

Abstract
Extracellular matrix remodeling occurs during development, tissue repair, and in a number of pathologies, including fibrotic disorders, hypertension, and atherosclerosis. Extracellular matrix remodeling involves the complex interplay between extracellular matrix synthesis, deposition, and degradation. Factors that control these processes are likely to play key roles in regulating physiological and pathological extracellular matrix remodeling. Our data show that fibronectin polymerization into the extracellular matrix regulates the deposition and stability of other extracellular matrix proteins, including collagen I and thrombospondin-1 (Sottile and Hocking, 2002. Mol. Biol. Cell 13, 3546). In the absence of continual fibronectin polymerization, there is a loss of fibronectin matrix fibrils, and increased levels of fibronectin degradation. Fibronectin degradation occurs intracellularly after endocytosis and can be inhibited by chloroquine, an inhibitor of lysosomal degradation, and by caveolae-disrupting agents. Down-regulation of caveolin-1 by RNAi inhibits loss of fibronectin matrix fibrils, fibronectin internalization, and fibronectin degradation; these processes can be restored by reexpression of caveolin-1. These data show that fibronectin matrix turnover occurs through a caveolin-1–dependent process. Caveolin-1 regulation of fibronectin matrix turnover is a novel mechanism regulating extracellular matrix remodeling.
INTRODUCTION
Extracellular matrix (ECM) remodeling is a dynamic, cell-mediated process that occurs during development, tissue repair, and in a variety of pathological events including atherosclerosis, hypertension, and ischemic injury (Clark, 1996; Prescott et al., 1999; Streuli, 1999; Newby and Zaltsman, 2000; Intengan and Schiffrin, 2001). Furthermore, abnormal matrix remodeling is associated with fibrosis, arthritis, reduced angiogenesis, and developmental abnormalities (Liu et al., 1995; Vu et al., 1998; Holmbeck et al., 1999). During tissue repair, the precise deposition of ECM molecules, including collagen I and fibronectin, is required to preserve the structural and functional integrity of tissues (Clark, 1996). Excessive or inappropriate deposition of ECM molecules, as occurs during fibrosis, disrupts normal tissue architecture, leading to impaired organ function (Mutsaers et al., 1997; Zeisberg et al., 2000). The mechanisms that control ECM organization and homeostasis are incompletely understood. We have recently shown that fibronectin matrix polymerization is essential for the organization, as well as the maintenance of ECM architecture (Sottile and Hocking, 2002). Our data show that the cell-dependent process of polymerizing fibronectin into the ECM is required for the deposition and maintenance of fibrillar fibronectin, collagen-I, and thrombosponin-1 (Sottile and Hocking, 2002). These data are consistent with other studies showing that collagen I and collagen III deposition into the ECM are regulated by fibronectin (McDonald et al., 1982; Velling et al., 2002). Fibronectin has also been implicated in regulating the deposition of tenascin C (Chung and Erickson, 1997), fibulin (Roman and McDonald, 1993; Godyna et al., 1995b; Sasaki et al., 1996), and fibrinogen (Pereira et al., 2002) in the ECM. Hence, fibronectin plays a key role in regulating ECM composition and stability.
ECM remodeling is controlled by a combination of matrix synthesis, deposition, and degradation. Extracellular proteases such as plasmin, plasminogen activators, and matrix metalloproteinases (MMPs) can degrade ECM proteins. ECM turnover is also regulated by endocytosis. ECM proteins such as thrombosponin-1 and vitronectin are known to be internalized by receptor-mediated endocytosis and degraded in the lysosomes (McKeown-Longo et al., 1984; Murphy-Ullrich and Mosher, 1987; Godyna et al., 1995a; Pijuan-Thompson and Gladson, 1997; Memmo and McKeown-Longo, 1998). Recent studies also indicate that collagen I can be endocytosed by the Endo180 receptor (East et al., 2003; Engelholm et al., 2003; Wienke et al., 2003). We previously showed that the loss of ECM fibronectin fibrils could not be inhibited by a variety of protease inhibitors (Sottile and Hocking, 2002), suggesting that turnover of ECM fibronectin may also involve endocytosis and intracellular degradation. Recently published data (Salicioni et al., 2002) also support a role for fibronectin endocytosis in regulating the degradation of soluble fibronectin.
We have developed a model system using fibronectin-null myofibroblasts (FN-null MF) to examine the role of fibronectin polymerization in regulating ECM turnover. The fibronectin-null background has proven to be a valuable tool for determining cell behavior in the complete absence of fibronectin and for distinguishing the effects of ECM fibronectin from the effects of soluble fibronectin (Sottile et al., 1998; Hocking et al., 2000; Sottile and Hocking, 2002; Hocking and Chang, 2003). In this article, we have used FN-null MF to determine the mechanism(s) that regulate the turnover of ECM fibronectin. Our data show that turnover of matrix fibronectin involves caveolin-1–mediated endocytosis and intracellular degradation.
MATERIALS AND METHODS
Immunological Reagents and Chemicals
Polyclonal antifibronectin antibody was a generous gift from Dr. Deane Mosher (University of Wisconsin, Madison, WI). Antibodies to LAMP-1, EEA-1, and caveolin-1 were from BD Biosciences (San Jose, CA). Chloroquine, β-cyclodextrin, genistein, and staurosporin were from Sigma (St. Louis, MO).
Proteins
Human fibronectin was purified from Cohn's fractions 1 and 2 (a generous gift from Dr. Ken Ingham, American Red Cross, Bethesda, MD) as previously described (Miekka et al., 1982). Thrombospondin-1 was purchased from Hematologic Technologies (Essex Junction, VT). Human RAP was purchased from Molecular Innovations (Southfield, MI). A bacterial expression vector containing GST-RAP (Herz et al., 1991) was a kind gift of Dr. Herz (University of Texas Southwestern Medical Center, Dallas, TX). GST-RAP was purified on a glutathione-agarose column as described (Herz et al., 1991). pUR4 was a kind gift of Dr. Hanski (Ozeri et al., 1996) and was provided to us by Dr. Mosher. pUR4 is a 49-mer peptide derived from Streptococcus pyogenes adhesin F1, that binds to fibronectin, and inhibits fibronectin matrix polymerization (Tomasini-Johansson et al., 2001). A carboxyl terminal fragment (68mer) of fibronectin's III-11 module (III-11C) was produced with a his tag and expressed in bacteria as described (Morla et al., 1994). Both pUR4 and the control III-11C peptides were purified from bacterial lysates on nickel-agarose columns as described (Tomasini-Johansson et al., 2001). Vitronectin was purified from human serum as described previously (Yatohgo et al., 1988). Recombinant vitronectin was produced in bacteria with (Wojciechowski et al., 2004) or without a GST tag and purified on a heparin-agarose column as described (Wojciechowski et al., 2004). Some recombinant vitronectin was provided by Dr. Hocking (University of Rochester, Rochester, NY)
Cell Culture
We previously described the isolation of fibronectin-null cells from fibronectin-null embryos (Sottile et al., 1998). These cells were adapted to grow in defined media to establish a model system in which all cell- and serum-derived fibronectin was eliminated (Sottile et al., 1998). The integrin repertoire of these cells was previously described (Sottile et al., 1998). We characterized these cells as myofibroblasts (FN-null MF) based on their expression of some smooth muscle cell (SMC) marker proteins (SM calponin and SM α-actin) but not others (SM22 and desmin; unpublished data) and on their ability to contract collagen gels (Hocking et al., 2000). Rat and human aortic SMCs were obtained from Cell Applications (San Diego, CA) and maintained in serum containing media (Cell Applications).
Fibronectin Pulse-Chase Experiments
FN-null MF were plated onto glass coverslips precoated with 5 μg/ml vitronectin or 10 μg/ml recombinant vitronectin. Cells were grown to 80–90% confluence in defined medium at 37°C and were then incubated (“pulsed”) overnight with 20 nM fibronectin. Cells were then either processed for immunofluorescence, or were washed, and then incubated (“chased”) with culture medium containing or lacking 20 nM fibronectin. In some experiments, the fibronectin used was conjugated to either Texas Red (Molecular Probes Inc, Eugene, OR) or to FITC (Cappel, West Chester, PA) as described (Sottile and Mosher, 1997). The chase medium was supplemented with various inhibitors, as described in the figure legends. Cells were fixed with paraformaldehyde, permeabilized with 0.5% Triton X-100 and then mounted in glycerol gel (Sigma) or in 90% glycerol/10% 0.05 M carbonate buffer, pH 9, with 2 mg/ml ascorbate. After fixing and permeabilizing, cells were incubated with antibodies to caveolin-1, EEA-1, or LAMP-1, followed by Texas-Red– or FITC-conjugated secondary antibodies. Cells were examined using an Olympus BX60 microscope (Melville, NY) equipped with epifluorescence. For some experiments, images were obtained with an Olympus scanning confocal microscope.
