cells (red; white arrow). LSM510 (magnification, 25×). Cells also were stained with the apical marker gp135 for luminal polarity, and the nuclei counterstained with 4,6-diamidino-2-phenylindole. Bottom, cartoon illustration of the different structures and single invading cells.
HGF and NeuNT Synergize to Enhance Cell Invasion
Malignant cells acquire the ability to invade the extracellular matrix and survive in the bloodstream in an anchorage-independent manner, allowing them to metastasize to distant organs. Using Matrigel-coated filters, we show that MDCK cells in the absence of HGF show no invasion over the time period assayed (16 h), whereas pretreatment of these cells with HGF (15 d) induces a modest invasive capacity (18 cells per average field; Figure 5, A and B). MDCK cells expressing NeuNT show increased levels of invasion (40 cells per field; Figure 5, A and B) compared with HGF-treated MDCK cells (18 cells per field; Figure 5, A and B). Notably, the most dramatic effect is observed after pretreatment of NeuNT-expressing MDCK cells with HGF, which increases their invasive potential by sixfold (250 cells per field; Figure 5, A and B) compared with untreated NeuNT cells. In contrast, EGF promotes invasion of parental MDCK cells when seeded as single cells. However, EGF pretreatment did not enhance the invasion of NeuNT-expressing cells (Figure 5C), demonstrating synergy between HGF and NeuNT for cell invasion but not between EGF and NeuNT.
Figure 5.
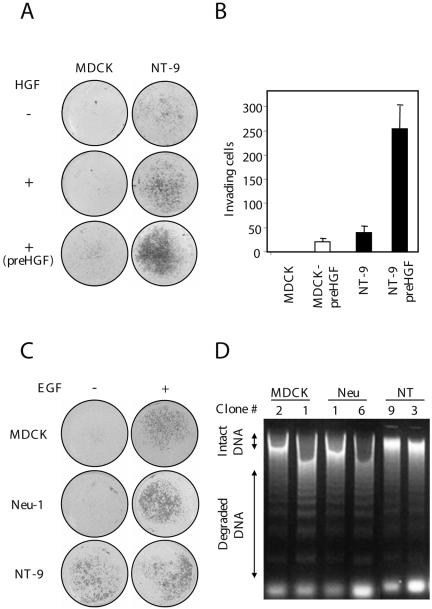
Synergy between HGF and deregulated ErbB2/Neu promotes Matrigel invasion. (A) Matrigel assay was performed in the presence or absence of HGF (5 U/ml), as described in Materials and Methods. In one case, NeuNT lines were maintained in HGF-containing medium for 15 d before seeding them on Matrigel (labeled as pre-HGF). Representative filters were scanned using a digital scanner. (B) Quantitation of the invasive response. Filters were photographed and cells counted using the Northern Eclipse image processing system. Values represent the average of three filters. (C) EGF promotes MDCK cell invasion. EGF (50 ng/ml) was added to parental MDCK cells or cells expressing WT or activated Neu in a Matrigel invasion assay for 24 h. Digital scans of representative filters are shown. (D) NT protects cells from anoikis. Stable MDCK cells expressing ErbB2/Neu receptor mutants were treated with different doses of HGF. Treated cells were then incubated at 37°C in suspension for 2 h and genomic DNA was extracted and separated on a 1.5% agarose gel.
HGF increases cell survival by protecting epithelial cells from anoikis or cell death induced by detachment from the extracellular matrix. Anoikis can be monitored by increased genomic DNA degradation (Frisch, 2000). To assess whether deregulated NeuNT protects MDCK cells from anoikis, we isolated genomic DNA from NeuNT-expressing cells grown in suspension and followed DNA degradation on agarose gels. We show that under these conditions, wild-type Neu fails to prevent DNA degradation, whereas expression of NeuNT prevents DNA degradation in a similar manner to HGF pretreatment (Figure 5D; our unpublished results). Hence, activated ErbB2/Neu confers a cell survival advantage to epithelial cells in an anchorage-independent manner.
HGF and NeuNT Synergize to Break Cell-Cell Junctions
The development of invasive cancers correlates with the loss of epithelial organization, referred to as EMT (Boyer et al., 1996; Thiery, 2002). This process involves, in addition to increased cell invasion and motility, the disruption of cell-cell junctions and cell dispersal. To assess the effect of HGF and NeuNT on the composition of cell-cell junctions, we have examined the levels of adherens (E-cadherin) and tight junction (claudin-1, occludin, and ZO-1) proteins in NeuNT-expressing cells grown in the presence or absence of HGF. E-cadherin and claudin-1 protein levels are consistently reduced in HGF-treated NeuNT cells, whereas levels of occludin or ZO-1 are not altered (Figure 6). This decrease occurs in a time-dependent manner, because prolonged treatment with HGF (5 d to 2 wk) enhances the reduction in E-cadherin and claudin-1 protein levels (Figure 6; our unpublished results). The decrease in E-cadherin and claudin-1 is specific to NeuNT-expressing cells, because HGF treatment of MDCK cells promotes no detectable change in the levels of E-cadherin or claudin-1. In addition, MDCK cells that have undergone an epithelial-mesenchymal transition after the expression of activated c-Src show a reduction in protein levels of occludin and ZO-1 but not E-cadherin or claudin-1 (Figure 6), indicating that these changes are not a general response to the breakdown of cell-cell junctions.
