

Abstract
Activation of G-protein-coupled chemoattractant receptors triggers dissociation of Gα and Gβγ subunits. These subunits induce intracellular responses that can be highly polarized when a cell experiences a gradient of chemoattractant. Exactly how a cell achieves this amplified signal polarization is still not well understood. Here, we quantitatively measure temporal and spatial changes of receptor occupancy, G-protein activation by FRET imaging, and PIP3 levels by monitoring the dynamics of PHCrac-GFP translocation in single living cells in response to different chemoattractant fields. Our results provided the first direct evidence that G-proteins are activated to different extents on the cell surface in response to asymmetrical stimulations. A stronger, uniformly applied stimulation triggers not only a stronger G-protein activation but also a faster adaptation of downstream responses. When naïve cells (which have not experienced chemoattractant) were abruptly exposed to stable cAMP gradients, G-proteins were persistently activated throughout the entire cell surface, whereas the response of PHCrac-GFP translocation surprisingly consisted of two phases, an initial transient and asymmetrical translocation around the cell membrane, followed by a second phase producing a highly polarized distribution of PHCrac-GFP. We propose a revised model of gradient sensing, suggesting an important role for locally controlled components that inhibit PI3Kinase activity.
INTRODUCTION
Chemotaxis, the directed movement of cells along gradients of chemoattractants, plays an important role in many physiological processes such as neuronal patterning, the recruitment of leukocytes to sites of infection, and cell aggregation in the life cycle of the social amoebae, Dictyostelium discoideum (Zigmond, 1978; Devreotes and Zigmond, 1988; Murphy, 1994; Segall, 1999; Meinhardt, 1999; Chung et al., 2001; Iijima et al., 2002; Postma et al., 2004a, 2004b). Many of the key molecules and biochemical events regulating such responses have been identified. Chemoattractant signaling is mediated by seven-transmembrane receptors that are linked to heterotrimeric G-proteins (GPCR) (Devreotes and Zigmond, 1988; Murphy, 1994; Thelen, 2001; Devreotes and Janetopoulos, 2003). Ligand binding to the receptors induces the dissociation of the G-proteins into Gα and Gβγ subunits. Free Gβγ plays a role in the activation of PI3K, which phosphorylates inositol phospholipids of the inner plasma membrane (Stoyanov et al., 1995; Hazeki et al., 1998; Segall, 1999; Hirsch et al., 2000; Li et al., 2000; Sasaki et al., 2000; Rickert et al., 2000; Stephens et al., 2002). Phosphatidylinositol-4,5-diphosphate (PIP2) is highly abundant and plays a key role in downstream effects of GPCR signaling because the product of its 3-phosphorylation by PI3K, PI(3,4,5)P3 (PIP3), stimulates cellular processes through its ability to bind proteins with pleckstrin homology (PH) domains and thereby recruit them to the plasma membrane (Kavran et al., 1998), such as Cytosolic Regulator of Adenylyl Cyclase (CRAC) and protein kinase B (Akt/PKB; Insall et al., 1994; Parent et al., 1998; Meili et al., 1999; Servant et al., 2000). The phosphatase PTEN acts as a direct antagonist of PI3K, dephosphorylating PIP3 to generate PIP2 (Liliental et al., 2000; Iijima and Devreotes, 2002; Funamoto et al., 2002; Comer and Parent, 2002).
The biochemical responses can show a high degree of spatial polarization if a cell experiences asymmetrical activation of chemoattractant receptors (Parent and Devreotes, 1999; Servant et al., 2000; Meili and Firtel, 2003). Translation of small asymmetries in receptor occupancy into strong intracellular signaling polarity enables the cells to respond to very shallow gradients, as little as a 2% difference in chemoattractant concentration between the front and back of a migrating cell (Chung et al., 2001; Iijima et al., 2002; Devreotes and Janetopoulos, 2003; Postma et al., 2004a, 2004b). Many proteins and lipids involved in chemotactic responses are distributed asymmetrically in chemotaxing D. discoideum amoebae and neutrophils that display clear morphological polarity (Zigmond et al., 1981; Comer and Parent, 2002; Xu et al., 2003). In these cells, chemoattractant receptors are uniformly distributed on the cell surface (Xiao et al., 1997; Servant et al., 1999), the G-protein βγ subunits are localized in a shallow anterior to posterior gradient (Jin et al., 2000), PI3K accumulates at the leading edge (Funamoto et al., 2001, 2002; Wang et al., 2002), PTEN is enriched at the trailing end (Funamoto et al., 2002; Iijima and Devreotes, 2002), PH domain-containing proteins translocate from the cytosol to the plasma membrane at the leading front (Parent et al., 1998; Meili et al., 1999; Servant et al., 2000), and some small G-proteins localize in either the leading edge or the trailing end (Xu et al., 2003). As a consequence, the front of a polarized cell shows greater responsiveness to attractant than do the sides and the trailing end (Zigmond et al., 1981; Jin et al., 2000; Funamoto et al., 2002; Iijima and Devreotes, 2002), and uniform stimulations can trigger localized PIP3 increases (Postma et al., 2003, 2004a, 2004b).
Treatment with latrunculin, an inhibitor of actin polymerization, eliminates preexisting morphological polarity and also prevents cell movement. However, the ability to detect chemoattractant gradients is preserved (Parent et al., 1998; Jin et al., 2000). This allows analysis of the gradient sensing machinery in the absence of preexisting asymmetries in the distribution of key signaling components. Studies in D. discoideum amoebae have shown that receptors and G-proteins are uniformly distributed around the perimeter of latrunculin-treated cells and all points around the perimeter are almost equally sensitive to cAMP, a chemoattractant for D. discoideum. On spatially uniform cAMP exposure, PH domain-containing proteins evenly translocate to the plasma membrane and then quickly return to the cytosol (a process termed adaptation). In a cAMP gradient, although distributions of the receptors and G-proteins remain the same, some PH domain-containing proteins are localized to the membrane in a polarized manner (Xiao et al., 1997; Parent et al., 1998; Jin et al., 2000; Janetopoulos et al., 2004).
Models have been proposed to explain how cells achieve such adaptation to uniform increases in chemoattractant concentration while displaying persistent, spatially polarized responses to gradients (Parent and Devreotes, 1999; Postma and Van Haastert, 2001; Iijima et al., 2002; Levchenko and Iglesias, 2002; Rappel et al., 2002; Devreotes and Janetopoulos, 2003). Two key questions remain to be answered: At which step of the signal transduction pathway are slight differences in concentration of a chemoattractant across the cell body amplified into sharply localized biochemical responses, and what is the dynamic process of the signal amplification when a cell initially expose to a gradient?
In this study, we analyzed the dynamics of key aspects of the gradient sensing pathway in single living cells using confocal fluorescence microscopy with high spatio-temporal resolution. We found that cAMP stimulation induced a local and concentration-dependent activation of G-proteins around the cell membrane and that G-proteins remained dissociated at a steady-state level as long as cAMP was present. Analysis of cells suddenly exposed to a steady cAMP gradient revealed a two-step process of gradient sensing, consisting of first rapidly “switching on” the gradient sensing machinery and then more slowly “amplifying” differences in receptor occupancy to achieve a highly polarized PHCrac-GFP distribution. These new findings lead to a modified scheme of gradient sensing that can account for the observed temporal and spatial dynamics of PHCrac-GFP and provides the foundation for further quantitative modeling.
MATERIALS AND METHODS
Cell Lines and Cell Growth and Differentiation
The D. discoideum cell line expressing both Gα2 CFP and YFPGβ subunits, which was described previously (Janetopoulos et al., 2001), was cloned by limiting dilution to obtain high CFP- and YFP-expressing cell lines. Newly obtained cell lines designated as G cells were cultured axenically in D3-T Basic Media containing 20 μg/ml geneticin (Sigma, Steinheim, Germany). To establish a PHCrac-GFP-expressing cell line, wild-type (AX2) cells were transformed with a linearized integration vector carrying a cassette encoding PHCrac-GFP and the blasticidin S resistance gene. Transformants were selected in D3-T Basic Media (KD Medical, Columbia, MD) containing 10 μg/ml blasticidin S sulfate (Invitrogen, Carlsbad, CA) and cloned by limiting dilution. Cell clones with a high level of cytosolic PHCrac-GFP expression were selected, and their functionality and experimental utility were examined by cAMP-mediated chemotaxis and cAMP-trigged PHCrac-GFP translocation. The resultant cell lines were designated as JAX and called PH cells in this study. Cell maintenance, transformation and development to the chemotactic stage were carried out as previously described (Jin et al., 2000). Before being observed, the cells were treated with 2.0 μM latrunculin B (Molecular Probes, Eugene, OR).
Live Cell Imaging and Microscopic Analyses with LSM 510 META
Differentiated cells (2 × 104) were plated on a four-well chamber for uniformly applied stimulation and a one-well chamber for the microinjector delivered cAMP stimulation (Nalge Nunc International, Naperville, IL), allowed to adhere to the cover glass for 10 min and then covered with DB buffer. Live cells were imaged using a Zeiss Laser Scanning Microscope (Thornwood, NY), LSM 510 META, with a 40× NA 1.3 or 60× NA 1.4 oil Dic Plan-Neofluar objective. To monitor cAMP and PHCrac-GFP, the specimen were excited with two laser lines, 488 nm for GFP and 543 nm for Alexa 594, a water-soluble fluorescence dye. Images were simultaneously recorded in three channels: channel one: fluorescent emissions from 505 to 530 nm for GFP (green); channel two: emissions from 580 to 650 nm for Alexa 594 (red); channel three: DIC.
Generation and Measurement of Applied cAMP Stimulations
Live cells were imaged during stimulations with a uniform concentration, an asymmetrical increase, or a steady gradient of cAMP. The temporal-spatial intensity changes of Alexa 594 and PHCrac-GFP in cells were directly imaged using a confocal microscope with Z-axis resolution of ∼1-2 μm. Fluorescence intensities of Alxea 594 and GFP within the focal plane were simultaneously recorded in two different channels. To apply a uniform stimulation, 100 μl of a mixture of cAMP and Alexa 594 hydrazide sodium salt (0.1 μg/μl; Molecular Probes) was dropped on top of cells placed in a four-well chamber (Nalge Nunc International) and covered with 400 μl DB buffer. Under this experimental condition, during the first minute, cells were uniformly exposed to a cAMP concentration that was about 2.5-fold of the final concentration. For other stimulations, cells were plated in a one-well chamber and covered with 6 ml DB buffer. To establish a steady gradient, we set FemtoJet (FemtoJet and micromanipulator 5171, Eppendorf, Germany) with Pc = 70 and Pi = 70 to ensure the injection of a constant and small volume of cAMP and Alxea 594 into a one-well chamber. Under this condition, a stable gradient was established within 100 μm around the tip of the micropipette (Supplementary Figure S2). To suddenly expose a cell to a stable gradient, a micropipette filled with a mixture of cAMP and 0.1 g/μl Alexa 594 linked with a FemtoJet was positioned 1000 μm away from the cells and then was quickly moved to a position within 100 μm to the cells. During the experiments, we only changed the distance between the micropipette and the cells. The speed of the movement determines how fast a stable gradient can form around a cell. To generate a brief asymmetrical stimulation, a small volume of cAMP mixture was injected into the chamber by applying a pressure on the microinjector for 3 s. In the fluorescence resonance energy transfer (FRET) experiments, where the cAMP was not monitored, we applied the same conditions to ensure similar stimulations.
FRET Measurement
FRET allows us to examine the activity of biologically active proteins by detecting changes in conformation of a protein or in the interaction of two proteins that have been tagged with a FRET donor/acceptor pair, such as CFP and YFP (Adams et al., 1991; Miyawaki, 2003; Sekar and Periasamy, 2003). Using a spectral confocal fluorescence microscope, we used two methods to measure FRET that indicate dissociation of Gα2CFP and YFPGβ: first, measuring intensity decrease of acceptor (YFP) and increase of donor (CFP) in response to stimuli. This method measures cAMP-induced temporal G-protein dissociations in single living cells. We monitored intensity changes of CFP (donor) and (YFP) acceptor after a stimulation using a time-lapse acquisition of Lambda Stacks. The cells were excited with a 454-nm laser-line, and the spectral emissions in each pixel of the fluorescence images were simultaneously recorded in eight channels, each with a 10-nm width, from 464 to 544 nm. To separate multifluorescence signals, each of the fluorescence images was collected using Lambda Stack acquisition. The spectral emissions of fluorescence images were simultaneously recorded in a CHS-1 from 464 to 544 nm. The spectra of the cells expressing CFP, YFP, or GFP only were obtained and used as the references for the Linear Unmixing Function. The digitally separated images of CFP and YFP of the G cells and GFP of the PH cells were obtained. The intensities of each fluorophore in the regions of interest in the time-lapse experiments were measured, normalized, and expressed as a function of time in responses to cAMP stimulations, using the software of LSM510 META. Second, measuring intensity increase of the donor (CFP) flowing photobleaching the acceptor (YFP). This method measures FRET efficiency that reflects relative levels of heterotrimeric (inactive) G-proteins in the front and back of cells that are exposed to cAMP gradients. After photobleaching YFP, the increase, or dequenching, of CFP emission is a direct measure of FRET efficiency. If FRET occurs, CFP emission will be quenched by YFP before it is photobleached, and the real CFP intensity can only be measured after photobleaching YFP. The efficiency of energy transfer E can be determined from the relative fluorescence intensity of the energy donor (CFP) in the absence, postbleaching (Ipost), and the presence, prebleaching (Ipre), of the energy acceptor (YFP). E = 1-(Ipre/Ipost). By photobleaching acceptor (YFP), we can record Ipost after and Ipre before bleaching and calculate FRET efficiency. In the time series, the cells were first excited with the 454-nm laser at ∼7.5% power to limit photobleaching. After recording fluorescence intensities of the cells three times, the entire G cell was illuminated with 100% 514-nm laser power 20-30 times to photobleach YFP and then excited with the 454-nm laser to record spectral images three more times. To determine the contribution of CFP and YFP in each pixel of the image and to obtain fluorescence images of each fluorophore, the spectrally resolved images of each time-lapse acquisition of Lambda Stack were processed using the Linear Unmixing Function of LSM510 META. The intensities of each fluorophore in the regions of interest in the time-lapse experiments were measured, normalized, and expressed as a function of time using the software of LSM510 META.
Imaging and Data Processing
Images were processed and analyzed by the LSM 510 META software and converted to TIFF files by the Adobe Photoshop software (San Jose, CA). All frames of any given series were processed identically. Selected frames of the series were assembled as montages using the Photoshop 7.0. Quantification of fluorescence intensities of Alexa 594, GFP, CFP, and YFP in the regions of interest was performed using the LSM 510 META software.
RESULTS
Simultaneously Visualizing cAMP Stimulation and PHCrac-GFP Translocation
An increase in Dictyostelium cAMP receptor occupancy activates a signal transduction pathway leading to a transient membrane translocation of PHCrac-GFP (Lilly and Devreotes, 1994; Parent et al., 1998). PIP3 serves as the binding site for PHCrac-GFP, with the levels of inner membrane leaflet PIP3 regulated by local opposing activities of PI3K and PTEN (Huang et al., 2003). The cAMP-triggered PHCrac-GFP translocation is fast and transient, peaking within a few seconds and returning to prestimulus levels in 30 s (Parent et al., 1998). However, cAMP concentrations across the cell and PHCrac-GFP translocation had never been simultaneously imaged, and therefore the relationship between the amount of cAMP that reaches various regions of the cell surface and the temporal responses of PHCrac-GFP in these regions was not known. We simultaneously measured both cAMP concentration and PHCrac-GFP membrane translocation in single living cells, using a confocal fluorescence microscope in fast time-lapse experiments (Figure 1).
Figure 1.
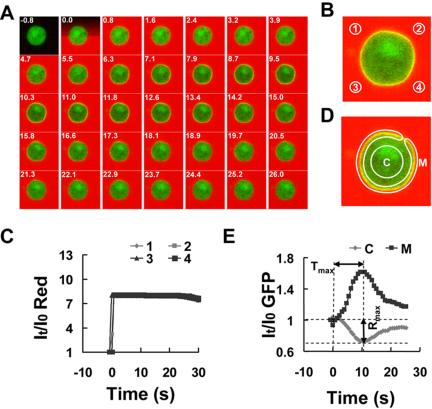
Membrane translocation of PHCrac-GFP in a living cell exposed to a uniform chemoattractant field. (A) PHCrac-GFP translocation (green) after exposure to a uniform field of cAMP chemoattractant (red). A cell expressing PHCrac-GFP (PH cell) was stimulated with 10 nM cAMP at time 0. The stimulation was imaged and quantified by inclusion of a fluorescence dye, Alexa 594, in the cAMP solution. Time (seconds) after cAMP exposure is shown in the top left corner of each image. Supplementary Figure 1 and Supplementary Video video1.avi show a complete sequence of GFP and Alexa intensity changes. Images were captured at 0.8-s intervals and replayed at 5 frames/s. (B) Regions of interest (1-4) used to assess concentration changes of cAMP with time in the vicinity of the cell. (C) Quantitative measurement over time of the increase in cAMP in the four regions of interest. (D) Regions of interest designated as the membrane (M) and the cytosol (C) of the cell and used for assessing cAMP-triggered redistribution of PHCrac-GFP, detected as intensity changes of green fluorescence within a specified region. (E) PHCrac-GFP membrane translocation between the cytosol (C) and membrane (M) over time after exposure to a uniform field of chemoattractant. Similar results were observed in more than 20 independent experiments.
Cells expressing PHCrac-GFP, designated as PH cells, were first allowed to differentiate to the chemotactic-competent stage, and were then treated with latrunculin to eliminate morphological cell polarization and migration. To directly visualize and measure the spatial and temporal distribution of the applied chemoattractant, we mixed cAMP with Alexa 594, a hydrophilic fluorescence dye with similar diffusion properties as cAMP and determined cAMP concentrations based on the fluorescence intensity in medium surrounding the cells. On the addition of cAMP to a cell chamber, the red signal reached the cell surface from all directions in less than 1 s and the intensity was uniform in the field. Thus, addition of cAMP resulted in an immediate and uniform exposure of the cell to cAMP (10 nM; Figure 1, A-C). The cAMP induced a transient translocation of PHCrac-GFP from the cytosol to the membrane. The translocation of PHCrac-GFP started upon exposure to extracellular cAMP, reached its maximum in ∼10 s, and declined to the basal level in less than 30 s (Figure 1A). A quantitative analysis demonstrated that the kinetics of intensity changes in the membrane-associated and cytosolic PHCrac-GFP pools were inversely related (Figure 1E), indicating that there is a transient and quantitative translocation of PHCrac-GFP from the cytosol to the membrane. PHCrac-GFP returned to the cytosol while the cell was still exposed to cAMP and the receptors were still occupied (Figure 1, A and C). The time from the addition of the stimulus to the peak of the response was designated as Tmax, and the maximal level of a transient response was measured as maximum difference Rmax between the levels of cytosolic PHCrac-GFP before and after application of the stimulus (Figure 1E and Supplementary Figure S1).
Covisualizing cAMP-triggered Activation of G-proteins and PHCrac-GFP Translocation in Single Living Cells
Dissociation of heterotrimeric G-proteins triggered by the binding of cAMP to a cAR1 receptor is the first excitation event leading to PHCrac-GFP translocation (Jin et al., 1998; Parent et al., 1998). G-protein dissociation can be monitored using FRET changes between Gα2CFP and YFPGβ (Janetopoulos et al., 2001). To relate the spatio-temporal changes in cAMP exposure to the resulting G-protein dissociation, and to PIP3 accumulation (as reflected by PHCrac-GFP relocalization), it would be ideal to measure these events in a single cell. However, because of the technical difficulty in accurately and simultaneously measuring intensity changes of CFP, YFP, and Alexa 594, we first determined the dynamics of PHCrac-GFP translocation to the membrane while also analyzing the field of applied cAMP (Figure 1). We then measured both G-protein dissociation by FRET and PHCrac-GFP translocation, using the dynamics of PHCrac-GFP as an indicator of the cAMP stimulation (Figure 2). Because the FRET signal between Ga2CFP and YFPGβγ is weak and the GFP spectrum overlaps the spectra of CFP and YFP, it was necessary to image the two signaling steps in separate cells in order to obtain precise FRET measurements. FRET changes between Gα2CFP and YFPGβγ were revealed in one latrunculin-treated cell (G cell), and the membrane translocation of a GFP-tagged PH domain fusion protein was monitored in a nearby latrunculin-treated cell (PH cell), which was within 10 μm of the G cell and thus exposed to the same cAMP stimulus (Figure 2, B and C). In these cells, both the cAMP receptors and the G-protein subunits are uniformly distributed on the cell membrane (Figure 2B; Xiao et al., 1997; Jin et al., 2000). We excited the cells with a 454-nm laser line and acquired spectral images in 8 channels from 464 to 534 nm with approximately a 1-s interval between each successive image. Each pixel of the image contains data corresponding to an emission spectrum resulting from both CFP and YFP fluorescence in the G cell, and from GFP in the PH cell. Spectrally resolved time-lapse images of a G cell and a neighboring PH cell were acquired upon stimulation by a uniform field of cAMP added at time 0. Under these conditions, PHCrac-GFP transiently translocated from the cytosol to the plasma membrane (Figure 2, B and E). G-protein dissociation was assessed as a loss of FRET, detected as a (donor) CFP intensity increase and a simultaneous (acceptor) YFP intensity decrease (Figure 2, B, F, and G). The digitally separated CFP and YFP channels of the G cell showed a clear increase in the CFP signal and a corresponding decrease in the YFP signal around the entire cell membrane with time after exposure to cAMP (Figure 2B). The emission spectra across the entire membrane demonstrated a cAMP-triggered FRET loss (Figure 2D), which was similar in magnitude to the spectral changes measured previously in a population of cells using a spectral fluorometer (Janetopoulos et al., 2001). G-protein dissociation started upon exposure of the cell to cAMP and reached a plateau (Figure 2, F and G), and the response of PHCrac-GFP membrane translocation was transient (Figure 2E), whereas Gα and Gβγ remained dissociated, indicating that an inhibitory process affecting the binding sites of PHCrac-GFP is regulated by a mechanism other than G-protein reassociation.
Figure 2.
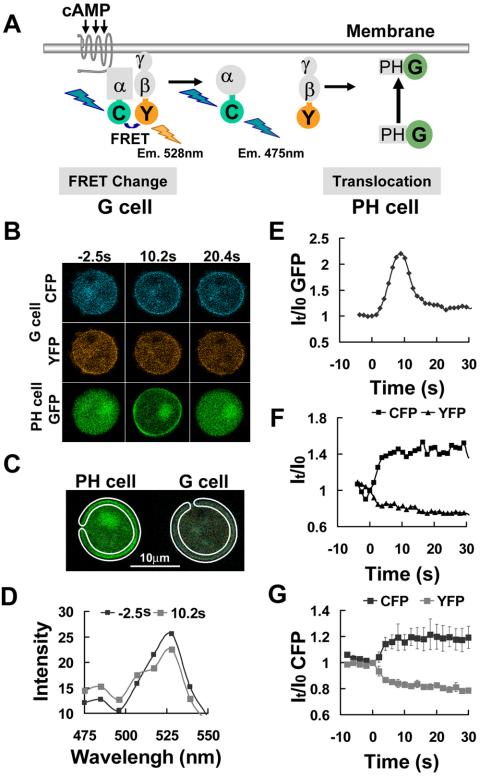
Single-cell FRET measurement of heterotrimeric G-protein dissociation in a uniform cAMP field. (A) Diagram shows how G-protein dissociation induced by cAMP binding to the receptor can be monitored by the loss of FRET between CFP and YFP tagged to the Gα and Gβ subunits, respectively. It also shows the membrane translocation of PHCrac-GFP to monitor cAMP stimulation. (B) cAMP, 1 μM, was uniformly applied at time 0. Fluorescence images of cAMP-triggered G-protein dissociation in the G cell and PHCrac-GFP translocation in the PH cell. Increased CFP and decreased YFP signal intensities around the G cell membrane at 10.2 and 20.4 s indicate G-protein subunit dissociation that simultaneously reduces quenching of CFP and excitation of YFP. Transient PHCrac-GFP translocation to all regions of the plasma membrane was clearly observed at 10.2 s in a nearby PH cell. Supplementary Videos video2.avi and video3.avi show the full time course of CFP and YFP intensity changes in a single living cell after cAMP stimulation, respectively. Images were captured at 1.1-s intervals and are replayed at five frames/s. (C) Regions of interest used for quantitative analysis of G-protein activation and membrane translocation of PHCrac-GFP in a uniform field of cAMP. (D) Combined emission spectra of the membrane region of the G cell before and after the addition of cAMP at time 0. On uniform stimulation with cAMP, a significant increase in the CFP emission signal near 475 nm and a reciprocal decrease in the YFP emission near 528 nm were observed, consistent with a loss of FRET upon subunit dissociation. (E) Temporal changes in membrane associated PHCrac-GFP after exposure of the cell to the same uniform field of cAMP. (F) Temporal changes in the G-protein dissociation at the cell membrane after stimulation, reflected as a CFP (M-CFP, black) signal intensity increase and a paralleled YFP (M-YFP, gray) signal decrease. (F) Uniformly applied cAMP stimulation triggered G-protein dissociation, reflected as CFP signal intensity increase and YFP signal decrease. Means and SEs for each time points are shown as temporal changes in the G-protein dissociation at the membrane after stimulation by 2 μM cAMP (n = 6).
Relationship between cAMP Concentration and the Time to Maximum PHCrac-GFP Translocation and G-protein Activation
To relate how different degrees of cAMP receptor activation affect the duration and the magnitude of the transient PIP3 levels on the cell membrane, we determined the kinetics of PHCrac-GFP translocation responses of cells stimulated with a high (1 μM) and a low (10 nM) cAMP concentrations (Figure 3A). The time required for reaching the maximal response, Tmax, and the maximal level of a response, Rmax, were further determined in multiple experiments with multiple cells exposed to three final concentrations of cAMP: 1, 10, and 100 nM that is close to the Kd for cAMP binding to the receptor (Johnson et al., 1992). Means of Tmax for stimulation of 1 nM (n = 24), 10 nM (n = 28) and 100 nM (n = 29) are 11.2; 10.13, 7.04 s, respectively; and means of Rmax are 0.68, 0.64, 0.66, respectively (Supplementary Table 1). These results demonstrated that a higher degree of receptor activation resulted in a transient response that reaches its maximal level faster. The smaller Tmax for the higher concentration is consistent with the faster adaptation observed in Figure 3A, i.e., a faster return of PHCrac-GFP to cytosol after membrane translocation. Interestingly, different doses of uniformly applied cAMP triggered similar levels of maximal PIP3 production on the membrane.
Figure 3.
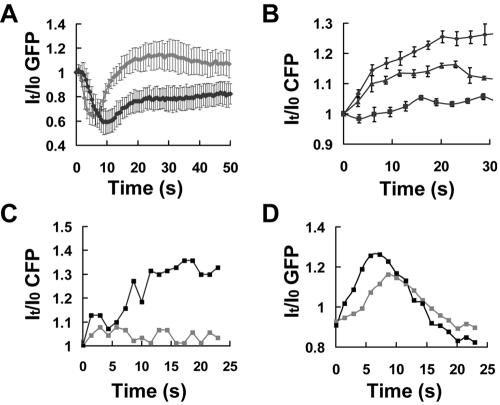
Concentration-dependent changes in the rate of PHCrac-GFP recruitment to the membrane and G-protein activation. (A) The kinetics of intensity changes in cytosolic PHCrac-GFP pools in response to two doses of cAMP stimulation. Means and SDs for each time point are shown as temporal changes in cytosolic PHCrac-GFP in response to a high-dose (1 μM, gray line, n = 6) and a low-dose (10 nM, black line, n = 8) cAMP stimulation. (B) Dose-dependent G-protein activation. Means ± SE are shown as temporal changes in the G-protein dissociation at the cell membrane after stimulation by 1 nM (▪, n = 6), 100 nM (▴, n = 3), and 10 μM (•, n = 5). (C) and (D) One PH and one G cell were first stimulated with 1 nM cAMP and then exposed to 100 nM cAMP. Before the second stimulation, the cells were washed with buffer to remove previously added cAMP and were allowed to recover for 10 min. (C) The graph shows CFP intensity changes on the membrane after stimulation with a low dose (1 nM, gray) or a high dose (100 nM, black) of cAMP. (D) PHCrac-GFP association with the entire cell surface membrane in response to a low dose (1 nM, gray) and a high dose (100 nM, black). Similar results were obtained in more than 10 experiments (another example is shown in Supplementary Figure S1, B and C).
We then measured G-protein activation in response to different chemoattractant concentrations in entire membranes of individual cells by monitoring cAMP-triggered FRET losses between the Gα2CFP and YFPGβ. Stimulation with increasing concentrations of cAMP (1 nM, 100 nM, and 10 μM) led to increased FRET loss, indicating that a higher degree of receptor activation resulted in a higher level of G-protein activation in single living cells (Figure 3B). To further confirm our results, we measured G-protein activation in one G cell and PHCrac-GFP translocation in one PH cell in response to two concentrations of uniformly applied cAMP. The cells were first stimulated with a low dose (1 nM), washed with buffer to remove cAMP, settled for at least 6 min to allow for complete recovery, and then exposed to a higher concentration of cAMP (100 nM; Figure 3, C and D). cAMP, 1 nM, triggered very little detectable G-protein dissociation, whereas stimulation with 100 nM cAMP robustly activated G-proteins (Figure 3C). Both cAMP concentrations triggered PHCrac-GFP translocation responses, and the higher dose stimulated a slightly greater peak and a considerably faster rise in PHCrac-GFP recruitment to the membrane (Figure 3D). These results showed that receptor occupancy regulates the level of G-protein activation and a higher activation of G-proteins leads to a smaller Tmax of PHCrac-GFP translocation, suggesting that a larger increase in receptor occupancy results not only in a higher level of an excitation process but also a more rapid and robust elevation of an inhibitory process, causing the transient response to reach its maximal level faster (see Discussion for more detailed interpretation).
A Wave of cAMP Stimulation Induces a Transient and Asymmetrical PHCrac-GFP Membrane Translocation and a Local and Dose-dependent Activation of G-proteins
Cells usually encounter asymmetrical stimulation. What is the initial response when a naïve, unpolarized cell is exposed to a sudden asymmetrical stimulation? We first determined the kinetics of PHCrac-GFP membrane translocation in different regions of the cell while simultaneously monitoring the applied stimulus. A micropipette filled with a mixture of cAMP and dye (Alexa 594) was placed next to PH cells. By applying pressure to a microinjector linked to the micropipette, a small volume of the mixture of cAMP and the dye was released at time 0, and a wave of chemoattractant swept across the chamber. A cAMP wave induced transient and asymmetrical translocations of PHCrac-GFP in PH cells (Figure 4 and Supplementary Video video8.avi). PHCrac-GFP membrane translocations were analyzed in the selected membrane regions PH-F, PH-M, and PH-B as the front, middle, and back, respectively, of a single cell (Figure 4B), and the extent of cAMP exposure to these regions was determined as the intensity changes of the dye in the same front, middle, and back regions (DF, DM, and DB; Figure 4, B and C). Quantitative analyses showed the peak value of DF was about twice that of DB (Figure 4C), indicating that the maximal cAMP concentration at the front region of the cell was nearly twice that found at the back region. PHCrac-GFP translocated to the front, the middle, and the back regions of the membrane, and the maximal level of PHCrac-GFP membrane association in each region of the cell membrane (the peak value of PH-F, PH-M, and PH-B in Figure 4C). We further quantified the relationship between the peak value of cAMP stimulation and the peak value of the PHCrac-GFP membrane association in different regions of the cell membrane (Figure 4D). The results demonstrated that a directional stimulation of a cAMP wave triggered a crescent-shaped transient membrane pattern of PHCrac-GFP and the crescent can be used as a directional indicator for the on-coming cAMP wave.
Figure 4.
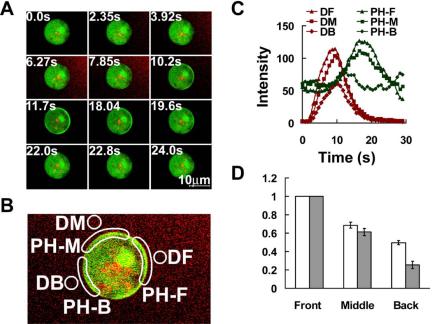
PHCrac-GFP translocation in response to acute exposure to a cAMP gradient. (A) A pulse of cAMP was released by applying a pressure increase from a nearby micropipette and sequential fluorescence images were captured to monitor PHCrac-GFP distribution and cAMP concentration represented as Alexa 594 intensity. Numbers in the top left corner are seconds after cAMP release of the selected frames. Supplementary Video video5.avi presents the complete sequence from this experiment. Frames were captured at 785-ms intervals and are replayed at five frames/s. (B) DF, DM, and DB represent the selected front, middle, and back regions surrounding the cell used for quantitative measurement of dynamic changes of cAMP concentration. PH-F, PH-M, and PH-B show the membrane regions for measuring PHCrac-GFP translocation responses to this asymmetrical cAMP stimulation. (C) Time course of changing cAMP concentration and of membrane translocation of PHCrac-GFP in the different regions of a cell. (D) Quantitative analyses of the relationship between the peak value of cAMP stimulation and the peak value of the PHCrac-GFP membrane association in each region of response of PHCrac-GFP translocation was normalized by dividing the peak value of PH-F, the maximal local response. Data were obtained from 16 independent experiments. Means ± SE are shown.
Using the same experimental design, we measured the spatial and temporal activation of G-proteins in a G cell stimulated with a transient directional stimulus of cAMP by FRET changes and related the activation of G-proteins to the recruitment of PHCrac-GFP in a nearby PH cell (Figure 5). Transient membrane accumulation of PHCrac-GFP in the PH cell was used to monitor the cAMP stimulation (Figure 5A). The dissociation of G-proteins in regions of the front and back of the G cell was quantified (Figure 5, A and B), and the responses of PHCrac-GFP membrane translocation were also simultaneously measured in the front and back regions of the neighboring PH cell (Figure 5, A and C). The front and back regions of the cells that were analyzed were selected to show similar mean intensities at time 0: CFP for G cells and GFP for PH cells. The cAMP-triggered responses were normalized as the ratio of the mean intensity at any given time (I) to that of time 0 (I0), It/I0CFP for G-protein dissociation (Figure 5B) and It/I0GFP for PHCrac-GFP translocation (Figure 5C). The kinetics of the PHCrac-GFP translocation responses in the front and back regions of the PH cell indicated that the cells were exposed to a transient and asymmetrical cAMP stimulation. This stimulation resulted in G-protein activation in both the front and back regions of the G cell (Figure 5, B and D). The extent of G-protein dissociation was higher in the front than in the back region (Figure 5, B and D), as expected from the asymmetric cAMP concentration at the two poles of the cell. These data provide direct evidence that G-proteins are activated to different extents within distinct domains of a cell's perimeter in response to a transient and directional stimulus, validating the local G-protein excitation hypothesis that is an essential component of current gradient sensing models (Parent and Devreotes, 1999; Iijima et al., 2002).
Figure 5.
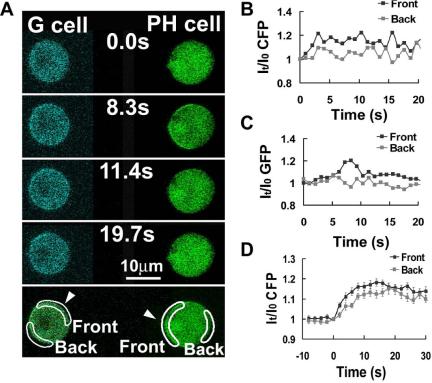
G-protein dissociation in the front and back regions of cells in response to an acute exposure to a cAMP gradient. (A) G-protein dissociation in a single living cell upon an acute stimulation by a directional source of cAMP. Only CFP images of the G cell are shown. (B) G-protein dissociation over time in the front and back regions of the G cell, expressed as a ratio of the CFP signal at each time point to the CFP signal at time 0. (C) Temporal changes in PHCrac-GFP translocation to the front side of the cell. Similar results were obtained in eight experiments. (D). Kinetics of G-protein dissociation in the front and back regions of cells upon stimulation of a cAMP wave. Cells were stimulated by a cAMP wave at 0 s. Time course of G-protein dissociation, expressed as a ratio of the CFP signal at each time point to the CFP signal at 0 s, are shown as means ± SE (n = 22) at each time point.
The Sensing Machinery Spatially Amplifies a Stable cAMP Gradient into Localized PHCrac-GFP Membrane Association
Previous studies have demonstrated that when unpolarized cells are exposed to a stable gradient for several minutes, the differences in receptor occupancy across the cell surface lead to persistently higher PHCrac-GFP membrane association at the side facing the source of cAMP (Parent et al., 1998; Jin et al., 2000). To quantitatively relate the local strength of cAMP to the levels of membrane-associated PHCrac-GFP in the front and back regions of a stably polarized cell, we placed the latrunculin-treated PH cells into a gradient for several minutes and then measured both the extracellular cAMP gradient and the intracellular distribution of PHCrac-GFP (Figure 6, A and B). Our analyses showed that when previously unpolarized cells were exposed to a cAMP gradient (steepness: 23 ± 1.4% difference across the cell diameter, n = 6, in Figure 6C), the front membrane region accumulated more than twice of the amount of PHCrac-GFP as did the back (ratio between front and back is 2.5 ± 0.27, n = 6, difference between the front and back is around 150 ± 27% shown in Figure 5C). The data indicate that there is a substantial spatial amplification from the difference in cAMP concentration to PHCrac-GFP membrane localization in a latrunculin-treated cell positioned in a chemoattractant gradient (with a steepness of ∼23%).
Figure 6.
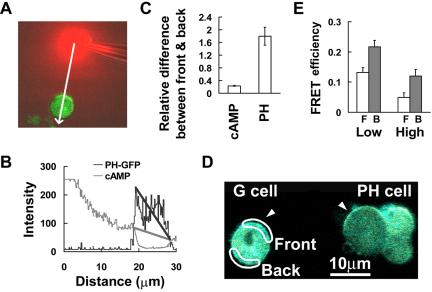
Asymmetrical subcellular distributions of PHCrac-GFP and inactive G-protein heterotrimers in a nonpolarized cell exposed to a steady cAMP gradient. (A) PHCrac-GFP distribution in a cell exposed to a steady cAMP gradient visualized as the red Alexa 594 fluorescence signals. (B) Quantitative measurement of the cAMP gradient and intracellular distribution of PHCrac-GFP along the line starting from the position of the dispensing micropipette and through the central part of the cell in A. The gray line reflects the relative concentration of cAMP. Assuming the maximal intensity equals 1 μM cAMP, we estimated that the concentration is ∼320 nM at the front side and 240 nM at the back side of the cell. The black line plots the PHCrac-GFP distribution from the front to the back side of the cell. These results are typical of six experiments. (C) Membrane-associated PHCrac-GFP at the front and back regions of nonpolarized cells in response to the difference in cAMP concentration (cAMP). cAMP concentration (cAMP) and membrane associated PHCrac-GFP in the front and back regions of nonpolarized cells were directly measured from six independent experiments. (D) G and PH cell images in a steady gradient. The selected front and back regions for FRET measurement of the distribution of inactive heterotrimeric G-proteins are shown. The entire area of the G cell was illuminated to photobleach YFP, and FRET was monitored as increased CFP emission in the selected front and back regions after photobleaching. FRET efficiency was calculated as [IntensityCFP(post) - IntensityCFP(pre)]/IntensityCFP(post). (E) FRET efficiency that reflects the proportion of inactive G-proteins in the front and back regions of cells in steady gradients with similar steepness (∼20%) but different cAMP concentrations. Means and SEs of FRET efficiency show the proportion of inactive G-proteins in the back (▪) is higher than that in the front (□) in response to both low (1 μM cAMP in the micropipette, n = 16 and p < 0.002) and high (3 μM in the micropipette, n = 21 and p < 0.002) cAMP concentration.
We next measured the steady-state level of inactive (heterotrimeric) G-proteins in latrunculin-treated G cells residing in stable cAMP gradients with similar steepness (∼20%) but different cAMP concentrations (Figure 6, D and E). The G cells were mixed with PH cells, which were used as indicators of the gradient (Figure 6 D). We previously measured cAMP-induced G-protein dissociation by monitoring the FRET donor (CFP) intensity increase and the acceptor (YFP) intensity decrease in response to stimulation. To compare the difference in the steady-state levels of G-protein activation in the front and back regions of a G cell, we applied acceptor photobleaching (or donor dequenching) method to measure the level of stable, associated Gα2CFP and YFPGβγ, the inactive form of the G-proteins by measuring the intensity increase of the donor (CFP) after photobleaching the acceptor (YFP). FRET efficiency is defined as 1 - (Ipre/Ipost), where Ipost is photobleaching the intensity of the donor (CFP) after and Ipre is the intensity of the donor (CFP) before photobleaching (Lippincott-Schwartz et al., 2001; Gu et al., 2004). FRET efficiency reflects the amount of inactive G-proteins in the selected front (□) and back (▪) regions of G cells (Figure 6E). After photobleaching YFP in the entire G cell, CFP emission increases were detected in both the front and back regions of the G cell (Figure 6D). Our quantitative analysis showed that the average FRET efficiency was lower in the front surface region than in the back region in cells residing in stable gradients of both cAMP concentrations; and the proportion of inactive G-protein decrease in both the front and back of cells in a gradient with similar steepness by a higher concentration of cAMP (Figure 6E). Because G-protein subunits are uniformly distributed on the membrane of latrunculin-treated cells exposed to a cAMP gradient, the amount of active G-protein is inversely related to the amount of inactive G-protein in a region. Our results indicated that there is a difference in the steady state of active G-proteins between the front and back of cells exposed to gradients.
Dynamic Process of Signal Amplification in a Cell Exposed to a Stable cAMP Gradient
To investigate the dynamics of the amplification of differences in receptor occupancy between the front and back of the cell, we abruptly exposed naïve cells to a stable cAMP gradient and measured the dynamics of PHCrac-GFP translocation and G-protein activation in the front and back regions of the cell membrane. To do so, a micropipette filled with a mixture of cAMP (1 μM) and dye was first placed roughly 1 mm away from latrunculin-treated cells and then quickly moved to within 10 μm of the cells to rapidly establish a steady gradient. We measured the fluorescence intensity of the dye in the front and back regions of the cell and found that a stable gradient was quickly established around the cell (Figure 7, B and C). After the cell was exposed to the gradient, membrane translocations of PHCrac-GFP occurred initially in both the front and the back regions of the cell surface, reached maximal levels within 11 s and then declined by 40 s (Figure 7, A and D). The peak intensity in the front region was higher than in the back region of the cell and correlated with the magnitude of the difference in the cAMP concentration at the two poles of the cell (Figure 7, A and D). Surprisingly, continued exposure of the cell to the gradient for about a minute led to a second increase in the amount of membrane-associated PHCrac-GFP at the front of the cell that exceeded the initial response at this location. In contrast, membrane-associated PHCrac-GFP at the back of the cell showed no second increase and remained at a low steady state level (Figure 7, A and D); The slight decrease of GFP intensity over time is caused by photobleaching). The experiments, which were repeated nine times (Supplementary Figure S3 and Supplementary Video video6.avi show two other examples), revealed that previously not documented two-step process is involved in establishing the highly asymmetric steady-state distribution of PHCrac-GFP in a cell exposed to a gradient. Interestingly, a gradient with a similar steepness but a lower cAMP concentration triggered a dynamic process of PHCrac-GFP response with different temporal-spatial characteristics (Figure 8). Membrane translocations of PHCrac-GFP occurred in the front but barely in the back. Therefore, the maximal levels of PHCrac-GFP membrane association of the initial response in the front and back displayed a bigger difference. In addition, the membrane associated PHCrac-GFP at the front hardly declined after the initial response and the second increase started earlier (Figure 8A) compared with that generated under a higher concentration of cAMP (Figure 8B).
Figure 7.
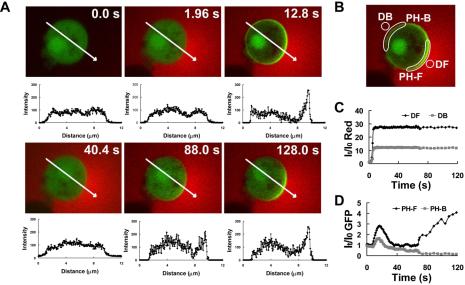
Dynamics of PHCrac-GFP translocation in a cell suddenly exposed to a static cAMP gradient. (A) A PH cell (green) is exposed to a sudden gradient (red). Membrane translocation of PHCrac-GFP shows a peak, then a decline, and a second peak. Green fluorescence intensity along the white line across the cell is shown under each image, indicating the distribution of PHCrac-GFP in the cell. Images were captured at 0.96-s intervals, and the frames at selected time points are shown here. (B) Front (DF) and back (DB) regions used to evaluate quantitative changes of Alexa 594 fluorescence intensity as a measure of cAMP concentration. PH-F and PH-B were selected membrane regions used for monitoring the response of PHCrac-GFP translocation to the front and back of the cell relative to the cAMP gradient, respectively. (C) Rapid generation of a stable cAMP gradient. (D) Dynamic changes in PHCrac-GFP membrane translocation at the front (PH-F) and the back (PH-B) sides of the cell. The slight decrease of GFP intensity over time is caused by photobleaching. Data are representative of nine experiments. Supplementary Video video6.avi shows a full set of images from one of nine experiments.
Figure 8.
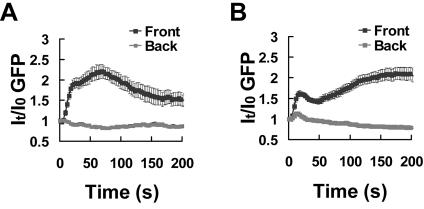
Kinetics of PHCrac-GFP membrane translocation in the front and back of cells when they were suddenly exposed to stable cAMP gradients with similar steepness but different cAMP concentrations. (A) and (B) Naive cells were suddenly exposed to a stable gradient of a low and a high concentration of cAMP, respectively. Micropipettes filled with 100 nM or 1 μM of cAMP were moved from far to near the cells at 0 s. Dynamics changes in membrane associated PHCrac-GFP in the front and back regions of cells are shown as means ± SE (n = 26 and 23 for A and B, respectively).
Using the same experimental design, we measured G-protein activation in a G cell and simultaneously monitored PHCrac-GFP response in the front region of a nearby PH cell as an indicator of the cAMP gradient (Figure 9). A micropipette filled with cAMP was quickly moved to the upper middle of the two cells at time 0 s. The amount of membrane-associated PHCrac-GFP in the front region of the PH cell increased, reached a peak in 20 s, declined, started a second phase of increase at 45 s, and reached another high steady state at about 80 s, as observed in the previous experiment. In response to the same stimulation, G-protein activation occurred in both the front and back regions of the G cell and the activation was slightly higher in the front than in the back of the cell after a 20-s exposure to the gradient (Figure 9, A and C). Dissociated G-proteins remained at steady levels in both the front and back of the G cell (Figure 9C) during the second increase of PHCrac-GFP, in which additional binding sites for PHCrac-GFP were only generated in the front of the PH cell experiencing the highest concentration of cAMP (Figure 9B). Similar results were obtained from five independent experiments (Supplementary Figure S4 shows another example). We measured G-protein activation in cells that were suddenly exposed to cAMP gradients with different steepness but similar cAMP concentrations in the front of the cells (Figure 9, D and E). In responses to similar cAMP concentrations, G-proteins in the front regions were activated to similar levels (Figure 9, D and E). However, in the back regions, G-protein activation was clearly lower, after reaching steady states, in the steeper gradient (with a steepness of ∼100%) because of a much lower local cAMP concentration (Figure 9D). Therefore, in a steeper gradient, the difference in G-protein activation between the front and back of cells was bigger (estimated ∼130%; Figure 9D) than that difference (estimated ∼19%) measured by shallower gradients (with a steepness of ∼20%; Figure 9E). These results suggested that extent of G-protein activation in difference regions of a cells surface was determined by the local concentration of cAMP.
Figure 9.
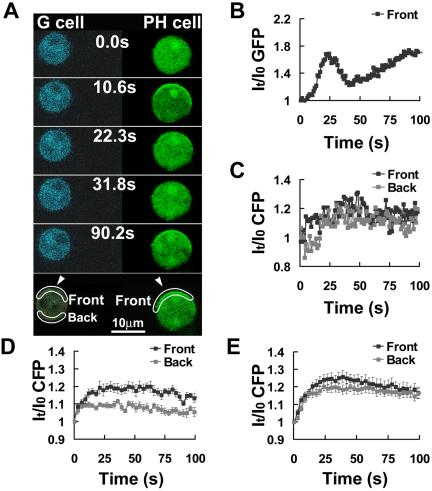
G-protein activation after a sudden exposure to a steady cAMP gradient. (A) Comparison of G-protein activation (CFP images) and PHCrac-GFP translocation. Frames were captured at 1.06-s intervals and selected frames were shown. Regions of interest for the data reported in B and C are also shown. (B) Dynamics of the PHCrac-GFP membrane association in the front of the PH cell. (C) G-protein activation in the front (black) and back (gray) of the G cell, measured as the increase of CFP intensity. Similar results were obtained five times. (D) and (E) Kinetics of G-protein dissociation in the front and back of cells in response to cAMP gradients with different steepness but similar cAMP concentration in the front of the cells. Micropipette filled with 1 or 3 μM of cAMP was moved from 1500 μm away to ∼10 μm (D) or 50 μm (E) from the cells at 0 s. These movement generated gradients with similar cAMP concentration in the front of the cells but different steepness of ∼100% (D) or 20% (E), respectively, which were estimated from the measurement of a stable gradient shown in Supplementary Figure S2. G-protein activation in the front (black) and back (gray) of G cells, measured as the increase of CFP intensity, are shown. Means ± SE of each time point (n = 10 and 18 for D and E, respectively) are shown as temporal changes in the G-protein dissociation in the front and back after stimulation. To estimate the relative difference in G-protein activation, after reaching the steady states, between the front and back side of cells, we first calculated Means, ∑It/I0CFP/n, where n is the number of the time points; the first time point is 27s and the last time point is 100 s. (D) 118.4 ± 1.3% (front) and 108 ± 1.7% (back); (E) 121.5 ± 2.7% (front) and 118 ± 1.4% (back). Relative difference in D: (118.4% - 1)/(108% - 1) - 1 = 130%; in E: (121.5% - 1)/(118% - 1) - 1 = 19.3%.
DISCUSSION
The central question driving chemotaxis research is how an external chemoattractant gradient is translated into a steep intracellular gradient of certain signaling components, leading to morphological cell polarization and directional cell movement. Determining the detailed spatiotemporal patterns of intracellular localization and activation of the components of the chemotactic signaling pathway is essential for answering this question. Here, we have reported advances in live-cell fluorescence microscopy that allowed us to measure the applied chemoattractant concentration, the degree of G-protein activation by FRET imaging, and changes in the level and distribution of PIP3 in single living cells with the needed high temporal and spatial resolution. Our study is consistent with the expectation that the extent of G-protein activation in different regions of the cell surface reflects the local extracellular cAMP concentration. We found that a higher level of uniformly applied cAMP stimulation triggers not only a stronger G-protein activation but also a faster adaptation. Using a new method that allowed us to abruptly expose naïve cells (which have not experienced chemoattractant) to stable cAMP gradients in a controlled manner, we found that G-proteins were persistently activated at the entire cell surface under this condition, whereas PIP3 accumulation in the front of the cell displayed an unexpected biphasic temporal pattern. Details of the behavior of PIP3 suggest modifications of current gradient sensing models (Postma and Van Haastert, 2001; Iglesias and Levchenko, 2002; Iijima et al., 2002; Rappel et al., 2002; Devreotes and Janetopoulos, 2003), and we propose a modified model involving signal-dependent locally recruited inhibitors of PI3K activity to account for the observed dynamics of PIP3.
G-protein Activation on the Cell Surface Depends on the Strength of Local Extracellular Stimuli
Previous studies have shown that both cAMP receptors and G-protein β subunits are uniformly distributed along the perimeter of unpolarized cells (Xiao et al., 1997; Servant et al., 1999; Jin et al., 2000) and that receptor occupancy reflects the local external concentration of chemoattractant (Ueda et al., 2001). cAMP-dose-dependent G-protein activation had previously only been measured as FRET changes in a population of cells (Janetopoulos et al., 2001). Based on the kinetics of FRET change, it was proposed that G-protein activation reflects the local extracellular cAMP concentration (Iijima et al., 2002; Devreotes and Janetopoulos, 2003). This assumption has been a major component in models of chemotactic gradient sensing, but direct measurement of G-protein activation had not been carried out at the single cell level. In this study, temporal and spatial activation of G-proteins in single living cells exposed to various chemoattractant fields was directly visualized and measured for the first time. We found that the level of G-protein activation on membrane of the uniformly stimulated cells depends on cAMP concentrations (Figure 3). In response to asymmetrical cAMP stimulations, a higher degree of G-protein activation occurs in the front of a cell where the local receptor occupancy is higher than that in the back (Figures 5, 6, and 9). We measured G-protein activities of single cells that were exposed to gradients with similar steepness but two different cAMP concentrations (Figure 6) and gradients with similar concentration but two different steepness (Figure 9). When cells were exposed to a gradient (with a steepness of ∼20%), the degree of G-protein dissociation was ∼19% higher in the front than in the back side of the cells (Figure 9E), and the relative difference in PHCrac-GFP distribution between the front and back was ∼150% (Figure 6), which is independent of cAMP concentration (Xu and Jin, unpublished data and Janetopoulos et al., 2004), indicating that cells are able to spatially amplify the difference in G-protein activation into the PHCrac-GFP membrane localization. Furthermore, by determining spatial-temporal dynamics of G-protein activation and PHCrac-GFP membrane translocation in cells that were suddenly exposed to gradients, our study revealed that the process that is responsible for the spatial signal amplification occurs with a significant time lag after initial changes in receptor occupancy and G-protein activation, suggesting that activation of G-proteins provides a simple, intracellular translation of the external gradient and the amplification of the external gradient must hence be achieved further downstream in the signaling pathway. Our results are consistent with the idea that the extent of G-protein activation in different regions of the cell surface reflects the local cAMP receptor occupancy (Iijima et al., 2002; Devreotes and Janetopoulos, 2003).
A Higher Dose of Homogeneously Applied Chemoattractant Leads to Faster Adaptation
Models of chemotactic gradient sensing need to account for two aspects of cellular behavior: i) transient activation, followed by adaptation when cells are exposed to uniform increases in chemoattractant concentration (as seen in the transient nature of PHCrac-GFP translocation to the plasma membrane) and ii) development of strong intracellular biochemical asymmetry in response to chemoattractant gradients. A “local excitation, global inhibition” model has been proposed to explain these behaviors (Parent and Devreotes, 1999), according to which the extent of receptor activation determines the strengths of two opposing processes: a local excitatory one and a global inhibitory one. The balance of these two processes is assumed to control the activities of two enzymes with opposing chemical functions, PI3K (a kinase) and PTEN (a phosphatase). Changes in this balance lead to alterations in PIP3 levels on the inner cell membrane. Local excitation, reflecting local levels of receptor occupancy, increases the recruitment and activation of PI3K and decreases membrane-bound PTEN, whereas the proposed global inhibition, determined by the cell's average receptor occupancy, deactivates PI3K and promotes reassociation of PTEN with the membrane (Devreotes and Janetopoulos, 2003, Janetopoulos et al., 2004).
Although there seems to be general agreement that the primary intracellular stimulus is provided through G-protein activation and that excitation involves activation of PI3K, the biochemical components responsible for the inhibitory process are currently unknown. Consequently, we can only indirectly draw inferences about the nature of those components by measuring the effects of their activities. To investigate the relationship between stimulus strength and the speed at which inhibition induces adaptation, we exposed cells to different doses of homogeneously applied cAMP. We reasoned that if excitation rises quickly and plateaus, and the effects of inhibition increase more slowly after receptor activation, then initially PIP3 levels would increase until at some point (Tmax) the activities of PI3K and PTEN would be equally strong and the net change in the level of PIP3 would be zero. Further increase of the inhibitory components would then cause PIP3 to decrease. A smaller Tmax after a stronger uniform stimulus would therefore occur if the stronger stimulus not only led to a higher excitation but also to a faster induction of strong inhibitory processes. We found that exposing cells to a higher concentration of cAMP increased the level of G-protein activation and indeed shortened Tmax. This finding, together with the fact that the amplitude of the PIP3 response showed only a weak dependence on the strength of the applied stimulus, reveals the cells' ability to robustly respond and quickly adapt to a wide range of stimulus strengths due to a fine-tuned interplay between excitation and inhibition.
A Gradient-induced Biphasic Response of PIP3 in the Front Side of a Cell Suggests Asymmetrical Recruitment of Inhibitors to the Membrane
Several attempts have been made to predict the spatiotemporal patterns of G-protein activation and PIP3 accumulation at the front and back sides of a cell that is abruptly exposed to a stable cAMP gradient (Postma and Van Haastert, 2001; Iglesias and Levchenko, 2002; Iijima et al., 2002; Rappel et al., 2002; Devreotes and Janetopoulos, 2003). Temporal changes in PIP3 levels are predicted to display a simple peak in both the front and back of the cell according to these recent models: After exposure to a gradient, PIP3 accumulation is assumed to first occur at both ends, to reach a maximum, and then to decline to reach different steady-state levels in distinct parts of the cell. A global, stimulus-dependent inhibition, is assumed to act equally everywhere on the cell membrane suppressing the response in the back, but much less in the front of the cell where the excitatory stimulus is stronger. In contrast to these predictions, we found that PIP3 accumulation at the front of cells exposed to such a sudden, stable gradient did not persistently remain at a high level after the onset of stimulation. Instead, it decayed after an initial peak and subsequently went through a slower, longer-lasting second phase of increase, followed by a second, attenuated decay. This secondary rise did not occur at the back side of the cell. Recent studies have shown that a uniform cAMP stimulation induces a temporal biphasic translocation response of PHCrac-GFP to the plasma membrane in polarized and even in latrunculin-treated D. discoideum cells (Postma et al., 2003, 2004a, 2004b). After a rapid and transient translocation of PHCrac-GFP to nearly the entire plasma membrane, multiple self-organizing PHCrac-GFP patches form in various regions of the membrane in response to a uniform stimulation, suggesting that cells can respond in various directions without an extracellular directional cue from a gradient. We found that a cAMP gradient induced a persistent and graded G-protein activation on the inner membrane, which provides spatial cues to generate a polarized PHCrac-GFP translocation through the temporal biphasic pattern that specifically occurred in the front side, indicating that the gradient sensing machinery is able to determine the preferred direction of a polarized response independent of cell polarity and the actin cytoskeleton in D. discoideum cells. We interpret a gradient-induced PHCrac-GFP translocation response as a two-step process consisting of an “activation and adaptation” step, during which a cell transiently responds to a sudden increase in receptor occupancy, followed by an “amplification” step, during which PIP3 reaccumulates only at the front of the cell.
When a cell is exposed to a gradient, PI3K is enriched at the membrane in the front whereas membrane-bound PTEN accumulates in the back (Funamoto et al., 2002; Janetopoulos et al., 2004). However, this simple redistribution of PTEN and PI3K could not fully explain the details of the biphasic response observed in our study. Several possible models are discussed below.
In our view, the “activation and adaptation” step is similar to the response to uniform stimulation: G-proteins dissociate, PI3K is recruited and activated (Funamoto et al., 2002; Huang et al., 2003), and PTEN dissociates from the membrane (Funamoto et al., 2002; Iijima and Devreotes, 2002). As a result, PIP3 levels increase along the entire perimeter of the cell in proportion to the cAMP level at each point along the membrane. PI3K activity is then downregulated because of inhibitor recruitment to the membrane, a process that must be strong enough to result in a decrease of PIP3 levels even though the amount of membrane-associated PTEN has not yet returned to its prestimulus level (Funamoto et al., 2002). The gradual redistribution of PTEN away from the front of the cell, which has recently been observed in latrunculin-treated D. discoideum cells (Iijima et al., 2004), is presumed to result in the second rise of PIP3 in that region. In response to a gradient with lower absolute concentration of cAMP, we observed a smaller decline after the first peak (Figure 8). The biphasic response reported here, which we suggest arises from differential rates of PI3K inactivation and redistribution of PTEN at the front of the cell, had been predicted by a computational model before the experiment (Meier-Schellersheim, unpublished results). Interestingly, the second increase of PIP3 levels in the front of the cell reaches a peak and declines a little after some time and PIP3 in the back of the cell does not decay indefinitely in spite of the highly polarized distribution of PTEN. If we assume that the loss of PTEN from the front is not accompanied by a loss of PI3K, then the PI3K-inhibiting activity we postulate must be regulated by local, rather than global, feedback mechanisms; this would result in a stronger suppression of PI3K activity (as opposed to concentration) in the front and a much weaker suppression of its activity in the back, accounting for the limitation of PIP3 accumulation in the front and the stabilization of PIP3 levels in the back.
The putative inhibitor(s) remain(s) to be identified. Among the candidates are Ras GTPase-activating proteins (RasGAP). RasGAP binds PIP3 on the cell membrane following activation of PI3K (Lockyer et al., 1999). The recruitment of this molecule would contribute to a negative feedback loop by deactivating Ras, an activator of PI3K (Insall et al., 1996; Tuxworth et al., 1997; Funamoto et al., 2002, Li et al., 2003; Meili and Firtel 2003; Xu et al., 2003). Biochemical identification of the proposed PI3K inhibitor, whether Ras-GAP or another molecule, would allow for new fluorescence microscopy experiments that assess G-protein activation, PIP3 dynamics, PTEN and PI3K distribution, and PI3K inhibitor location in a single cell to rigorously test our model.
Other models may also explain the biphasic response. For example, if receptor activation would elicit a transient increase in PTEN activity (as opposed to membrane localization), this could result in a transient decrease in PIP3 level on the whole cell membrane, and redistribution of PTEN to the back side of the cell could result in a PIP3 reaccumulation only at the front of the cell. When PIP3 levels in both the front and back of a cell reach the steady-states and the cell becomes biochemically polarized, the level of PTEN in the front is low (Funamoto et al., 2002; Iijima and Devreotes, 2002), whereas the PI3K amount in the front remains very high (Funamoto et al., 2002). It is possible that PIP3 level in the front do not keep raising indefinitely because diffusion of PIP3 is fast enough to bring it to PTEN that then degrades it. Alternatively, a reduction in the level of PIP2, the substrate of PI3K, on the membrane may limit the rise of PIP3 in the front. However, this seems unlikely because PIP2 is unlikely a limiting factor because the level of PIP2 is several hundred fold higher than that of PIP3 in the plasma membrane (Huang et al., 2003).
Gradient Sensing, Cell Polarity, and Chemotaxis
Although we investigated gradient sensing in the absence of preexisting cell polarity, chemotaxing cells are polarized, and many signaling components are asymmetrically distributed in such cells. In neutrophils and D. discoideum, Rac and CDC42 play different roles in the regulation of PIP3 in various regions of polarized cells (Butty et al., 2002; Li et al., 2003; Meili and Firtel, 2003; Xu et al., 2003; Park et al., 2004). It has been proposed that PIP3 and Rac serve as signals in a positive feedback loop that enhances PIP3 production at the leading edge of a polarized chemotaxing cell (Weiner et al., 2002; Welch et al., 2002; Srinivasan et al., 2003). Another mechanism leading to intracellular polarization in mammalian cells is the generation of divergent signals by different receptor-activated heterotrimeric G-proteins in the leading and trailing edges of a cell: in the front, GPCR-activated Gi lead to production of PIP3, activate Rac, and induce polymerization of actin; in the back, the same receptor stimulates G12 and G13, which mediate the inhibition of the responses occurring at the leading edge (Xu et al., 2003). Both mechanisms require a polarized morphology before a cell is able to amplify chemoattractant gradients. The actin-independent biphasic response we observed allows a cell to localize biochemical responses before it becomes morphologically polarized. After morphological polarization occurs, other mechanisms may further enhance a cell's gradient sensing ability during chemotaxis.
Supplementary Material
Acknowledgments
We thank Ronald N. Germain, Susan K. Pierce, Dale Hereld, Alan Kimmel, and Corale Parent for discussions and critical readings of the manuscript.
Article published online ahead of print in MBC in Press on November 24, 2004 (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-07-0544).
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Adams, S. R., Harootunian, A. T., Buechler, Y. J., Taylor, S. S., and Tsien, R. Y. (1991). Fluorescence ratio imaging of cyclic AMP in single cells. Nature 349, 694-697. [DOI] [PubMed] [Google Scholar]
- Butty, A. C., Perrinjaquet, N., Petit, A., Jaquenoud, M., Segall, J. E., Hofmann, K., Zwahlen, C., and Peter, M. (2002). A positive feedback loop stabilizes the guanine-nucleotide exchange factor Cdc24 at sites of polarization. EMBO J. 21, 1565-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, C. Y., Funamoto, S., and Firtel, R. A. (2001). Signaling pathways controlling cell polarity and chemotaxis. Trends Biochem. Sci. 26, 557-566. [DOI] [PubMed] [Google Scholar]
- Comer, F. I., and Parent, C. A. (2002). PI 3-kinases and PTEN: how opposites chemoattract. Cell 109, 541-544. [DOI] [PubMed] [Google Scholar]
- Devreotes, P., and Janetopoulos, C. (2003). Eukaryotic chemotaxis: distinctions between directional sensing and polarization. J. Biol. Chem. 278, 20445-20448. [DOI] [PubMed] [Google Scholar]
- Devreotes, P. N., and Zigmond, S. H. (1988). Chemotaxis in eukaryotic cells: a focus on leukocytes and Dictyostelium. Annu. Rev. Cell Biol. 4, 649-686. [DOI] [PubMed] [Google Scholar]
- Funamoto, S., Meili, R., Lee, S., Parry, L., and Firtel, R. A. (2002). Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell 109, 611-623. [DOI] [PubMed] [Google Scholar]
- Funamoto, S., Milan, K., Meili, R., and Firtel, R. A. (2001). Role of phosphatidylinositol 3′ kinase and a downstream pleckstrin homology domain-containing protein in controlling chemotaxis in Dictyostelium. J. Cell Biol. 153, 795-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, Y., Di, W. L., Kelsell, D. P., and Zicha, D. (2004). Quantitative fluorescence resonance energy transfer (FRET) measurement with acceptor photobleaching and spectral unmixing. J. Microsc. 21, 162-173. [DOI] [PubMed] [Google Scholar]
- Hazeki, O., Okada, T., Kurosu, H., Takasuga, S., Suzuki, T., and Katada, T. (1998). Activation of PI 3-kinase by G protein betagamma subunits. Life Sci. 62, 1555-1559. [DOI] [PubMed] [Google Scholar]
- Hirsch, E., Katanaev, V. L.,. Garlanda, C., Azzolino, O., Pirola, L., Silengo, L., Sozzani, S., Mantovani, A., Altruda, F., and Wymann, M. P. (2000). Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287, 1049-1053. [DOI] [PubMed] [Google Scholar]
- Huang, Y. E., Iijima, M., Parent, C. A., Funamoto, S., Firtel, R. A., and Devreotes, P. (2003). Receptor-mediated regulation of PI3Ks confines PI(3,4,5)P3 to the leading edge of chemotaxing cells. Mol. Biol. Cell 14, 1913-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias, P. A., and Levchenko, A. (2002). Modeling the cell's guidance system. Sci. STKE 2002, RE12. [DOI] [PubMed] [Google Scholar]
- Iijima, M., and Devreotes, P. (2002). Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell 109, 599-610. [DOI] [PubMed] [Google Scholar]
- Iijima, M., Huang, Y. E., and Devreotes, P. (2002). Temporal and spatial regulation of chemotaxis. Dev. Cell 3, 469-478. [DOI] [PubMed] [Google Scholar]
- Iijima, M., Huang, Y. E., Luo, H.R., Vazquez, F., and Devreotes, P. N. (2004). Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis. J. Biol. Chem. 279, 16606-16613. [DOI] [PubMed] [Google Scholar]
- Insall, R., Kuspa, A., Lilly, P. J., Shaulsky, G., Levin, L. R., Loomis, W. F., and Devreotes, P. (1994). CRAC, a cytosolic protein containing a pleckstrin homology domain, is required for receptor and G. protein-mediated activation of adenylyl cyclase in Dictyostelium. J. Cell Biol. 126, 1537-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insall, R. H., Borleis, J., and Devreotes, P. N. (1996). The aimless RasGEF is required for processing of chemotactic signals through G-protein-coupled receptors in Dictyostelium. Curr. Biol. 6, 719-729. [DOI] [PubMed] [Google Scholar]
- Janetopoulos, C., Jin, T., and Devreotes, P. (2001). Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science 291, 2408-2411. [DOI] [PubMed] [Google Scholar]
- Janetopoulos, C., Ma, L., Devreotes, P. N., and Iglesias, P. A. (2004). Chemoattractant-induced phosphatidylinositol 3,4,5-trisphosphate accumulation is spatially amplified and adapts, independent of the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 101, 8951-8956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, T., Amzel, M., Devreotes, P. N., and Wu, L. (1998). Selection of gbeta subunits with point mutations that fail to activate specific signaling pathways in vivo: dissecting cellular responses mediated by a heterotrimeric G protein in Dictyostelium discoideum. Mol. Biol. Cell 9, 2949-2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, T., Zhang, N., Long, Y., Parent, C. A., and Devreotes, P. N. (2000). Localization of the G protein betagamma complex in living cells during chemotaxis. Science 287, 1034-1036. [DOI] [PubMed] [Google Scholar]
- Johnson, R. L., Van Haastert, P. J., Kimmel, A. R., Saxe, C. L., 3rd, Jastorff, B., and Devreotes, P. N. (1992). The cyclic nucleotide specificity of three cAMP receptors in Dictyostelium. J. Biol. Chem. 267, 4600-4607. [PubMed] [Google Scholar]
- Kavran, J. M., Klein, D. E., Lee, A., Falasca, M., Isakoff, S. J., Skolnik, E. Y., and Lemmon, M. A. (1998). Specificity and promiscuity in phosphoinositide binding by pleckstrin homology domains. J. Biol. Chem. 273, 30497-30508. [DOI] [PubMed] [Google Scholar]
- Levchenko, A., and Iglesias, P. A. (2002). Models of eukaryotic gradient sensing: application to chemotaxis of amoebae and neutrophils. Biophys. J. 82, 50-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., Hannigan, M., Mo, Z., Liu, B., Lu, W., Wu, Y., Smrcka, A. V., Wu, G., Li, L., Liu, M., Huang, C. K., and Wu, D. (2003). Directional sensing requires G beta gamma-mediated PAK1 and PIX alpha-dependent activation of Cdc42. Cell 114, 215-227. [DOI] [PubMed] [Google Scholar]
- Li, Z., Jiang, H., Xie, W., Zhang, Z., Smrcka, A. V., and Wu, D. (2000). Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science 287, 1046-1049. [DOI] [PubMed] [Google Scholar]
- Liliental, J., Moon, S. Y., Lesche, R., Mamillapalli, R., Li, D., Zheng, Y., Sun, H., and Wu, H. (2000). Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr. Biol. 10, 401-404. [DOI] [PubMed] [Google Scholar]
- Lilly, P. J., and Devreotes, P. N. (1994). Identification of CRAC, a cytosolic regulator required for guanine nucleotide stimulation of adenylyl cyclase in Dictyostelium. J. Biol. Chem. 269, 14123-14129. [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., Snapp, E., and Kenworthy, A. (2001). Studying protein dynamics in living cells. Nat. Rev. 2, 444-456. [DOI] [PubMed] [Google Scholar]
- Lockyer, P. J., Wennstrom, S., Kupzig, S., Venkateswarlu, K., Downward, J., and Cullen, P. J. (1999). Identification of the ras GTPase-activating protein GAP1(m) as a phosphatidylinositol-3,4,5-trisphosphate-binding protein in vivo. Curr. Biol. 9, 265-268. [DOI] [PubMed] [Google Scholar]
- Miyawaki, A. (2003). Visualization of the spatial and temporal dynamics of intracellular signaling. Dev. Cell 4, 295-305. [DOI] [PubMed] [Google Scholar]
- Meili, R., Ellsworth, C., Lee, S., Reddy, T. B., Ma, H., and Firtel, R. A. (1999). Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium. EMBO J. 18, 2092-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meili, R., and Firtel, R. A. (2003). Two poles and a compass. Cell 114, 153-156. [DOI] [PubMed] [Google Scholar]
- Meinhardt, H. (1999). Orientation of chemotactic cells and growth cones: models and mechanisms. J. Cell Sci. 112(Pt 17), 2867-2874. [DOI] [PubMed] [Google Scholar]
- Murphy, P. M. (1994). The molecular biology of leukocyte chemoattractant receptors. Annu. Rev. Immunol. 12, 593-633. [DOI] [PubMed] [Google Scholar]
- Parent, C. A., Blacklock, B. J., Froehlich, W. M., Murphy, D. B., and Devreotes, P. N. (1998). G protein signaling events are activated at the leading edge of chemotactic cells. Cell 95, 81-91. [DOI] [PubMed] [Google Scholar]
- Parent, C. A., and Devreotes, P. N. (1999). A cell's sense of direction. Science 284, 765-770. [DOI] [PubMed] [Google Scholar]
- Park, K. C., Rivero, F., Meili, R., Lee, S., Apone, F., and Firtel, R. A. (2004). Rac regulation of chemotaxis and morphogenesis in Dictyostelium. EMBO J. 23, 4177-4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma, M., Bosgraaf, L., Loovers, H. M., and Van Haastert, P. J. (2004a). Chemotaxis: signalling modules join hands at front and tail. EMBO Rep. 5, 35-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma, M., Roelofs, J., Goedhart, J., Gadella, T. W., Visser, A. J., and Van Haastert, P. J. (2003). Uniform cAMP stimulation of Dictyostelium cells induces localized patches of signal transduction and pseudopodia. Mol. Biol. Cell 14, 5019-5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma, M., Roelofs, J., Goedhart, J., Loovers, H. M., Visser, A.J., and Van Haastert, P. J. (2004b). Sensitization of Dictyostelium chemotaxis by phosphoinositide-3-kinase-mediated self-organizing signalling patches. J. Cell Sci. 117, 2925-2935. [DOI] [PubMed] [Google Scholar]
- Postma, M., and Van Haastert, P. J. (2001). A diffusion-translocation model for gradient sensing by chemotactic cells. Biophys. J. 81, 1314-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappel, W. J., Thomas, P. J., Levine, H., and Loomis, W. F. (2002). Establishing direction during chemotaxis in eukaryotic cells. Biophys. J. 83, 1361-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert, P., Weiner, O. D., Wang, F., Bourne, H. R., and Servant, G. (2000). Leukocytes navigate by compass: roles of PI3Kgamma and its lipid products. Trends Cell Biol. 10, 466-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki, T. et al. (2000). Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 287, 1040-1046. [DOI] [PubMed] [Google Scholar]
- Segall, J. E. (1999). Cell polarization: chemotaxis gets CRACKing. Curr. Biol. 9, R46-R48. [DOI] [PubMed] [Google Scholar]
- Sekar, R. B., and Periasamy, A. (2003). Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 160, 629-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant, G., Weiner, O. D., Herzmark, P., Balla, T., Sedat, J. W., and Bourne, H. R. (2000). Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science 287, 1037-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant, G., Weiner, O. D., Neptune, E. R., Sedat, J.W., and Bourne, H. R. (1999). Dynamics of a chemoattractant receptor in living neutrophils during chemotaxis. Mol. Biol. Cell 10, 1163-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan, S., Wang, F., Glavas, S., Ott, A., Hofmann, F., Aktories, K., Kalman, D., and Bourne, H. R. (2003). Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 160, 375-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, L., Ellson, C., and Hawkins, P. (2002). Roles of PI3Ks in leukocyte chemotaxis and phagocytosis. Curr. Opin. Cell Biol. 14, 203-213. [DOI] [PubMed] [Google Scholar]
- Stoyanov, B. et al. (1995). Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269, 690-693. [DOI] [PubMed] [Google Scholar]
- Thelen, M. (2001). Dancing to the tune of chemokines. Nat. Immunol. 2, 129-134. [DOI] [PubMed] [Google Scholar]
- Tuxworth, R. I., Cheetham, J. L., Machesky, L. M., Spiegelmann, G. B., Weeks, G., and Insall, R. H. (1997). Dictyostelium RasG is required for normal motility and cytokinesis, but not growth. J. Cell Biol. 138, 605-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda, M., Sako, Y., Tanaka, T., Devreotes, P., and Yanagida, T. (2001). Single-molecule analysis of chemotactic signaling in Dictyostelium cells. Science 294, 864-867. [DOI] [PubMed] [Google Scholar]
- Wang, F., Herzmark, P., Weiner, O. D., Srinivasan, S., Servant, G., and Bourne, H. R. (2002). Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat. Cell Biol. 4, 513-518. [DOI] [PubMed] [Google Scholar]
- Weiner, O. D., Neilsen, P. O., Prestwich, G. D., Kirschner, M.W., Cantley, L. C., and Bourne, H. R. (2002). A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat. Cell Biol. 4, 509-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, H. C., Coadwell, W. J., Ellson, C. D., Ferguson, G. J., Andrews, S. R., Erdjument-Bromage, H., Tempst, P., Hawkins, P. T., and Stephens, L. R. (2002). P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell 108, 809-821. [DOI] [PubMed] [Google Scholar]
- Xiao, Z., Zhang, N., Murphy, D. B., and Devreotes, P. N. (1997). Dynamic distribution of chemoattractant receptors in living cells during chemotaxis and persistent stimulation. J. Cell Biol. 139, 365-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J., Wang, F., Van Keymeulen, A., Herzmark, P., Straight, A., Kelly, K., Takuwa, Y., Sugimoto, N., Mitchison, T., and Bourne, H. R. (2003). Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell 114, 201-214. [DOI] [PubMed] [Google Scholar]
- Zigmond, S. H. (1978). Chemotaxis by polymorphonuclear leukocytes. J. Cell Biol. 77, 269-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond, S. H., Levitsky, H. I., and Kreel, B. J. (1981). Cell polarity: an examination of its behavioral expression and its consequences for polymorphonuclear leukocyte chemotaxis. J. Cell Biol. 89, 585-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.