Protein Colocalization
Confocal z-sections were analyzed using Fluoview software (Olympus). Plots of fluorescence intensity versus position were generated from several separate fields of view. Background fluorescence was determined for each image. Colocalization was defined as those areas where the fluorescent peaks of the two fluors coincided and were above background. Peaks lacking one or the other fluor were defined as areas lacking colocalization. One hundred or more peaks were analyzed per field of view. Data represents the average percent overlap ± SEM of three or more separate images.
Iodination of Proteins and Binding Assays
Fibronectin and thrombospondin were iodinated using the chloramine T method as described (McKeown-Longo and Mosher, 1985). Labeled proteins were separated from unincorporated iodine by gel filtration on Pharmacia (Piscataway, NJ) PD-10 columns (fibronectin) or by using a heparin-Sepharose column (thrombospondin). Iodinated proteins were dialyzed against phosphate-buffered saline (PBS) at room temperature for 3 h. The specific activity of iodinated fibronectin was: 2.5–5.7 × 1011 μCi/mol. The specific activity of iodinated thrombospondin was 1.56 × 1012 μCi/mol. Binding assays were performed essentially as described (Sottile and Wiley, 1994). Briefly, FN-null MF were seeded into 24-well cluster dishes in Cellgro:Aim V (1:1) and allowed to grow to 80–90% confluence for 2–3 d. The cell culture medium was then replaced with medium containing iodinated fibronectin. Rat SMCs were grown to confluence in serum containing media. Binding assays with SMCs were done in either 5% fibronectin-depleted fetal bovine serum/DMEM or in 0.2% bovine serum albumin (BSA)/DMEM. After an overnight incubation with iodinated fibronectin, cells were either processed as described below or were washed with Cellgro:Aim V and then incubated in culture medium containing or lacking unlabeled fibronectin for 6–24 h. After this incubation period, the cell culture supernatant was collected to determine the amount of released fibronectin. The cell layers were washed and then solubilized in 0.2 N NaOH to determine the amount of cell- and matrix-associated counts. The culture supernatant was precipitated with 13% trichloroacetic acid (TCA) at 4°C for ≥1 h. Samples were then centrifuged at 4°C at 12,000 × g for 15 min, and the amount of TCA-soluble and -insoluble counts determined using a gamma counter. The amount of TCA-soluble counts is expressed as a percentage of the total counts present in each well. Binding studies with thrombospondin were done by culturing the cells in media containing 125I-labeled thrombospondin for 1–8 h in the presence and absence of inhibitors as described in the figure legends. Nonspecific degradation was determined by incubating 125I-labeled thrombospondin in the absence of cells at 37°C and was subtracted from each data point.
Western Blotting
Cells lysates were prepared as described (Sottile and Hocking, 2002). Equal amounts of proteins were analyzed under reducing conditions by SDS-PAGE, transferred to nitrocellulose paper (Towbin et al., 1979), and probed with antibodies that recognize caveolin-1. To ensure equal protein loading, the blots were also probed with antibodies that recognize vinculin. Blots were imaged and analyzed using an Odyssey Infrared Imaging System (Li-Cor Biosciences, Lincoln, NE).
siRNA
Pairs of oligonucleotides based on the mouse caveolin-1 sequence were designed using the RNA interference (RNAi) OligoRetriever program (http://katahdin.cshl.org:9331/RNAi). The short hairpin (sh) was targeted to nucleotides 429–459 of mouse caveolin-1. Oligonucleotides used were: Primer A: GACGTAGATGGAGTAGACGCGGCTGATGGAAGCTTGCGTCGGCTGCGTCTACTCCGTCTACGTCCATTTTTTT; Primer B: GATCAAAAAAATGGACGTAGACGGAGTAGACGCAGCCGACGCAAGCTTCCATCAGCCGCGTCTACTCCATCTACGTCCG. Oligonucleotide pairs were annealed and ligated into the vector pSHAG (Paddison et al., 2002), a generous gift from Dr. Hannon (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). In this vector, shRNA is under the control of RNA polymerase III promoter, U6. A HincII-PvuII fragment containing the U6-shRNA cassette was subcloned from pSHAG into pCDNA3.1/Zeo (Invitrogen, Carlsbad, CA) that had been previously digested with NruI and EcoRV to remove the CMV promoter region. The resulting construct expresses shRNA under the direction of the U6 promoter, and also contains a zeomycin resistance gene. shcav/pCDNA was transfected into FN-null MF using FuGENE (Roche Diagnostics, Indianapolis, IN). Zeocin-resistant cell lines were cloned and expanded. The sequence of mouse and human caveolin-1 in the region that is targeted by the siRNA varies by three bases (two of which are internal). Control cells were transfected with pCDNA containing an shRNA based on the firefly luciferase gene (shffluc). The U6-shffluc cassette was obtained from pSHAG-Ff1 (Paddison et al., 2002) (a gift from Dr. Hannon) as described for shcav. Flow cytometry experiments demonstrate that cells expressing caveolin-1 siRNA (shcav cells) and parental FN null MF express very similar amounts of α5, αv, β1, and β3 integrins (unpublished data).
Adenoviruses
The coding sequence of human caveolin-1 was PCR amplified from a construct containing human caveolin-1 cDNA (a gift from Dr. Mundy, University of Texas Southwestern, Dallas, TX) and inserted into the vector pENTR1A (Invitrogen). A cav-1/pENTR1A clone was recombined with the adenoviral vector pAD/pLDEST (Invitrogen) using Gateway LR Clonase according to the manufacturer's protocol. The resulting cav-1/pAD plasmid was transfected into HEK293 cells and amplified according to the manufacturer's protocol. An adenovirus containing the tetracycline regulatable transactivator was a kind gift of Dr. Schmid (Scripps Research Institute, La Jolla, CA) (Fish et al., 2000) and was used as a negative control virus. Viruses were purified and titered using kits from BD Biosciences.
RESULTS
Fibronectin Is Internalized and Degraded During Fibronectin Matrix Turnover
Turnover of ECM proteins is an important mechanism to remove biologically active proteins from the extracellular environment. We previously reported that the maintenance of ECM fibronectin fibrils requires the continual polymerization of a fibronectin matrix (Sottile and Hocking, 2002). Cells cultured in the absence of fibronectin, or in the presence of fibronectin polymerization inhibitors, showed a dramatic loss of fibronectin matrix fibrils (Sottile and Hocking, 2002). The loss of fibronectin matrix fibrils could not be inhibited with a variety of protease inhibitors (Sottile and Hocking, 2002). Hence, we examined the possibility that fibronectin was internalized and degraded intracellularly. We conducted pulse-chase studies with 125I-fibronectin in cells that were actively polymerizing a fibronectin matrix and in cells where fibronectin polymerization was inhibited. We tested whether 125I-fibronectin was degraded by precipitating the cell culture supernatant with TCA. FN-null MF were incubated with 125I-fibronectin overnight and allowed to establish a fibronectin matrix. The cells were washed and then incubated (chased) in media containing or lacking unlabeled fibronectin. We previously showed that >85% of the cell-associated fibronectin was incorporated into the ECM at the start of the chase, indicating that the great majority of the fibronectin turnover that occurs during the chase involves matrix fibronectin (Sottile and Hocking, 2002). As shown in Figure 1, there was a time-dependent increase in the amount of degraded (TCA soluble) fibronectin in the cell culture supernatant during the chase. As much as 56% of the total counts in cells that were chased in the absence of fibronectin represent degraded fibronectin. This represents a 1.8–2× increase in fragment production compared with cells that were chased in the presence of fibronectin. The ability of unlabeled fibronectin to inhibit 125I-fibronectin degradation was dependent on its ability to become polymerized into the ECM, because cells coincubated with fibronectin and the fibronectin polymerization inhibitor, pUR4, degraded fibronectin at levels similar to that of cells incubated in the absence of fibronectin. The control peptide had no effect on fibronectin degradation. These data are consistent with our previously published data showing that fibronectin polymerization inhibitors prevent fibronectin from stabilizing preexisting fibronectin matrix fibrils (Sottile and Hocking, 2002). The increased levels of TCA-soluble counts in cells in which fibronectin polymerization is inhibited suggests that there is increased endocytosis and degradation of fibronectin under conditions where the fibronectin matrix is turned over.
Figure 1.