Figure 6.
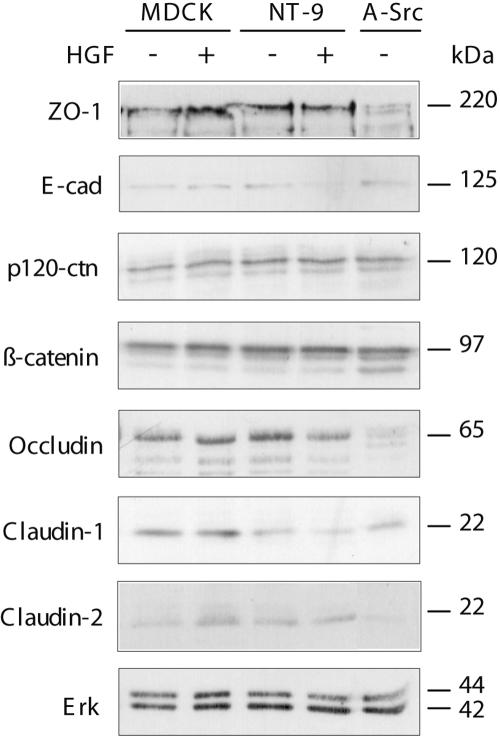
HGF and deregulated ErbB2/Neu synergize to down-modulate components of adherens and tight junctions. (A) Cells were maintained in the presence or absence of HGF for 5 d as indicated. RIPA cell lysates (25-50 μg) were separated on 8% polyacrylamide gels, transferred to nitrocellulose membranes, and probed for components of the adherens (E-cadherin, β-catenin, and p120 catenin) and tight junctions (ZO-1, occludin, claudin-1, and claudin-2) as indicated.
The down-regulation of E-cadherin at the transcriptional level has been observed after epithelial mesenchymal transitions (Thiery, 2002). No significant alterations of E-cadherin mRNA levels after long-term treatment with HGF were observed by quantitative real-time PCR analyses (our unpublished results), suggesting that posttranslational mechanisms might be involved in the observed reduction of E-cadherin protein levels. Consistent with this, immunofluorescence staining of NeuNT cells in three-dimensional collagen cultures reveals a reduction in E-cadherin protein levels, although E-cadherin is retained at cell-cell junctions. However, after stimulation with HGF, E-cadherin is redistributed from cell-cell junctions as observed in HGF-stimulated MDCK cells, to a cytosolic dot-like distribution in NeuNT-expressing cells (Figure 7). Similarly, indirect immunofluorescence shows that ZO-1 is redistributed from an apical junctional compartment localization in MDCK or NeuNT-expressing MDCK cells, to the cytosol in NeuNT-expressing cells in response to HGF (Figure 7). These data demonstrate that HGF and NT cooperate to destabilize junctional complexes in MDCK epithelial cells and that the expression of NeuNT alone is insufficient to promote the breakdown of cell-cell junctions in three-dimensional cultures.
Figure 7.
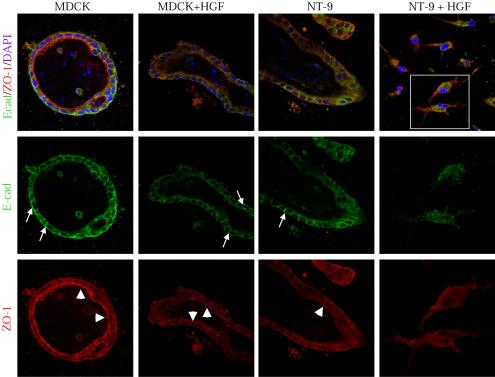
HGF induces E-cadherin internalization in MDCK cells expressing deregulated NeuNT. Collagen cultures of NT-expressing MDCK cells treated or not with HGF (5 U/ml) were labeled with anti E-cadherin (green) and anti ZO-1 (red). White arrows indicate basolateral distribution of E-cadherin. Arrowheads refer to the apical distribution of ZO-1. Confocal LSM510 (magnification 63×).
The MEK/MAPK Pathway Is Required for HGF-Induced Invasive Dispersal of NeuNT-expressing Cells
The breakdown of epithelial cell-cell junctions in response to HGF requires PI3K-, MEK-, Ras-, and Rho family-dependent signals (Royal and Park, 1995; Potempa and Ridley, 1998; Royal et al., 2000). Moreover, it has been suggested that c-Src activity is required for HGF-induced cell dispersal (Elliott et al., 2002). To examine the role of the MEK-Erk, PI3K, Rho, and Src pathways in HGF-induced disruption of tubular structures, NeuNT-expressing cells were treated in a dose-dependent manner with a MEK1 inhibitor (UO126), an inhibitor of PI3K (LY294002), A RhoA inhibitor (Exoenzyme C3), and a Src inhibitor (PP2). Pretreatment of NeuNT-expressing cells with low levels of UO126 (5 μM) before stimulation with HGF prevents the disruption of organized tubules, whereas pretreatment with LY294002, C3, or PP2 fails to prevent cell dispersal (Figure 8B; our unpublished results). After pretreatment with UO126, NeuNT branching tubules are retained in response to HGF, and E-cadherin and ZO-1 are maintained at cell-cell contacts as revealed by indirect immunofluorescence on three-dimensional structures (Figure 8C). However, when the NeuNT-expressing cells are pretreated with HGF before UO126 treatment, UO126 fails to block HGF-induced breakdown of tubular structures (Figure 8B). Consistent with a requirement for MEK-dependent pathways for a synergistic response, HGF stimulation of NeuNT-expressing MDCK cells induces further activation of extracellular signal-regulated kinase (Erk), which is downstream from MEK (Figure 8D). Erk phosphorylation is abolished upon pretreatment of the cells with UO126 before HGF stimulation. Together, these results demonstrate that MEK-dependent signals are required for the HGF-dependent breakdown of cell-cell junctions and cell invasion observed in NeuNT-expressing cells.
Figure 8.

MEK inhibitor UO126 prevents HGF-induced epithelial disruption in MDCK cells expressing deregulated ErbB2/Neu. Stable lines of MDCK cells were grown in collagen I. Five days later, HGF (5 U/ml) and UO126 (5 and 20 μM) were added either simultaneously as in A, or at a 2-d interval where pretreatment was required, as in B. Seven days later, cultures were fixed in 4% paraformaldehyde. Representative pictures were taken using bright field (2.5×) and phase contrast (10×) objectives. Live/dead staining was performed on cultures after treatment with UO126. A representative picture is shown as an insert. (C) Representative collagen cultures from B stained for E-cadherin and ZO-1 show restored tubular structures with E-cadherin and ZO-1 staining at cell-cell junctions. Confocal LSM510 (magnification, 63×). (D) HGF and NeuNT synergize to enhance Erk phosphorylation. MDCK cells expressing deregulated ErbB2/Neu (NT9) were serum starved for 18 h, and then stimulated with HGF (10 U/ml) for the indicated times. Cells were pretreated with the MEK inhibitor UO126 (10 μM) 1 h before HGF stimulation where indicated. Proteins from total cell lysate (50 μg) were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and blotted for phospho-Erk and total Erk.
DISCUSSION
HGF induces transient morphological changes in MDCK epithelial cells that can be reversed after HGF withdrawal (Weidner et al., 1993; Zhu et al., 1994; Khoury et al., 2001). Similarly, we have shown that a constitutively activated ErbB2/Neu receptor (NT) induces an EM-like transition in MDCK cells and promotes the inherent morphogenic program of these cells in a collagen matrix (Khoury et al., 2001). However, the same NeuNT receptor induces metastatic mammary tumors in transgenic mice with long latency (Muller et al., 1988), suggesting that other factors may play a role in the metastatic process.
We show in this article that signals downstream from the HGF/Met receptor convert a morphogenic signal, induced by an activated NeuNT receptor in MDCK cells, to an invasive response. This is specific to HGF and activated ErbB2/Neu. Neither EGF nor heregulin promote cell dispersal in MDCK cells, and HGF does not induce a similar phenotype in MDCK cells expressing the wild-type ErbB2/Neu receptor (Figures 2A and 3B). HGF fails to synergize with ErbB2/Neu for cell invasion in an immortalized mammary epithelial cell line (MCF10A), whereas TGFβ induces cell invasion in the same cell model (Seton-Rogers et al., 2004). This difference probably reflects differences between cell lines. For example, MDA435 breast cancer cells transfected with HGF form tumors with metastasis to the lung in the absence of E-cadherin, but on the reintroduction of E-cadherin HGF promotes the formation of three-dimensional epithelial structures (Meiners et al., 1998).
Loss of differentiation during cancer progression is accompanied by the breakdown of cell-cell junctions, a prerequisite for cell dissociation (Thiery, 2002). Several mechanisms lead to the breakdown of cell-cell junctions. For example, E-cadherin, a major component of adherens junctions, is down-regulated through transcriptional repression, genomic deletion and perhaps protein degradation, in many human cancers and in established cancer cell lines (Birchmeier and Behrens, 1994; Ji et al., 1997). In addition, components of the tight junctions are destabilized during EMT and in some cancer cells (Li and Mrsny, 2000; Liebner et al., 2000; Blanco et al., 2004; Ohkubo and Ozawa, 2004; Tobioka et al., 2004). Expression of claudins and occludin is repressed by the transcription factor Snail (Ikenouchi et al., 2003).
In three-dimensional cultures, NeuNT-expressing MDCK cells retain E-cadherin at adherens junctions and ZO-1 at tight junctions (Figure 7). However, exposure to HGF disrupts the three-dimensional epithelial structures of NeuNT-expressing cells, promoting their conversion into single invading cells (Figure 7). In these cells, E-cadherin and ZO-1 seem internalized, as revealed by the vesicle-like staining pattern (Figure 7). Although the scatter factor, HGF or expression of NeuNT induces the redistribution of E-cadherin from the cell membrane into a cytoplasmic-soluble compartment in two-dimensional culture (Royal and Park, 1995; Khoury et al., 2001), E-cadherin and ZO-1 are retained at cell-cell junctions in three-dimensional culture (Figure 7) (Pollack et al., 1998). Protein levels of E-cadherin and claudin-1, a component of tight junctions, are consistently reduced when NeuNT-expressing cells, but not MDCK cells, are maintained in HGF-containing medium (Figure 6). The reduction in E-cadherin, a key component of adherens junctions, and claudin-1, which is important for the recruitment of ZO-1 to tight junctions, would promote the destabilization of both adherens and tight junctions. The reduction in E-cadherin is not due to transcriptional repression as revealed by quantitative real-time PCR (our unpublished results), and is consistent with previous reports where protein internalization and subsequent degradation have been reported as a mechanism for the down-modulation of E-cadherin (Fujita et al., 2002). In this model, Src-mediated tyrosine phosphorylation leads to the association of E-cadherin with a ubiquitin ligase and subsequent endocytosis and degradation of the protein (Fujita et al., 2002). Importantly, activated c-Src-transformed MDCK cells do not show altered levels of E-cadherin (Figure 6), suggesting that other pathways downstream from HGF and deregulated ErbB2/Neu might be involved in the loss of E-cadherin.
The stable breakdown of cell-cell junctions, observed in NeuNT-expressing cells in response to HGF, is inhibited by a pharmacological inhibitor of MEK, UO126 (Figure 8). Moreover, pretreatment of NeuNT cells with UO126 prevents the internalization of E-cadherin and ZO-1 observed in untreated cells in response to HGF (Figure 8C). HGF synergizes with NeuNT to enhance Erk phosphorylation (Figure 8D), which is blocked by UO126. Consistent with this, sustained Erk activity is required for ErbB2-TGFβ cooperation for the induction of cell invasion in the MCF-10A mammary epithelial cell model (Seton-Rogers et al., 2004), and the inhibition of the MEK pathway restores tight junctions in HGF-stimulated and Ras-transformed MDCK cells (Potempa and Ridley, 1998; Maroun et al., 1999; Chen et al., 2000; Khoury et al., 2001). Furthermore, deregulated activation of the MAPK pathway alone is sufficient to induce permanent loss of epithelial structures in MDCK cells in collagen (Khwaja et al., 1998), although Neu add-back mutants capable of activating Erk (YB, YC, YD, and YE) (Dankort et al., 2001) are not sufficient, suggesting that a sustained threshold of Erk activation may be required. Erk activation is necessary to trigger the initial stage of HGF-induced tubulogenesis during which cell depolarization and remodeling occur without the loss of cell-cell contacts (Pollack et al., 1998; O'Brien et al., 2004). Consistent with our model, prolonged Erk activation prevented the formation of tubular structures in MDCK cells expressing activated Raf, an upstream activator of MEK/Erk; hence, synergistic Erk activation by HGF and deregulated ErbB2/Neu acts to promote the loss of cell-cell contacts, thus preventing cell redifferentiation. Sustained and prolonged Erk activation might be required to induce changes in gene expression resulting in the secretion of matrix degrading metalloproteases that promote cleavage of junctional proteins, hence destabilizing cell-cell junctions (McCawley et al., 1998; Liang and Chen, 2001; Tanimura et al., 2002).
In conclusion, we provide evidence that synergy between HGF/Met and ErbB2/Neu promotes progression of the malignant phenotype of polarized MDCK epithelial cells through the destabilization of cell-cell junctions. A similar synergy between TGFβ and ErbB2/Neu was demonstrated where TGFβ was shown to promote pulmonary metastasis in transgenic mouse models of ErbB2/Neu-induced mammary tumorigenesis (Siegel et al., 2003), as well as migration and invasion in mammary cell models in culture (Seton-Rogers et al., 2004; Ueda et al., 2004). Moreover, synergy between TGFβ and Ras or EGF has been reported to induce EMT in primary cultured cell models (Grande et al., 2002; Janda et al., 2002). HGF-induced cell dispersal correlates with metastatic behavior of cancer cells and highlights the importance of the cross-talk between normal and deregulated signals in the progression of invasive malignancies. This observation is particularly significant for breast cancer where overexpression of HER2/Neu and HGF/Met are independent markers for poor prognosis.
Acknowledgments
This research was supported by operating grants from the Canadian Breast Cancer Research Initiative with funds from the Canadian Cancer Society to M.P. and W.J.M. M.P. and W.J.M. are Scientists of the Canadian Institutes of Health Research, and W.J.M. is supported by the Canadian Research Council (CRC) Chair in Molecular Oncology. Financial support was provided by the Royal Victoria Hospital Research Institute and McGill's Faculty of Medicine Studentship program to H.K., the Canadian Institutes of Health Research M.D.-Ph.D. studentship program to S.P., and the Cancer Research Society postdoctoral fellowship to V.S.
Article published online ahead of print in MBC in Press on November 17, 2004 (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-07-0567).
References
- Andrechek, E. R., Hardy, W. R., Girgis-Gabardo, A. A., Perry, R. L., Butler, R., Graham, F. L., Kahn, R. C., Rudnicki, M. A., and Muller, W. J. (2002). ErbB2 is required for muscle spindle and myoblast cell survival. Mol. Cell. Biol. 22, 4714-4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balda, M. S., and Matter, K. (2003). Epithelial cell adhesion and the regulation of gene expression. Trends Cell Biol. 13, 310-318. [DOI] [PubMed] [Google Scholar]
- Birchmeier, W., and Behrens, J. (1994). Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim. Biophys. Acta 1198, 11-26. [DOI] [PubMed] [Google Scholar]
- Bissell, M. J., Radisky, D. C., Rizki, A., Weaver, V. M., and Petersen, O. W. (2002). The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation 70, 537-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco, D., Vicent, S., Elizegi, E., Pino, I., Fraga, M. F., Esteller, M., Saffiotti, U., Lecanda, F., and Montuenga, L. M. (2004). Altered expression of adhesion molecules and epithelial-mesenchymal transition in silica-induced rat lung carcinogenesis. Lab. Investig. 84, 999-1012. [DOI] [PubMed] [Google Scholar]
- Boyer, B., Valles, A. M., and Thiery, J. P. (1996). Model systems of epithelium-mesenchyme transitions. Acta Anat. 156, 227-239. [DOI] [PubMed] [Google Scholar]
- Brandt, B. H., et al. (1999). c-erbB-2/EGFR as dominant heterodimerization partners determine a motogenic phenotype in human breast cancer cells. FASEB J. 13, 1939-1949. [DOI] [PubMed] [Google Scholar]
- Chan, R., Hardy, W. R., Laing, M. A., Hardy, S. E., and Muller, W. J. (2002). The catalytic activity of the ErbB-2 receptor tyrosine kinase is essential for embryonic development. Mol. Cell. Biol. 22, 1073-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chausovsky, A., Tsarfaty, I., Kam, Z., Yarden, Y., Geiger, B., and Bershadsky, A. D. (1998). Morphogenetic effects of neuregulin (neu differentiation factor) in cultured epithelial cells. Mol. Biol. Cell 9, 3195-3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., Lu, Q., Schneeberger, E. E., and Goodenough, D. A. (2000). Restoration of tight junction structure and barrier function by down-regulation of the mitogen-activated protein kinase pathway in ras-transformed Madin-Darby canine kidney cells. Mol. Biol. Cell 11, 849-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort, D., Maslikowski, B., Warner, N., Kanno, N., Kim, H., Wang, Z., Moran, M. F., Oshima, R. G., Cardiff, R. D., and Muller, W. J. (2001). Grb2 and Shc adapter proteins play distinct roles in Neu (ErbB-2)-induced mammary tumorigenesis: implications for human breast cancer. Mol. Cell. Biol. 21, 1540-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort, D. L., Wang, Z., Blackmore, V., Moran, M. F., and Muller, W. J. (1997). Distinct tyrosine autophosphorylation sites negatively and positively modulate neu-mediated transformation. Mol. Cell. Biol. 17, 5410-5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath, J., Mills, K. R., Collins, N. L., Reginato, M. J., Muthuswamy, S. K., and Brugge, J. S. (2002). The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 111, 29-40. [DOI] [PubMed] [Google Scholar]
- Debnath, J., Muthuswamy, S. K., and Brugge, J. S. (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256-268. [DOI] [PubMed] [Google Scholar]
- Ebens, A., Brose, K., Leonardo, E. D., Hanson, M. G., Jr., Bladt, F., Birchmeier, C., Barres, B. A., and Tessier-Lavigne, M. (1996). Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron 17, 1157-1172. [DOI] [PubMed] [Google Scholar]
- Elliott, B. E., Hung, W. L., Boag, A. H., and Tuck, A. B. (2002). The role of hepatocyte growth factor (scatter factor) in epithelial-mesenchymal transition and breast cancer. Can. J. Physiol. Pharmacol. 80, 91-102. [DOI] [PubMed] [Google Scholar]
- Fournier, T. M., Kamikura, D., Teng, K., and Park, M. (1996). Branching tubulogenesis but not scatter of Madin-Darby canine kidney cells requires a functional Grb2 binding site in the Met receptor tyrosine kinase. J. Biol. Chem. 271, 22211-22217. [DOI] [PubMed] [Google Scholar]
- Frisch, S. M. (2000). Anoikis. Methods Enzymol. 322, 472-479. [DOI] [PubMed] [Google Scholar]
- Fujita, Y., Krause, G., Scheffner, M., Zechner, D., Leddy, H. E., Behrens, J., Sommer, T., and Birchmeier, W. (2002). Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 4, 222-231. [DOI] [PubMed] [Google Scholar]
- Ghoussoub, R. A., Dillon, D. A., D'Aquila, T., Rimm, E. B., Fearon, E. R., and Rimm, D. L. (1998). Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer 82, 1513-1520. [DOI] [PubMed] [Google Scholar]
- Grande, M., Franzen, A., Karlsson, J. O., Ericson, L. E., Heldin, N. E., and Nilsson, M. (2002). Transforming growth factor-beta and EGF synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J. Cell Sci. 115, 4227-4236. [DOI] [PubMed] [Google Scholar]
- Guy, C. T., Cardiff, R. D., and Muller, W. J. (1996). Activated neu induces rapid tumor progression. J. Biol. Chem. 271, 7673-7678. [DOI] [PubMed] [Google Scholar]
- Guy, C. T., Webster, M. A., Schaller, M., Parsons, T. J., Cardiff, R. D., and Muller, W. J. (1992). Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc. Natl. Acad. Sci. USA 89, 10578-10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenouchi, J., Matsuda, M., Furuse, M., and Tsukita, S. (2003). Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J. Cell Sci. 116, 1959-1967. [DOI] [PubMed] [Google Scholar]
- Janda, E., Lehmann, K., Killisch, I., Jechlinger, M., Herzig, M., Downward, J., Beug, H., and Grunert, S. (2002). Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J. Cell Biol. 156, 299-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, X., Woodard, A. S., Rimm, D. L., and Fearon, E. R. (1997). Transcriptional defects underlie loss of E-cadherin expression in breast cancer. Cell Growth Differ. 8, 773-778. [PubMed] [Google Scholar]
- Jones, F. E., and Stern, D. F. (1999). Expression of dominant-negative ErbB2 in the mammary gland of transgenic mice reveals a role in lobuloalveolar development and lactation. Oncogene 18, 3481-3490. [DOI] [PubMed] [Google Scholar]
- Kang, J. Y., Dolled-Filhart, M., Ocal, I. T., Singh, B., Lin, C. Y., Dickson, R. B., Rimm, D. L., and Camp, R. L. (2003). Tissue microarray analysis of hepatocyte growth factor/Met pathway components reveals a role for Met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast cancer. Cancer Res. 63, 1101-1105. [PubMed] [Google Scholar]
- Khoury, H., Dankort, D. L., Sadekova, S., Naujokas, M. A., Muller, W. J., and Park, M. (2001). Distinct tyrosine autophosphorylation sites mediate induction of epithelial mesenchymal like transition by an activated ErbB-2/Neu receptor. Oncogene 20, 788-799. [DOI] [PubMed] [Google Scholar]
- Khwaja, A., Lehmann, K., Marte, B. M., and Downward, J. (1998). Phosphoinositide 3-kinase induces scattering and tubulogenesis in epithelial cells through a novel pathway. J. Biol. Chem. 273, 18793-18801. [DOI] [PubMed] [Google Scholar]
- Kuperwasser, C., Chavarria, T., Wu, M., Magrane, G., Gray, J. W., Carey, L., Richardson, A., and Weinberg, R. A. (2004). Reconstruction of functionally normal and malignant human breast tissues in mice. Proc. Natl. Acad. Sci. USA 101, 4966-4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. F., Simon, H., Chen, H., Bates, B., Hung, M. C., and Hauser, C. (1995). Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 378, 394-398. [DOI] [PubMed] [Google Scholar]
- Leu, M., Bellmunt, E., Schwander, M., Farinas, I., Brenner, H. R., and Muller, U. (2003). Erbb2 regulates neuromuscular synapse formation and is essential for muscle spindle development. Development 130, 2291-2301. [DOI] [PubMed] [Google Scholar]
- Li, D., and Mrsny, R. J. (2000). Oncogenic Raf-1 disrupts epithelial tight junctions via downregulation of occludin. J. Cell Biol. 148, 791-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, C. C., and Chen, H. C. (2001). Sustained activation of extracellular signal-regulated kinase stimulated by hepatocyte growth factor leads to integrin alpha 2 expression that is involved in cell scattering. J. Biol. Chem. 276, 21146-21152. [DOI] [PubMed] [Google Scholar]
- Liebner, S., Fischmann, A., Rascher, G., Duffner, F., Grote, E. H., Kalbacher, H., and Wolburg, H. (2000). Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 100, 323-331. [DOI] [PubMed] [Google Scholar]
- Maroun, C. R., Holgado-Madruga, M., Royal, I., Naujokas, M. A., Fournier, T. M., Wong, A. J., and Park, M. (1999). The Gab1 PH domain is required for localization of Gab1 at sites of cell-cell contact and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol. Cell. Biol. 19, 1784-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCawley, L. J., O'Brien, P., and Hudson, L. G. (1998). EGF- and scatter factor/hepatocyte growth factor (SF/HGF)-mediated keratinocyte migration is coincident with induction of matrix metalloproteinase (MMP)-9. J. Cell Physiol. 176, 255-265. [DOI] [PubMed] [Google Scholar]
- Meiners, S., Brinkmann, V., Naundorf, H., and Birchmeier, W. (1998). Role of morphogenetic factors in metastasis of mammary carcinoma cells. Oncogene 16, 9-20. [DOI] [PubMed] [Google Scholar]
- Muller, W. J., Sinn, E., Pattengale, P. K., Wallace, R., and Leder, P. (1988). Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 54, 105-115. [DOI] [PubMed] [Google Scholar]
- Niemann, C., Brinkmann, V., Spitzer, E., Hartmann, G., Sachs, M., Naundorf, H., and Birchmeier, W. (1998). Reconstitution of mammary gland development in vitro: requirement of c-met and c-erbB2 signaling for branching and alveolar morphogenesis. J. Cell Biol. 143, 533-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien, L. E., Tang, K., Kats, E. S., Schutz-Geschwender, A., Lipschutz, J. H., and Mostov, K. E. (2004). ERK and MMPs sequentially regulate distinct stages of epithelial tubule development. Dev. Cell 7, 21-32. [DOI] [PubMed] [Google Scholar]
- O'Brien, L. E., Zegers, M. M., and Mostov, K. E. (2002). Opinion: building epithelial architecture: insights from three-dimensional culture models. Nat. Rev. Mol. Cell. Biol. 3, 531-537. [DOI] [PubMed] [Google Scholar]
- Ohkubo, T., and Ozawa, M. (2004). The transcription factor Snail downregulates the tight junction components independently of E-cadherin downregulation. J. Cell Sci. 117, 1675-1685. [DOI] [PubMed] [Google Scholar]
- Ojakian, G. K., Nelson, W. J., and Beck, K. A. (1997). Mechanisms for de novo biogenesis of an apical membrane compartment in groups of simple epithelial cells surrounded by extracellular matrix. J. Cell Sci. 110, 2781-2794. [DOI] [PubMed] [Google Scholar]
- Pegram, M. D., Pauletti, G., and Slamon, D. J. (1998). HER-2/neu as a predictive marker of response to breast cancer therapy. Breast Cancer Res. Treat. 52, 65-77. [DOI] [PubMed] [Google Scholar]
- Pollack, A. L., Runyan, R. B., and Mostov, K. E. (1998). Morphogenetic mechanisms of epithelial tubulogenesis: MDCK cell polarity is transiently rearranged without loss of cell-cell contact during scatter factor/hepatocyte growth factor-induced tubulogenesis. Dev. Biol. 204, 64-79. [DOI] [PubMed] [Google Scholar]
- Potempa, S., and Ridley, A. J. (1998). Activation of both MAP kinase and phosphatidylinositide 3-kinase by Ras is required for hepatocyte growth factor/scatter factor-induced adherens junction disassembly. Mol. Biol. Cell 9, 2185-2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal, I., Lamarche-Vane, N., Lamorte, L., Kaibuchi, K., and Park, M. (2000). Activation of cdc42, rac, PAK, and rho-kinase in response to hepatocyte growth factor differentially regulates epithelial cell colony spreading and dissociation. Mol. Biol. Cell 11, 1709-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal, I., and Park, M. (1995). Hepatocyte growth factor-induced scatter of Madin-Darby canine kidney cells requires phosphatidylinositol 3-kinase. J. Biol. Chem. 270, 27780-27787. [DOI] [PubMed] [Google Scholar]
- Savagner, P. (2001). Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays 23, 912-923. [DOI] [PubMed] [Google Scholar]
- Schmeichel, K. L., and Bissell, M. J. (2003). Modeling tissue-specific signaling and organ function in three dimensions. J. Cell Sci. 116, 2377-2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, C., Bladt, F., Goedecke, S., Brinkmann, V., Zschiesche, W., Sharpe, M., Gherardi, E., and Birchmeier, C. (1995). Scatter factor/hepatocyte growth factor is essential for liver development. Nature 373, 699-702. [DOI] [PubMed] [Google Scholar]
- Selli, C., Amorosi, A., Vona, G., Sestini, R., Travaglini, F., Bartoletti, R., and Orlando, C. (1997). Retrospective evaluation of c-erbB-2 oncogene amplification using competitive PCR in collecting duct carcinoma of the kidney. J. Urol. 158, 245-247. [DOI] [PubMed] [Google Scholar]
- Seton-Rogers, S. E., Lu, Y., Hines, L. M., Koundinya, M., LaBaer, J., Muthuswamy, S. K., and Brugge, J. S. (2004). Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc. Natl. Acad. Sci. USA 101, 1257-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, P. M., Shu, W., Cardiff, R. D., Muller, W. J., and Massague, J. (2003). Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc. Natl. Acad. Sci. USA 100, 8430-8435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasiri, A., and Roskelley, C. D. (1999). Cell shape and integrin signaling regulate the differentiation state of mammary epithelial cells. Methods Mol. Biol. 129, 271-283. [DOI] [PubMed] [Google Scholar]
- Tanimura, S., Nomura, K., Ozaki, K., Tsujimoto, M., Kondo, T., and Kohno, M. (2002). Prolonged nuclear retention of activated extracellular signal-regulated kinase 1/2 is required for hepatocyte growth factor-induced cell motility. J. Biol. Chem. 277, 28256-28264. [DOI] [PubMed] [Google Scholar]
- Thiery, J. P. (2002). Epithelial-mesenchymal transitions in tumour progression. Nat Rev. Cancer 2, 442-454. [DOI] [PubMed] [Google Scholar]
- Tobioka, H., Isomura, H., Kokai, Y., Tokunaga, Y., Yamaguchi, J., and Sawada, N. (2004). Occludin expression decreases with the progression of human endometrial carcinoma. Hum. Pathol. 35, 159-164. [DOI] [PubMed] [Google Scholar]
- Tuck, A. B., Park, M., Sterns, E. E., Boag, A., and Elliott, B. E. (1996). Coexpression of hepatocyte growth factor and receptor (Met) in human breast carcinoma. Am. J. Pathol. 148, 225-232. [PMC free article] [PubMed] [Google Scholar]
- Ueda, Y., Wang, S., Dumont, N., Yi, J. Y., Koh, Y., and Arteaga, C. L. (2004). Overexpression of HER2 (erbB2) in human breast epithelial cells unmasks transforming growth factor beta-induced cell motility. J. Biol. Chem. 279, 24505-24513. [DOI] [PubMed] [Google Scholar]
- Uehara, Y., Minowa, O., Mori, C., Shiota, K., Kuno, J., Noda, T., and Kitamura, N. (1995). Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 373, 702-705. [DOI] [PubMed] [Google Scholar]
- Weidner, K. M., Hartmann, G., Sachs, M., and Birchmeier, W. (1993). Properties and functions of scatter factor/hepatocyte growth factor and its receptor c-Met. Am. J. Respir. Cell. Mol. Biol. 8, 229-237. [DOI] [PubMed] [Google Scholar]
- Wrobel, C. N., Debnath, J., Lin, E., Beausoleil, S., Roussel, M. F., and Brugge, J. S. (2004). Autocrine CSF-1R activation promotes Src-dependent disruption of mammary epithelial architecture. J. Cell Biol. 165, 263-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita, J., Ogawa, M., Yamashita, S., Nomura, K., Kuramoto, M., Saishoji, T., and Shin, S. (1994). Immunoreactive hepatocyte growth factor is a strong and independent predictor of recurrence and survival in human breast cancer. Cancer Res. 54, 1630-1633. [PubMed] [Google Scholar]
- Yang, Y., Spitzer, E., Meyer, D., Sachs, M., Niemann, C., Hartmann, G., Weidner, K. M., Birchmeier, C., and Birchmeier, W. (1995). Sequential requirement of hepatocyte growth factor and neuregulin in the morphogenesis and differentiation of the mammary gland. J. Cell Biol. 131, 215-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G. J., Tsuda, H., Adachi, I., Fukutomi, T., Yamamoto, H., and Hirohashi, S. (1997). Prognostic indicators for breast cancer patients with one to three regional lymph node metastases, with special reference to alterations in expression levels of bcl-2, p53 and c-erbB-2 proteins. Jpn. J. Clin. Oncol. 27, 371-377. [DOI] [PubMed] [Google Scholar]
- Zhu, H., Naujokas, M. A., and Park, M. (1994). Receptor chimeras indicate that the met tyrosine kinase mediates the motility and morphogenic responses of hepatocyte growth/scatter factor. Cell Growth Differ. 5, 359-366. [PubMed] [Google Scholar]