

Abstract
Tubulogenesis by epithelial cells regulates kidney, lung, and mammary development, whereas that by endothelial cells regulates vascular development. Although functionally dissimilar, the processes necessary for tubulation by epithelial and endothelial cells are very similar. We performed microarray analysis to further our understanding of tubulogenesis and observed a robust induction of regulator of G protein signaling 4 (RGS4) mRNA expression solely in tubulating cells, thereby implicating RGS4 as a potential regulator of tubulogenesis. Accordingly, RGS4 overexpression delayed and altered lung epithelial cell tubulation by selectively inhibiting G protein-mediated p38 MAPK activation, and, consequently, by reducing epithelial cell proliferation, migration, and expression of vascular endothelial growth factor (VEGF). The tubulogenic defects imparted by RGS4 in epithelial cells, including its reduction in VEGF expression, were rescued by overexpression of constitutively active MKK6, an activator of p38 MAPK. Similarly, RGS4 overexpression abrogated endothelial cell angiogenic sprouting by inhibiting their synthesis of DNA and invasion through synthetic basement membranes. We further show that RGS4 expression antagonized VEGF stimulation of DNA synthesis and extracellular signal-regulated kinase (ERK)1/ERK2 and p38 MAPK activation as well as ERK1/ERK2 activation stimulated by endothelin-1 and angiotensin II. RGS4 had no effect on the phosphorylation of Smad1 and Smad2 by bone morphogenic protein-7 and transforming growth factor-β, respectively, indicating that RGS4 selectively inhibits G protein and VEGF signaling in endothelial cells. Finally, we found that RGS4 reduced endothelial cell response to VEGF by decreasing VEGF receptor-2 (KDR) expression. We therefore propose RGS4 as a novel antagonist of epithelial and endothelial cell tubulogenesis that selectively antagonizes intracellular signaling by G proteins and VEGF, thereby inhibiting cell proliferation, migration, and invasion, and VEGF and KDR expression.
INTRODUCTION
Biological tubes comprise a major component of multicellular organisms and function in the delivery of gases and nutrients to tissues as well as the removal of their metabolic by-products (Hogan and Kolodziej, 2002). Tubulogenesis by epithelial cells gives rise to highly branched tubule networks of the lung, kidney, mammary, and other tissues, whereas that by endothelial cells gives rise to the vascular network. Although tubes formed by epithelial and endothelial cells perform a variety of distinct and specialized functions, the cellular processes necessary for tubule formation by either cell type are surprisingly similar (Hogan and Kolodziej, 2002). In particular, tubulation by epithelial and endothelial cells is coupled to their acquisition of polarity and to their proliferation, invasion, and migration toward the site of new tubule formation (Carmeliet, 2000; Hogan and Kolodziej, 2002; Kerbel and Folkman, 2002).
Endothelial cell tubulogenesis (i.e., angiogenesis) is a highly regulated process whereby new blood vessels form from preexisting vessels. Angiogenesis is essential to many biological processes, including embryonic development, wound repair, and the female reproductive cycle (Carmeliet, 2000). Conversely, uncoordinated or inappropriate angiogenesis is vital to the pathogenicity of many human diseases, such as arthritis, diabetic retinopathy, and cancer (Folkman, 1995; Carmeliet and Jain, 2000). Given the importance of angiogenesis to carcinogenesis (Folkman, 1995; Carmeliet and Jain, 2000; Kerbel and Folkman, 2002), a basic knowledge of the mechanisms and molecules that regulate endothelial cell tubulogenesis are important for the development of effective antiangiogenic treatments (Kerbel and Folkman, 2002). In particular, molecules that promote the resolution phase of angiogenesis may one day be exploited to inhibit neovascularization.
The role of growth factors and cytokines, particularly vascular endothelial growth factor (VEGF) and basis fibroblast growth factor (bFGF), in endothelial cell tubulogenesis (Carmeliet, 2000; Carmeliet and Jain, 2000; Kerbel and Folkman, 2002) and hepatocyte growth factor in epithelial cell tubulogenesis (Matsumoto and Nakamura, 2001; Hogan and Kolodziej, 2002) is firmly established. In comparison, the role of G proteins and G protein-coupled receptors (GPCRs) in epithelial and endothelial tubulogenesis is relatively unexplored. Recent studies have shown that stimulators of GPCRs, such as thrombin, angiotensin II (Ang II), endothelin-1 (ET-1), and prokineticin I and II couple to regulation of angiogenesis (Williams et al., 1995; Richard et al., 2001; Bagnato and Spinella, 2002; Lin et al., 2002; Masuda et al., 2002; Spinella et al., 2002). In addition, endothelial cell G proteins, particularly Gαq and Gα11, interact with and mediate intracellular signaling stimulated by VEGF KDR receptors (Zeng et al., 2002, 2003). Therefore, molecules that regulate GPCR activity also may function as regulators of tubulogenesis. Members of the regulator of G protein signaling (RGS) family of proteins activate the intrinsic GTP hydrolysis activity of Gαi and Gαq subunits, thus modulating G protein signaling by shortening the duration that GTP-bound Gα subunits remain active (Berman and Gilman, 1998; Ross and Wilkie, 2000; Wieland and Mittmann, 2003). The RGS family is comprised of at least 25 proteins that share a conserved ∼120-aa RGS motif. Through their GAP and effector activities, RGS proteins regulate a wide range of cellular functions, including cell migration, proliferation, and mitogen-activated protein (MAP) kinase activities (Berman and Gilman, 1998; Ross and Wilkie, 2000; Wieland and Mittmann, 2003).
Despite recent advance in deciphering tubulogenic mechanisms, our understanding of this process remains incomplete. We therefore conducted a microarray-based screen to identify the mRNAs that were differentially expressed during epithelial cell tubulogenesis, and subsequently compared these findings with those obtained in tubulating endothelial cells. We observed that RGS4 mRNA was up-regulated significantly solely in tubulating epithelial and endothelial cells compared with nontubulating control cells, suggesting an involvement of RGS4 during tubulogenesis. We show that RGS4 is a pleiotropic inhibitor of epithelial and endothelial cell tubulogenesis, doing so by inhibiting G protein- and VEGF-mediated activation of MAP kinases (e.g., ERK1/ERK2 and p38 MAPK), which results in diminished cell proliferation, migration, and invasion. Moreover, we demonstrate that epithelial cell expression of constitutively active MKK6, an activator of p38 MAPK, rescues the tubulogenic defects and repressed expression of VEGF mediated by RGS4. Finally, we show that constitutive RGS4 expression inhibits VEGF-stimulated DNA synthesis in endothelial cells by inhibiting VEGF receptor-2 (KDR) expression. Collectively, our findings establish RGS4 as a novel, general antagonist of epithelial and endothelial cell tubulogenesis.
MATERIALS AND METHODS
Materials
Recombinant human VEGF165, transforming growth factor-β1 (TGF-β1), and bone morphogenic protein-7 (BMP-7) were obtained from R&D Systems (Minneapolis, MN), whereas Ang II, ET-1, and mastoparan and its inactive analog were purchased from Calbiochem (San Diego, CA). Recombinant human epidermal growth factor (EGF) and anisomycin were obtained from Upstate Biotechnology (Charlottesville, VA) and Sigma-Aldrich (St. Louis, MO), respectively. cDNAs encoding constitutively active MKK6 (MKK6-EE) and VEGF receptor-2 (VEGFR2 or KDR) were generously provided by Drs. Lynn Heasley (University of Colorado Health Sciences Center) and Jacques Huot (Laval University, Quebec, Canada), respectively. Wild-type (pVEGF/K) and HIF-1α-deficient (pVEGF/P) luciferase constructs were kindly supplied by Dr. J. Silvio Gutkind (National Institute of Dental and Craniofacial Research, Bethesda, MD). The constitutively active Gαq and Gα11 cDNAs were purchased from Guthrie Research Institute (Sayre, PA). Mouse brain microvascular MB114 endothelial cells were kindly provided by Dr. Michael Hart (University of Wisconsin). All additional supplies or reagents were routinely available.
Cell Culture
Mink lung Mv1Lu epithelial cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). Murine brain microvascular MB114 endothelial cells were cultured in DMEM supplemented with 10% FBS, 1× essential and nonessential amino acids, 50 μM β-mercaptoethanol, and 100 mM HEPES, pH 7.3.
Plasmids
A full-length human RGS4 cDNA was polymerase chain reaction (PCR) amplified from an expressed sequence tag (IMAGE clone 3920860) by using oligonucleotides that incorporated unique KpnI (N terminus) and SacII (C terminus) restriction sites for subcloning into the pcDNA3.1/Myc-His B vector (Invitrogen, Carlsbad, CA) to C-terminally tag RGS4 with Myc and His6 tags. A retroviral RGS4 vector was generated by PCR amplification of the full-length Myc-His6-tagged RGS4 cDNA with oligonucleotides containing unique XhoI (N terminus) and HpaI (C terminus) restriction sites. The resulting PCR product was subcloned into identical sites immediately upstream of the internal ribosome entry site (IRES) in the bicistronic retroviral vector pMSCV-IRES-GFP (Schiemann et al., 2002, 2003).
A retroviral vector for constitutively-active MKK6 [MKK6-EE, which contains S207E/T211E substitutions (Raingeaud et al., 1996)] was constructed by first shuttling a PCR-amplified MKK6-EE cDNA fragment through the pcDNA3.1/Myc-His B vector to C-terminally Myc-His6 tag MKK6-EE. The resulting tagged MKK6-EE cDNA fragment was PCR amplified using oligonucleotides containing BglII (N terminus) and HpaI (C terminus) restriction sites and subsequently ligated into corresponding sites in pMSCV-IRES-YFP.
All RGS4 and MKK6-EE inserts were sequenced on an Applied Biosystems 377A DNA sequencing machine.
Microarray Analysis
To identify genes expressed differentially during tubulogenesis, log phasegrowing Mv1Lu cultures were seeded onto 6-cm plates (3 × 106 cells/plate) supplemented with or without 4 ml of solidified Matrigel (diluted 3:1 in serum-free media [SFM]). Six hours later, the cells were gently washed twice with ice-cold phosphate-buffered saline (PBS) and were immediately solubilized in RNAzol (Tel-Test, Friendswood, TX; 1 ml for control and 20 ml for Matrigel) to isolate total RNA. 33P-Labeled cDNA probes were generated by reverse transcription of 2.8 μg of total RNA isolated from cells cultured either on plastic or Matrigel according to NIA Array unit protocols. Microarrays containing 1152 human cDNAs (generously provided by Dr. John Cambier, National Jewish Medical and Research Center) were prehybridized for 6 h at 42°C in MicroHyb solution (Research Genetics, Huntsville, AL) supplemented with 2.5 μg/ml human Cot-1 DNA and 8 μg/ml polyA; they were subsequently hybridized overnight at 42°C in 4 ml of prehybridization buffer containing radiolabeled cDNA probes (2 × 106 cpm/microarray). The next morning, the microarrays were washed in 2× SSC/0.1% SDS for 10 min at 50°C and then for 10 min at room temperature. Microarrays were exposed to a phosphor screen for 72 h and subsequently were scanned on an Amersham Biosciences Storm PhosphorImager. cDNA signal intensities were measured using ImageQuant software (Amersham Biosciences).
mRNAs differentially expressed in cells cultured on Matrigel compared with those cultured on plastic were identified as follows. First, the average cDNA signal intensity was determined by adding all cDNA signals per microarray and dividing this sum by the total number of cDNAs present in the microarray. Second, individual cDNA signals were divided by the corresponding array average cDNA signal intensity to yield individual cDNA signal ratios. Finally, the overall ratios of Matrigel versus plastic cDNA signal intensity were calculated by dividing a given individual Matrigel cDNA signal ratio by its corresponding plastic cDNA signal ratio. Overall ratios ≥2 were considered significant. To reduce false positives, the microarrays were performed three times and genes expressed differentially in at least two experiments were considered true positives.
Retroviral Infections
Control (i.e., pMSCV-IRES-GFP or pMSCV-IRES-YFP), RGS4, or MKK6-EE retroviral supernatants were produced by EcoPack2 retroviral packaging cells (BD Biosciences Clontech, Palo Alto, CA) and used to infect Mv1Lu or MB114 cells as described in Schiemann et al. (2003). Infected cells were analyzed 48 h postinfection and the highest 10% of green fluorescent protein (GFP)-, yellow fluorescent protein (YFP)-, or GFP/YFP-expressing cells were collected on a MoFlo cell sorter (DakoCytomation Colorado, Fort Collins, CO). Isolated cells were subsequently expanded to yield stable polyclonal populations of control, RGS4-, or RGS4/MKK6-EE-expressing cells. The resulting populations of Mv1Lu and MB114 cells were ≥90% positive for transgene expression and were used to analyze the effects of RGS4 and MKK6-EE on tubule development and cell proliferation, migration, and invasion.
Northern Blotting
Mv1Lu cells were cultured on plastic or Matrigel for 6 h and subsequently were harvested in RNAzol (Tel-Test) to isolate total RNA. Afterward, 1.5 μg of total RNA was fractionated through 1.7% agarose/formaldehyde gels and transferred to nylon membrane. Immobilized RNA was probed with a random primed 32P-labeled human RGS4 cDNA for 1 h at 68°C in ExpressHyb (BD Biosciences Clontech) and subsequently was washed according to the manufacturer's recommendations. RGS4 mRNA was visualized by autoradiography.