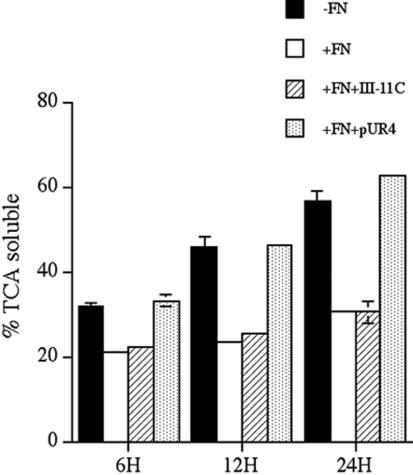
Fibronectin degradation is inhibited by fibronectin polymerization. FN-null MF were incubated overnight with 125I-labeled fibronectin. Cells were washed and then incubated for the indicated time with culture medium containing (+FN) or lacking unlabeled fibronectin (-FN) and containing or lacking pUR4 or the control his-tagged peptide, III-11C for the indicated times. The cell culture supernatant was collected and precipitated with TCA as described in Materials and Methods. The amount TCA soluble counts is expressed as a percentage of the total counts present in each well. Error bars represent the range of duplicate determinations.
Endocytosis is inhibited by culturing cells at 4°C. Therefore, we asked whether incubating cells at 4°C would prevent the turnover of matrix fibronectin fibrils. Cells were allowed to establish a FITC-fibronectin matrix at 37°C (Figure 2A). These cells were then cultured in the absence of fibronectin at 37 or 4°C. The fibronectin matrix was extensively disrupted in cells chased in the absence of fibronectin at 37°C (Figure 2B). As shown previously (Sottile and Hocking, 2002), cell monolayers were intact, and the cells spread despite disruption of the fibronectin matrix (Figure 2E). Incubation of cells at 4°C (Figure 2C) prevented the loss of the preestablished fibronectin matrix. These data are consistent with the idea that endocytosis is an important component regulating the turnover of matrix fibronectin.
Figure 2.
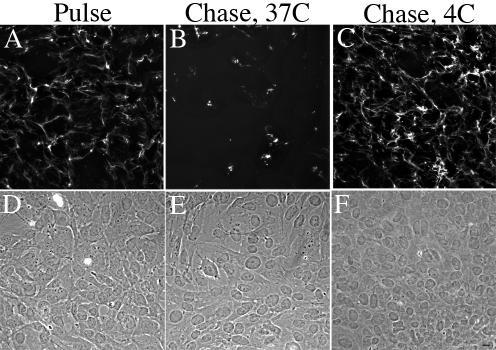
Fibronectin matrix turnover does not occur at 4°C. FN-null MF were incubated overnight with 20 nM FITC-conjugated fibronectin. Cell were then either processed for immunofluorescence (Pulse, A) or were washed, and then incubated with culture medium lacking fibronectin (Chase) at 37°C (B) or 4°C (C) for 8 h. Cells were fixed and permeabilized. FITC-fibronectin staining (A–C) and the corresponding phase images (D–F) are shown. Bar, 10 μm.
Other ECM proteins such as thrombospondin, vitronectin, and heparan sulfate proteoglycans are internalized and degraded in the lysosomes (McKeown-Longo et al., 1984; Yanagishita and Hascall, 1984; Murphy-Ullrich and Mosher, 1987; Hausser et al., 1992; Godyna et al., 1995a; Pijuan-Thompson and Gladson, 1997; Memmo and McKeown-Longo, 1998). To determine whether fibronectin degradation occurs in the lysosomes, we tested whether fibronectin degradation could be inhibited by the lysosomotropic agent chloroquine, which increases the pH of lysosomes, and inhibits the activity of lysosomal hydrolases (de Duve et al., 1974). Fibronectin pulse-chase studies were conducted with 125I-fibronectin in the presence and absence of chloroquine. Addition of chloroquine to cells resulted in a dose-dependent inhibition of fibronectin degradation. The highest dose of chloroquine blocked >90% of the TCA-soluble counts (Figure 3), suggesting that the major route of fibronectin degradation is in the lysosomes. To directly test whether extracellular fibronectin is targeted to the lysosomes, we used confocal microscopy to ask whether fibronectin colocalizes with the lysosomal marker protein, LAMP-1. FN-null MF were incubated with Texas Red-fibronectin and allowed to establish a fibronectin matrix. The cells were then incubated in the absence of soluble fibronectin for 24 h. Internalized fibronectin (TR-fibronectin) was readily detected in permeabilized cells that have been treated with chloroquine (Figure 4B). A comparison of panels A and B show that fibronectin (B) and LAMP-1 (A) partially colocalize within cells. Some areas of colocalization can be seen as yellow/orange staining in the merged image in panel C. Quantitative analysis of confocal z-sections showed that >50% of the internalized fibronectin colocalized with Lamp-1. These data provide further supporting evidence that extracellular fibronectin is internalized by cells and targeted to the lysosomes for degradation.
Figure 3.
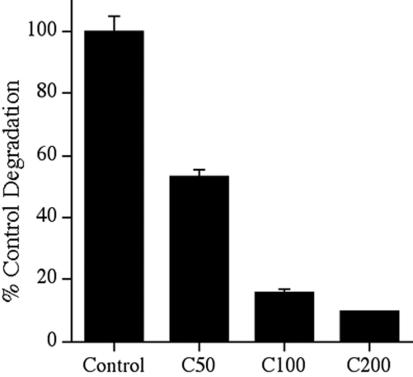
Fibronectin degradation is inhibited by chloroquine. FN-null MF were incubated overnight with 125I-labeled fibronectin. Cells were washed and then incubated with culture medium lacking fibronectin and lacking (control) or containing 50–200 μM chloroquine (C50, C100, C200) for 10 h. The cell culture supernatant was collected, and precipitated with TCA as described in the legend to Figure 1. The amount of degradation that occurred in the absence of chloroquine was set equal to 100%. Error bars represent the range of duplicate determinations.
Figure 4.
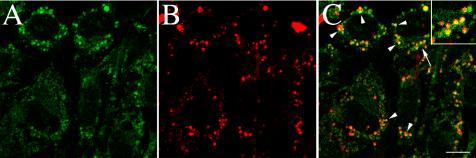
Colocalization of fibronectin with the lysosomal marker, LAMP-1. FN-null MF were incubated with Texas Red–conjugated fibronectin (20 nM) overnight. Cells were washed and then incubated for 25 h in cell culture media lacking fibronectin, but containing 50 μM chloroquine. Chloroquine was used to inhibit lysosomal degradation and enhance the levels of intracellular fibronectin. Cells were then fixed, permeabilized, and incubated with an antibody to LAMP-1. Localization of Texas-Red-fibronectin (B) and LAMP-1 (A) are shown. (C) An overlay of the fibronectin and LAMP-1 images shown in A and B. Some areas of colocalization (arrowheads) of fibronectin and LAMP-1 are shown in yellow. It should be noted that yellow will only be seen if the intensity of the red and green fluors are approximately equal. The area marked by the arrow in C was magnified and is shown in the inset. The images are optical sections collected from a confocal z-series scan obtained with an Olympus confocal microscope. Bar, 10 μm.
To determine whether fibronectin matrix degradation occurs by a similar mechanism in fibronectin producing cells, we asked whether inhibition of fibronectin polymerization induces the loss of fibronectin matrix fibrils, and up-regulates fibronectin degradation in SMC. Incubation of SMC with pUR4 resulted in a marked reduction in the amount of fibronectin matrix fibrils (Figure 5A, panel B) in comparison with cells before pUR4 treatment (panel A) or cells incubated with a control peptide (panel C). To determine whether fibronectin is degraded by SMC in a manner that depends on fibronectin polymerization, SMC were incubated overnight with 125I-fibronectin. The cells were washed and then incubated in culture medium containing the fibronectin polymerization inhibitor, pUR4, or control peptide. As shown in Figure 5B, addition of pUR4 to SMC resulted in a 1.7–2.3× increase in fibronectin degradation in comparison with control cells. In addition, chloroquine inhibited ∼80% of fibronectin degradation in SMC (unpublished data). These data indicate that fibronectin matrix turnover in fibronectin-producing cells involves intracellular lysosomal degradation that is regulated by fibronectin polymerization.
Figure 5.
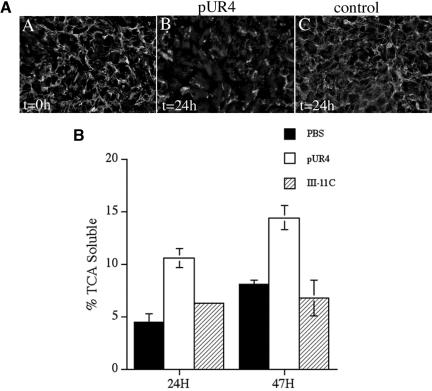
(A) Fibronectin matrix stability in SMCs. Human aortic SMCs (passage 4) were seeded in serum-containing medium and grown to confluence. SMCs were then incubated with 250 nM pUR4 (B) or 250 nM control peptide, III-11C (C) for 24 h. At the time of protein addition, some cells were processed for immunofluorescence (A) to determine the amount of matrix deposited by the cells before pUR4 addition (t = 0). Cells were permeabilized and then incubated with a polyclonal antibody to fibronectin, followed by a FITC anti-rabbit antibody. Bar, 20 μm. (B) Fibronectin degradation in SMC. Confluent cultures of rat SMCs were incubated overnight with 125I-fibronectin. Cells were washed and then incubated with culture medium lacking (PBS) or containing 250 nM pUR4 or the control III-11C peptide for the indicated times. The amount of TCA-soluble counts was determined as described in Materials and Methods and is expressed as a percentage of the total counts present in each well. Error bars represent the average of duplicate samples, and the error bars the range.
Fibronectin Degradation in FN-null MF Is Not Lipoprotein Receptor–related Protein Dependent
Others have shown that fibronectin can be degraded in certain cell types but not in others (Godyna et al., 1995a; Salicioni et al., 2002) by a low density lipoprotein receptor–related protein (LRP)-dependent mechanism. These studies examined the degradation of soluble fibronectin; the fate of ECM fibronectin was not tested. We asked whether turnover of matrix fibronectin in FN-null MF depends on LRP by examining whether the LRP inhibitor, receptor-associated protein (RAP), could inhibit loss or degradation of matrix fibronectin. RAP binds to LRP and competes for binding with LRP ligands, thus preventing their internalization and degradation (Herz et al., 1991; Godyna et al., 1995a; Lazic et al., 2003). As shown in Figure 6A, cells incubated with RAP degraded similar levels of fibronectin as control cells. To demonstrate that functional LRP exists on the surface of FN-null MF, we tested whether RAP could inhibit 125I-thrombospondin degradation, because thrombospondin degradation is LRP dependent (Godyna et al., 1995a; Chen et al., 1996a). Thrombospondin degradation was inhibited by RAP in FN-null MF (Figure 6B). These data demonstrate that turnover of matrix fibronectin in FN-null MF is not LRP dependent.
Figure 6.
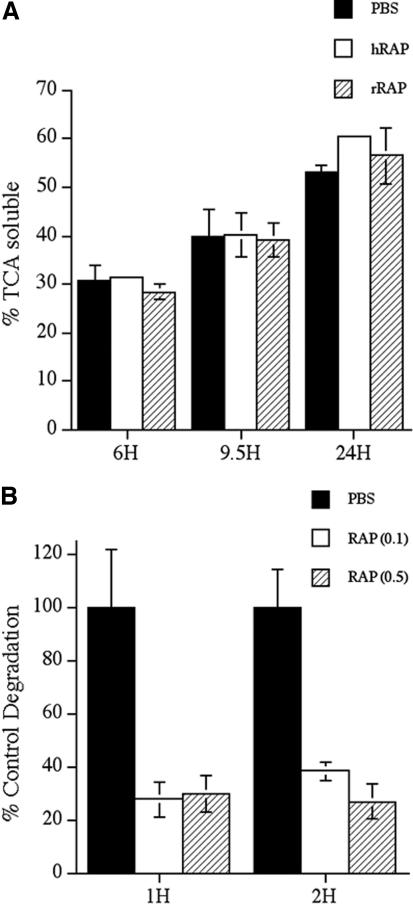
(A) Fibronectin degradation is not inhibited by RAP. FN-null MF were incubated overnight with 125I-labeled fibronectin. Cells were washed and then incubated with culture medium lacking fibronectin and lacking (PBS) or containing 1 μM human RAP (hRAP) or recombinant GST-RAP (rRAP) for the indicated times. The amount of TCA soluble counts was determined as described in Materials and Methods and is expressed as a percentage of the total counts present in each well. Similar results were obtained when RAP was added at the time of 125I-labeled fibronectin addition. (B) Thrombospondin degradation is inhibited by RAP. FN-null MF were incubated with 125I-labeled thrombospondin in the presence or absence of 0.1–0.5 μM RAP for the indicated times. The amount of degraded thrombosponin was determined as described in Materials and Methods. Error bars represent the range of duplicate determinations.
Agents That Disrupt Caveolae or Interfere with Caveolae-mediated Endocytosis Block Fibronectin Degradation
Fibronectin internalization and degradation could occur via clathrin- or caveolin-mediated endocytosis. Others have shown that a portion of ECM fibronectin colocalizes with caveolin in lipid rafts (Hocking and Kowalski, 2002). Hence, we tested whether β-cyclodextrin, a cholesterol-binding agent that disrupts caveolae (Smart and Anderson, 2002), can inhibit fibronectin degradation. As shown in Figure 7, the presence of cyclodextrin in the chase media partially inhibited fibronectin degradation in FN-null MF. Others have shown that addition of staurosporin (a general kinase inhibitor) or genistein (a tyrosine kinase inhibitor) can block caveolae-mediated endocytosis (Pelkmans et al., 2002; Pang et al., 2004). To determine whether these kinase inhibitors block fibronectin matrix turnover, fibronectin pulse-chase experiments were done with FN-null MF in the presence and absence of genistein or staurosporin. Addition of either genistein or staurosporin prevented the loss of fibronectin matrix fibrils (Figure 8) and caused a dramatic reduction in fibronectin degradation (Figure 7). Caveolae-mediated endocytosis has also been reported to be blocked by jasplakinolide, an actin filament stabilizing agent (Pelkmans et al., 2002). Our previous data demonstrated that fibronectin turnover was also blocked by jasplakinolide (Sottile and Hocking, 2002).
Figure 7.
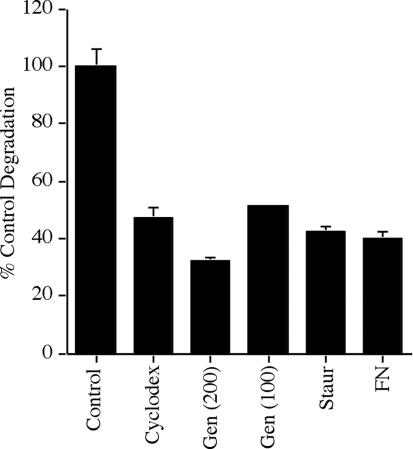
Fibronectin degradation is inhibited by agents that disrupt caveolae or that interfere with caveolae-mediated endocytosis. FN-null MF were incubated overnight with 125I-fibronectin. Cells were washed, and then incubated with culture medium lacking fibronectin and lacking (control) or containing 10 mM β-cyclodexdrin, 200 μM genistein, 100 μM genistein, 0.1 μM staurosporin, or 20 nM fibronectin. Cells were incubated for 6 h (cyclodextrin) or 24 h (all other treatments). The cell culture supernatant was collected and precipitated with TCA as described in the legend to Figure 1. The amount of fibronectin degraded in control FN null MF was 11.4% of the total counts at 6 h. The amount of degradation that occurred in the absence of inhibitors at each time point was set equal to 100%. Error bars represent the range of duplicate or triplicate determinations.
Figure 8.
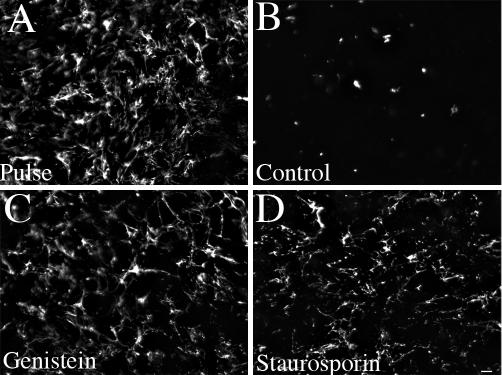
Kinase inhibitors prevent the loss of fibronectin matrix fibrils. FN-null MF were incubated overnight with 20 nM FITC-conjugated fibronectin. Cell were then either processed for immunofluorescence (Pulse, A) or were washed and then incubated with culture medium lacking fibronectin and lacking (B, control) or containing 200 μM genistein (C) or 0.1 μM staurosporin (D) for 24 h. Cells were fixed and permeabilized. FITC-fibronectin staining. Bar, 20 μm.
Others have shown that caveolin-containing vesicles do not contain markers of early endosomes and hence are distinct from the early endosomes that are formed during clathrin-mediated endocytosis (Pelkmans et al., 2001; Nichols, 2002). It should be noted that intracellular fibronectin must be derived from extracellular fibronectin by endocytosis, because FN-null MF do not synthesize fibronectin (Sottile et al., 1998). Internalized fibronectin did not colocalize with the early endosome marker, EEA1 (Figure 9C). Quantitative analysis of confocal z-sections indicated that <5% of the internalized fibronectin colocalized with EEA-1. Fibronectin also failed to colocalize with the transferrin receptor, another marker of classical early endosomes (unpublished data). These data suggest that endocytosis of fibronectin may involve caveolin-mediated endocytosis.
Figure 9.
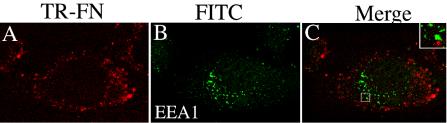
Fibronectin does not colocalize with EEA-1-containing vesicles. FN-null MF were incubated with Texas Red–conjugated fibronectin (20 nM) overnight. Cells were washed and then incubated for 24 h in cell culture media lacking fibronectin, but containing 30 μM chloroquine. Chloroquine was used to inhibit lysosomal degradation and enhance the levels of intracellular fibronectin. Cells were fixed, permeabilized, and then incubated with antibodies to EEA-1. Localization of Texas-Red-FN (A) and EEA-1 (B) are shown. (C) An overlay of the images in A and B. Little or no colocalization of fibronectin and EEA-1 was seen (C). The boxed area in C was magnified and is shown by the inset. The images are optical sections collected from a confocal z-series scan obtained with an Olympus confocal microscope. Bar, 10 μm.
Down-regulation of Caveolin-1 by RNAi Stabilizes Fibronectin Matrix Fibrils
Our data show that agents that disrupt caveolae or that interfere with caveolae-mediated endocytosis partially prevent fibronectin degradation (Figure 7). These data suggest that fibronectin turnover may be regulated by caveolin-1. This could be due to decreased endocytosis via caveolae and/or to caveolin-1–dependent signaling event that is necessary for fibronectin internalization and degradation. To test whether caveolin-1 is important for fibronectin matrix turnover, we used RNAi to generate a stable FN-null cell line in which caveolin-1 was down-regulated. Caveolin-1 was decreased ∼70% in cells stably expressing caveolin-1 siRNA (shcav) in comparison with the parental FN-null MF and with control cell lines expressing luciferase shRNA (shluc; Figure 10A). To determine whether down-regulation of caveolin-1 affects fibronectin matrix stability, we performed fibronectin pulse-chase studies with FITC-fibronectin in FN-null MF and shcav cells. Both the parental FN-null MF and FN-null MF expressing caveolin-1 siRNA polymerized a robust fibronectin matrix (Figure 10B, panels A and G) when cultured in the presence of fibronectin. When soluble fibronectin was removed from the culture media, parental FN-null MF showed a dramatic loss of fibronectin matrix fibrils (Figure 10B, panels B and C). In contrast, there was a striking retention of fibronectin fibrils in caveolin siRNA-expressing cells (Figure 10B, panels H and I). These data are the first to show that down-regulating caveolin-1 levels stabilizes fibronectin matrix fibrils.
Figure 10.
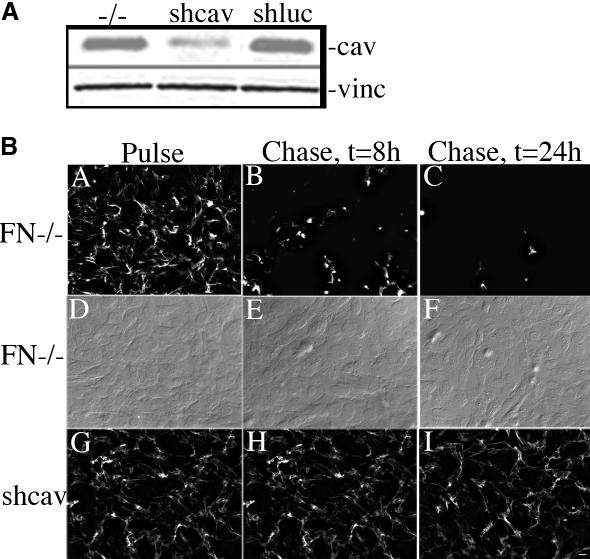
Expression of caveolin-1 siRNA reduces caveolin-1 levels and stabilizes matrix FN fibrils. (A) Cell lysates were prepared from confluent cultures of FN-null MF or FN-null MF-expressing caveolin-1 siRNA (shcav) or luciferase siRNA (shluc). Equal amounts of total protein were run on an SDS-PAGE gel, transferred to nitrocellulose paper, and then incubated with antibodies to caveolin-1 and vinculin. (B) FN-null MF or FN-null MF-expressing caveolin-1 siRNA (shcav) were incubated with 20 nM Texas Red-FN overnight. Cells were then either processed for immunofluorescence (Pulse, A, D, and G), or were washed and then incubated with culture medium lacking fibronectin (Chase, B, C, E, F, H, and I) for 8 (B, E, and H) or 24 h (C, F, and I). Cells were fixed and permeabilized, then examined with an Olympus microscope). (D–F) Nomarski images of cells in A–C. Bar, 20 μm.
To quantitate the levels of fibronectin degradation in shcav and control cell lines, we compared the levels of 125I-fibronectin degradation using pulse-chase studies. There was a dramatic 3.7–4-fold reduction in fibronectin degradation in cells expressing caveolin siRNA (shcav) in comparison with the parental FN-null MF (FN-/-) or with a control cell line expressing luciferase siRNA (shluc) (Figure 11). The effect on fibronectin degradation was specific, because the three cell lines showed no difference in thrombospondin degradation (Figure 12).
Figure 11.
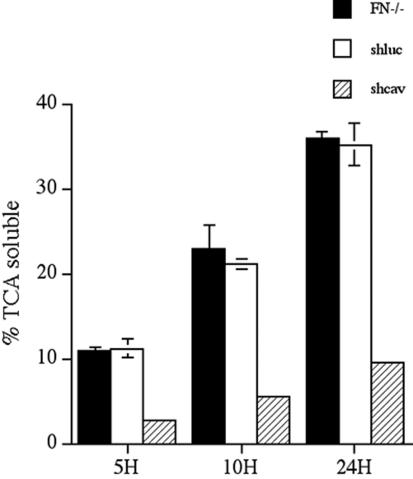
Expression of caveolin-1 siRNA reduces fibronectin degradation. FN-null MF, or FN-null MF-expressing caveolin-1 siRNA (shcav) or ffluc siRNA (shluc) were incubated overnight with 125I-fibronectin. Cells were washed and then incubated with culture medium lacking fibronectin for the indicated times. The cell culture supernatant was collected and precipitated with TCA as described in Materials and Methods. The amount TCA-soluble counts is expressed as a percentage of the total counts present in each well. Error bars represent the range of duplicate determinations.
Figure 12.
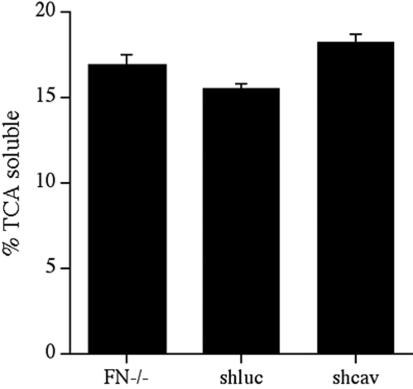
Expression of caveolin-1 siRNA does not effect thrombospondin degradation. FN-null MF, or FN-null MF-expressing caveolin-1 siRNA (shcav) or ffluc siRNA (shluc) were grown to confluence and then incubated with 125I-labeled thrombospondin for 8 h. The amount of degraded thrombospondin was determined by collecting the cell culture supernatant and precipitating with 13% TCA. The amount TCA-soluble counts is expressed as a percentage of the total counts present in each well. Error bars represent the range of duplicate determinations. Similar results were obtained when degradation assays were performed for 2 or 4 h.
The data shown in Figures 10 and 11 indicate that there is a dramatic reduction in fibronectin matrix turnover and degradation in cells expressing caveolin siRNA. Our data also show that the majority of fibronectin degradation occurs in the lysosomes. Hence, decreased fibronectin degradation in caveolin-1 siRNA-expressing cells could result from a reduction in fibronectin endocytosis. To determine whether differences in the levels of intracellular fibronectin can be detected in shcav and control cells, we performed fibronectin pulse-chase studies with TR-labeled fibronectin and examined the levels of intracellular fibronectin at different times during the chase using confocal microscopy. As shown in Figure 13A, intracellular vesicular fibronectin was readily detected in the control shluc cells within 5 h of the start of the chase period. In contrast, little or no intracellular fibronectin staining was seen in the shcav cells at this time point, although fibronectin fibrils were readily detected (Figure 13B). At 10 h, increased levels of vesicular fibronectin staining was seen in the control shluc cells (Figure 13C). Some vesicular fibronectin staining was also detected in the shcav cells, although this staining was less intense and less extensive than in the control cells (Figure 13D). Thus, intracellular accumulation of fibronectin is delayed in cells expressing caveolin-1 siRNA.
Figure 13.
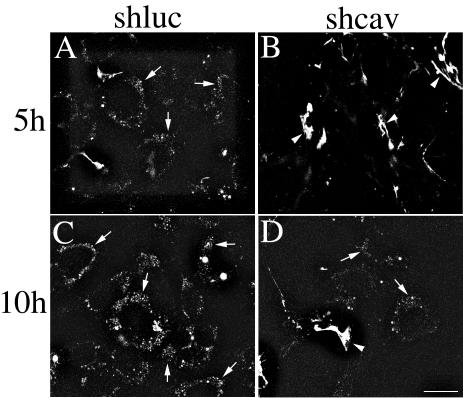
Cells expressing caveolin-1 siRNA show reduced levels of intracellular fibronectin. FN-null MF-expressing caveolin-1 siRNA (shcav) or ffluc siRNA (shluc) were incubated overnight with TR-labeled fibronectin. Cells were washed and then incubated in culture medium lacking fibronectin and containing 30 μM chloroquine. Chloroquine was used to inhibit lysosomal degradation and enhance the levels of intracellular fibronectin. Cells were fixed at the indicated times. The images are optical sections collected from a confocal z-series scan obtained with an Olympus confocal microscope. Bar, 20 μm.
To demonstrate that down-regulation of caveolin-1 in the siRNA-expressing cells (shcav) is responsible for decreased turnover of matrix fibronectin, we reexpressed caveolin-1 in these cells using an adenovirus containing caveolin-1 (Adcav-1). As shown in Figure 14A, caveolin-1 levels are increased 14-fold in shcav cells transduced with Ad-cav-1 in comparison with cells transduced with control adenovirus. Increased levels of caveolin-1 persisted for >60 h after transduction (unpublished data). To determine the effect of caveolin-1 overexpression on fibronectin matrix fibril stability and fibronectin degradation in shcav cells, fibronectin pulse-chase experiments were performed using either FITC- or 125I-labeled fibronectin. Reexpression of caveolin-1 resulted in an increased loss of fibronectin matrix fibrils (Figure 14B, panel C) in comparison with control cells (Figure 14B, panels B and D). In addition, there was a 46% increase in fibronectin degradation in shcav cells treated with Adcav-1 in comparison with cells treated with a control adenovirus (Figure 15). Fibronectin degradation in shcav cells was restored to 86% of the levels of the parental FN null MF upon reexpression of caveolin-1. Addition of unlabeled fibronectin to the chase media reduced fibronectin degradation in both control and Ad-cav-1–treated cells. Taken together, these data indicate that caveolin-1 is a key player in regulating ECM remodeling by a novel pathway that involves endocytosis and intracellular degradation of fibronectin.
Figure 14.
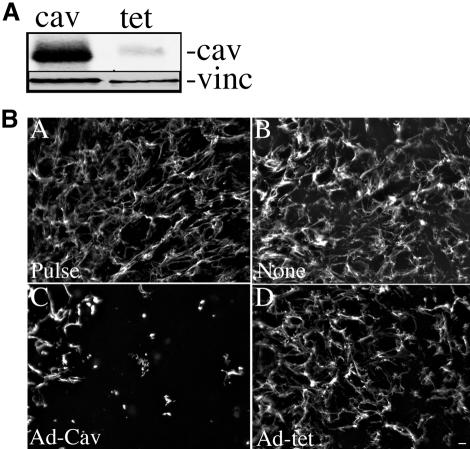
(A) Caveolin levels in shcav cells transduced with Ad-Cav-1. FN-null MF-expressing caveolin-1 siRNA were incubated with Ad-cav-1 (cav) or Ad-tet TA (tet) at an MOI of 300 for 19 h. The cells were washed and then incubated in culture medium lacking virus. Cell lysates were prepared at 24 h after virus addition. Equal amounts of total protein were run on an SDS PAGE gel, transferred to nitrocellulose paper, and then incubated with antibodies to caveolin-1 (cav) and vinculin (vinc). (B) Reexpression of caveolin-1 in shcav cells increases the turnover of fibronectin fibrils. shcav cells were incubated with Ad-cav-1 (C) or Ad-tet TA (D) at an MOI of 300. Control cells were not infected with virus (none). Four hours after addition of virus, 20 nM FITC-FN was added, and the cells were incubated for an additional 16 h. Cells were then either processed for immunofluorescence (Pulse, A) or were washed and then incubated with culture medium lacking fibronectin (Chase, B–D) for 24 h. Cells were fixed and permeabilized and then examined with an Olympus microscope. Bar, 20 μm.
Figure 15.
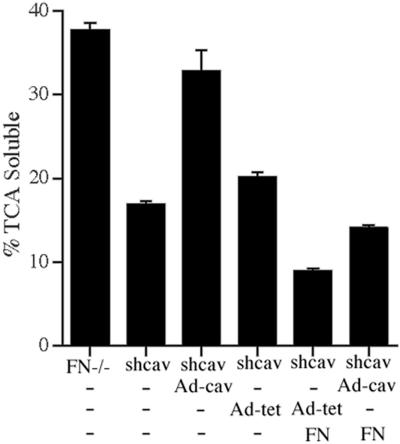
Reexpression of caveolin-1 in shcav cells increases fibronectin degradation. FN null MF (FN-/-) or FN null MF-expressing caveolin-1 siRNA (shcav) were incubated without virus (-) or with Ad-cav-1 or Ad-tet TA at an MOI of 300 for 19 h. The virus was removed, and the cells were incubated for 12 h with 125I-labeled fibronectin. Cells were washed and then incubated with culture medium containing (FN) or lacking (-FN) unlabeled fibronectin for 24 h. The cell culture supernatant was collected, and precipitated with TCA as described in Materials and Methods. The amount TCA-soluble counts is expressed as a percentage of the total counts present in each well. Error bars, SEM of triplicate determinations.
DISCUSSION
We and others have shown that fibronectin polymerization is a key regulator of ECM architecture (Sottile and Hocking, 2002), cell proliferation (Clark et al., 1997; Bourdoulous et al., 1998; Sechler and Schwarzbauer, 1998; Sottile et al., 1998, 2000; Christopher et al., 1999), and cell migration (Darribere and Schwarzbauer, 2000; Hocking and Chang, 2003), all critically important components of tissue repair and fibrosis. ECM remodeling can occur through extracellular proteolysis by the action of MMPs, and/or other proteases, or by intracellular proteolysis, after endocytosis. We previously showed that fibronectin matrix polymerization regulates the stability of fibronectin and collagen I matrix fibrils (Sottile and Hocking, 2002). In this article, we show that loss of fibronectin matrix fibrils is accompanied by increased endocytosis and degradation of ECM fibronectin. Further, our data demonstrate that caveolin-1 is an important regulator of fibronectin endocytosis and degradation. Down-regulation of caveolin-1 by siRNA in FN null MF results in stabilization of fibronectin matrix fibrils and a reduction in fibronectin degradation. Reexpression of caveolin-1 in the siRNA-expressing cells promotes increased turnover of fibronectin matrix fibrils and increases fibronectin degradation. These data are the first to demonstrate a role for caveolin-1 in regulating fibronectin matrix turnover. Because fibronectin is an important regulator of ECM remodeling (Sottile and Hocking, 2002), these data suggest that caveolin-1 plays a key role in modulating ECM composition and stability.
Others have shown that fibronectin can be degraded in peritoneal cells by chymases (Tchougounova et al., 2000). However, this process may be specific for peritoneal cells, because chymase production appears to be restricted to mast cells (Yong, 1997; Krishnaswamy et al., 2001). In addition, chymase inhibitors do not block loss of fibronectin matrix fibrils in FN null MF (unpublished data). LRP is involved in the endocytosis and intracellular degradation of a number of extracellular proteins including thrombospondin-1, thrombosponin-2, and plasminogen activators (Grobmyer et al., 1993; Godyna et al., 1995a; Chen et al., 1996b; Herz and Strickland, 2001). It has previously been reported that soluble fibronectin can be degraded in mouse embryo fibroblasts (MEF) by an LRP-dependent pathway (Salicioni et al., 2002). It should be emphasized that these studies examined the degradation of soluble fibronectin; the turnover of ECM fibronectin was not examined. In addition, the LRP inhibitor, RAP, does not inhibit degradation of matrix fibronectin in FN null MF (Figure 6A) or in SMC (Godyna et al., 1995a). Hence, LRP-mediated fibronectin degradation may also be restricted to a subset of cells. Our data demonstrating turnover and degradation of ECM fibronectin in vitro is consistent with in vivo data indicating that tissue fibronectin is turned over relatively rapidly (Rebres et al., 1995). In addition, evidence suggests that turnover of matrix fibronectin in the liver may involve receptor-mediated endocytosis (Rotundo et al., 1999).
Our data show that extracellular fibronectin is endocytosed and degraded in both FN-null MF and in SMCs. Differences in the kinetics of degradation in FN-null MF and SMCs are likely due to the presence of cell- and serum-derived fibronectin in SMC. Because soluble fibronectin inhibits matrix fibronectin degradation (Figure 1), reduced fibronectin degradation would be expected to occur in cells that produce fibronectin. Our data show that fibronectin degradation in FN-null MF and in SMC is enhanced by blocking fibronectin polymerization (Figures 1 and 5) and inhibited by blocking lysosomal degradation (Figure 3). Thus, endocytosis and intracellular degradation is an important pathway that regulates fibronectin matrix stability.
Receptor-mediated endocytosis can occur via two major pathways, one involving clathrin-coated vesicles, and the other involving caveolae (Schmid et al., 1998; Pelkmans and Helenius, 2002; Nabi and Le, 2003). Our data indicate that internalized fibronectin does not colocalize with a marker of classical early endosomes (Figure 9) and that agents that disrupt caveolae and/or block caveolae-mediated endocytosis partially inhibit fibronectin degradation (Figure 7). Moreover, down-regulation of caveolin-1 results in stabilization of fibronectin matrix fibrils, a 4× reduction in fibronectin degradation, and a reduction in the levels of intracellular fibronectin. These data suggest that fibronectin internalization proceeds by a caveolin-1–dependent process. Interestingly, caveolin-1, caveolin-3, and caveolin 1/3 double knockout mice develop fibrosis in the lungs and/or heart (Drab et al., 2001; Razani et al., 2001; Park et al., 2002). Cells from caveolin-1 null mice are also defective in endocytosis of albumin (Schubert et al., 2001). These data suggest the interesting possibility that fibrosis in mice lacking functional caveolae may, in part, be due to defects in ECM endocytosis and turnover. Recent reports indicate that the levels of caveolin-1 can be modulated by growth factors (Liu et al., 2002; Peterson et al., 2003). Addition of platelet-derived growth factor (PDGF) to smooth muscle cells resulted in down-regulation of caveolin-1 and caveolae (Peterson et al., 2003). PDGF treatment also results in increased levels of fibronectin matrix fibrils (Sarkissian and Lafyatis, 1998). Hence, regulation of caveolin by growth factors may be a physiological mechanism that controls the rate and/or extent of fibronectin matrix turnover. Most proteins that are degraded in the lysosomes are endocytosed in clathrincoated vesicles (Cavalli et al., 2001). However, FGF degradation has been shown to occur after caveolae-mediated endocytosis (Gleizes et al., 1996). Our data are the first to demonstrate that fibronectin polymerization controls ECM fibronectin turnover by a novel mechanism involving caveolin-1–dependent endocytosis.
The role of integrins in regulating turnover of matrix fibronectin is unknown. Integrins can be endocytosed by receptor mediated endocytosis and recycled to the cell surface (Bretscher, 1989, 1992; Ng et al., 1999). In addition, integrins are involved in the internalization of other ECM proteins, such as vitronectin (Pijuan-Thompson and Gladson, 1997; Memmo and McKeown-Longo, 1998). In one report, fibronectin was shown to colocalize with integrins in intracellular vesicles in cells overexpressing protein kinase C (PKC)α (Ng et al., 1999). Our flow cytometry data indicate that the cell surface levels of β1, β3, and αv integrins are unchanged when fibronectin polymerization is inhibited by removal of soluble fibronectin from the cell culture media or when fibronectin degradation is inhibited by chloroquine (unpublished data). Of the integrins tested, the only integrin whose levels change is the α5 integrin, which increases after removal of soluble fibronectin from the cell culture media, and decreases upon addition of chloroquine. Hence, conditions that promote fibronectin degradation do result in consistent changes in α5 integrin levels. Endocytosis of most integrins occurs through a clathrin-mediated process (Raub and Kuentzel, 1989; Van Nhieu et al., 1996). However, a recent report indicates that the α2β1 integrin is endocytosed via caveolae (Upla et al., 2004). It will be interesting to determine whether other integrins can be endocytosed by a similar pathway and whether these integrins regulate fibronectin endocytosis.
ECM fibronectin is comprised of high-molecular-weight multimers that are stabilized by disulfide bonds (Ali and Hynes, 1978; Choi and Hynes, 1979; McKeown-Longo and Mosher, 1984). It is unlikely that these large fibrils are directly endocytosed by cells. A more likely scenario is that matrix fibronectin may be proteolyzed before endocytosis. In addition, we previously showed that multimerization of fibronectin may involve a disulfide isomerase activity (Langenbach and Sottile, 1999). Hence, it is also possible that matrix fibronectin may be dissociated into monomers or dimers by an isomerase activity before endocytosis. Future studies will be directed at understanding the mechanism by which extracellular matrix fibronectin fibrils are internalized by cells before degradation.
ECM molecules play critical roles in regulating tissue repair by providing a scaffold for cell migration and by regulating cell growth and cell contractility. Our data demonstrate that the maintenance and structural organization of ECM fibronectin depends on the continuous polymerization of a fibronectin matrix (Sottile and Hocking, 2002). Furthermore, fibronectin matrix polymerization regulates the organization of other ECM molecules, including collagen I (Sottile and Hocking, 2002; Velling et al., 2002), and enhances cell contractility (Hocking et al., 2000). Thus, factors that regulate fibronectin matrix turnover likely play a critical role in regulating ECM remodeling. A variety of agents have been shown to stimulate or inhibit the ability of cultured cells to polymerize fibronectin into fibrils in the ECM. Transforming growth factor-β (Ignotz and Massague, 1986; Allen-Hoffmann et al., 1988), PDGF (Sarkissian and Lafyatis, 1998), PKC (Sommers and Mosher, 1993), integrin-linked kinase (Wu et al., 1998), and agents that stimulate cell contractility (such as lysophophatidic acid, LPA; Burridge et al., 1997; Zhang et al., 1997; Zhong et al., 1998) up-regulate fibronectin matrix polymerization. LPA binds to G-protein–coupled receptors and triggers intracellular signaling pathways that lead to activation of Rho-A (Moolenaar et al., 1997). In contrast, H-Ras, Raf-1 (Hughes et al., 1997), cAMP, and agents that inhibit cell contractility and Rho activation (Allen-Hoffmann and Mosher, 1987; Burridge et al., 1997; Zhang et al., 1997; Zhong et al., 1998) inhibit fibronectin polymerization. Hence, several physiological mediators have the potential to alter the rate of fibronectin polymerization and thus, the rate of fibronectin turnover. Our data show that fibronectin matrix turnover is regulated by both fibronectin polymerization and by caveolin-1. In addition, our published data show that fibronectin polymerization is a key regulator of ECM remodeling (Sottile and Hocking, 2002). Hence, these data define a novel pathway for controlling ECM remodeling through the caveolin-1–dependent regulation of fibronectin metabolism. Determining the mechanism(s) by which caveolin-1 regulates fibronectin-dependent ECM remodeling will provide important insights into factors that contribute to the development of fibrosis and into mechanisms that could lead to reversal of excess matrix accumulation during fibrosis.
Acknowledgments
We thank Drs. Hannon, Herz, Hanski, Hocking, Mosher, Mundy, and Schmid for providing reagents used in this study; Mr. Jeffrey Streb (University of Rochester) for help with the RNAi experiments; Dr. Chen Yan (University of Rochester) for help generating adenoviruses; Dr. Fujiwara for advice on imaging; Dr. Hocking for useful discussions, and Dr. LaFlamme (Albany Medical College, Albany, NY) for critically reading the manuscript. This research was supported by grants HL03971 and GM069729 from the National Institutes of Health and 0255621N from the American Heart Association.
Article published online ahead of print in MBC in Press on November 24, 2004 (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-08-0672).
Abbreviations used: BSA, bovine serum albumin; ECM, extracellular matrix; GST, glutathione S-transferase; LPA, lysophophatidic acid; LRP, low-density lipoprotein receptor–related protein; MFI, mean fluorescence intensity; MMP, matrix metalloproteinase; short hairpin RNA (shRNA); SMC, smooth muscle cell; RAP, receptor-associated protein.
References
- Ali, I. U., and Hynes, R. O. (1978). Role of disulfide bonds in the attachment and function of large, external, transformation-sensitive glycoprotein at the cell surface. Biochem. Biophys. Acta 510, 140-150. [DOI] [PubMed] [Google Scholar]
- Allen-Hoffmann, B. L., Crankshaw, C. L., and Mosher, D. F. (1988). Transforming growth factor beta increases cell surface binding and assembly of exogenous (plasma) fibronectin by normal human fibroblasts. Mol. Cell. Biol. 8, 4234-4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen-Hoffmann, L., and Mosher, D. F. (1987). Matrix assembly sites for exogenous fibronectin are decreased on human fibroblasts after treatment with agents which increase intracellular cAMP. J. Biol. Chem. 262, 14361-14365. [PubMed] [Google Scholar]
- Bourdoulous, S., Orend, G., MacKenna, D. A., Pasqualini, R., and Ruoslahti, E. (1998). Fibronectin matrix regulates activation of RHO and CDC42 GTPases and cell cycle progression. J. Cell Biol. 143, 267-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher, M. S. (1989). Endocytosis and recycling of the fibronectin receptor in chinese hamster ovary (CHO) cells. EMBO J. 8, 1341-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher, M. S. (1992). Circulating integrins: alpha 5 beta 1, alpha 6 beta 4 and Mac-1, but not alpha 3 beta 1, alpha 4 beta 1 or LFA-1. EMBO J. 11, 405-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge, K., Chrzanowska-Wodnicka, M., and Zhong, C. (1997). Focal adhesion assembly. Trends Cell Biol. 7, 342-347. [DOI] [PubMed] [Google Scholar]
- Cavalli, V., Corti, M., and Gruenberg, J. (2001). Endocytosis and signaling cascades: a close encounter. FEBS Lett. 498, 190-196. [DOI] [PubMed] [Google Scholar]
- Chen, H., Sottile, J., Strickland, D. K., and Mosher, D. F. (1996a). Binding and degradation of thrombospondin-1 mediated through heparan sulphate proteoglycans and low-density-lipoprotein receptor-related protein: localization of the functional activity to the trimeric N-terminal heparin-binding region of thrombospondin-1. Biochem. J. 318, 959-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H., Strickland, D. K., and Mosher, D. F. (1996b). Metabolism of thrombospondin 2. Binding and degradation by 3T3 cells and glycosaminoglycan-variant CHO cells. J. Biol. Chem. 271, 15993-15999. [DOI] [PubMed] [Google Scholar]
- Choi, M. G., and Hynes, R. O. (1979). Biosynthesis and processing of fibronectin in NIL8 hamster cells. J. Biol. Chem. 23, 12050-12055. [PubMed] [Google Scholar]
- Christopher, R. A., Judge, S. R., Vincent, P. A., Higgins, P. J., and McKeown-Longo, P. J. (1999). The amino-terminal matrix assembly domain of fibronectin stabilizes cell shape and prevents cell cycle progression. J. Cell Sci. 112, 3225-3235. [DOI] [PubMed] [Google Scholar]
- Chung, C. Y., and Erickson, H. P. (1997). Glycosaminoglycans modulate fibronectin matrix assembly and are essential for matrix incorporation of tenascin-C. J. Cell Sci. 110, 1413-1419. [DOI] [PubMed] [Google Scholar]
- Clark, R. A. F. (1996). Wound repair: overview and general considerations. In: The Molecular and Cellular Biology of Wound Repair, ed. R. A. F. Clark, New York: Plenum Press, 3-50.
- Clark, R. A. F., McCoy, G. A., Folkvord, J. M., and McPherson, J. M. (1997). TGF-β1 stimulates cultured human fibroblasts to proliferate and produce tissue-like fibroplasia: a fibronectin matrix-dependent event. J. Cell. Physiol. 170, 69-80. [DOI] [PubMed] [Google Scholar]
- Darribere, T., and Schwarzbauer, J. E. (2000). Fibronectin matrix composition and organization can regulate cell migration during amphibian development. Mech. Dev. 92, 239-250. [DOI] [PubMed] [Google Scholar]
- de Duve, C., de Barsy, T., Poole, B., Trouet, A., Tulkens, P., and Van Hoof, F. (1974). Commentary. Lysosomotropic agents. Biochem. Pharmacol. 23, 2495-2531. [DOI] [PubMed] [Google Scholar]
- Drab, M. et al. (2001). Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293, 2449-2452. [DOI] [PubMed] [Google Scholar]
- East, L., McCarthy, A., Wienke, D., Sturge, J., Ashworth, A., and Isacke, C. M. (2003). A targeted deletion in the endocytic receptor gene Endo180 results in a defect in collagen uptake. EMBO Rep. 4, 710-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelholm, L. H. et al. (2003). uPARAP/Endo180 is essential for cellular uptake of collagen and promotes fibroblast collagen adhesion. J. Cell Biol. 160, 1009-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish, K. N., Schmid, S. L., and Damke, H. (2000). Evidence that dynamin-2 functions as a signal-transducing GTPase. J. Cell Biol. 150, 145-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleizes, P. E., Noaillac-Depeyre, J., Dupont, M. A., and Gas, N. (1996). Basic fibroblast growth factor (FGF-2) is addressed to caveolae after binding to the plasma membrane of BHK cells. Eur. J. Cell Biol. 71, 144-153. [PubMed] [Google Scholar]
- Godyna, S., Liau, G., Popa, I., Stefansson, S., and Argraves, W. S. (1995a). Identification of the low density lipoprotein receptor-related protein (LRP) as an endocytic receptor for thrombospondin-1. J. Cell Biol. 129, 1403-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godyna, S., Mann, D. M., and Argraves, W. S. (1995b). A quantitative analysis of the incorporation of fibulin-1 into extracellular matrix indicates that fibronectin assembly is required. Matrix Biol. 14, 467-477. [DOI] [PubMed] [Google Scholar]
- Grobmyer, S. R., Kuo, A., Orishimo, M., Okada, S. S., Cines, D. B., and Barnathan, E. S. (1993). Determinants of binding and internalization of tissue-type plasminogen activator by human vascular smooth muscle and endothelial cells. J. Biol. Chem. 268, 13291-13300. [PubMed] [Google Scholar]
- Hausser, H., Ober, B., Quentin-Hoffmann, E., Schmidt, B., and Kresse, H. (1992). Endocytosis of different members of the small chondroitin/dermatan sulfate proteoglycan family. J. Biol. Chem. 267, 11559-11564. [PubMed] [Google Scholar]
- Herz, J., Goldstein, J. L., Strickland, D. K., Ho, Y. K., and Brown, M. S. (1991). 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor. J. Biol. Chem. 266, 21232-21238. [PubMed] [Google Scholar]
- Herz, J., and Strickland, D. K. (2001). LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 108, 779-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking, D. C., and Chang, C. H. (2003). Fibronectin matrix polymerization regulates small airway epithelial cell migration. Am. J. Physiol. 285, L169-L179. [DOI] [PubMed] [Google Scholar]
- Hocking, D. C., and Kowalski, K. (2002). A cryptic fragment from fibronectin's III1 module localizes to lipid rafts and stimulates cell growth and contractility. J. Cell Biol. 158, 175-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking, D. C., Sottile, J., and Langenbach, K. J. (2000). Stimulation of integrin-mediated cell contractility by fibronectin polymerization. J. Biol. Chem. 275, 10673-10682. [DOI] [PubMed] [Google Scholar]
- Holmbeck, K. et al. (1999). MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 99, 81-92. [DOI] [PubMed] [Google Scholar]
- Hughes, P. E., Renshaw, M. W., Pfaff, M., Forsyth, J., Keivens, V. M., Schwartz, M. A., and Ginsberg, M. H. (1997). Supression of integrin activation: a novel function of a ras/raf-initiated MAP kinase pathway. Cell 88, 521-530. [DOI] [PubMed] [Google Scholar]
- Ignotz, R. A., and Massague, J. (1986). Transforming growth factor-β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 261, 4337-4345. [PubMed] [Google Scholar]
- Intengan, H. D., and Schiffrin, E. L. (2001). Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension 38, 581-587. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy, G., Kelley, J., Johnson, D., Youngberg, G., Stone, W., Huang, S. K., Bieber, J., and Chi, D. S. (2001). The human mast cell: functions in physiology and disease. Front. Biosci. 6, D1109-D1127. [DOI] [PubMed] [Google Scholar]
- Langenbach, K. J., and Sottile, J. (1999). Identification of protein disulfide isomerase activity in fibronectin. J. Biol. Chem. 274, 7032-7038. [DOI] [PubMed] [Google Scholar]
- Lazic, A., Dolmer, K., