

Abstract
cAR1, a G protein-coupled receptor (GPCR) for cAMP, is required for the multicellular development of Dictyostelium. The activation of multiple pathways by cAR1 is transient because of poorly defined adaptation mechanisms. To investigate this, we used a genetic screen for impaired development to isolate four dominant-negative cAR1 mutants, designated DN1-4. The mutant receptors inhibit multiple cAR1-mediated responses known to undergo adaptation. Reduced in vitro adenylyl cyclase activation by GTPγS suggests that they cause constitutive adaptation of this and perhaps other pathways. In addition, the DN mutants are constitutively phosphorylated, which normally requires cAMP binding and possess cAMP affinities that are ∼100-fold higher than that of wild-type cAR1. Two independent activating mutations, L100H and I104N, were identified. These residues occupy adjacent positions near the cytoplasmic end of the receptor's third transmembrane helix and correspond to the (E/D)RY motif of numerous mammalian GPCRs, which is believed to regulate their activation. Taken together, these findings suggest that the DN mutants are constitutively activated and block development by turning on natural adaptation mechanisms.
INTRODUCTION
G protein-coupled receptors (GPCRs) number in the thousands in humans and are responsible for physiological responses to diverse extracellular signals including light, odorants, hormones, chemoattractants, and neurotransmitters. On activation by their ligands (or photons in the case of rhodopsin), these seven transmembrane domain-containing receptors typically activate heterotrimeric G proteins that, in turn, regulate the activities of a variety of downstream effectors. Ligand binding also triggers various desensitization mechanisms which diminish the cell's responsiveness to subsequent stimulation (Strader et al., 1995; Bourne, 1997; Ferguson and Caron, 1998).
These systems are highly conserved and found in all eukaryotes including the social amoeba Dictyostelium discoideum, which uses extracellular cAMP signaling and cAMP-specific GPCRs to orchestrate its multicellular development. When deprived of nutrients, this free-living soil amoeba ceases growth and embarks on a 24-h program in which some 105 cells aggregate and differentiate to yield a sporeladen fruiting body. The first of four highly homologous cAMP receptors (cARs) expressed during development, cAR1, is essential for aggregation and subsequent development for three principal reasons. First, by transiently activating an adenylyl cyclase (ACA), cAR1 mediates “cAMP relay,” the amplification and propagation of cAMP waves, which arise spontaneously every 6 min at aggregation centers. Second, cAR1 plays a direct role in aggregation as it mediates chemotaxis of cells up the cAMP gradient inherent in each oncoming wave. Lastly, cAR1 is responsible for the induction of various aggregation-stage genes including those encoding cAR1 itself, ACA, and Gα2, the G protein α-subunit through which cAR1 signals (Parent and Devreotes, 1996).
cAR1 governs these diverse cellular responses by activating multiple G protein-dependent and G protein-independent pathways, resulting in the uptake of Ca2+, polymerization of actin and myosin, and activation of a host of downstream effectors including adenylyl and guanylyl cyclases, a phosphatidylinositol-specific phospholipase C (PI-PLC), PI 3-kinase (PI3K), and protein kinases PKA, Akt/PKB, and ERK2 (Brzostowski and Kimmel, 2001). Interestingly, all of these responses are terminated within seconds to minutes of persistent cAMP stimulation by poorly understood adaptation mechanisms. Cells adapted to a subsaturating concentration of cAMP retain the ability to mount a response to a higher dose, which is proportional to the change in receptor occupancy (Dinauer et al., 1980a; Nebl and Fisher, 1997). These observations suggest that ligand-occupied cAR1 sends out both an excitatory signal that activates effectors such as ACA as well as a delayed inhibitory signal that terminates the response (Dinauer et al., 1980a). Removal of cAMP initiates a process of deadaptation whereby cells become resensitized to cAMP challenge with a half-time of 3-4 min (Dinauer et al., 1980b).
Sequential ACA activation, adaptation, clearance of secreted cAMP by a cell surface phosphodiesterase (PDE), and deadaptation result in the oscillation of cells between cAMP-responsive and adapted states (Parent and Devreotes, 1996). Thus, adaptation both accounts for the oscillatory nature of the cAMP signal during aggregation and necessitates such a signal to achieve continued activation of adapting responses. For instance, a number of so-called “pulse-induced” genes, including those encoding cAR1 and Gα2, are induced by periodic but not constant cAMP stimulation, indicating that they are regulated by an adaptation mechanism (Kimmel, 1987; Mann and Firtel, 1987). This adaptation permits these genes to be expressed during aggregation when they are required and represses them after aggregation when cAMP is persistently elevated. In chemotaxis, adaptation prevents cells from chasing after passing cAMP waves and is thought to facilitate the perception of shallow chemoattractant gradients and restrict pseudopod extension and uropod retraction to the poles of cells undergoing chemotaxis (Chung and Firtel, 2002; Iijima et al., 2002).
Because of the multiplicity of adapting responses mediated by cAR1, slowly hydrolyzed cAMP analogs or high concentrations of cAMP block aggregation and subsequent development (Wallraff et al., 1984). We hypothesized that constitutively active cAR1 mutants would similarly promote adaptation and thus interfere with wild-type cAR1 function and development. Such mutant receptors could be useful for delineating adaptation and other regulatory mechanisms extrinsic to cAR1 as well as mechanisms intrinsic to cAR1 that regulate its activation. Therefore, we undertook a largescale genetic screen for dominant-negative point mutants of cAR1 capable of blocking the development of wild-type cells. In this report, we describe four such mutants that exhibit properties expected of constitutively active receptor mutants and point to a common mechanism of regulating the activation of highly divergent GPCRs.
MATERIALS AND METHODS
Cell Line Maintenance and Preparation
Dictyostelium cells were cultivated in HL5 medium and those designated “wild-type” in this study are strain AX3 (Sussman, 1987). DNA constructs were introduced into cells by electroporation and the resulting transformants were selected with 20 μg/ml G418 (Pang et al., 1999). Cells were harvested and washed by centrifugation at 600 × g for 5 min.
Pulse-developed cells were prepared by shaking washed cells (120 rpm) at a density of 2 × 107 cells/ml in PB (5 mM KH2PO4, 5 mM Na2HPO4, pH 6.9) containing 2 mM MgSO4 and 0.2 mM CaCl2 for 5 h (unless noted otherwise) with the addition of cAMP to 50 nM every 6 min, mimicking natural cAMP oscillations. Where indicated, pulse-developed cells (1 × 107/ml) were “basalated” by shaking at 200 rpm in the presence of 2 mM caffeine for 30 min to interrupt spontaneous cAMP oscillations (Theibert and Devreotes, 1986). The cells were then washed twice with and resuspended in the indicated buffer at 4°C.
Mutant Library Construction
Plasmid pJK53 (Kim et al., 1997a), which contains a full-length cAR1 cDNA, was used as the template for PCR under conditions designed to induce misincorporation of nucleotides (Cadwell and Joyce, 1992). The amplification reaction (30 μl) contained pJK53 (5 ng), T3 and T7 primers (0.5 μM each), dNTPs (1 mM each), 6 mM MgCl2, 0.5 mM MnCl2, 50 mM KCl, Taq polymerase (5 U; Promega, Madison, WI), and 10 mM Tris, pH 9.0, and was subjected to 27 amplification cycles, each consisting of 40 s at 94°C, 60 s at 48°C, and 140 s at 72°C. After restriction digestion with NdeI and NcoI, the 560-base pair mutagenized fragment encoding the third through seventh transmembrane domains (i.e., TM3-TM7) was cloned into pJK53, replacing the corresponding wild-type fragment. After limited amplification of the mutated plasmid library in Escherichia coli, the mutant sequences were moved en masse to cAR1 expression plasmid pMZ62 by simple replacement of its BglII-BstXI fragment, which includes the 5′-UTS and all seven TM domains. pMZ62 was constructed by excising the cAR1 cDNA from pJK53 with BglII and BamHI and inserting it in the sense orientation into the BglII site of pJK1 (Kim et al., 1997b), a multicopy extrachromosomal vector for expression under the control of the actin-15 promoter.
Other Plasmid Constructs
A version of cAR1 mutant DN4 lacking phosphorylation sites (designated DN4ΔP) was created by replacing its C-terminal cytoplasmic domain with that of a previously described cAR1 mutant, cm1234, which lacks all 18 serines of this domain (Hereld et al., 1994). Briefly, a BglII-NcoI fragment encoding the receptor's N-terminal transmembrane region was excised from the DN4-encoding plasmid isolated in the genetic screen (this report). In addition, an NcoI-EcoRI fragment encoding the C-terminal cytoplasmic domain of cm1234 was excised from a previously described plasmid (Hereld et al., 1994). These two fragments were simultaneously ligated into pJK53 digested with BglII and EcoRI, yielding pMZ68. For coexpression, the cAR1 and DN4ΔP sequences of pJK53 and pMZ68 were each moved to pCV5 (Janetopoulos et al., 2001) in the same manner described for pJK1, yielding pMZ80 and pMZ81. pCV5 has the same actin-15 promoter and other properties described for pJK1 but is better suited for stable cotransformation.
cAMP-binding Assays
cAMP binding in the presence of ammonium sulfate was assayed essentially as described by van Haastert (1985a). In brief, 2.5 × 106 PB-washed vegetative cells were combined on ice with 18 nM [3H]cAMP, 100 μg bovine serum albumin (BSA), and ammonium sulfate (90% saturated at 22°C) in a final volume of 560 μl. After incubating on ice for 5 min, the cells were recovered by centrifugation at 14,000 × g for 2 min at 4°C, washed once with ice-cold saturated ammonium sulfate (500 μl), and dissolved in 0.1% SDS (80 μl) for scintillation counting. Nonspecific binding was determined with binding reactions containing 0.1 mM unlabeled cAMP.
To determine receptor affinities under physiological conditions, cAMP binding measurements were made in PB essentially as described by Caterina et al. (1995a). Vegetative cells were washed twice in ice-cold PB and resuspended at a density of 108/ml in PB. The binding of 10 nM [3H]cAMP (24 Ci/mmol) in the presence of unlabeled cAMP, ranging from 0 to 10-6 M, was determined in duplicate. Cell-bound cAMP was resolved from unbound ligand by pelleting the cells through a 1:1 mixture of silicone oils AR20 and AR200 (Wacker Silicones, Adrian, MI) and quantitated by scintillation counting. Nonspecific signal, determined in the presence of 0.1 mM unlabeled cAMP, was subtracted. The resulting data were analyzed using Prism software (GraphPad, San Diego, CA).
For measuring the GTPγS sensitivity of binding, pulse-developed cells at 108 cells/ml in ice-cold PMB (PB containing 2 mM MgSO4) were lysed by forcing them through the 5-μm pores of a polycarbonate filter (Millipore, Bedford, MA) using a syringe. The membrane fraction was recovered by centrifugation at 14,000 × g for 5 min at 4°C, washed once, and resuspended at 108 cell equivalents/ml in PMB. cAMP-binding reactions (100 μl) containing 5 nM [3H]cAMP, membranes from 7 × 106 cells, and 30 μM GTPγS where indicated, were incubated for 5 min on ice and then centrifuged at 14,000 × g for 2 min at 4°C. The pelleted membranes were dissolved in 100 μl of 1% SDS and counted with 1 ml of scintillant.
cGMP and cAMP Accumulation Assays
cAMP-stimulated cGMP and cAMP accumulation was measured essentially as previously described (van Haastert, 1985b; Kim et al., 1997b). Pulse-developed cells at a density of 5 × 107 cells/ml were stimulated with 1 μM cAMP. Samples (50 μl) were withdrawn at various time points, combined with 50 μl of 3.5% HClO4 to stop the reactions and lyse the cells, and then neutralized with 25 μl of 50%-saturated KHCO3. After centrifugation, cGMP in 25 μl of clarified sample was determined by competitive radioimmunoassay (Amersham, Piscataway, NJ; TRK500). Accumulated cAMP in similarly prepared samples was measured by competitive binding (Amersham, TRK432). In this case, stimulation was with 10 μM 2′-deoxy-cAMP, a cAR1 agonist that does not interfere with the measurement of cAMP. Results were normalized to total cellular protein, determined by Bradford assay (Bio-Rad, Richmond, CA) using BSA as a standard.
In Vitro Adenylyl Cyclase Assays
Adenylyl cyclase activity was assayed essentially as described by Theibert and Devreotes (1986). Pulse-developed cells (8 × 107 cells/ml in ice-cold PMB) were mixed with an equal volume of ice-cold 2× lysis buffer (20 mM Tris, pH 8.2, 2 mM MgSO4) and immediately lysed on ice through filters as described above. Without delay, 200 μl of cell lysate was combined with 20 μl of 10× reaction mix (0.1 M Tris, pH 8.0, 1 mM ATP, 5 mM cAMP, 0.1 M dithiothreitol [DTT]) containing ∼2-10 × 106 cpm [α-32P]ATP. Reactions were incubated at 22°C for 1.5 min and terminated by the addition of 200 μl of stop mix (1% SDS, 22.5 mM ATP, 0.65 mM cAMP). To assess adenylyl cyclase activity independent of G protein influences, 5 mM MnSO4 was included in reactions where indicated. To determine GTPγS-stimulated activity, GTPγS (40 μM), and cAMP (1 μM) were added to cells just before their lysis and the resulting lysates were incubated on ice for 5 min before being assayed. Background was determined by adding stop mix before lysate addition. [32P]cAMP synthesized during the reaction was purified by Dowex and alumina chromatography and measured in a scintillation counter as previously described (Theibert and Devreotes, 1986).
RT-PCR Analysis of Gene Expression
For RT-PCR analysis of gene expression, total RNA was isolated using Trizol reagent (Gibco BRL, St. Louis, MO), treated with DNase, extracted with phenol, precipitated, and quantified by absorbance at 260 nm. For reverse transcription, RNA (2 μg) and random hexanucleotides (0.5 μg) were combined in 14 μl, denatured at 70°C for 5 min, and chilled on ice. AMV reverse transcriptase (9 U), buffer, and dNTPs (0.4 μM each) were then added and the volume was adjusted to 25 μl. Reactions were incubated 10 min at 20°C followed by 50 min at 42°C. The resulting cDNA was serially diluted and amplified by 30 cycles of PCR using TaqDNA polymerase and the following gene-specific primer pairs (product sizes in parentheses): cAR1, CACCTATTTGAGTGTATCCC, CAGTCTGTTTTTCTTCATCAGATG (398 base pairs); Gα2, ATGGGTATTTGTGCATCATC, GAACACCTCTTGTCATAACACGAGTATG (565 base pairs); ACA, ACAATCGATCACACAACAAAGGG, CAACCGAGTGCATATAGACAAC (564 base pairs); and mitochondrial large subunit rRNA encoded by rrn, GTACTGTGAAGGAAAGATGAAAAGG, CCTATGGACCTTAGCGCTC (560 base pairs). In the case of Gα2 and ACA, the primers flank an intron to rule out the possibility that contaminating genomic DNA was being amplified. The rRNA primers, included for normalization, were used with 105-fold dilutions of the cDNA. This dilution yielded 16% of the maximal amount of PCR product achieved with lesser dilutions. For all other primer pairs, 103-fold dilutions were used, yielding amounts of product in the responsive range for this assay established with the rRNA.
RESULTS
Identification of Dominant-negative cAR1 Mutants
Error-prone PCR was used to generate a library of cAR1 mutants having, on average, a single amino acid substitution within the 186-amino-acid region spanning the third through the seventh transmembrane domains. To identify dominant-negative mutant receptors, the library was expressed in wild-type AX3 cells from a multicopy, extrachromosomal vector under the control of the Dictyostelium actin-15 promoter. This results in essentially constitutive expression of cAR1 protein at levels typically three- to five-fold higher than peak endogenous cAR1 expression, which occurs some 5 h after the onset of starvation (Johnson et al., 1991). Approximately 19,000 transformants were grown clonally on bacterial lawns and screened visually for developmental abnormalities, leading to the isolation of seven transformants exhibiting impaired development. The plasmids were retrieved from each of these cell lines and retransformed into fresh wild-type cells. In four of the seven cases, the developmental phenotypes could be recapitulated, confirming that they were indeed caused by the plasmidencoded cAR1 mutants. These four dominant-negative mutant receptors were designated DN1, DN2, DN3, and DN4.
The developmental phenotypes caused by the four dominant-negative mutants were verified by starving them on nutrient-free agar. As shown in Figure 1, the parental AX3 cells and cAR1 cells (i.e., AX3 cells expressing plasmidencoded cAR1) aggregate and develop efficiently to yield normal fruiting bodies in 24 h. In contrast, DN1, DN3, and DN4 cells are largely defective in aggregation but occasionally form minute fruiting bodies. DN2 cells exhibit a less severe phenotype, forming aggregates that give rise to clusters of small to moderately sized fruiting bodies.
Figure 1.
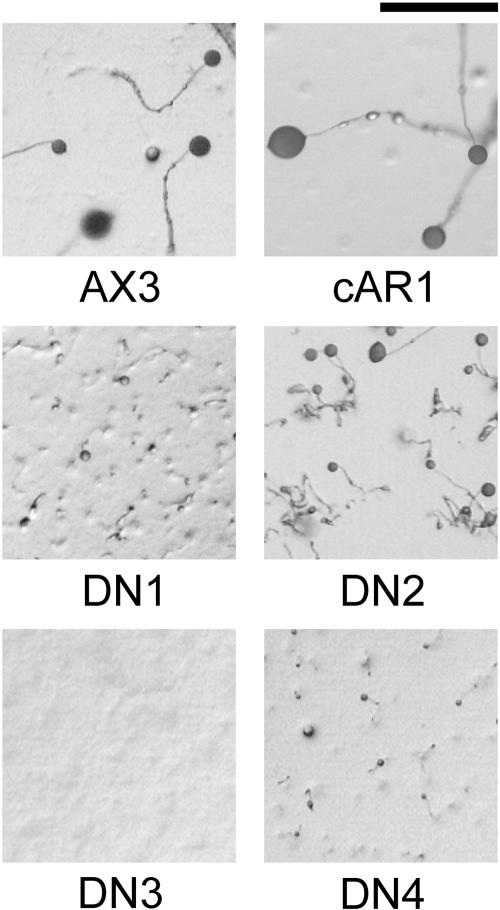
Developmental phenotypes of dominant-negative mutants. Wild-type AX3 cells and AX3 cells expressing cAR1, DN1, DN2, DN3, or DN4 cells were starved on nutrient-free agar plates and photographed after 25 h at 22°C. Scale bar, 1 mm.
The vast majority of wild-type cAR1 is at the cell surface based on microscopic localization by immunofluorescence and GFP-tagging (Wang et al., 1988; Xiao et al., 1997). To determine if the dominant mutants are appropriately expressed on the cell surface, cAMP binding to intact cells (Figure 2A) and cAR1 immunoblotting (Figure 2B), a measure of total receptors, were performed simultaneously. The mutant receptors were all able to bind cAMP. Furthermore, the numbers of cell surface binding sites detected for wild-type and mutant receptors alike were found to be proportional to the amount of receptor protein expressed in each case (compare Figure 2A with Figure 2B, c lanes), indicating that the mutant receptors, like wild-type cAR1, are efficiently displayed on the cell surface. Because DN1 and DN4 share a common mutation (see below) and DN2 has a relatively weak developmental phenotype, further characterization focused primarily on DN3 and DN4.
Figure 2.
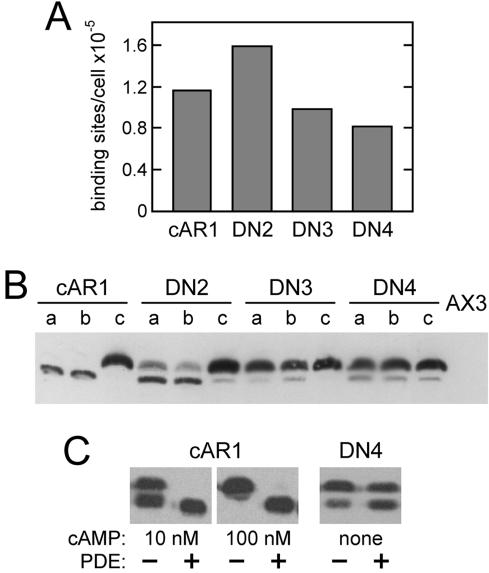
Surface expression and phosphorylation of dominant-negative cAR1 mutants. (A) Receptors on the surface of vegetative cAR1 cells or the indicated DN cells were measured by the binding of 20 nM [3H]cAMP in ammonium sulfate. Wild-type cAR1 is nearly saturated under these conditions. (B) To assess total receptor amounts and phosphorylation, the same cells (2 × 106/ml in PB) were shaken at 22°C for 15 min with no addition (a lanes), 5 mM caffeine (b lanes), or 1 μM cAMP (c lanes). Treated cells were extracted with CHAPS as previously described (Hereld et al., 1994) and cAR1 in the insoluble fraction from 2 × 106 cells was immunoblotted using cAR1 antiserum R4 (gift of P. Devreotes) and chemiluminescent detection. Parental AX3 cells (far right lane) show that little endogenous cAR1 is expressed under these conditions. Comparison of cAR1 lanes b and c indicates that the lower cAR1 band is inefficiently detected in this blot. (C) Effect of phosphodiesterase (PDE) treatment on receptor phosphorylation. Vegetative cAR1 cells (2 × 106/ml in PB) were stimulated with cAMP as indicated for 5 min to induce phosphorylation and were subsequently incubated 15 min without or with PDE. Unstimulated DN4 cells were similarly treated without and with PDE for 20 min. The cells were shaken constantly at 22°C throughout these treatments. Partially purified PDE, included at 3.2 μg protein/ml, was prepared from Dictyostelium as described by van Haastert and van der Heijden (1983). Samples (106 cells each) were CHAPS-extracted on ice and immunoblotted for cAR1 as described above.
DN Receptors Are Constitutively Phosphorylated
On cAMP binding, cAR1 becomes phosphorylated on several serine residues within its C-terminal cytoplasmic domain, resulting in a discrete mobility retardation on SDS-PAGE (Hereld et al., 1994). Site-directed mutagenesis indicated that the mobility shift is specifically due to the phosphorylation of either Ser303 or Ser304 (Caterina et al., 1995b). Ligand-induced cAR1 phosphorylation persists as long as cAMP is bound but is rapidly reversed upon cAMP removal with a half-time of ∼2 min, returning the receptor to its original electrophoretic mobility state (Vaughan and Devreotes, 1988). Interestingly, all of the DN mutants were found to be constitutively phosphorylated in the absence of added cAMP (Figure 2B, a lanes). After correcting for the preferential detection of the upper, phosphorylated cAR1 band apparent in Figure 2B, ∼20% of DN2 and roughly 60-70% of DN3 and DN4 were determined to be in the up-shifted phosphorylated state. Thus, the degree of constitutive phosphorylation of the DN mutants correlates with their disruptive effects on development.
To assess whether the constitutive phosphorylation of the mutant receptors is spontaneous or the consequence of binding secreted cAMP, the cells were treated with caffeine to block cAMP synthesis by ACA, the major aggregation-stage adenylyl cyclase. Under these conditions, residual cAMP should be degraded by the extracellular PDE, allowing any cAR1 phosphorylated because of cAMP binding to undergo dephosphorylation. As shown in Figure 2B (b lanes), the mutant receptors remained phosphorylated despite caffeine treatment. In a separate experiment, cells were treated with a concentrated and purified preparation of the secreted PDE. As shown in Figure 2C, cAMP pretreatment of cAR1 cells resulted in phosphorylation that could be completely reversed by the PDE treatment. In the case of 100 nM cAMP, complete dephosphorylation of the largely up-shifted cAR1 indicates that PDE treatment rapidly reduced cAMP levels by a factor of 103 or more based on the known cAMP dose-dependence of cAR1 phosphorylation (Kim et al., 1997a; see Figure 3C, for example), leaving sufficient time for cAR1 dephosphorylation to occur. In contrast, the constitutive phosphorylation of DN3 (unpublished data) and DN4 was largely refractory to PDE treatment, indicating that it is not caused by cAMP. Taken together, these results suggest that the DN mutant receptors mimic the ligand-bound conformation of cAR1 in the absence of extracellular cAMP and are consequently recognized and phosphorylated by the endogenous receptor kinase at natural sites of ligand-induced phosphorylation.
Figure 3.
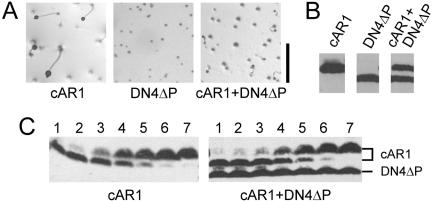
Effects of expressing DN4ΔP alone or with wild-type cAR1. AX3 cells expressing cAR1, DN4ΔP, or both cAR1 and DN4ΔP were obtained by transformation with pMZ80, pMZ81, or both plasmids. The stability of the cotransformant was verified by recloning and cAR1 immunoblotting (unpublished data). (A) Development on nonnutrient agar as in Figure 1. Scale bar, 1 mm. (B) Comparable cAR1 immunoblots of the cell lines depicted in A. The cells were caffeine-treated for 10 min so that cAR1 would run discretely in its nonphosphorylated form. DN4ΔP runs below cAR1 due to a 9-amino-acid deletion. (C) Dose-dependence of cAMP-induced phosphorylation of wild-type cAR1 expressed alone (left) or with DN4ΔP (right). Cells shaking at 22°C were exposed to 0, 1, 3.2, 10, 32, 100, and 1000 nM cAMP (lanes 1-7, respectively) for 10 min in the presence of 10 mM DTT and 5 mM caffeine to inhibit cAMP breakdown by PDE or amplification by ACA, respectively. Whole cell samples were immunoblotted for cAR1.
Dominance of DN Receptors Does Not Result from Their Constitutive Phosphorylation
To examine the possibility that DN receptor phosphorylation somehow causes the dominant effects on development, a version of DN4 lacking all 18 C-terminal-domain serines (designated DN4ΔP) was generated and expressed in wild-type cells. Elimination of these serines in an otherwise wild-type receptor was previously shown to abolish receptor phosphorylation but have little impact on the receptor's ability to support aggregation (Kim et al., 1997b). However, DN4ΔP was found to be similar to DN4 in its ability to impair aggregation (Figure 3A). Thus, the constitutive phosphorylation of DN4 (and presumably also DN1-3) is not required for its dominant-negative effect on development but instead is merely indicative of the mutant receptor's constitutively activated conformation. Therefore, a separate phosphorylation-independent activity of the mutant receptor must account for the observed developmental phenotype.
DN4ΔP Is Dominant over Coexpressed Wild-type cAR1 without Altering Its Ability to Bind and Respond to cAMP
Because the DN receptors were constitutively overexpressed in contrast to the endogenous cAR1 gene, which is weakly expressed at the outset of development, we wanted to examine the properties of cells expressing comparable levels of a DN receptor and the wild-type receptor. Cotransformation of AX3 cells with pCV5-derived plasmids encoding cAR1 and DN4ΔP yielded clonal transformants that stably expressed nearly equal amounts of the two receptors (Figure 3B). Development of these coexpressers was markedly impaired and only slightly better than that of DN4ΔP cells. Similar to DN4ΔP cells, many cells in the coexpresser population failed to aggregate, whereas others coalesced into small, loose-appearing mounds, which occasionally gave rise to small fruiting bodies (Figure 3A). On average, these mounds were slightly larger, better defined, and more likely to give rise to small fruiting bodies than those of DN4ΔP cells.
To examine the possibility that DN4ΔP might directly impair the function of coexpressed wild-type cAR1, the dose-dependence for cAMP-induced phosphorylation of wild-type cAR1 was determined in the presence and absence of coexpressed DN4ΔP (Figure 3C). In both cases, all of the cAR1 was phosphorylated (i.e., up-shifted) in response to 10-6 M cAMP and a half-maximal response was elicited by ∼10-8 M in agreement with published results (Kim et al., 1997a). These results indicate that DN4ΔP has little, if any, impact on the cell surface localization of wild-type cAR1, its affinity for cAMP, or its ability to respond to bound cAMP. Furthermore, the fact that phosphorylation of the wild-type receptor retains its strict ligand dependence in the context of constitutively phosphorylated DN4ΔP suggests the wild-type receptor must be occupied to be a suitable substrate for phosphorylation.
DN Receptors Have Elevated cAMP Affinity
Agonists are believed to activate receptors by preferentially binding to their active conformation, thereby perturbing the equilibrium between inactive (R) and active (R*) conformations in favor of the latter. Mutation of residues that stabilize the R state can similarly alter this equilibrium in favor of the R* state, resulting in receptors with both increased affinity and constitutive activity (Samama et al., 1993; Parnot et al., 2002). Given the constitutive phosphorylation of the DN receptors, we wanted to ascertain whether their affinities are elevated as would be expected if they are constitutively activated. Therefore, we measured the dose-dependent binding of cAMP to intact cells expressing either the wild-type or mutant receptors (Figure 4A). In agreement with previous results (Johnson et al., 1991), wild-type cAR1 exhibited a complex binding pattern consistent with the existence of two affinity states with Kd's of ∼600 and 10 nM that are populated by ∼90 and 10% of the receptors, respectively. Interestingly, each of the dominant-negative mutants examined (DN1, DN3, and DN4) exhibited only a single high-affinity (Kd's ranging from 3 to 16 nM) comparable to that of the wild-type receptor's high-affinity state and nearly 100-fold higher than the average affinity of the wild-type receptor.
Figure 4.
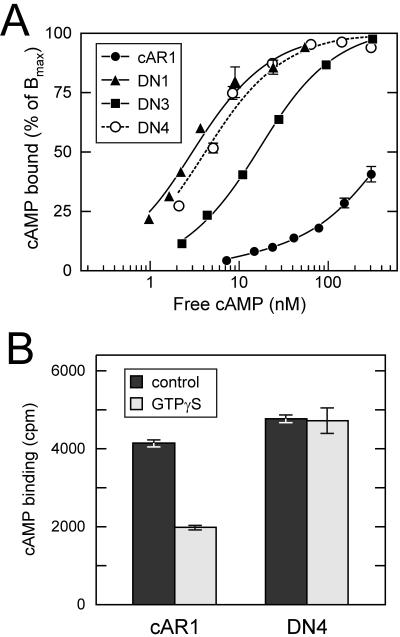
cAMP-binding properties of DN receptors. (A) cAMP binding to wild-type and mutant receptors on vegetative cells in PB was determined by [3H]cAMP binding in the presence of competing doses of unlabeled cAMP. Data points represent the means of duplicate determinations. The plots reflect single-site models for DN1, DN3, and DN4 with Kd values of 3.0, 15.7, and 4.2 nM and Bmax values of 8.8 × 104, 2.9 × 105, and 2.2 × 105 sites/cell, respectively. The wild-type cAR1-binding data best fit a two-site model consisting of 4.5 × 104 high-affinity sites/cell and 5 × 105 low-affinity sites with Kd values of 10 and 560 nM, respectively. (B) Effect of GTPγS on cAMP binding. The binding of 5 nM [3H]cAMP to membranes of cAR1 and DN4 cells was determined with and with-out 30 μM GTPγS. Means of triplicate determinations are plotted.
The high-affinity state exhibited by wild-type cAR1 and many GPCRs is thought to reflect an activated receptor conformation stabilized by its association with a G protein based on its sensitivity to nonhydrolyzable GTP analogs such as GTPγS (Wu et al., 1995). To assess the possibility that the high affinities observed for the DN mutants result from interactions with G proteins, we tested the influence of GTPγS on the binding of cAMP to membranes bearing wild-type or dominant-negative cAMP receptors (Figure 4B). Although GTPγS diminished the binding of 5 nM cAMP to wild-type cAR1 by ∼50%, it had no appreciable effect on the binding of DN4. Thus, the high-affinity of DN4 (and presumably also DN1-DN3) is not due to an enhanced tendency to associate with G proteins but instead is apparently an intrinsic property of the mutant receptor. Taken together, these results suggest that the DN mutants can adopt a high-affinity conformation resembling that of the ligand-occupied active receptor in agreement with their constitutive phosphorylation.
DN Receptors Interfere with Multiple cAR1-mediated Responses
To better understand the mechanism by which the dominant-negative receptors impair development, several cAR1-mediated responses were examined. As shown in Figure 5, AX3 cells overexpressing wild-type cAR1 (cAR1 cells) exhibit normal chemotaxis to both cAMP and another chemoattractant, folate. In contrast, DN4 cells are unable to undergo chemotaxis to cAMP. However, they did exhibit normal chemotaxis to folate, indicating that the chemotaxis machinery is functional in DN4 cells and that their chemotaxis defect is receptor specific. Next, the cAMP-stimulated activation of guanylyl and adenylyl cyclases was evaluated indirectly by measuring the accumulation of their respective products, cGMP and cAMP. In contrast to wild-type cells, DN4 cells displayed only a weak increase in cGMP upon cAMP stimulation (Figure 6A). As was the case for chemotaxis, folate stimulated normal accumulation of cGMP by DN4 cells (unpublished data). Similar results were obtained for cAMP accumulation (Figure 6B). In this case, the cAR1 agonist 2′-deoxy-cAMP evoked the accumulation of markedly reduced amounts of cAMP by both DN3 and DN4 cells in contrast to the robust response of wild-type cells.
Figure 5.
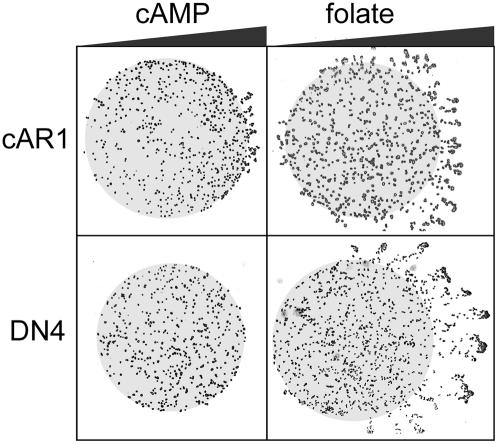
Chemotaxis to cAMP and folate. Droplets (∼0.5 μl) containing either cAR1 or DN4 cells were spotted on agar (0.8% in PB) ∼1-2 mm from paper discs (not depicted) soaked with 50 μM cAMP or 0.5 mM folate as indicated. Pulse-developed cells and PB-washed vegetative cells were used for cAMP and folate chemotaxis, respectively. After 3-5 h, the cells were photographed using an inverted microscope. The resulting images were contrast-enhanced to improve cell visibility and overlaid on gray circles indicating the boundaries of the cell droplets. Triangles symbolize the expected chemoattractant gradient.
Figure 6.
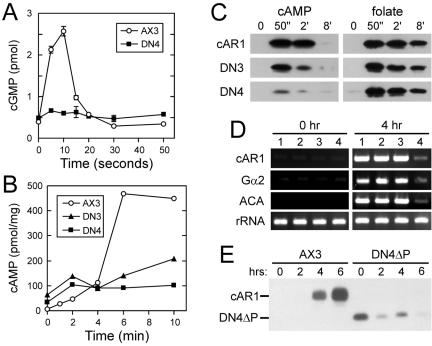
Effect of DN receptors on cAR1-mediated responses. (A) cGMP and (B) cAMP accumulation were measured at the indicated times after stimulation with cAMP (1 μM) and 2′-deoxy-cAMP (10 μM), respectively. Results for 6-h-pulse-developed wild-type cells (AX3, ○), DN3 cells (▴), and DN4 cells (▪) are shown. cGMP per 106 cells was determined in duplicate; the mean and range are plotted. cAMP determinations were normalized to total cellular protein. (C) Stimulation of ERK2 in 40-min-starved cAR1, DN3, and DN4 cells by 2 μM cAMP or 50 μM folate was demonstrated by immunoblotting with phospho-MAPK-specific antibodies (Cell Signaling Technology, Beverly, MA; 9101). (D) RT-PCR analysis of endogenous cAR1, Gα2, and ACA transcripts in wild-type AX3 (lane 1), empty vector (pJK1)-transformed (lane 2), cm1234-expressing (lane 3), and DN4ΔP-expressing (lane 4) cells before and after pulsing with cAMP for 4 h. (E) Endogenous cAR1 protein expression. Samples of 106 pulse-developed wild-type and DN4ΔP cells were withdrawn at the indicated times, treated for 10 min with 10 μM cAMP so that any endogenous cAR1 would migrate as a single phosphorylated species, and subjected to cAR1 immunoblotting as in Figure 3. The positions of endogenous cAR1 and plasmid-derived DN4ΔP are indicated.
The above-mentioned responses to cAMP depend on the Gα2βγ heterotrimer. To determine whether the dominant-negative receptors also impair G protein-independent responses mediated by cAR1, we examined the stimulation of mitogen-activated protein kinase (MAPK) activity by cAMP and, for comparison, folate. Both ligands activate primarily the ERK2 isoform of MAPK in a transient manner with peak activation occurring after ∼50 s of stimulation (Maeda et al., 1996; Maeda and Firtel, 1997). As shown by immunoblotting with antibodies specific for activated phospho-MAPK (Figure 6C), robust activation was obtained with folate for cAR1 cells as well as DN3 and DN4 cells. On the other hand, the ability of cAMP to activate ERK2 in DN4 cells was greatly impaired, whereas the response of DN3 cells was diminished to a lesser extent.
We next examined the influence a DN receptor on pulse-induced gene expression. Low levels of cAR1 and Gα2 expressed early in development mediate the induction of their own genes and other pulse-induced genes (Kumagai et al., 1991). mRNA levels for cAR1, Gα2, and ACA were assessed by RT-PCR in cells expressing DN4ΔP, which made the selective assay of endogenous cAR1 transcripts and protein possible. As shown in Figure 6D, the induction of all three transcripts after 4 h of cAMP pulsing was robust in parental AX3 cells, empty vector-transformed cells, and cells expressing cm1234, which like DN4ΔP lacks all C-terminal-domain serines but is otherwise wild-type in sequence (lanes 1-3, respectively). In contrast, all three transcripts were only modestly induced in DN4ΔP cells (lanes 4). To evaluate the impact of the impaired transcript induction on protein levels, similarly pulsed cells were subjected to cAR1 immunoblotting (Figure 6E). A nine-amino-acid deletion in DN4ΔP, which increases its electrophoretic mobility, made it possible to resolve DN4ΔP from the wild-type cAR1 derived from the endogenous gene. As expected, cAR1 protein levels in AX3 cells increased dramatically after 6 h of cAMP pulsing. However, in the case of DN4ΔP cells, only the plasmid-derived DN4ΔP protein was detected. In other experiments (unpublished data), trace amounts of endogenous cAR1 were occasionally observed in DN4ΔP cells after 4 h of pulsing.
These results indicate that DN receptors interfere broadly with G proteindependent and -independent signaling by cAR1. Impairment of responses dependent on pulse-induced levels of Gα2 (i.e., cAMP chemotaxis, cAMP and cGMP accumulation) can be explained by the markedly diminished induction of Gα2 as was observed with DN4ΔP cells. More difficult to explain are the general impairment of pulse-induced gene expression in DN4ΔP cells, despite detectable levels of cAR1 and Gα2 transcripts (Figure 6D), and the diminished activation of ERK2 in DN3 and DN4 cells. The activated conformations of the DN receptors suggested by their constitutive phosphorylation and high affinities raise the possibility that they trigger the adaptation mechanisms that normally regulate these responses.
DN Receptors Cause Adaptation of the ACA Pathway
Although Gα2 is essential for cAR1-mediated activation of ACA, it is not required for ACA activation by GTPγS in cell lysates presumably because identical Gβγ dimers liberated by GTPγS from other G proteins in lysates can activate the cyclase (Pupillo et al., 1992; Parent and Devreotes, 1996). Furthermore, ACA activation by GTPγS is reduced 70-80% by adapting wild-type or gα2- cells to cAMP before lysis, demonstrating the utility of this assay for assessing adaptation of the ACA pathway as well as the Gα2-independence of this adaptation (Theibert and Devreotes, 1986; Pupillo et al., 1992). To assess the possibility that the dominant-negative receptors cause adaptation, we measured the ability of GTPγS to stimulate ACA activity in lysates of naive DN1, DN3, DN4, and wild-type AX3 cells. As shown in Figure 7A, ACA activity in a wild-type cell lysate was stimulated by GTPγS ∼8.6-fold over basal levels. As expected, ACA activation by GTPγS was substantially diminished by prior adaptation of the intact wild-type cells with cAMP. In contrast to the nonadapted wild-type cells, activity was stimulated only 2.3-, 4.1-, and 3.4-fold in lysates of DN1, DN3, DN4 cells, respectively. We also assayed Mn2+-stimulated ACA activity, which provides a useful measure of available ACA as this activity is insensitive to the influence of G proteins (Ross and Gilman, 1980; Theibert and Devreotes, 1986). Normalization of the GTPγS-stimulated activities to those measured with Mn2+ gave a similar and perhaps more accurate assessment of the state of adaptation in these cells (Figure 7B). For wild-type cells, the stimulation was 4.2-fold relative to the Mn2+ value, whereas for the three mutants, the stimulation was at most 1.5-fold. These data are consistent with the existence of an adapted state in cells expressing the DN receptors. The finding that the DN cell lysates were activated by GTPγS to a greater extent than was the adapted wild-type cell lysate suggests that the DN cells are not fully adapted perhaps because the mutant receptors are not maximally activated.
Figure 7.
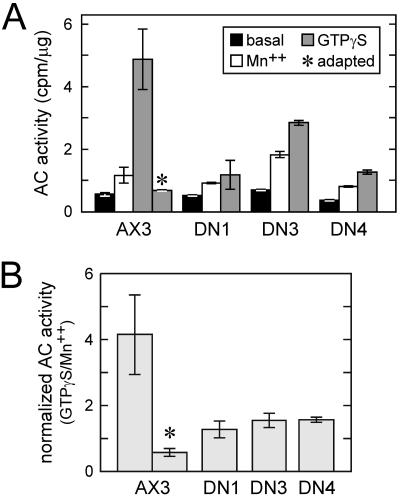
In vitro adenylyl cyclase activation. (A) Lysates of pulse-developed, basalated wild-type, DN1, DN3, and DN4 cells were assayed in duplicate for adenylyl cyclase activity in the absence (“basal”) or presence of 5 mM MnSO4. GTPγS-stimulated activity was determined in triplicate in the presence of 40 μM GTPγS and 1 μM cAMP. (B) For each cell line, GTPγS-stimulated adenylyl cyclase activity is normalized to the activity obtained with Mn2+ (data from A). Adenylyl cyclase activity measure in the presence of Mn2+ is G protein-independent and, thus, reflects the amount of available cyclase. Basal activity was not used for normalization because it is sensitive to variations in G protein activity. In A and B, asterisks (*) identify results from adapted wild-type cells that had been pre-treated with 10 μM cAMP for 10 min.
Mutations in DN Receptors Are Clustered in a Conserved Region of TM3
The mutations in DN1-4 were identified by sequencing. Surprisingly, all of the mutant receptors had mutations at the cytoplasmic end of the third transmembrane helix (TM3), suggesting that this region plays a critical role in receptor activation (Figure 8, A and B). Two of these, L100H and I104N, are spaced four amino acids apart and would therefore be expected to be adjacent to each other on one face of the helix. Mutants DN3 and DN4 have single amino acid changes at these two positions, respectively, leaving no doubt that these substitutions cause the dominance. In DN1, the I104N substitution occurred along with a presumably inconsequential second mutation, V136A. The occurrence of the I104N mutation in both DN1 and DN4, which were derived from independent PCR mutagenesis reactions, suggests that our screen for dominant-negative mutants was nearly exhaustive. In addition, both the L100H and I104N substitutions resulted from relatively rare transversions, suggesting that substitutions resulting from the more likely transitions (i.e., L100F, L100P, I104T, and I104V) were present in our screen but are not activated to the same extents as I104N and L100H.
Figure 8.
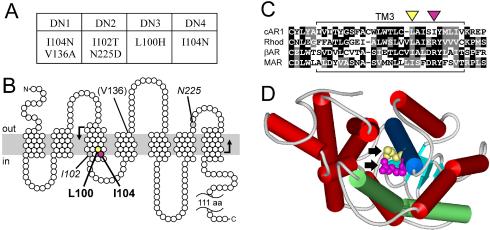
Activating mutations are clustered in a conserved region of the third transmembrane helix. (A) Summary of mutations present in cAR1 mutants DN1-4. (B) Topological model of cAR1 (based on Klein et al., 1988) indicating the amino acids mutated in DN1-4. Those causing dominant receptor activation are shown in bold, those whose role is ambiguous are italicized, and those unlikely to have a role are in parentheses. The plasma membrane is depicted as a gray bar. The region mutagenized is bounded by bent arrows. (C) Amino acid sequence alignment of the third transmembrane domains of cAR1 (residues 79-113), bovine rhodopsin (residues 110-144), bovine β-adrenergic receptor (residues 131-165), and human M1 muscarinic ACh receptor (residues 98-132). Black and gray boxes emphasize identical and similar residues, respectively. Hyphens indicate gaps introduced for optimal alignment. Yellow and magenta triangles identify L100 and I104 in cAR1, respectively. (D) Rhodopsin's structure (Palczewski et al., 2000) viewed from cytoplasm, highlighting TM3 (blue) and TM6 (green). Arrows indicate residues corresponding to L100 (yellow) and I104 (magenta) of cAR1. WebLab ViewerLite software (Accelrys, San Diego, CA) was used for structure modeling.
In the case of DN2, it is unclear which of its two mutations, I102T or N225D, is responsible for its dominant-negative activity. However, cells expressing a site-directed mutant having only the I102T substitution aggregated and developed normally, suggesting that N225D acting either alone or in concert with I102T is responsible for the phenotype of DN2 cells (unpublished data).
DISCUSSION
We describe here the isolation and characterization of four dominant-negative cAR1 mutants, designated DN1-4, which impair the aggregation and subsequent development of Dictyostelium. Dominant-negative GPCRs acting by several distinct mechanisms have been described in other systems. Dominant-negative rhodopsin mutants have been described that cause autosomal-dominant retinitis pigmentosa by interfering with the transport of wild-type rhodopsin to the cell surface (Saliba et al., 2002). In another example, Overton and Blumer (2000) attributed the dominance of a yeast pheromone receptor mutant (Ste2-M250I) to its ability to oligomerize with wild-type Ste2 and directly inhibit its function. Our DN mutants appear not to act by either of these mechanisms because they are expressed efficiently on the cell surface and DN4ΔP did not alter either the surface localization or cAMP-induced phosphorylation of coexpressed wild-type cAR1. In addition, G protein-sequestering mutants of Ste2 and mammalian α1B-adrenergic receptor have been described (Dosil et al., 1998; Chen et al., 2000). Although we have not formally ruled out this mechanism for our DN mutants, it seems unlikely as it does not readily account for their constitutive phosphorylation. In light of the fact that the phosphorylation of wild-type cAR1 is G protein-independent (Milne et al., 1995), a tightly associated G protein is probably more likely to interfere with phosphorylation by impeding the kinase's access to the receptor.
The properties of the DN cAR1 mutants strongly suggest that they are constitutively activated. Although wild-type cAR1 exhibits an electrophoretic mobility shift because of phosphorylation only upon cAMP binding, the DN mutant proteins are constitutively phosphorylated to varying extents in the absence of cAMP. The severity of developmental impairment caused by the DN mutants is correlated with the extent to which they are constitutively phosphorylated, suggesting that they impair development by the same mechanism and that this mechanism is related in some way to their phosphorylation. However, analysis of DN4ΔP, a nonphosphorylatable version of DN4, showed that the dominant activity and phosphorylation are independent consequences of the acquired mutation. These findings suggest that the mutant receptors, in the absence of cAMP, can assume an active conformation resembling that of cAMP-bound cAR1, which triggers phosphorylation as well as a separate activity responsible for their dominance. The dramatically elevated affinities of the DN receptors and the localization of the causative mutations to a region of TM3 implicated in regulating the activation of other GPCRs (discussed below) provide additional independent evidence that the DN receptors are constitutively active. This conclusion raises the possibility that the DN receptor exert their dominant-negative influence on development by persistently activating the natural adaptation mechanisms that normally regulate cAR1 signaling.
An early and critical function of cAR1 is the feed-forward induction of the cAMP signaling apparatus required for aggregation. cAR1 and Gα2, initially expressed at low levels, maximally induce their own expression and that of other pulse-induced genes by triggering the elevation of intracellular cAMP and activation of PKA (Iranfar et al., 2003). cAR1 controls intracellular cAMP levels through the simultaneous activation of ACA and ERK2-mediated inhibition of the cAMP-specific PDE RegA (Maeda et al., 2004), which are both subject to adaptation.
ACA activation by cAR1 is mediated by the PH-domaincontaining protein CRAC (cytosolic regulator of adenylyl cyclase; Insall et al., 1994). Gβγ dimers, derived from activation of the Gα2βγ heterotrimer, together with a Ras-like GTPase stimulate PI3K to transiently produce PI-3,4,5-trisphosphate (PIP3), which prompts CRAC translocation to the plasma membrane where it can initiate ACA activation (Parent et al., 1998; Meili et al., 1999; Funamoto et al., 2002; Huang et al., 2003). The PIP3 increase also causes the membrane translocation of other PH-domain-containing proteins, PKB and PhdA, which have important roles in chemotaxis (Meili et al., 1999). Although the role of PI3K in ACA activation remains to be verified, its role in chemotaxis is well established (Funamoto et al., 2002). Thus, the cAR1-PI3K pathway and its adaptation are likely to underlie the regulation of pulse-induced gene expression, cAMP relay, and chemotaxis.
We model the adaptation of the cAR1-PI3K pathway as the consequence of a Gα2-independent inhibitory signal that emanates from cAR1 and acts at a site in the excitatory pathway below the G protein and at or above PI3K (Figure 9A, right). This model is consistent with the persistent dissociation and presumed activation of Gα2βγ in adapted cells (Janetopoulos et al., 2001), the diminished ability of GTPγSto stimulate ACA activity in lysates of adapted wild-type and gα2- lysates but not those of car1- cells (Theibert and Devreotes, 1986; Pupillo et al., 1992), and the transient activation of PI3K (Huang et al., 2003).
Figure 9.
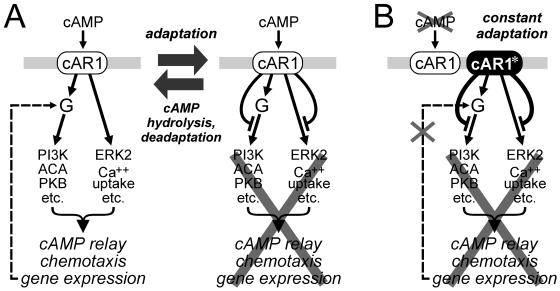
Model of periodic activation and adaptation of cAR1-mediated responses and proposed mechanism of DN mutant dominant-negative activity. (A) cAR1 is normally stimulated by cAMP waves every 6 min, triggering the excitation of multiple Gα2βγ (“G” in figure)-dependent as well as multiple G protein-independent pathways (depicted by single arrows for clarity), which collectively result in cAMP relay, chemotaxis, and pulse-induced gene expression (left). The latter is depicted for Gα2 (dashed line). After a delay, inhibitory signals (flat-headed curved arrows) from cAR1 block transmission of the excitatory signal, resulting in adaptation (right). PDE-mediated cAMP hydrolysis initiates deadaptation, allowing the cell to respond to the next cAMP wave. (B) The dominant-negative mutants described are proposed to be constitutively activated (cAR1*) and as such are expected to cause persistent adaptation, thereby interfering with pulse-induced gene expression, cAMP relay, and chemotaxis. Failure to express high levels of Gα2 due to impaired pulse-induced gene expression would contribute to the impairment of cAMP relay, chemotaxis, and other Gα2-mediated processes required for aggregation and development. See text for additional details.
Our findings suggest that the dominant-negative effects of the DN mutants result either directly or indirectly from their ability to cause constitutive adaptation of the cAR1-ACA pathway (Figure 9B). This adaptation is supported by the relatively weak activation of ACA by GTPγS in lysates of cells expressing DN mutants and can explain the impairment of pulse-induced gene expression. The reduced ability of cAMP to activate ERK2 in these cells, possibly due to adaptation of the cAR1-ERK2 pathway, may also contribute to defective pulse-induced gene expression. The failure to induce pulse-induced genes, particularly that of Gα2, to high levels in DN cells is likely an important reason for the profound impairment of other Gα2-dependent responses (i.e., chemotaxis, cAMP and cGMP accumulation) and consequently aggregation, which depends on these responses. However, the possibility that persistent adaptation contributes to the impairment of these responses cannot be dismissed.
Much of what is known about GPCR structure and activation comes from studies of rhodopsin. The only available GPCR structure is that of rhodopsin's inactive state (Palczewski et al., 2000), which shows that the seven transmembrane helices are organized in a bundle roughly perpendicular to membrane. Site-directed spin labeling, intramolecular cross-linking, and residue accessibility studies indicate that activation entails an opening of the helical bundle at its cytoplasmic end due to outward rigid-body movements of primarily TM6 and to a lesser extent TM3, exposing residues critical for activating the G protein (Meng and Bourne, 2001). Consistent findings with β2-adrenergic receptors suggest that separation of the cytoplasmic ends of TM3 and TM6 is a general mechanism of GPCR activation (Gether et al., 1997).
Mutations could conceivably act locally to activate a GPCR by introducing kinks into helices or by some other means exposing residues that interact with and activate the G protein. However, constitutively active mutants frequently exhibit elevated affinities for agonists, suggesting a more global impact of these mutations on receptor conformation. Such mutations are thought to affect residues that normally constrain the unoccupied receptor in its inactive state and, consequently, perturb the equilibrium between inactive (R) and active (R*) states in favor of R* to which agonists preferentially bind (Samama et al., 1993). Constitutively active mutants have been observed for many GPCRs and have resulted from mutations in any of the transmembrane domains and less often in the interconnecting loops, suggesting that a network of interhelical interactions constrains the inactive state (Parnot et al., 2002). A large proportion of these mutations are in TM3 and TM6, consistent with their central role in activation.
The elevated affinities of the DN mutants suggest that the responsible mutations altered the R↔ R* equilibrium in favor of the active R* conformation of cAR1. All four of the dominant cAR1 mutants had substitutions at the cytoplasmic end of TM3. Substitution of L100 or I104 of cAR1 accounts for the dominant activities of DN1, DN3, and DN4. Based on the alignment of Klein et al. (1988), these cAR1 residues correspond to residues L131 and R135 of rhodopsin (Figure 8C), which protrude from the base of TM3 into the pocket formed by the bundle of the seven transmembrane helices (Figure 8D). R135 of rhodopsin is involved in a hydrogen bonded network bridging TM3 and TM6 and is in the (D/E)RY motif of many mammalian GPCRs, which is believed to play a key role in their activation (Palczewski et al., 2000; Scheer et al., 2000). Lu and Hulme (1999) also identified a patch of hydrophobic TM3 residues adjacent to the DRY sequence of the M1 muscarinic acetylcholine (ACh) receptor, whose mutation increased receptor affinity and constitutive activity. Similarly, Parrish et al. (2002) provided genetic evidence for a hydrogen bond between Q149 at the cytoplasmic end of TM3 and N84 of TM2 in Ste2 of Saccharomyces cerevisiae that promotes this receptor's inactivation. Therefore, it seems likely that L100 and I104 of cAR1 constitute a patch of residues involved directly or indirectly in interhelical interactions that constrain cAR1 in an inactive conformation. Alternatively, rather than participating in interactions that stabilize the R state, L100 and I104 might destabilize R* because of the energetic cost of exposing these hydrophobic residues to the aqueous cytoplasm, a cost that is normally offset by the energy of ligand binding. If so, their mutation to the hydrophilic residues His and Asn, respectively, may have mitigated this energetic cost and hence the requirement for ligand.
Our findings suggest that the role of the cytoplasmic end of TM3 in regulating GPCR activation has been conserved in evolutionarily distant species. Thus, deliberate mutagenesis of the corresponding region of poorly characterized GPCRs such as orphan receptors revealed by genome sequencing might be a fruitful approach for elucidating their functions and regulation. To our knowledge, the constitutively active GPCR mutants described in this report are the first to be identified based on their activation of innate adaptation mechanisms. These mutants should be useful for elucidating intrinsic mechanisms of GPCR regulation as well as the mechanisms of adaptation that govern the chemotaxis of Dictyostelium and are likely shared by human immune cells capable of GPCR-mediated chemotaxis.
Acknowledgments
We are indebted to Drs. Katherine Borkovich, Richard Clark, Tian Jin, Adam Kuspa, William Margolin, Steven Norris, and John Spudich for helpful discussions and suggestions regarding this work and to Dr. Chii-Shen Yang for his assistance with molecular modeling. This work was supported by American Heart Association grant 9630188N awarded to D.H.
Article published online ahead of print in MBC in Press on December 1, 2004 (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-06-0456).
Abbreviations used: CHAPS, 3-[(cholamidopropyl)-dimethylammonio]-1-propanesulfonate; CRAC, cytosolic regulator of adenylyl cyclase; GFP, green fluorescent protein; GPCR, G protein-coupled receptor; GTPγS, guanosine 5′-[γ-thio]triphosphate; PDE, phosphodiesterase; PH, pleckstrin-homology; PI3K, phosphatidylinositol 3-kinase; PI-PLC, phosphatidylinositol-specific phospholipase C; PKA, protein kinase A; PKB, protein kinase B; TM, transmembrane domain.
References
- Bourne, H. R. (1997). How receptors talk to trimeric G proteins. Curr. Opin. Cell Biol. 9, 134-142. [DOI] [PubMed] [Google Scholar]
- Brzostowski, J. A., and Kimmel, A. R. (2001). Signaling at zero G: G-protein-independent functions for 7-TM receptors. Trends Biochem. Sci. 26, 291-297. [DOI] [PubMed] [Google Scholar]
- Cadwell, R. C., and Joyce, G. F. (1992). Randomization of genes by PCR mutagenesis. PCR Methods Appl. 2, 28-33. [DOI] [PubMed] [Google Scholar]
- Caterina, M. J., Hereld, D., and Devreotes, P. N. (1995a). Occupancy of the Dictyostelium cAMP receptor, cAR1, induces a reduction in affinity which depends upon COOH-terminal serine residues. J. Biol. Chem. 270, 4418-4423. [DOI] [PubMed] [Google Scholar]
- Caterina, M. J., Devreotes, P. N., Borleis, J., and Hereld, D. (1995b). Agonist-induced loss of ligand binding is correlated with phosphorylation of cAR1, a G protein-coupled chemoattractant receptor from Dictyostelium. J. Biol. Chem. 270, 8667-8672. [DOI] [PubMed] [Google Scholar]
- Chen, S., Lin, F., Xu, M., Hwa, J., and Graham, R. M. (2000). Dominant-negative activity of an α1B-adrenergic receptor signal-inactivating point mutation. EMBO J. 19, 4265-4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, C. Y., and Firtel, R. A. (2002). Signaling pathways at the leading edge of chemotaxing cells. J. Muscle Res. Cell Motil. 23, 773-779. [DOI] [PubMed] [Google Scholar]
- Dinauer, M. C., Steck, T. L., and Devreotes, P. N. (1980a). Cyclic 3′,5′-AMP relay in Dictyostelium discoideum. V. Adaptation of the cAMP signaling response during cAMP stimulation. J. Cell Biol. 86, 554-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinauer, M. C., Steck, T. L., and Devreotes, P. N. (1980b). Cyclic 3′,5′-AMP relay in Dictyostelium discoideum. IV. Recovery of the cAMP signaling response after adaptation to cAMP. J. Cell Biol. 86, 545-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosil, M., Giot, L., Davis, C., and Konopka, J. B. (1998). Dominant-negative mutations in the G-protein-coupled α-factor receptor map to the extracellular ends of the transmembrane segments. Mol. Cell. Biol. 18, 5981-5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson, S. S., and Caron, M. G. (1998). G protein-coupled receptor adaptation mechanisms. Semin. Cell Dev. Biol. 9, 119-127. [DOI] [PubMed] [Google Scholar]
- Funamoto, S., Meili, R., Lee, S., Parry, L., and Firtel, R. A. (2002). Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell 109, 611-623. [DOI] [PubMed] [Google Scholar]
- Gether, U., Ballesteros, J. A., Seifert, R., Sanders-Bush, E., Weinstein, H., and Kobilka, B. K. (1997). Structural instability of a constitutively active G protein-coupled receptor. Agonist-independent activation due to conformational flexibility. J. Biol. Chem. 272, 2587-2590. [DOI] [PubMed] [Google Scholar]
- Hereld, D., Vaughan, R., Kim, J. Y., Borleis, J., and Devreotes, P. (1994). Localization of ligand-induced phosphorylation sites to serine clusters in the C-terminal domain of the Dictyostelium cAMP receptor, cAR1. J. Biol. Chem. 269, 7036-7044. [PubMed] [Google Scholar]
- Huang, Y. E., Iijima, M., Parent, C. A., Funamoto, S., Firtel, R. A., and Devreotes, P. (2003). Receptor-mediated regulation of PI3Ks confines PI(3,4,5)P3 to the leading edge of chemotaxing cells. Mol. Biol. Cell 14, 1913-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima, M., Huang, Y. E., and Devreotes, P. (2002). Temporal and spatial regulation of chemotaxis. Dev. Cell 3, 469-478. [DOI] [PubMed] [Google Scholar]
- Insall, R., Kuspa, A., Lilly, P. J., Shaulsky, G., Levin, L. R., Loomis, W. F., and Devreotes, P. (1994). CRAC, a cytosolic protein containing a pleckstrin homology domain, is required for receptor and G protein-mediated activation of adenylyl cyclase in Dictyostelium. J. Cell Biol. 126, 1537-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iranfar, N., Fuller, D., and Loomis, W. F. (2003). Genome-wide expression analyses of gene regulation during early development of Dictyostelium discoideum. Eukaryot. Cell 2, 664-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetopoulos, C., Jin, T., and Devreotes, P. (2001). Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science 291, 2408-2411. [DOI] [PubMed] [Google Scholar]
- Johnson, R. L., Vaughan, R. A., Caterina, M. J., van Haastert, P.J.M., and Devreotes, P. N. (1991). Overexpression of the cAMP receptor 1 in growing Dictyostelium cells. Biochemistry 30, 6982-6986. [DOI] [PubMed] [Google Scholar]
- Kim, J. Y., Caterina, M. J., Milne, J.L.S., Lin, K. C., Borleis, J. A., and Devreotes, P. N. (1997a). Random mutagenesis of the cAMP chemoattractant receptor, cAR1, of Dictyostelium. Mutant classes that cause discrete shifts in agonist affinity and lock the receptor in a novel activational intermediate. J. Biol. Chem. 272, 2060-2068. [DOI] [PubMed] [Google Scholar]
- Kim, J. Y., Soede, R.D.M., Schaap, P., Valkema, R., Borleis, J. A., van Haastert, P.J.M., Devreotes, P. N., and Hereld, D. (1997b). Phosphorylation of chemoattractant receptors is not essential for chemotaxis or termination of G-protein-mediated responses. J. Biol. Chem. 272, 27313-27318. [DOI] [PubMed] [Google Scholar]
- Kimmel, A. R. (1987). Different molecular mechanisms for cAMP regulation of gene expression during Dictyostelium development. Dev. Biol. 122, 163-171. [DOI] [PubMed] [Google Scholar]
- Klein, P. S., Sun, T. J., Saxe, C. L., 3rd, Kimmel, A. R., Johnson, R. L., and Devreotes, P. N. (1988). A chemoattractant receptor controls development in Dictyostelium discoideum. Science 241, 1467-1472. [DOI] [PubMed] [Google Scholar]
- Kumagai, A., Hadwiger, J. A., Pupillo, M., and Firtel, R. A. (1991). Molecular genetic analysis of two Gα protein subunits in Dictyostelium. J. Biol. Chem. 266, 1220-1228. [PubMed] [Google Scholar]
- Lu, Z. L., and Hulme, E. C. (1999). The functional topography of transmembrane domain 3 of the M1 muscarinic ACh receptor, revealed by scanning mutagenesis. J. Biol. Chem. 274, 7309-7315. [DOI] [PubMed] [Google Scholar]
- Maeda, M., Aubry, L., Insall, R., Gaskins, C., Devreotes, P. N., and Firtel, R. A. (1996). Seven helix chemoattractant receptors transiently stimulate mitogen-activated protein kinase in Dictyostelium. Role of heterotrimeric G proteins. J. Biol. Chem. 271, 3351-3354. [DOI] [PubMed] [Google Scholar]
- Maeda, M., and Firtel, R. A. (1997). Activation of the mitogen-activated protein kinase ERK2 by the chemoattractant folic acid in Dictyostelium. J. Biol. Chem. 272, 23690-23695. [DOI] [PubMed] [Google Scholar]
- Maeda, M., Lu, S., Shaulsky, G., Miyazaki, Y., Kuwayama, H., Tanaka, Y., Kuspa, A., and Loomis, W. F. (2004). Periodic signaling controlled by an oscillatory circuit that includes protein kinases ERK2 and PKA. Science 304, 875-878. [DOI] [PubMed] [Google Scholar]
- Mann, S.K.O., and Firtel, R. A. (1987). Cyclic AMP regulation of early gene expression in Dictyostelium discoideum: mediation via the cell surface cyclic AMP receptor. Mol. Cell Biol. 7, 458-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meili, R., Ellsworth, C., Lee, S., Reddy, T. B., Ma, H., and Firtel, R. A. (1999). Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium. EMBO J. 18, 2092-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, E. C., and Bourne, H. R. (2001). Receptor activation: what does the rhodopsin structure tell us? Trends Pharmacol. Sci. 22, 587-593. [DOI] [PubMed] [Google Scholar]
- Milne, J. L., Wu, L., Caterina, M. J., and Devreotes, P. N. (1995). Seven helix cAMP receptors stimulate Ca2+ entry in the absence of functional G proteins in Dictyostelium. J. Biol. Chem. 270, 5926-5931. [DOI] [PubMed] [Google Scholar]
- Nebl, T., and Fisher, P. R. (1997). Intracellular Ca2+ signals in Dictyostelium chemotaxis are mediated exclusively by Ca2+ influx. J. Cell Sci. 110, 2845-2853. [DOI] [PubMed] [Google Scholar]
- Overton, M. C., and Blumer, K. J. (2000). G-protein-coupled receptors function as oligomers in vivo. Curr. Biol. 10, 341-344. [DOI] [PubMed] [Google Scholar]
- Palczewski, K. et al. (2000). Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739-745. [DOI] [PubMed] [Google Scholar]
- Pang, K. M., Lynes, M. A., and Knecht, D. A. (1999). Variables controlling the expression level of exogenous genes in Dictyostelium. Plasmid 41, 187-197. [DOI] [PubMed] [Google Scholar]
- Parent, C. A., and Devreotes, P. N. (1996). Molecular genetics of signal transduction in Dictyostelium. Annu. Rev. Biochem. 65, 411-440. [DOI] [PubMed] [Google Scholar]
- Parent, C. A., Blacklock, B. J., Froehlich, W. M., Murphy, D. B., and Devreotes, P. N. (1998). G protein signaling events are activated at the leading edge of chemotactic cells. Cell 95, 81-91. [DOI] [PubMed] [Google Scholar]
- Parnot, C., Miserey-Lenkei, S., Bardin, S., Corvol, P., and Clauser, E. (2002). Lessons from constitutively active mutants of G protein-coupled receptors. Trends Endocrinol. Metab. 13, 336-343. [DOI] [PubMed] [Google Scholar]
- Parrish, W., Eilers, M., Ying, W., and Konopka, J. B. (2002). The cytoplasmic end of transmembrane domain 3 regulates the activity of the Saccharomyces cerevisiae G-protein-coupled α-factor receptor. Genetics 160, 429-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupillo, M., Insall, R., Pitt, G. S., and Devreotes, P. N. (1992). Multiple cyclic AMP receptors are linked to adenylyl cyclase in Dictyostelium. Mol. Biol. Cell 3, 1229-1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, E. M., and Gilman, A. G. (1980). Biochemical properties of hormonesensitive adenylate cyclase. Annu. Rev. Biochem. 49, 533-564. [DOI] [PubMed] [Google Scholar]
- Saliba, R. S., Munro, P. M., Luthert, P. J., and Cheetham, M. E. (2002). The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J. Cell Sci. 115, 2907-2918. [DOI] [PubMed] [Google Scholar]
- Samama, P., Cotecchia, S., Costa, T., and Lefkowitz, R. J. (1993). A mutation-induced activated state of the β2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 268, 4625-4636. [PubMed] [Google Scholar]
- Scheer, A., Costa, T., Fanelli, F., De Benedetti, P. G., Mhaouty-Kodja, S., Abuin, L., Nenniger-Tosato, M., and Cotecchia, S. (2000). Mutational analysis of the highly conserved arginine within the Glu/Asp-Arg-Tyr motif of the α1b-adrenergic receptor: effects on receptor isomerization and activation. Mol. Pharmacol. 57, 219-231. [PubMed] [Google Scholar]
- Strader, C. D., Fong, T. M., Graziano, M. P., and Tota, M. R. (1995). The family of G-protein-coupled receptors. FASEB J. 9, 745-754. [PubMed] [Google Scholar]
- Sussman, M. (1987). Cultivation and synchronous morphogenesis of Dictyostelium under controlled experimental conditions. Methods Cell Biol. 28, 9-29. [DOI] [PubMed] [Google Scholar]
- Theibert, A., and Devreotes, P. N. (1986). Surface receptor-mediated activation of adenylate cyclase in Dictyostelium. Regulation by guanine nucleotides in wild-type cells and aggregation deficient mutants. J. Biol. Chem. 261, 15121-15125. [PubMed] [Google Scholar]
- van Haastert, P.J.M. (1985a). The modulation of cell surface cAMP receptors from Dictyostelium discoideum by ammonium sulfate. Biochim. Biophys. Acta 845, 254-260. [DOI] [PubMed] [Google Scholar]
- van Haastert, P.J.M. (1985b). cAMP activates adenylate and guanylate cyclase of Dictyostelium discoideum cells by binding to different classes of cell-surface receptors. A study with extracellular Ca2+. Biochim. Biophys. Acta 846, 324-333. [DOI] [PubMed] [Google Scholar]
- van Haastert, P.J.M., and van der Heijden, P. R. (1983). Excitation, adaptation, and deadaptation of the cAMP-mediated cGMP response in Dictyostelium discoideum. J. Cell Biol. 96, 347-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan, R. A., and Devreotes, P. N. (1988). Ligand-induced phosphorylation of the cAMP receptor from Dictyostelium discoideum. J. Biol. Chem. 263, 14538-14543. [PubMed] [Google Scholar]
- Wallraff, E., Welker, D.L., Williams, K. L., and Gerisch, G. (1984). Genetic analysis of a Dictyostelium discoideum mutant resistant to adenosine 3′:5′-cyclic phosphorothioate, an inhibitor of wild-type development. J. Gen. Microbiol. 130, 2103-2114. [Google Scholar]
- Wang, M., van Haastert, P.J.M., Devreotes, P. N., and Schaap, P. (1988). Localization of chemoattractant receptors on Dictyostelium discoideum cells during aggregation and down-regulation. Dev. Biol. 128, 72-77. [DOI] [PubMed] [Google Scholar]
- Wu, L., Valkema, R., van Haastert, P.J.M., and Devreotes, P. N. (1995). The G protein β subunit is essential for multiple responses to chemoattractants in Dictyostelium. J. Cell Biol. 129, 1667-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, Z., Zhang, N., Murphy, D. B., and Devreotes, P. N. (1997). Dynamic distribution of chemoattractant receptors in living cells during chemotaxis and persistent stimulation. J. Cell Biol. 139, 365-374. [DOI] [PMC free article] [PubMed] [Google Scholar]