Quantitative Real-Time PCR Assay
Three-dimensional MB114 tubule cultures were prepared by mixing 2.4 × 106 cells in 1.5 ml of type I collagen, which then was allowed to solidify in six-well plates. Once solidified, the three-dimensional endothelial cell cultures were overlaid with media and incubated for varying times at 37°C, whereupon total RNA was isolated using the RNAqueous kit (Ambion, Austin, TX). Isolated total RNA was further purified by phenol/chloroform extraction and ethanol precipitation. cDNAs were synthesized by reverse transcribing total RNA (0.5 μg/reaction) with random hexamers and iScript reverse transcriptase according to the manufacturer's recommendations (Bio-Rad, Hercules, CA). Afterward, the reverse transcription reactions were diluted 10-fold and subjected to real-time PCR analysis (25 μl/reaction) by using the SYBR Green PCR system (Applied Biosystems, Foster City, CA) containing 2.5 μl of cDNA template and 0.1 μM of the following RGS oligonucleotides pairs: 1) RGS4, forward: 5′GAAGAAGATTTTCAACCTGATGG; reverse: 5′GAACTCTTGGCTCCTTTCTGC; 2) RGS5, forward: 5′GCGGAGAAGGCAAAGCAAA; reverse: 5′CGGTTCCACCAGGTTCTTCAT; 3) RGS7, forward: 5′GGCACCTTCTACCGGTTTCAG; reverse: 5′ GCCTTGCCTTGTTTTGCATT; and 4) RGS10, forward: 5′GGAAGCAGATGCAGGAAAAGG; reverse: 5′TGGTCCTGGAGCTTTTGGAA. Quantitative real-time PCR reactions were performed and analyzed on an ABI Prism 7000 sequence detection system. Relative RGS4 expression levels were determined according to the manufacturer's recommendations and subsequently normalized to corresponding 18S or GAPDH RNA signals.
[3H]Thymidine Incorporation Assay
The effect of RGS4 on cell proliferation was measured essentially as described previously (Schiemann et al., 2002). Briefly, control or RGS4-expressing cells were cultured onto 96-well plates at a density of 10,000 cells/well (Mv1Lu cells) or at 5000 cells/well (MB114 cells) in DMEM containing 10% FBS. Newly synthesized DNA was radiolabeled 24 h later by adding [3H]thymidine to the wells for 4 h. Afterward, the cells were washed twice with ice-cold PBS, precipitated with ice-cold 5% trichloroacetic acid, and subsequently solubilized with 0.5 N NaOH before scintillation counting to determine radionucleotide incorporation into DNA.
The effects of RGS4 expression in DNA synthesis stimulated by recombinant human VEGF165 was determined by culturing control and RGS4 expressing MB114 cells as described above. After adhering overnight, the cells were washed 2× in PBS and subsequently cultured for 24 h in SFM supplemented with increasing concentrations of VEGF165 (0 → 35 ng/ml). Newly synthesized DNA was radiolabeled and quantified as described above.
Migration and Invasion Assays
The effect of RGS4 on cell migration was measured using a modified Boydenchamber assay as described previously (Schiemann et al., 2002). Briefly, the underside of a porous membrane (8-μm pore, 24-well format; BD Biosciences, San Jose, CA) was coated overnight at 4°C in PBS containing 50 μg/ml fibronectin (Invitrogen). Afterward, the fibronectin solution was removed and replaced with 750 μl of serum-free DMEM containing 0.1% bovine serum albumin (BSA) (SFM/0.1% BSA). Control or RGS4-expressing cells were cultured in the upper chambers at a density of 100,000 cells/well in SFM/0.1% BSA for 18-24 h at 37°C. Afterward, the cells were washed twice in ice-cold PBS and immediately fixed for 15 min with 95% ethanol. Cells remaining in the upper chambers were removed with a cotton swab, whereas those remaining in the lower chamber were stained with crystal violet. Migrating cells were enumerated by manual counting under a light microscope.
The effect of RGS4 on MB114 cell invasion was performed as described previously (Schiemann et al., 2002). Briefly, upper chambers were coated with 100 μl of diluted Matrigel (1:50 in SFM), which was allowed to evaporate to dryness overnight at room temperature. Matrigel mixtures were rehydrated the next morning in 500 μl of serum-free DMEM. Afterward, control and RGS4-expressing cells were prepared and cultured in upper chambers as for migration assays. Cellular invasion was induced by adding 2% serum to lower chambers and was allowed to proceed for 48 h at 37°C. Subsequently, Matrigel-invading cells were fixed and stained with crystal violet as described above.
Protein Kinase Activation
Control and RGS4-expressing Mv1Lu or MB114 cells were cultured onto six-well plates. On reaching ∼90% confluence, cells were washed twice with PBS and serum starved in DMEM for 1.5 h (Mv1Lu) or overnight (MB114). Mv1Lu cells were stimulated for 15 min with either 5% serum, 50 μM mastoparan, or 50 μM mastoparan 17. MB114 cells were stimulated with VEGF165 (50 ng/ml), ET-1 (0.1 μM), or Ang II (1 μM) for 0-60 min. After agonist stimulation, the cells were washed with ice-cold PBS and lysed with buffer H/1% Triton X-100 (Schiemann et al., 2002). After incubation on ice for 30 min, whole cell lysates were clarified by microcentrifugation, fractionated through 10% SDS-PAGE, and transferred electrophoretically to nitrocellulose. Protein kinase activation was measured by Western blotting with antibodies recognizing either phosphorylated p38 MAPK (1:500 dilution) or ERK1/2 (1:1000 dilution) (Cell Signaling Technology, Beverly, MA). Differences in protein loading were monitored by stripping and reprobing Western blots with antibodies (1:1000 dilution) against either p38 MAPK or ERK1/2 (Santa Cruz Biotechnology, Santa Cruz, CA). Western blots were developed by enhanced chemiluminescence.
p38 MAPK phosphotransferase activity was determined using an in vitro protein kinase assay that measured the ability of immunoprecipitated p38 MAPK to phosphorylate recombinant ATF-2 essentially as described previously (Schiemann et al., 1997, 1999). Briefly, control or RGS4-expressing Mv1Lu cells (1.2 × 106 cell/well) were cultured onto Matrigel cushions in six-well plates for 5 h at 37°C. Afterward, the cells were washed twice in ice-cold PBS and lysed in buffer H/1% Triton X-100 (Schiemann et al., 1997; Perlman et al., 2001). p38 MAPK was immunoprecipitated from clarified whole cell extracts and the resulting immunocomplexes were incubated with 0.5 μg of ATF-2 (1-96; Santa Cruz Biotechnology) in 30 μl of protein kinase assay buffer (Schiemann et al., 1997; Perlman et al., 2001) for 30 min at 30°C. The reactions then were fractionated through 10% SDS-PAGE and transferred to nitrocellulose before visualizing phosphorylated ATF-2 by autoradiography. In some experiments, ATF-2 phosphorylation was detected using phospho-specific ATF-2 antibodies according to the manufacturer's recommendations (Cell Signaling Technology). Protein loading differences were monitored by immunoblotting with anti-p38 MAPK antibodies as described above.
Smad Phosphorylation
Analysis of Smad1 and Smad2 phosphorylation was performed as described previously (Schiemann et al., 2003). Briefly, control and RGS4-expressing Mv1Lu or MB114 cells were cultured onto 24-well plates at a density of 200,000 cells/well and allowed to adhere overnight. The next morning, the cells were washed twice in PBS and serum-starved for 2 h before stimulation with TGF-β1 (5 ng/ml) or recombinant BMP-7 (1 μg/ml) as indicated. Phosphorylation of Smad1 and Smad2 was determined by immunoblotting with a 1:500 dilution of either anti-phospho-Smad1 or -Smad2 polyclonal antibodies (Cell Signaling Technology). The resulting immunocomplexes were visualized by enhanced chemiluminescence. Differences in protein loading were monitored by reprobing stripped membranes with anti-ERK1 antibodies as described above.
Luciferase Reporter Gene Assay
VEGF expression in Mv1Lu cells was analyzed by measuring luciferase activity driven by full-length (pVEGF/K) and truncated (pVEGF/P) VEGF promoters (Sodhi et al., 2001). Briefly, control, MKK6-, RGS4-, or MKK6/RGS4-expressing Mv1Lu cells were cultured onto 24-well plates at a density of 45,000 cells/well and allowed to adhere overnight. The cells were transiently transfected by overnight exposure to LT1 liposomes (Mirus, Madison, WI) containing 300 ng/well of either pVEGF/K- or pVEGF/P-luciferase, and 100 ng/well of CMV-β-gal, which was used to control for differences in transfection efficiency. The next morning, the cells were washed twice with PBS and incubated in serum-free media for an additional 24 h. Afterward, luciferase and β-gal activities contained in detergent-solubilized cell extracts were determined. Data are the mean (± SEM) luciferase activities of three independent experiments normalized to corresponding untreated GFP cells.
Secretory Protein Expression Assay
Cos-7 cells were cultured onto six-well plates at a density of 400,000 cells/well. The cells were transiently transfected the next day by overnight exposure to LT1 liposomes containing 2 μg/well of either pcDNA3-VEGFR2 (KDR), pcDNA1-TGF-β type II receptor (TβR-II; Lin et al., 1992), or pcDNA3.1-Fibulin-5 (FBLN-5; (Schiemann et al., 2002), together with or with-out an equivalent quantity of pcDNA3.1-RGS4-Myc-His. Forty-eight hours posttransfection, the effects of RGS4 on secretory protein expression were determined as follows: VEGFR2 (KDR) by immunoprecipitation and immunoblotting with anti-KDR antibodies (1:1000 dilution; Santa Cruz Biotechnology); TβR-II by iodinated TGF-β1 binding and cross-linking assay as described previously (Schiemann et al., 2004); and FBLN-5 by Ni2+-affinity chromatography and Myc immunoblotting as described previously (Schiemann et al., 2002).
RESULTS
Tubulogenesis Induces RGS4 Expression in Mv1Lu Epithelial Cells
To identify genes expressed differentially during epithelial cell tubulation, we developed a rapid in vitro tubulogenesis assay that used the ability of mink lung Mv1Lu epithelial cells to form tubule-like structures when cultured onto the synthetic basement membrane Matrigel. As shown in Figure 1A, Mv1Lu cells seeded onto Matrigel migrated rapidly to establish cell-cell contacts (within 1.5 h), leading to the formation of well developed tubule-like structures by 6 h that seemed similar to those formed by other epithelial and endothelial cells. When examined over time, Mv1Lu cell tubule development proceeded for ∼12 h and then arrested (Figure 2). To identify genes operant in regulating Mv1Lu cell tubulogenesis, we isolated total RNA from Mv1Lu cells cultured for 6 h on either plastic (i.e., control) or Matrigel (i.e., tubulogenesis). We subsequently reverse transcribed these RNAs into radiolabeled cDNAs that were hybridized to microarrays containing 1152 distinct human cDNAs (Figure 1B).
Figure 1.
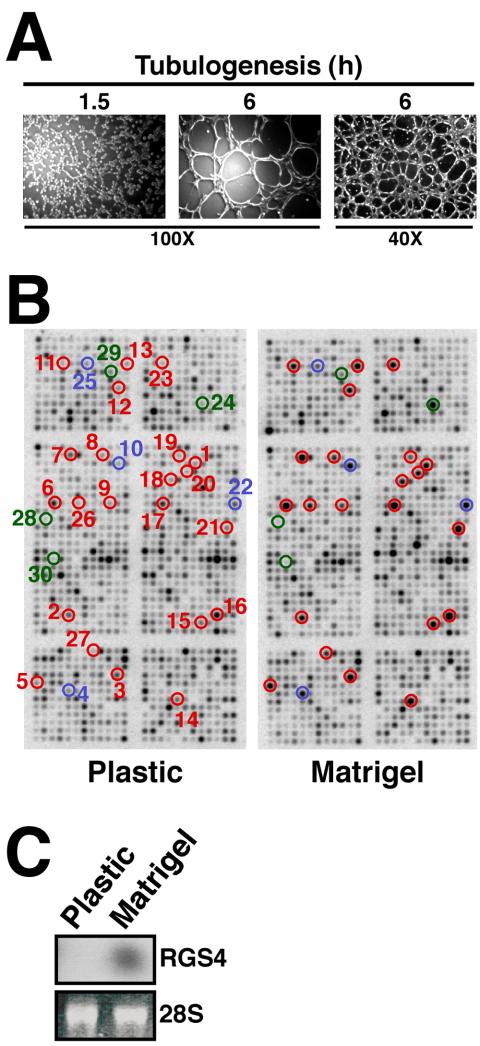
Tubulogenesis induces RGS4 expression in Mv1Lu epithelial cells. (A) Mv1Lu cells were plated onto Matrigel-coated wells, and tubule formation was monitored at varying times. Bright field pictures were captured on Nikon Diaphot microscope. (B) Total mRNA was extracted from Mv1Lu cells cultured for 6 h on plastic (i.e., control) or Matrigel and subsequently was used to synthesize cDNA probes that were hybridized to microarrays containing 1152 human genes. Individual gene identities, accession numbers, and fold-regulation are provided in Table 1. RGS4 and other angiogenesis-regulated genes are circled: blue, genes previously associated with angiogenesis; red, genes newly associated with angiogenesis; and green, RGS genes. (C) Total RNA obtained from 6 h cultures was fractioned and hybridized with a radiolabeled human RGS4 probe (top). Differences in mRNA loading were monitored by ethidium bromide staining of the 28S rRNA (bottom).
Figure 2.
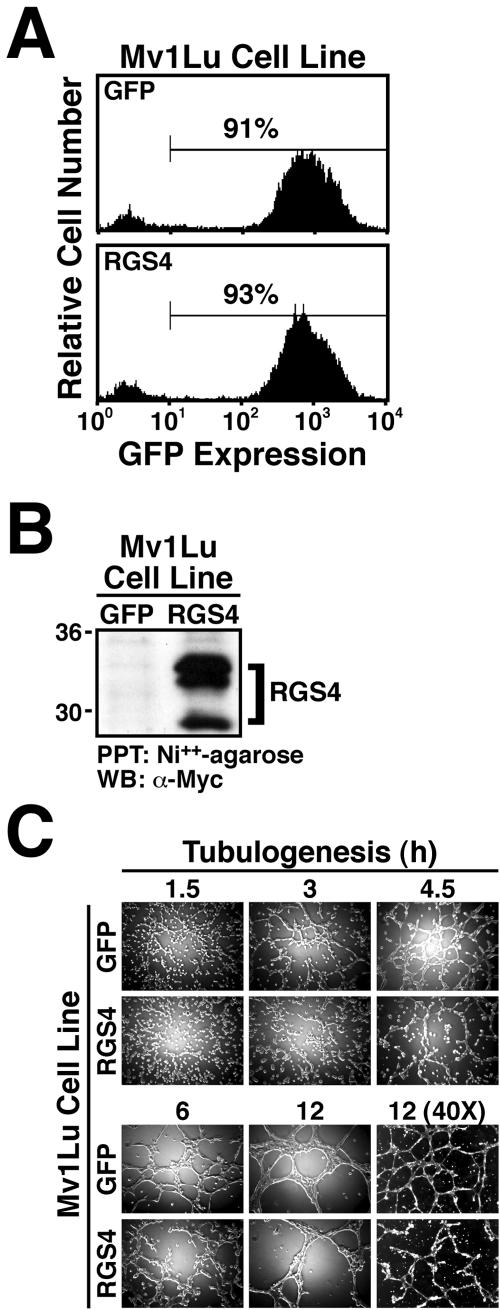
RGS4 expression inhibits Mv1Lu cell tubulogenesis. (A) Mv1Lu cells were infected with either GFP control or RGS4 retrovirus, and the infected cells were FACS-sorted by GFP expression (highest 10%). Shown are the resulting stable populations of control (top) and RGS4-expressing (bottom) Mv1Lu cells that expressed equivalent levels of GFP at a positivity rate of ≥90%. (B) Mycimmunoreactivity of proteins captured by nickel affinity chromatography from detergent-solubilized whole cell extracts demonstrates that Mv1Lu cells transduced with RGS4 retrovirus constitutively express recombinant RGS4 protein. (C) Mv1Lu cells stably expressing either GFP or RGS4 were seeded onto Matrigel. Tubule formation was monitored at varying times as indicated.
Using this approach, we identified 27 distinct Mv1Lu cell genes that were consistently up-regulated ≥2-fold during tubulogenesis (Table 1). Our results showed that a remarkable number of genes known to be differentially expressed during endothelial tubulogenesis also were differentially regulated during epithelial cell tubulogenesis. These genes include the Flt-1 VEGF receptor, hypoxia-inducible factor-1α (HIF-1α), the erythropoietin receptor, and the ET(A) endothelin receptor, Caspase 4, TRAIL (TNFSF10), and Gelsolin (Pedram et al., 1997; Bagnato and Spinella, 2002; Jaquet et al., 2002; Spinella et al., 2002; Yasuda et al., 2002) (Table 1 and Figure 1B). In addition to identifying these known angiogenic genes, we also identified several genes not previously associated with either epithelial or endothelial tubulogenesis (Table 1 and Figure 1B). Included in this group of novel tubulogenesis-regulated genes was the RGS4, whose expression was consistently up-regulated ≥8-fold in tubulating cells compared with control cells (Table 1 and Figure 1B). RGS4 protein expression was undetectable by immunoblot analysis of nontubulating and tubulating Mv1Lu cells (our unpublished data). Whether the poor immunoreactivity of our RGS4 antibodies results from species epitope differences remains to be determined. However, Northern blotting total RNA isolated from control and Matrigel-cultured Mv1Lu cells confirmed that RGS4 expression was indeed up-regulated during tubulogenesis (Figure 1C). Notably, three other RGS proteins present in the microarray (e.g., RGS1, 7, and 12) did not show differential expression in cells cultured on Matrigel compared with plastic (Figure 1B). Thus, tubulogenesis significantly induced RGS4 expression in lung epithelial cells.
Table 1.
Genes differentially expressed during Mv1Lu cell tubulogenesis
| Gene category | Array no. | Fold-up-regulation | Accession no. |
|---|---|---|---|
| Angiogenesis associated | |||
| HIF-1α | 4 | 4.8 | 29165 |
| Flt-1 | 10 | 7.3 | X51602 |
| ET(A) | 22 | 3.6 | X61950 |
| EPO-R | 25 | 2.0 | M60459 |
| Growth factors and cytokines/receptors/modulators | |||
| CRHBP | 2 | 2.2 | X58022 |
| IGFBP4 | 3 | 3.2 | M62403 |
| IGF2R | 6 | 3.5 | Y00285 |
| ADRβ3 | 14 | 5.6 | X70811 |
| IGF2 | 17 | 3.4 | AH002703 |
| BMP4 | 21 | 6.9 | U43842 |
| IRF2 | 23 | 7.6 | X15949 |
| Cell cycle/apoptosis | |||
| TNFSF10 | 9 | 2.4 | U37518 |
| CDK6 | 13 | 6.9 | H73724 |
| CASP4 | 20 | 5.2 | Z48810 |
| Signaling molecules/transcription factors | |||
| MAPK10 | 5 | 5.5 | U07620 |
| PKCβ1 | 7 | 2.2 | X06318 |
| GFRA2 | 8 | 3.2 | AF002700 |
| IFNαR2 | 11 | 6.5 | X77722 |
| JMJ | 15 | 3.6 | U57592 |
| PKCα | 19 | 2.2 | X52479 |
| RGS4 | 24 | 8.1 | U27768 |
| ATF2 | 27 | 4.5 | M86842 |
| Cytoskeletal proteins | |||
| GSN | 1 | 4.0 | X04412 |
| PFN1 | 12 | 3.9 | J03191 |
| Miscellaneous | |||
| UBL1 | 16 | 3.2 | U82117 |
| ACO2 | 18 | 3.9 | U87939 |
| NR4A2 | 26 | 4.3 | X75198 |
| Nonangiogenic regulated RGS proteins | |||
| RGS1 | 28 | 1.0 | X73427 |
| RGS7 | 29 | 1.0 | R43370 |
| RGS12 | 30 | 1.0 | NM002926 |
Radiolabeled cDNA probes prepared from total RNA extracted from Mv1Lu cells cultured on plastic or Matrigel for 6 h were hybridized to microarrays containing 1152 genes. This process was performed three times and genes differentially expressed ≥2-fold in two or more experiments are shown. In Figure 1, individual genes are numbered and circled: blue, genes previously associated with angiogenesis; red, genes newly associated with angiogenesis; and green, RGS genes.
RGS4 Expression Inhibits Mv1Lu Cell Tubulogenesis
Because RGS4 expression was up-regulated in tubulating Mv1Lu cells, we speculated that RGS4 functions to maintain the fidelity of tubule formation by Mv1Lu cells. A corollary is that constitutive RGS4 expression might negatively impact Mv1Lu cell tubulogenesis. To test this hypothesis, Mv1Lu cells expressing the ecotropic receptor (Liu et al., 1997) were infected with either control (i.e., pMSCV-IRES-GFP) or human RGS4 retrovirus. Subsequently, cells that expressed GFP were isolated by flow cytometry to establish stable polyclonal populations of GFP control and RGS4-expressing Mv1Lu cells. Figure 2A shows that the resulting Mv1Lu cell lines had purities ≥90% and expressed GFP indistinguishably. Moreover, Mv1Lu cells infected with RGS4 retrovirus expressed high levels of RGS4 protein, whereas those infected with GFP control retrovirus were negative for expression of recombinant RGS4 protein (i.e., Myc-immunoreactivity; Figure 2B). We attempted to assess the degree of RGS4 overexpression by monitoring RGS4 protein and mRNA levels in control and RGS4-overexpressing cells; however, endogenous RGS4 expression was undetectable by either Western (our unpublished data) or Northern blotting (Figure 1C), indicating that RGS4 is not expressed in nontubulating Mv1Lu cells. Although currently unknown, we suspect that the multiple recombinant RGS4 species observed in transduced Mv1Lu cells results from either 1) palmitylation of RGS4 within its “RGS box” (Tu et al., 1999); 2) N-terminal truncation and arginylation of RGS4 (Davydov and Varshavsky, 2000); or 3) translation initiation from an alternative RGS4 start codon (Davydov and Varshavsky, 2000). Nonetheless, these stable populations of Mv1Lu cells were used to examine the effects of RGS4 expression on Mv1Lu cell tubulogenesis.
As shown in Figure 2C, GFP-expressing Mv1Lu cells rapidly formed tubules when cultured onto Matrigel. For instance, GFP-expressing Mv1Lu cells seeded onto Matrigel established visible cell-cell contacts by 1.5 h and tubules by 3 h (Figure 2C). By 12 h, GFP-expressing Mv1Lu cells formed intricate evenly spaced, highly branched tubule networks that uniformly covered the Matrigel surface (Figure 2C). In stark contrast, constitutive RGS4 expression significantly delayed and altered the formation of tubules by Mv1Lu cells. Whereas GFP-expressing Mv1Lu cells clearly established cell-cell contacts and formed tubules by 1.5 and 3 h, respectively, their RGS4-expressing counterparts required twice as much time to exhibit comparable development (Figure 2C). Although Mv1Lu cells expressing RGS4 did eventually form tubules (i.e., by 12 h), their appearance was vastly different from those formed by control cells. Indeed, tubules of RGS4-expressing Mv1Lu cells were readily distinguished from those of control cells by their 1) rough appearance, 2) uneven spacing, 3) poor branching, and 4) incomplete networks (Figure 2C). Thus, these findings demonstrate that constitutive RGS4 expression does indeed negatively impact Mv1Lu cell tubulogenesis, raising the possibility that RGS4 functions to inhibit epithelial cell tubulation.
RGS4 Inhibits Mv1Lu Cell Proliferation and Migration
We thus far have shown that RGS4 expression was upregulated after initiation of epithelial cell tubulation and that constitutive RGS4 expression negatively impacted tubule formation by epithelial cells. Given these results, we hypothesized RGS4 as a novel antagonist of epithelial cell tubulogenesis. To test this hypothesis, we measured the effects of RGS4 on several key events operant during tubule formation by epithelial cells, including cell proliferation, migration, and invasion.
Cell proliferation is an essential component of tubulogenesis (Carmeliet, 2000; Hogan and Kolodziej, 2002; Kerbel and Folkman, 2002). We therefore performed a [3H]thymidine incorporation assay to measure changes in DNA synthesis elicited by RGS4 expression in Mv1Lu cells. In accordance with its inhibitory effect on tubule formation, RGS4 expression significantly reduced DNA synthesis in Mv1Lu cells (Figure 3A). Thus, one mechanism whereby RGS4 may antagonize tubulogenesis is by attenuating cell proliferation.
Figure 3.
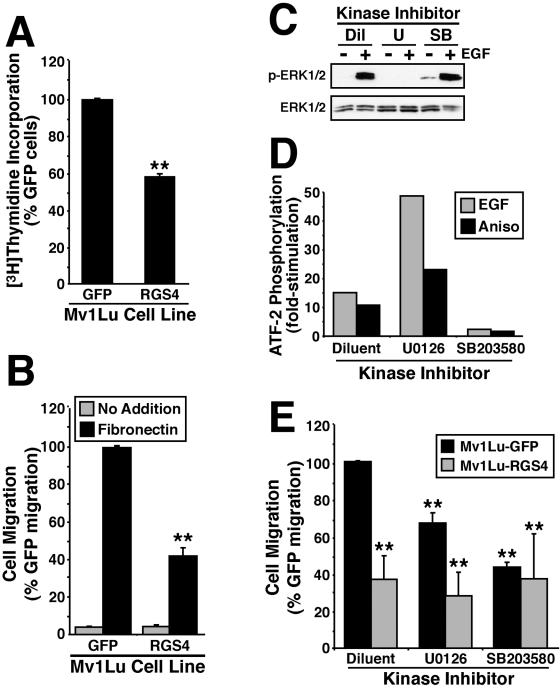
RGS4 inhibits Mv1Lu cell proliferation and migration. (A) Rates of DNA synthesis in Mv1Lu cells expressing either GFP or RGS4 were measured by a [3H]thymidine incorporation assay. The data are the means (± SEM) of three independent experiments presented as the percentage of [3H]thymidine incorporation relative to GFP-expressing cells. RGS4 expression significantly decreased DNA synthesis in Mv1Lu cells (**p < 0.05; Student's t test). (B) The migration of GFP- or RGS4-expressing cells to diluent (gray bars) or fibronectin (black bars) was performed in a modified Boyden-chamber for 24 h. Data are the mean (± SEM) of four independent experiments presented as the percentage of migration relative to GFP-expressing Mv1Lu cells. RGS4 expression significantly decreased Mv1Lu cell migration to fibronectin (**p < 0.05; Student's t test). (C) Quiescent Mv1Lu cells were incubated with diluent (Dil), 25 μM U0126 (U), or 10 μM SB203580 (SB) for 2 h before stimulation with EGF (100 ng/ml) for 10 min at 37°C. ERK1/ERK2 phosphorylation was determined by immunoblotting whole cell extracts with phospho-specific antibodies to ERK1/ERK2. Protein loading differences were monitored by reprobing stripped membranes with anti-ERK1 antibodies. Shown are representative immunoblots from single experiment that was repeated once with identical results. (D) Quiescent Mv1Lu cells were incubated with diluent, 25 μM U0126, or 10 μM SB203580 for 2 h before stimulation with EGF (100 ng/ml) or anisomycin (25 μg/ml; Aniso) for 40 min at 37°C. Active p38 MAPK was immunoprecipitated from whole cell extracts and used to phosphorylate recombinant ATF-2 in vitro, which was detected by immunoblotting with phospho-specific ATF-2 antibodies. Equal protein loading was monitored by immunoblotting fractionated whole cell extracts with anti-p38 MAPK antibodies. Data depict the fold-stimulations of ATF-2 phosphorylation induced by EGF or anisomycin in the absence or presence of MAP kinase inhibitors. (E) GFP- or RGS4-expressing Mv1Lu cells were allowed to migrate to fibronectin in the presence or absence of 25 μM U0126 or 10 μM SB203580 as indicated. Data are the mean (± SEM) of three (U0126) or two (SB203580) independent experiments presented as the percentage of migration relative to untreated GFP-expressing Mv1Lu cells. RGS4 expression and protein kinase inhibitors significantly decreased Mv1Lu cell migration to fibronectin (**p < 0.05; Student's t test).
Cell migration is also an essential component of tubulogenesis (Carmeliet, 2000; Hogan and Kolodziej, 2002; Kerbel and Folkman, 2002). We therefore performed a modified Boyden-chamber assay to measure changes in cell migration and invasion elicited by RGS4 expression in Mv1Lu cells. Figure 3B shows that RGS4 expression significantly reduced the migration of Mv1Lu cells to fibronectin. Because Mv1Lu cells fail to invade synthetic basement membranes (Albig and Schiemann, unpublished observation), we were unable to assess the effects of RGS4 expression on Mv1Lu cell invasion. To confirm that the antitubulogenic activity of RGS4 was not due to enhanced cell death, we stained control and RGS4-expressing Mv1Lu cells with 7-amino-actinomycin D (Via-Probe; BD Biosciences PharMingen, San Diego, CA) to monitor changes in cell viability by flow cytometry. As expected, RGS4 overexpression failed to affect Mv1Lu cell viability (our unpublished data).
Through their GTPase-activating activities, RGS super-family members terminate intracellular signals stimulated by G proteins (Berman and Gilman, 1998; Ross and Wilkie, 2000; Wieland and Mittmann, 2003), including those leading to MAP kinase activation. Mammalian MAP kinases (e.g., ERKs, JNKs, and p38 MAPKs) are essential to a variety of physiological processes, including the control of gene expression, programmed cell death, and cell proliferation (Garrington and Johnson, 1999; Chang and Karin, 2001). The activity of these protein kinases, particularly that of ERK1/ERK2 and p38 MAPK, are also important for cell migration (Rousseau et al., 1997; Cara et al., 2001; Chang and Karin, 2001). Our finding that RGS4-expressing Mv1Lu cells were defective in their migration to fibronectin (Figure 3B) lead us to hypothesize that RGS4 inhibits cell migration by attenuating MAP kinase activities. A corollary is that interventions designed to inhibit ERK1/ERK2 or p38 MAPK activity might elicit cell migration defects reminiscent of those imparted by RGS4 expression.
To test this hypothesis, we measured the migration of Mv1Lu cells to fibronectin in the absence or presence of 1) 25 μM U0126, which blocks ERK1/ERK2 activity by inhibiting their upstream activators MKK1/MKK2 (Figure 3C; Favata et al., 1998), or 2) 10 μM SB203580, which directly inhibits p38 MAPK activity (Figure 3D; Tong et al., 1997). Relative to control cells, treatment of Mv1Lu cells with either U0126 or SB203580 significantly reduced their migration to fibronectin (Figure 3E). Thus, ERK1/ERK2 and p38 MAPK activities are required for maximal migration of Mv1Lu cells. Moreover, the inhibition of MAP kinase activities, particularly that of p38 MAPK, elicited migration defects reminiscent of those produced by RGS4 expression. Interestingly, the migration defects of RGS4-expressing cells were not potentiated further by their treatment with either U0126 or SB203580 (Figure 3E). Thus, RGS4 functions as an upstream inhibitor of MAP kinases (e.g., ERK1/ERK2 and p38 MAPK) in Mv1Lu cells, resulting in their reduced ability to migrate to fibronectin.
Collectively, these findings identify RGS4 a pleiotropic inhibitor of epithelial cell tubulogenesis, doing so by inhibiting epithelial cell proliferation and migration. They also suggest that RGS4 targets MAP kinases when inhibiting Mv1Lu cell migration.
RGS4 Selectively Inhibits p38 MAPK Activation in Mv1Lu Cells
We next sought to determine whether RGS4 expression inhibits MAP kinase activity in Mv1Lu cells. To do so, GFP- or RGS4-expressing Mv1Lu cells were serum starved for 1.5 h before stimulation with either 5% serum or the G protein activator mastoparan (Higashijima et al., 1988), which preferentially activates Gαi and Gαo subunits (Higashijima et al., 1990). The activation status of MAP kinases was determined by immunoblot analysis by using phospho-specific anti-MAP kinase antibodies. As shown in Figure 4A, treatment of GFP-expressing Mv1Lu cells with serum or mastoparan stimulated p38 MAPK activation to varying extents. As expected, the inactive mastoparan analog mastoparan 17 (Kowluru et al., 1995) failed to stimulate p38 MAP activity in these same cells, thus demonstrating the specificity of mastoparan for stimulating MAP kinases in Mv1Lu cells. The immunoblot in Figure 4A also shows that p38 MAPK activation by serum was unaffected by RGS4 expression, whereas that stimulated by mastoparan was inhibited significantly. In stark contrast to its effects on p38 MAPK activity, RGS4 expression had no effect on the ability of serum or mastoparan to stimulate ERK1/ERK2 in Mv1Lu cells Figure 4B). Treating control and RGS4-expressing Mv1Lu cells with MAP kinase inhibitors further confirmed the specific activation of ERK1/ERK2 and p38 MAPK by serum and mastoparan as well as the ability of RGS4 to selectively inhibit p38 MAPK (Figure 4C). The inhibitory effects of RGS4 in Mv1Lu cells are specific to signaling events mediated by G proteins, because RGS4 expression had no effect on TGF-β- or BMP-7-stimulated phosphorylation of Smad2 or Smad1, respectively (Figure 4D). Together, these findings demonstrate that RGS4 expression selectively inhibited G protein-stimulated p38 MAPK activity without affecting their ability to stimulate ERK1/ERK2 activity in Mv1Lu cells. These findings further suggest that the ability of RGS4 to inhibit p38 MAPK activity results in defective cell migration, and consequently, in defective tubulogenesis (see below).
Figure 4.
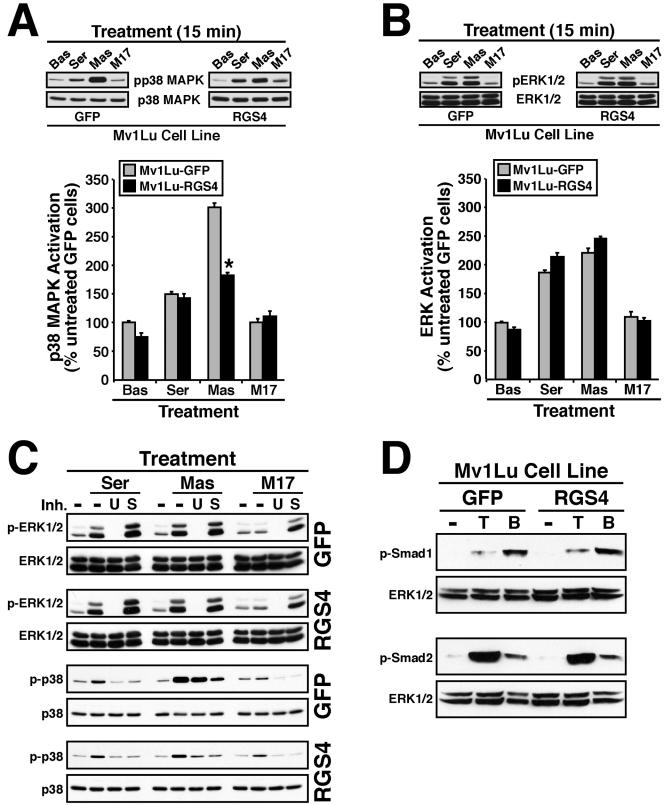
RGS4 selectively inhibits p38 MAPK activation in Mv1Lu cells. Serum-starved GFP- or RGS4-expressing Mv1Lu cells were stimulated for 15 min with 5% serum (Ser), 50 μM mastoparan (Mas), or 50 μM mastoparan 17 (M17) as indicated. The activation status of p38 MAPK (A) or ERK1/ERK2 (B) was determined by immunoblotting whole cell extracts with phospho-specific antibodies to p38 MAPK or ERK1/ERK2. Differences in protein loading were monitored by reprobing stripped membranes with either anti-p38 MAPK or -ERK1 antibodies. Accompanying graphs show the densitometric analysis of MAP kinase activation in GFP-(gray bars) or RGS4 (black bars)-expressing Mv1Lu cells normalized to untreated GFP-expressing cells. Data are the mean ± (SEM) of three independent experiments. RGS4 expression significantly reduced mastoparan-mediated activation of p38 MAPK in Mv1Lu cells (*p < 0.05; Student's t test). (C) Quiescent control or RGS4-expressing Mv1Lu cells were treated with 25 μM U0126 (U) or 10 μM SB203580 (S) for 30 min before stimulation with serum (Ser), mastoparan (Mas), or mastoparan M17 (M17) as described above. Phosphorylation of ERK1/ERK2 and p38 MAPK was determined by immunoblotting with phospho-specific antibodies as described above. (D) GFP- or RGS4-expressing Mv1Lu cells were stimulated with TGF-β1 (5 ng/ml) or BMP-7 (1 μg/ml) for 30 min at 37°C. The activation status of Smad2 or Smad1 was determined by immunoblotting whole cell extracts with phospho-specific Smad2 or Smad1 antibodies. Differences in protein loading were monitored by reprobing stripped membranes with anti-ERK1 antibodies. Shown are representative immunoblots from a single experiment that was repeated once with identical results.
Constitutively Active MKK6 Rescues RGS4 Defects in Mv1Lu Cells
The above-mentioned results indicated that RGS4 selectively inhibited the p38 MAPK signaling system. We speculated that RGS4 may antagonize tubulogenesis in part through its inhibition of p38 MAPK activity. To establish the necessity of p38 MAPK activity in Mv1Lu cell tubulation, we monitored Mv1Lu cell tubulogenesis in the absence or presence of the p38 MAPK inhibitor SB203580. Figure 5A shows that treatment of Mv1Lu cells with 20 μM SB203580 produced tubule defects similar of those observed in RGS4-expressing cells (Figure 2C), including the development of incomplete tubule networks having uneven spacing, poor branching, and a rough appearance. These results suggest that RGS4 expression may inhibit p38 MAPK activity in tubulating Mv1Lu cells. Accordingly, p38 MAPK immunoprecipitated from tubulating RGS4-expressing Mv1Lu cells was significantly less active in phosphorylating recombinant ATF-2 (by 29.8 ± 4.6%, n = 3, p = 0.002) compared with tubulating control cells. Thus, the inhibition of p38 MAPK activity in Mv1Lu cells elicited tubule defects reminiscent of RGS4 expression, which also inhibited p38 MAPK activity (Figure 4A).
Figure 5.
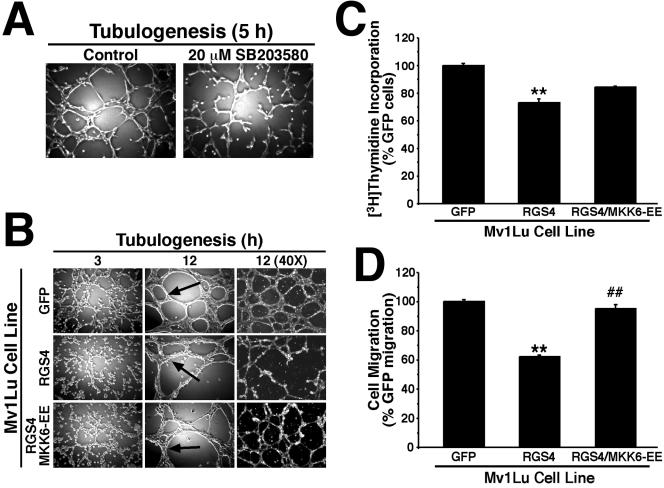
Constitutively active MKK6-EE rescues RGS4 defects in Mv1Lu cells. (A) Mv1Lu cell tubulogenesis was allowed to proceed for 5 h in the absence or presence of 20 μM SB203580. Bright field pictures were captured on Nikon Diaphot microscope. (B) Tubule formation by GFP-, RGS4-, and RGS4/MKK6-EE-expressing cells was monitored at varying times as indicated. Bright field pictures were captured on Nikon Diaphot microscope. (C) DNA synthesis rates in GFP-, RGS4-, and RGS4/MKK6-EE-expressing Mv1Lu cells were determined by a [3H]thymidine assay. Data are the means (± SEM) of three independent experiments presented as the percentage of [3H]thymidine relative to GFP-expressing cells. RGS4 expression significantly decreased DNA synthesis in Mv1Lu (**p <0.05; Student's t test). Although coexpression of MKK6-EE enhanced DNA synthesis by RGS4-expressing cells, this effect was not significantly different from that observed in RGS4-expressing cells. (D) The migration of GFP-, RGS4-, and RGS4/MKK6-EE-expressing Mv1Lu cells to fibronectin was allowed to proceed for 24 h. Data are the means (± SEM) of three independent experiments presented as the percentage of migration relative to GFP-expressing cells. RGS4 expression significantly inhibited Mv1Lu cell migration to fibronectin (**p < 0.05; Student's t test), whereas coexpression of MKK6-EE significantly rescued the migration defects imparted by RGS4 expression in Mv1Lu cells (##p < 0.05; Student's t test).
To confirm that the tubule defects observed in RGS4-expressing Mv1Lu cells arose as a consequence of their reduced capacity to stimulate p38 MAPK, we attempted to rescue their activation of p38 MAPK by coexpressing a constitutively active allele of MKK6 (i.e., MKK6-EE), which activates p38 MAPK. As discussed previously, RGS4-expressing Mv1Lu cells formed aberrant tubules characterized by their rough appearance, uneven spacing, poor branching, and incomplete networks (Figure 2C). Coexpression of MKK6-EE in RGS4-expressing Mv1Lu cells clearly rescued the tubule defects imparted by RGS4 expression in Mv1Lu cells. Indeed, RGS4/MKK6-EE-expressing cells formed complete, evenly spaced, and highly branched tubule networks when cultured onto Matrigel (Figure 5B). However, tubules formed by both RGS4-expressing Mv1Lu cell populations maintained a rough appearance that differed dramatically from the smooth tubules formed in control cultures (Figure 5B, arrows). We also assessed the ability of MKK6-EE to rescue cell migration and proliferation defects observed for RGS4-expressing Mv1Lu cells. Figure 5C shows that MKK6-EE coexpression partially rescued the proliferation defects associated with RGS4 expression in Mv1Lu cells but completely rescued their migration defects (Figure 5D). Thus, one mechanism whereby RGS4 inhibits epithelial cell tubulogenesis is by antagonizing G protein-mediated p38 MAPK activation, which reduces cell proliferation and migration.
Stimulation of p38 MAPK also has been linked to increases in VEGF expression through HIF-1α-dependent and -independent mechanisms (Kozawa et al., 2000; Jung et al., 2001; Xiong et al., 2001; Duyndam et al., 2003). Moreover, fetal and adult lung contain high levels of VEGF, which is produced by airway epithelial cells and stimulates their proliferation (Brown et al., 2001; Ohwada et al., 2003). In addition, pulmonary VEGF functions in directing lung alveolarization, vascularization, and epithelial branching morphogenesis (Acarregui et al., 1999; Compernolle et al., 2002; Akeson et al., 2003; Hosford and Olson, 2003), particularly in response to hypoxic conditions (Christou et al., 1998; Klekamp et al., 1999). Because p38 MAPK couples to VEGF expression, and because RGS4 inhibits p38 MAPK activity in Mv1Lu cells, we hypothesized RGS4 as a novel suppressor of VEGF expression in lung epithelial cells. To test this hypothesis, we measured changes in luciferase expression driven by the full-length VEGF promoter (i.e., VEGF/K) as well as by a truncated derivative that lacks HIF-1α responsiveness (i.e., VEGF/P). As expected, RGS4 expression significantly repressed VEGF expression in Mv1Lu cells (Figure 6). Although MKK6-EE expression exhibited a trend toward elevated VEGF expression, MKK6-EE completely rescued Mv1Lu cell expression of VEGF in a HIF-1α-independent manner (Figure 6). These findings suggest that Mv1Lu cells are subjected to autocrine VEGF signaling. Accordingly, we find that VEGF stimulates ERK1/ERK2 phosphorylation in Mv1Lu cells (our unpublished data). Thus, a second mechanism whereby RGS4 inhibits epithelial cell tubulogenesis is by repressing VEGF expression stimulated by p38 MAPK.
Figure 6.
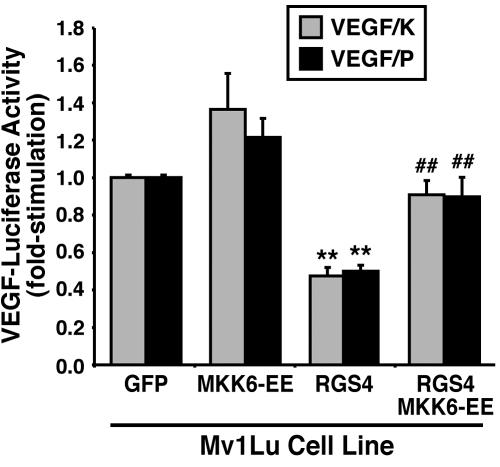
RGS4 inhibits p38 MAPK-mediated VEGF expression in Mv1Lu cells. Mv1Lu cells stably expressing either GFP, MKK6-EE, RGS4, or MKK6-EE/RGS4 were transiently transfected with either pVEGF/K- or pVEGF/P-luciferase and pCMV-β-gal. Forty-eight hours posttransfection, the cells were processed to measure luciferase and β-gal activities contained in detergent-solubilized whole cell extracts. Data are the mean (± SEM) luciferase activities of three independent experiments presented as the fold-stimulations relative to corresponding GFP-expressing cells. RGS4 expression significantly inhibited VEGF expression in Mv1Lu cells (**p < 0.05; Student's t test), whereas coexpression of MKK6-EE significantly rescued VEGF expression in RGS4-expressing Mv1Lu cells (##p < 0.05; Student's t test).
Tubulogenesis Induces RGS4 Expression in and Disrupts Angiogenic Sprouting by Endothelial Cells
Given the similar processes necessary for epithelial and endothelial cells to form tubules (Carmeliet, 2000; Hogan and Kolodziej, 2002; Kerbel and Folkman, 2002), we speculated that tubulating endothelial cells would up-regulate RGS4 expression analogous to that by epithelial cells. As such, we determined whether RGS4 was differentially expressed during endothelial cell tubulation (i.e., angiogenesis) and, if so, whether constitutive RGS4 expression could inhibit endothelial cell tubulogenesis. Using quantitative real-time PCR, we observed a robust increase in RGS4 expression in tubulating murine brain MB114 microvascular cells compared with control cells (Figure 7A). In addition, the synthesis of RGS5 and, to a lesser extent, RGS7 mRNA also was enhanced in tubulating MB114 cells, whereas that of RGS10 remained unchanged during tubulogenesis (Figure 7A). Thus, similar to Mv1Lu cells, tubulogenesis induced RGS4 expression in endothelial cells.
Figure 7.
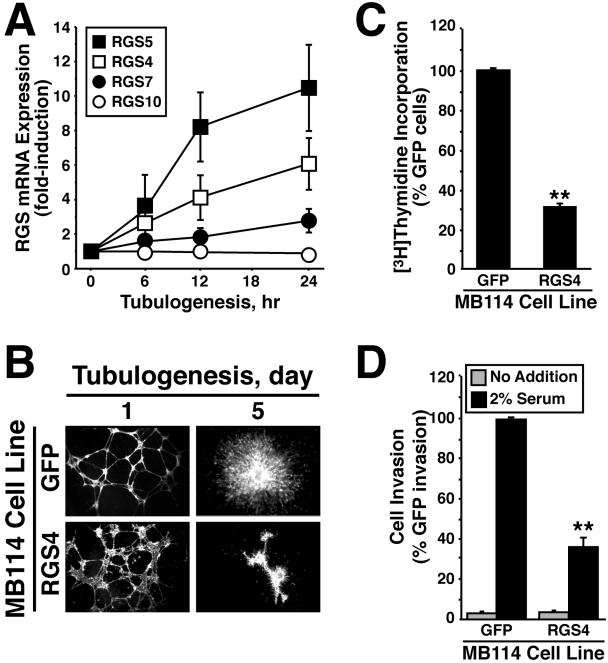
Endothelial cell tubulogenesis induces RGS4 expression and is inhibited by constitutive RGS4 expression. (A) MB114 cells were cultured in three-dimensional collagen matrices for varying times as indicated, whereupon total RNA was isolated and reverse transcribed before analyzing RGS4, RGS5, RGS7, and RGS10 expression by quantitative real-time PCR. Values are the means (± SEM) of three independent experiments and are normalized to transcript expression of cells grown on plastic. (B) MB114 endothelial cells stably expressing either GFP or RGS4 were seeded onto Matrigel. Tubule formation and endothelial cell sprouting was monitored on days 1 and 5. Bright field images were captured on Nikon Diaphot microscope. (C) DNA synthesis in GFP- or RGS4-expressing MB114 cells was measured by a [3H]thymidine incorporation assay. The data are the means (± SEM) of three independent experiments presented as the percentage of [3H]thymidine incorporation relative to GFP-expressing cells. RGS4 expression significantly decreased DNA synthesis in MB114 cells (**p < 0.05; Student's t test). (D) The invasion of GFP- or RGS4-expressing MB114 cells through Matrigel-coated membranes in the absence (gray bars) or presence (black bars) of chemoattractant was performed in a modified Boyden-chamber for 48 h. Data are the mean (± SEM) of four independent experiments presented as the percentage of invasion relative to GFP-expressing MB114 cells. RGS4 expression significantly reduced MB114 cell invasion through Matrigel (**p < 0.05; Student's t test).
Given the antitubulogenic activity of RGS4 in epithelial cells, we hypothesized that RGS4 expression would similarly impact tubulogenesis by endothelial cells. To test this hypothesis, we infected MB114 endothelial cells with control (i.e., GFP) or RGS4 retrovirus and cells expressing GFP were isolated by flow cytometry to establish stable polyclonal populations of control and RGS4-expressing MB114 cells as described above (our unpublished data). In addition, quantitative real-time PCR analyses demonstrated that RGS4 mRNA was overexpressed ∼20-fold in MB114 cells infected with RGS4 retrovirus compared with control cells (our unpublished data). We then compared the ability of these MB114 cell lines to form tubules when cultured onto Matrigel. Unlike Mv1Lu cells, the kinetics of tubule formation by RGS4-expressing MB114 cells was not significantly different from that of control cells (our unpublished data). However, RGS4 expression did elicit dramatic alterations in MB114 cell tubule morphology. For instance, tubules formed by RGS4-expressing MB114 cells seemed rougher, stunted, and more disorganized than those formed by control cells (Figure 7B). Moreover, RGS4 expression significantly inhibited angiogenic cell sprouting from MB114 cell clusters (Figure 7B) as well as blocked the formation of MB114 tubule networks in three-dimensional tubulogenesis assays (our unpublished data). Collectively, our findings establish RGS4 as a novel antagonist of tubulogenesis (i.e., angiogenesis) by endothelial cells.
RGS4 Inhibits Endothelial Cell Proliferation and Invasion
Similar to epithelial cells, tubulogenesis by endothelial cells is coupled to cell proliferation, migration, and invasion (Carmeliet, 2000; Hogan and Kolodziej, 2002; Kerbel and Folkman, 2002). Our findings that RGS4 inhibited epithelial cell proliferation and migration lead us to suspect that endothelial cell expression of RGS4 would similarly reduce their proliferation, migration, and invasion. As expected, RGS4 expression significantly inhibited DNA synthesis in MB114 cells (Figure 7C). In contrast to its effects on Mv1Lu cell migration, RGS4 expression had little effect on the migration of MB114 cells to fibronectin (our unpublished data). However, RGS4-expressing MB114 cells were significantly poorer in their ability to invade synthetic basement membranes as compared with control cells (Figure 7D). Thus, similar to epithelial cells, these findings establish RGS4 as a multifunctional inhibitor of endothelial cell tubulogenesis, doing so by reducing endothelial cell proliferation and invasion.
RGS4 Abrogates VEGF Signaling in MB114 Cells
In addition to its attenuation of GPCR signaling, RGS4 also may antagonize angiogenesis by blocking the activity of proangiogenic molecules such as VEGF. For instance, recent evidence indicates that Gα11 and Gαq interact physically with KDR VEGF receptors, and, consequently, induce endothelial cell migration stimulated by VEGF (Zeng et al., 2002, 2003). We therefore hypothesized that RGS4 may inhibit angiogenic sprouting by antagonizing VEGF signaling in endothelial cells. We tested this hypothesis by using a [3H]thymidine incorporation assay that measured the effects of RGS4 expression on MB114 cell DNA synthesis stimulated by VEGF165. Figure 8A shows that RGS4 abolished the ability of VEGF165 to stimulate DNA synthesis in MB114 endothelial cells. This finding indicates that RGS4 not only antagonizes GPCR-mediated signals but also inhibits VEGF signaling in endothelial cells.
Figure 8.
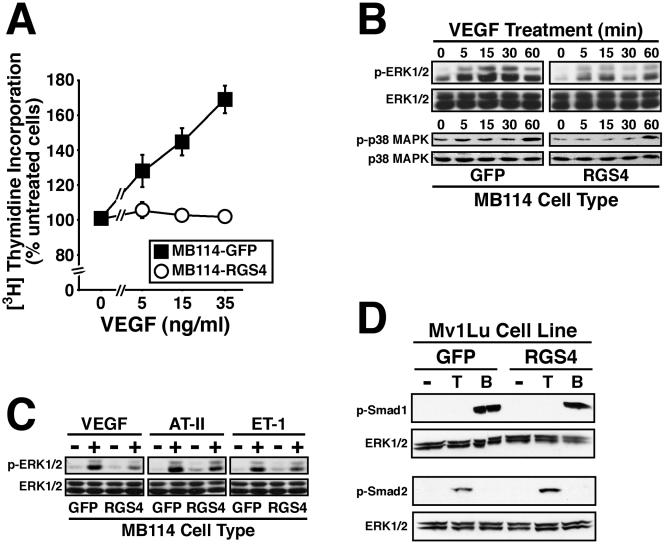
RGS4 abrogates VEGF signaling in MB114 cells. (A) Control and RGS4-expressing MB114 cells were incubated in the absence or presence of increasing concentrations of VEGF165 as indicated. Changes in DNA synthesis were measured by a [3H]thymidine incorporation. Data are the mean (± SEM) of three experiments and are presented as a percentage of [3H]thymidine incorporation relative to unstimulated cells. (B) Serum-starved MB114 endothelial cells were treated with VEGF165 (50 ng/ml) for 0-60 min. Alternations in protein kinase activation were monitored by immunoblotting with phospho-specific antibodies against ERK1/ERK2 (top) or p38 MAPK (bottom). Differences in protein loading were monitored by stripping and reprobing membranes with anti-ERK1 or anti-p38 MAPK polyclonal antibodies. Shown are representative immunoblots from a single experiment that was repeated twice with identical results. (C) Quiescent MB114 endothelial cells were stimulated with VEGF165 (50 ng/ml), angiotensin II (1 μM; AT-II), or endothelin-1 (0.1 μM; ET-1) for 5 min. ERK1/ERK2 phosphorylation was monitored by immunoblotting as described above. Shown are representative immunoblots from a single experiment that was repeated twice with identical results. (D) GFP- or RGS4-expressing MB114 cells were stimulated with TGF-β1 (5 ng/ml) or BMP-7 (1 μg/ml) for 30 min at 37°C. Smad2 and Smad1 phosphorylation was determined by immunoblotting whole cell extracts with phospho-specific Smad2 or Smad1 antibodies. Differences in protein loading were monitored by reprobing stripped membranes with anti-ERK1 antibodies. Shown are representative immunoblots from a single experiment that was repeated once with identical results.
We next investigated the effect of RGS4 on the ability of VEGF to stimulate MAP kinases in MB114 cells. To do so, we again performed immunoblot analysis on cell extracts obtained from control or RGS4-expressing MB114 cells before and after their stimulation with VEGF165. As shown in Figure 8B, untreated RGS4-expressing MB114 cells exhibit 40% less ERK1/ERK2 phosphorylation compared with control cells. Moreover, VEGF-stimulated ERK1/ERK2 phosphorylation was similarly inhibited in RGS4-expressing MB114 cells (Figure 8B, top). In addition, phosphorylation of p38 MAPK was reduced by 30% in untreated RGS4-expressing MB114 cells compared with control cells. In contrast to its effects on ERK1/ERK2 activity, RGS4 had only modest effects on p38 MAPK phosphorylation stimulated by VEGF in MB114 cells (Figure 8B, bottom). Finally, Cho et al. (2003) found that RGS4 expression inhibited ERK1/ERK2 activation stimulated by Ang II and ET-1, both of which couple to regulation of angiogenesis (Williams et al., 1995; Richard et al., 2001; Bagnato and Spinella, 2002; Spinella et al., 2002). Figure 8C shows that RGS4 expression in MB114 cells not only inhibited VEGF stimulation of ERK1/ERK2 but also that by Ang II and ET-1. Interestingly, RGS4 was slightly more effective in inhibiting ERK1/ERK2 activation by VEGF (i.e., receptor tyrosine kinase mediated) than that by Ang II or ET-1 (i.e., GPCR mediated), indicating efficient targeting and coupling of RGS4 to the VEGF signaling system in endothelial cells. Similar to Mv1Lu cells, RGS4 expression had no effect on TGF-β- or BMP-7-stimulated phosphorylation of Smad2 or Smad1, respectively (Figure 8D). Thus, RGS4 specifically targets and inhibits signaling events stimulated by VEGF and G proteins.
RGS4 Inhibits VEGF- and G Protein-induced Tyrosine Phosphorylation of KDR by Reducing KDR Expression
Zeng et al. (2002, 2003) recently showed that Gα11 and Gαq interact physically with KDR receptors and are essential for VEGF-stimulated KDR tyrosine phosphorylation. We therefore hypothesized that RGS4 may antagonize VEGF signaling by inhibiting KDR tyrosine phosphorylation mediated by VEGF, and by Gα11 and Gαq. We tested this hypothesis by transiently transfecting 293T cells with KDR, constitutively active Gα11 or Gαq, and RGS4 in all combinations. Figure 9A shows that VEGF stimulation of 293T cells induced significant tyrosine phosphorylation of ectopically expressed KDR, a response that was enhanced significantly by coexpression of either Gα11 or Gαq. Although the capacity of VEGF and G proteins to induce KDR tyrosine phosphorylation was blocked by RGS4, it did so predominantly by preventing KDR expression in 293T cells (Figure 9A). The inhibitory effect of RGS4 on KDR expression also occurred in Cos-7 cells (Figure 9B). However, in stark contrast to its effects on KDR expression, RGS4 had no effect on the expression of TβR-II and FBLN-5 in Cos-7 cells (Figure 9B). Collectively, these results identify RGS4 as a novel inhibitor of VEGF signaling (i.e., cell proliferation and MAP kinase activation) in MB114 endothelial cells. Our findings also suggest that RGS4 inhibits endothelial cell angiogenesis by abrogating the proangiogenic activities of VEGF, presumably by down-regulating KDR expression.
Figure 9.
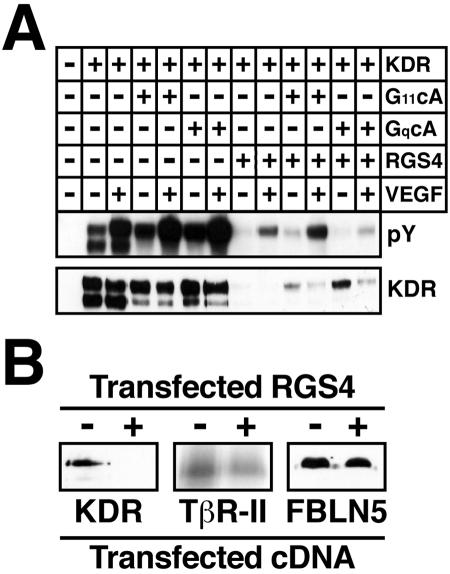
RGS4 inhibits VEGF- and G protein-stimulated KDR phosphorylation and KDR expression. (A) Human 293T cells were transiently transfected with 2 μg of KDR, constitutively active Gα11 or Gαq, and RGS4 as indicated. The transfectants were stimulated with VEGF (50 ng/ml) for 10 min and KDR activation was monitored by immunoblotting KDR immunocomplexes with anti-phosphotyrosine antibodies. Differences in KDR expression were monitored by reprobing stripped membranes with anti-KDR antibodies. Shown are representative immunoblots from a single experiment that was repeated three times with identical results. (B) COS-7 cells were transiently transfected with 2 μg of either KDR, TβR-II, or FBLN-5, together with or without an equivalent amount of RGS4. KDR expression was monitored by immunoprecipitation and immunoblotting with anti-KDR antibodies; TβR-II expression was monitored by iodinated TGF-β1 binding and cross-linking assay; and FBLN-5 expression was monitored by Ni2+-affinity chromatography and immunoblotting with anti-Myc antibodies. Shown is a representative experiment that was repeated once with similar results.
DISCUSSION
Organ and tissue development are critically dependent upon the formation of biological tubes, which provide a route for the efficient exchange of gases, nutrients, and metabolic waste (Hogan and Kolodziej, 2002). Although biological tubes derived from epithelial or endothelial cells fulfill distinct physiological functions, the molecular mechanisms operant in their formation are redundant. For instance, tubulation by epithelial and endothelial cells requires cell proliferation, migration, and invasion as well as the acquisition of cell polarity (Hogan and Kolodziej, 2002). Moreover, both cell types couple to similar signaling systems during tubulation. For example, epithelial c-Met receptors or endothelial VEGF receptors both stimulate MAP kinases (e.g., ERK1/ERK2 and p38 MAPK), PI3K, and AKT during tubulogenesis (Hogan and Kolodziej, 2002; Ferrara et al., 2003). Given the similarities between epithelial and endothelial cell tubulogenesis, we hypothesized the existence of general tubulogenic regulators capable of impacting tubulation by epithelial and endothelial cells. To this end, we identified genes that were expressed differentially in tubulating epithelial cells and then compared these findings with those obtained in tubulating endothelial cells. In accordance with our hypothesis, we observed that gene expression profiles in tubulating epithelial cells are strikingly similar to those described previously in tubulating endothelial cells. Indeed, analogous to endothelial cells, tubulating Mv1Lu cells differentially express the Flt-1 VEGF receptor HIF-1α, the erythropoietin receptor, and the ET(A) endothelin receptor, Caspase 4, TRAIL (TNFSF10), and Gelsolin (Pedram et al., 1997; Bagnato and Spinella, 2002; Jaquet et al., 2002; Spinella et al., 2002; Yasuda et al., 2002).
We also identified differentially expressed genes previously unassociated with epithelial and endothelial cell tubulogenesis. Indeed, we demonstrate herein that tubulogenesis induced RGS4 expression in epithelial (i.e., Mv1Lu cells) and endothelial (i.e., MB114 cells). Moreover, this response was specific for select RGS proteins because tubulogenesis robustly up-regulated RGS4 (and RGS5 in endothelial cells) expression, but it had little or no effect on epithelial cell expression of RGS1, 7, and 12 (Figure 1), and on endothelial cell expression of RGS7 and RGS10 (Figure 6). In contrast to our findings, Bell et al. (2001) reported down-regulation of RGS4 mRNA expression in human umbilical vein endothelial cells undergoing tubule morphogenesis. The reasons underlying this discrepancy are currently unknown, but they may be related to differences in the cell types studied, the kinetics of tubule formation, or the matrices used (i.e., Matrigel vs. collagen). In addition, Bell et al. (2001) failed to perform independent experiments (i.e., Northern blotting or reverse transcription-PCR) to confirm down-regulation of RGS4 mRNA during endothelial cell tubulation as well as to characterize the function of RGS4 during this process.
Functionally, we show for the first time that RGS4 antagonized tubulogenesis by epithelial and endothelial cells. We further show that RGS4 expression inhibited epithelial cell tubulation by reducing G protein-mediated p38 MAPK activation, resulting in diminished cell proliferation, migration, and VEGF expression. In endothelial cells, RGS4 expression was found to antagonize angiogenic sprouting by reducing cell proliferation and invasion as well as by reducing endothelial cell response to VEGF (e.g., ERK1/ERK2 activation) in part via down-regulation of KDR expression. Collectively, our study has established RGS4 as a novel tubulogenic antagonist in epithelial and endothelial cells.
Reports detailing the proangiogenic properties of growth factors and cytokines (e.g., VEGF, bFGF, and TGF-β) abound in the literature and have contributed greatly to our understanding of physiological and pathological angiogenesis (Carmeliet, 2000; Carmeliet and Collen, 2000; Ferrara, 2000; Kerbel and Folkman, 2002). Recently, however, G proteins and GPCRs also have emerged as important regulators of angiogenesis (Richard et al., 2001). For instance, the serine protease thrombin binds and activates members of the PAR family of GPCRs (i.e., PARs 1, 3, and 4), which promote angiogenesis by disrupting cell adhesion and ECM integrity and by stimulating cell permeability, proliferation, and invasion and migration (O'Brien et al., 2001; Richard et al., 2001). Similarly, ET-1 induces angiogenesis through direct and indirect mechanisms: 1) directly via its G protein-coupled ET(B) receptor, which stimulates cell proliferation, migration, and invasion; and 2) indirectly via its G protein-coupled ET(A) receptor, which induces VEGF expression by activating HIF-1α (Bagnato and Spinella, 2002). By activating its G protein-coupled AT-1 and AT-2 receptors, Ang II also stimulates HIF-1α-mediated induction of VEGF expression, leading to enhanced angiogenesis (Williams et al., 1995; Richard et al., 2000). And finally, gene targeting experiments further support the essential role of G protein signaling systems in angiogenesis. For instance, homozygous deletion of Gα13 in mice causes midgestation embryonic lethality due to abnormal vascular development and organization (Offermanns et al., 1997; Offermanns, 2001). In addition, primary endothelial cells of these mice are defective in their proliferation and migration as well as in their sprouting and ECM remodeling (Offermanns et al., 1997). Collectively, these studies highlight the general importance of G proteins and GPCRs to induce and maintain the fidelity of angiogenesis. These studies further suggest that mechanisms that positively and/or negatively modulate the signaling of these proangiogenic systems will likely play critical regulatory roles during the activation and resolution of normal and abnormal angiogenesis.
Given the emerging importance of G proteins and GPCRs in angiogenesis, it is not entirely surprising that RGS4 exerted an inhibitory effect on endothelial cell tubulogenesis. However, a similar role for G proteins and GPCRs in regulating epithelial cell tubulogenesis is largely unknown. Thus, our current findings showing that tubulating epithelial cells up-regulate RGS4 expression and that RGS4 expression antagonizes epithelial cell tubulogenesis identifies a novel and potentially important role for G proteins and GPCRs in epithelial cell tubulogenesis. In this way, RGS proteins are multifunctional signaling molecules that modulate the activity of Gα subunits by stimulating their intrinsic GTPase activity, thereby terminating intracellular signaling by G proteins (Berman and Gilman, 1998; Ross and Wilkie, 2000; Wieland and Mittmann, 2003). In addition to their GAP activity, RGS proteins also modulate G protein signaling by 1) enhancing G protein activation, 2) serving as effector molecules, and 3) acting as scaffolds linking GPCRs to G proteins and effector molecules (Berman and Gilman, 1998; Ross and Wilkie, 2000; Wieland and Mittmann, 2003). Although we cannot exclude the possibility that RGS4 possesses effector and/or scaffolding activities, the absence in RGS4 of identifiable protein-protein interaction domains suggests that RGS4 inhibits tubulogenesis primarily through its GAP activity, thus abrogating intracellular signaling downstream of targeted Gα subunits (e.g., p38 MAPK activation). Future studies clearly need to address this issue by determining which Gα subunits are targeted by RGS4 and, more importantly, what protubulogenic signals and/or GPCRs initially stimulated their activation. Because epithelial and endothelial cell tubulogenesis induces RGS4 expression, it also will be interesting to determine the molecular mechanisms/signals operant in stimulating RGS4 expression in tubulating epithelial and endothelial cells.
In addition to demonstrating that RGS4 antagonized GPCR-dependent angiogenic activities, we also found that RGS4 inhibited the proangiogenic activity of VEGF. Specifically, we determined that RGS4 negates endothelial cell proliferation stimulated by VEGF as well as its ability to activate ERK1/ERK2. Although the precise molecular mechanism(s) whereby RGS4 inhibits VEGF signaling in endothelial cells remains to be elucidated fully, recent findings by Zeng et al. (2002, 2003) have implicated coupling of VEGF receptors to heterotrimeric G proteins. Our findings also show that KDR tyrosine phosphorylation is enhanced by G proteins Gα11 and Gαq. However, instead of simply inhibiting G protein-coupled tyrosine phosphorylation of KDR, we found that RGS4 blocked this response by reducing KDR translation, and, consequently, cell surface expression of KDR (Figure 9). Moreover, we observed RGS4 to repress VEGF expression by inhibiting p38 MAPK activity. Thus, RGS4 provides a dual braking system designed to limit VEGF signaling by repressing the expression of VEGF and its receptor, KDR. As such, we propose that RGS4 inhibits angiogenesis through a bimodal mechanism that antagonizes signals downstream of GPCRs and signals upstream of VEGF and its receptors (i.e., Gα11 and Gαq, and KDR expression).
Last, the RGS superfamily is comprised of at least 25 proteins that primarily target and inhibit the activity of Gαi/o and Gαq/11 subunits when examined in vitro. Thus, the role redundancy plays during modulation of G protein signaling by RGS proteins must be resolved. Indeed, although our results have established RGS4 as a novel antagonist of tubulogenesis, other RGS proteins have been shown to 1) exhibit differential expression during tubule formation (e.g., RGS2, 3, and 5; Bell et al., 2001), and 2) regulate processes necessary for angiogenesis. We observed that RGS4 selectively inhibited the p38 MAPK signaling system in Mv1Lu cells, resulting in cell proliferation and migration defects that negatively impacted tubule formation by Mv1Lu cells; these defects were rescued in large part by coexpression of constitutively active MKK6, an upstream activator of p38 MAPK. Similar to RGS4, RGS16 blocks p38 MAPK activation by platelet-activating factor in Chinese hamster ovary cells (Zhang et al., 1999). In contrast, RGS1, 2, and 3 inhibit ERK1/ERK2 activation in 293T cells stimulated with IL-8 (Druey et al., 1996), whereas RGS5 inhibits their activation in aortic smooth muscle cells stimulated with Ang II (Wang et al., 2002). Moreover, RGS1 and RGS3 inhibit leukocyte migration to thrombin (Bowman et al., 1998); RGS3 also inhibits renal tubule cell migration to lysophosphatidic acid (Gruning et al., 1999). Although RGS4 failed to inhibit G protein-mediated activation of ERK1/ERK2 in Mv1Lu cells, ERK1/ERK2 inhibition did indeed reduce their migration to fibronectin (Figure 3). Thus, how these and other RGS proteins function in conjunction with RGS4 to regulate tubulogenesis remains to be clarified. We suspect that when governing complex biological processes (i.e., angiogenesis), RGS4 and other RGS proteins will be expressed in a spatiotemporal manner that curtails their redundant tendencies, thereby extending greater control over G protein and receptor tyrosine kinase signaling systems. In addition, the GAP activity of RGS4 and other RGS proteins may be tightly regulated such that perturbations to this system (e.g., overexpression) may eliminate an essential control mechanism necessary for proper tubule formation. Future studies will need to address these important issues.
As summarized in Figure 10, we have established RGS4 as a novel gene target for tubulogenesis in epithelial and endothelial cells. In epithelial cells, RGS4 expression antagonized tubule formation by selectively inhibiting p38 MAPK, which reduced cell proliferation and migration. In endothelial cells, RGS4 expression antagonized angiogenic sprouting by inhibiting VEGF-stimulated cell proliferation and ERK1/ERK2 activation as well as KDR phosphorylation by reducing KDR expression. We therefore propose RGS4 as a novel, general inhibitor of epithelial and endothelial cell tubulogenesis, doing so by antagonizing G protein-stimulated cell proliferation, migration, and invasion. Consequently, interventions designed to increase RGS4 expression and/or activity ultimately may be exploited to treat human diseases characterized by pathological tubulogenesis, such as cancer.
Figure 10.

Schematic of RGS4-mediated antagonism of epithelial and endothelial cell tubulogenesis. (Top) Tubulogenesis stimulates RGS4 expression in epithelial and endothelial cells. The induction of RGS4 expression during tubulogenesis occurs subsequent to primary tubule formation, suggesting involvement of RGS4 during the resolution of epithelial and endothelial tubulogenesis. (Bottom). Epithelial expression of RGS4 selectively inhibits G protein-mediated activation of p38 MAPK, thereby antagonizing epithelial cell tubulogenesis by inhibiting their proliferation, migration, and expression of VEGF. Endothelial expression of RGS4 inhibits G protein-mediated signaling (e.g., ET-1 and Ang II) as well as cell proliferation, ERK1/ERK2 activation, and KDR expression. Collectively, we show that RGS4 antagonizes tubulogenesis via a bimodal mechanism that inhibits GPCR and VEGF signaling, and likely additional alternative signaling pathways coupled to these signaling systems.
Acknowledgments
We thank Dr. John Cambier for providing the NIA microarrays. We also thank members of the Schiemann laboratory for critical reading of the manuscript, and William Townend and Shirley Sobus for expertise and help provided on studies performed in the Cytometry Core Facility at the National Jewish Medical and Research Center. This research was supported in part by a startup fund from the National Jewish Medical and Research Center and by grants from the American Cancer Society and the Elsa U. Pardee Foundation (to W.P.S.).
Article published online ahead of print in MBC in Press on November 17, 2004 (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-06-0479).
References
- Acarregui, M. J., Penisten, S. T., Goss, K. L., Ramirez, K., and Snyder, J. M. (1999). Vascular endothelial growth factor gene expression in human fetal lung in vitro. Am. J. Respir. Cell. Mol. Biol. 20, 14-23. [DOI] [PubMed] [Google Scholar]
- Akeson, A. L., Greenberg, J. M., Cameron, J. E., Thompson, F. Y., Brooks, S. K., Wiginton, D., and Whitsett, J. A. (2003). Temporal and spatial regulation of VEGF-A controls vascular patterning in the embryonic lung. Dev. Biol. 264, 443-455. [DOI] [PubMed] [Google Scholar]
- Bagnato, A., and Spinella, F. (2002). Emerging role of endothelin-1 in tumor angiogenesis. Trends Endocrinol. Metab. 14, 44-50. [DOI] [PubMed] [Google Scholar]
- Bell, S. E., Mavila, A., Salazar, R., Bayless, K. J., Kanagala, S., Maxwell, S. A., and Davis, G. E. (2001). Differential gene expression during capillary morphogenesis in 3D collagen matrices: regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell Sci. 114, 2755-2773. [DOI] [PubMed] [Google Scholar]
- Berman, D. M., and Gilman, A. G. (1998). Mammalian RGS proteins: barbarians at the gate. J. Biol. Chem. 273, 1269-1272. [DOI] [PubMed] [Google Scholar]
- Bowman, E. P., Campbell, J. J., Druey, K. M., Scheschonka, A., Kehrl, J. H., and Butcher, E. C. (1998). Regulation of chemotactic and proadhesive responses to chemoattractant receptors by RGS (regulator of G-protein signaling) family members. J. Biol. Chem. 273, 28040-28048. [DOI] [PubMed] [Google Scholar]
- Brown, K. R., England, K. M., Goss, K. L., Snyder, J. M., and Acarregui, M. J. (2001). VEGF induces airway epithelial cell proliferation in human fetal lung in vitro. Am. J. Physiol. 281, L1001-L1010. [DOI] [PubMed] [Google Scholar]
- Cara, D. C., Kaur, J., Forster, M., McCafferty, D. M., and Kubes, P. (2001). Role of p38 mitogen-activated protein kinase in chemokine-induced emigration and chemotaxis in vivo. J. Immunol. 167, 6552-6558. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P. (2000). Mechanisms of angiogenesis and arteriogenesis. Nat Med 6, 389-395. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P., and Collen, D. (2000). Molecular basis of angiogenesis. Role of VEGF and VE-cadherin. Ann. N.Y. Acad. Sci. 902, 249-262. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P., and Jain, R. K. (2000). Angiogenesis in cancer and other diseases. Nature 407, 249-257. [DOI] [PubMed] [Google Scholar]
- Chang, L., and Karin, M. (2001). Mammalian MAP kinase signalling cascades. Nature 410, 37-40. [DOI] [PubMed] [Google Scholar]
- Cho, H., Harrison, K., Schwartz, O., and Kehrl, J. H. (2003). The aorta and heart differentially express RGS (regulators of G-protein signalling) proteins that selectively regulate sphingosine 1-phosphate, angiotensin II and endothelin-1 signalling. Biochem. J. 371, 973-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christou, H., Yoshida, A., Arthur, V., Morita, T., and Kourembanas, S. (1998). Increased vascular endothelial growth factor production in the lungs of rats with hypoxia-induced pulmonary hypertension. Am. J. Respir. Cell. Mol. Biol. 18, 768-776. [DOI] [PubMed] [Google Scholar]
- Compernolle, V., et al. (2002). Loss of HIF-2a and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat. Med. 8, 702-710. [DOI] [PubMed] [Google Scholar]
- Davydov, I. V., and Varshavsky, A. (2000). RGS4 is arginylated and degraded by the N-end rule pathway in vitro. J. Biol. Chem. 275, 22931-22941. [DOI] [PubMed] [Google Scholar]
- Druey, K. M., Blumer, K. J., Kang, V. H., and Kehrl, J. H. (1996). Inhibition of G-protein-mediated MAP kinase activation by a new mammalian gene family. Nature 379, 742-746. [DOI] [PubMed] [Google Scholar]
- Duyndam, M. C., Hulscher, S. T., van der Wall, E., Pinedo, H. M., and Boven, E. (2003). Evidence for a role of p38 kinase in hypoxia-inducible factor 1-independent induction of vascular endothelial growth factor expression by sodium arsenite. J. Biol. Chem. 278, 6885-6895. [DOI] [PubMed] [Google Scholar]
- Favata, M. F., et al. (1998). Identification of a novel inhibitor of mitogenactivated protein kinase kinase. J. Biol. Chem. 273, 18623-18632. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. (2000). Vascular endothelial growth factor and the regulation of angiogenesis. Recent Prog. Horm. Res. 55, 15-35. [PubMed] [Google Scholar]
- Ferrara, N., Gerber, H. P., and LeCouter, J. (2003). The biology of VEGF and its receptors. Nat. Med. 9, 669-676. [DOI] [PubMed] [Google Scholar]
- Folkman, J. (1995). Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1, 27-31. [DOI] [PubMed] [Google Scholar]
- Garrington, T. P., and Johnson, G. L. (1999). Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol. 11, 211-218. [DOI] [PubMed] [Google Scholar]
- Gruning, W., Arnould, T., Jochimsen, F., Sellin, L., Ananth, S., Kim, E., and Walz, G. (1999). Modulation of renal tubular cell function by RGS3. Am. J. Physiol. 276, F535-F543. [DOI] [PubMed] [Google Scholar]
- Higashijima, T., Burnier, J., and Ross, E. M. (1990). Regulation of Gi and Go by mastoparan, related amphiphilic peptides, and hydrophobic amines. Mechanism and structural determinants of activity. J. Biol. Chem. 265, 14176-14186. [PubMed] [Google Scholar]
- Higashijima, T., Uzu, S., Nakajima, T., and Ross, E. M. (1988). Mastoparan, a peptide toxin from wasp venom, mimics receptors by activating GTP-binding regulatory proteins (G proteins). J. Biol. Chem. 263, 6491-6494. [PubMed] [Google Scholar]
- Hogan, B. L., and Kolodziej, P. A. (2002). Organogenesis: molecular mechanisms of tubulogenesis. Nat. Rev. Genet. 3, 513-523. [DOI] [PubMed] [Google Scholar]
- Hosford, G. E., and Olson, D. M. (2003). Effects of hyperoxia on VEGF, its receptors, and HIF-2a in the newborn rat lung. Am. J. Physiol. 285, L161-L168. [DOI] [PubMed] [Google Scholar]
- Jaquet, K., Krause, K., Tawakol-Khodai, M., Geidel, S., and Kuck, K. H. (2002). Erythropoietin and VEGF exhibit equal angiogenic potential. Microvasc. Res. 64, 326-333. [DOI] [PubMed] [Google Scholar]
- Jung, Y. D., Liu, W., Reinmuth, N., Ahmad, S. A., Fan, F., Gallick, G. E., and Ellis, L. M. (2001). Vascular endothelial growth factor is upregulated by interleukin-1b in human vascular smooth muscle cells via the P38 mitogenactivated protein kinase pathway. Angiogenesis 4, 155-162. [DOI] [PubMed] [Google Scholar]
- Kerbel, R., and Folkman, J. (2002). Clinical translation of angiogenesis inhibitors. Nat. Rev. Cancer 2, 727-739. [DOI] [PubMed] [Google Scholar]
- Klekamp, J. G., Jarzecka, K., and Perkett, E. A. (1999). Exposure to hyperoxia decreases the expression of vascular endothelial growth factor and its receptors in adult rat lungs. Am. J. Pathol. 154, 823-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru, A., Seavey, S. E., Rabaglia, M. E., and Metz, S. A. (1995). Nonspecific stimulatory effects of mastoparan on pancreatic islet nucleoside diphosphokinase activity: dissociation from insulin secretion. Biochem. Pharmacol. 49, 263-266. [DOI] [PubMed] [Google Scholar]
- Kozawa, O., Kawamura, H., Hatakeyama, D., Matsuno, H., and Uematsu, T. (2000). Endothelin-1 induces vascular endothelial growth factor synthesis in osteoblasts: involvement of p38 mitogen-activated protein kinase. Cell Signal. 12, 375-380. [DOI] [PubMed] [Google Scholar]
- Lin, D. C., Bullock, C. M., Ehlert, F. J., Chen, J. L., Tian, H., and Zhou, Q. Y. (2002). Identification and molecular characterization of two closely related G protein-coupled receptors activated by prokineticins/endocrine gland vascular endothelial growth factor. J. Biol. Chem. 277, 19276-19280. [DOI] [PubMed] [Google Scholar]
- Lin, H. Y., Wang, X. F., Ng-Eaton, E., Weinberg, R. A., and Lodish, H. F. (1992). Expression cloning of the TGF-b type II receptor, a functional transmembrane serine/threonine kinase. Cell 68, 775-785. [DOI] [PubMed] [Google Scholar]
- Liu, X., Sun, Y., Constantinescu, S. N., Karam, E., Weinberg, R. A., and Lodish, H. F. (1997). Transforming growth factor b-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc. Natl. Acad. Sci. USA 94, 10669-10674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda, Y., et al. (2002). Isolation and identification of EG-VEGF/prokineticins as cognate ligands for two orphan G-protein-coupled receptors. Biochem. Biophys. Res. Commun. 293, 396-402. [DOI] [PubMed] [Google Scholar]
- Matsumoto, K., and Nakamura, T. (2001). Hepatocyte growth factor: renotropic role and potential therapeutics for renal diseases. Kidney Int. 59, 2023-2038. [DOI] [PubMed] [Google Scholar]
- O'Brien, P. J., Molino, M., Kahn, M., and Brass, L. F. (2001). Protease activated receptors: theme and variations. Oncogene 20, 1570-1581. [DOI] [PubMed] [Google Scholar]
- Offermanns, S. (2001). In vivo functions of heterotrimeric G-proteins: studies in Gα-deficient mice. Oncogene 20, 1635-1642. [DOI] [PubMed] [Google Scholar]
- Offermanns, S., Mancino, V., Revel, J. P., and Simon, M. I. (1997). Vascular system defects and impaired cell chemokinesis as a result of Ga13 deficiency. Science 275, 533-536. [DOI] [PubMed] [Google Scholar]
- Ohwada, A., Yoshioka, Y., Iwabuchi, K., Nagaoka, I., Dambara, T., and Fukuchi, Y. (2003). VEGF regulates the proliferation of acid-exposed alveolar lining epithelial cells. Thorax 58, 328-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram, A., Razandi, M., Hu, R. M., and Levin, E. R. (1997). Vasoactive peptides modulate vascular endothelial cell growth factor production and endothelial cell proliferation and invasion. J. Biol. Chem. 272, 17097-17103. [DOI] [PubMed] [Google Scholar]
- Perlman, R., Schiemann, W. P., Brooks, M. W., Lodish, H. F., and Weinberg, R. A. (2001). TGF-b-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat. Cell Biol. 3, 708-714. [DOI] [PubMed] [Google Scholar]
- Raingeaud, J., Whitmarsh, A. J., Barrett, T., Derijard, B., and Davis, R. J. (1996). MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 16, 1247-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard, D. E., Berra, E., and Pouyssegur, J. (2000). Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1a in vascular smooth muscle cells. J. Biol. Chem. 275, 26765-26771. [DOI] [PubMed] [Google Scholar]
- Richard, D. E., Vouret-Craviari, V., and Pouyssegur, J. (2001). Angiogenesis and G-protein-coupled receptors: signals that bridge the gap. Oncogene 20, 1556-1562. [DOI] [PubMed] [Google Scholar]
- Ross, E. M., and Wilkie, T. M. (2000). GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 69, 795-827. [DOI] [PubMed] [Google Scholar]
- Rousseau, S., Houle, F., Landry, J., and Huot, J. (1997). p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 15, 2169-2177. [DOI] [PubMed] [Google Scholar]
- Schiemann, B. J., Neil, J. R., and Schiemann, W. P. (2003). SPARC inhibits epithelial cell proliferation in part through stimulation of the TGF-b-signaling system. Mol. Biol. Cell 14, 3977-3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann, W. P., Bartoe, J. L., and Nathanson, N. M. (1997). Box 3-independent signaling mechanisms are involved in leukemia inhibitory factor receptor a- and gp130-mediated stimulation of mitogen-activated protein kinase. Evidence for participation of multiple signaling pathways which converge at Ras. J. Biol. Chem. 272, 16631-16636. [DOI] [PubMed] [Google Scholar]
- Schiemann, W. P., Blobe, G. C., Kalume, D. E., Pandey, A., and Lodish, H. F. (2002). Context-specific effects of fibulin-5 (DANCE/EVEC) on cell proliferation, motility, and invasion: fibulin-5 is induced by TGF-b and affects protein kinase cascades. J. Biol. Chem. 277, 27367-27377. [DOI] [PubMed] [Google Scholar]
- Schiemann, W. P., Pfeifer, W. M., Levi, E., Kadin, M. E., and Lodish, H. F. (1999). A deletion in the gene for transforming growth factor b type I receptor abolishes growth regulation by transforming growth factor b in a cutaneous T-cell lymphoma. Blood 94, 2854-2861. [PubMed] [Google Scholar]
- Schiemann, W. P., Rotzer, D., Pfeifer, W., Levi, E., Rai, K. R., Knaus, P., and Kadin, M. E. (2004). TGF-b-resistant B-cells from chronic lymphocytic leukemia patients contain recurrent deletions in the signal sequence of TGF-b type I receptor. Cancer Detect. Prev. 28, 57-64. [DOI] [PubMed] [Google Scholar]
- Sodhi, A., Montaner, S., Miyazaki, H., and Gutkind, J. S. (2001). MAPK and Akt act cooperatively but independently on hypoxia inducible factor-1a in rasV12 upregulation of VEGF. Biochem. Biophys. Res. Commun. 287, 292-300. [DOI] [PubMed] [Google Scholar]
- Spinella, F., Rosano, L., Di Castro, V., Natali, P. G., and Bagnato, A. (2002). Endothelin-1 induces vascular endothelial growth factor by increasing hypoxia-inducible factor-1a in ovarian carcinoma cells. J. Biol. Chem. 277, 27850-27855. [DOI] [PubMed] [Google Scholar]
- Tong, L., Pav, S., White, D. M., Rogers, S., Crane, K. M., Cywin, C. L., Brown, M. L., and Pargellis, C. A. (1997). A highly specific inhibitor of human p38 MAP kinase binds in the ATP pocket. Nat. Struct. Biol. 4, 311-316. [DOI] [PubMed] [Google Scholar]
- Tu, Y., Popov, S., Slaughter, C., and Ross, E. M. (1999). Palmitoylation of a conserved cysteine in the regulator of G protein signaling (RGS) domain modulates the GTPase-activating activity of RGS4 and RGS10. J. Biol. Chem. 274, 38260-38267. [DOI] [PubMed] [Google Scholar]
- Wang, Q., Liu, M., Mullah, B., Siderovski, D. P., and Neubig, R. R. (2002). Receptor-selective effects of endogenous RGS3 and RGS5 to regulate mitogen-activated protein kinase activation in rat vascular smooth muscle cells. J. Biol. Chem. 277, 24949-24958. [DOI] [PubMed] [Google Scholar]
- Wieland, T., and Mittmann, C. (2003). Regulators of G-protein signalling: multifunctional proteins with impact on signalling in the cardiovascular system. Pharmacol. Ther. 97, 95-115. [DOI] [PubMed] [Google Scholar]
- Williams, B., Baker, A. Q., Gallacher, B., and Lodwick, D. (1995). Angiotensin II increases vascular permeability factor gene expression by human vascular smooth muscle cells. Hypertension 25, 913-917. [DOI] [PubMed] [Google Scholar]
- Xiong, S., Grijalva, R., Zhang, L., Nguyen, N. T., Pisters, P. W., Pollock, R. E., and Yu, D. (2001). Up-regulation of vascular endothelial growth factor in breast cancer cells by the heregulin-b1-activated p38 signaling pathway enhances endothelial cell migration. Cancer Res. 61, 1727-1732. [PubMed] [Google Scholar]
- Yasuda, Y., Fujita, Y., Masuda, S., Musha, T., Ueda, K., Tanaka, H., Fujita, H., Matsuo, T., Nagao, M., Sasaki, R., and Nakamura, Y. (2002). Erythropoietin is involved in growth and angiogenesis in malignant tumours of female reproductive organs. Carcinogenesis 23, 1797-1805. [DOI] [PubMed] [Google Scholar]
- Zeng, H., Zhao, D., and Mukhopadhyay, D. (2002). KDR stimulates endothelial cell migration through heterotrimeric G protein Gq/11-mediated activation of a small GTPase RhoA. J. Biol. Chem. 277, 46791-46798. [DOI] [PubMed] [Google Scholar]
- Zeng, H., Zhao, D., Yang, S., Datta, K., and Mukhopadhyay, D. (2003). Heterotrimeric Gaq/Ga11 proteins function upstream of vascular endothelial growth factor (VEGF) receptor-2 (KDR) phosphorylation in vascular permeability factor/VEGF signaling. J. Biol. Chem. 278, 20738-20745. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., Neo, S. Y., Han, J., Yaw, L. P., and Lin, S. C. (1999). RGS16 attenuates Gaq-dependent p38 mitogen-activated protein kinase activation by platelet-activating factor. J. Biol. Chem. 274, 2851-2857. [DOI] [PubMed] [Google Scholar]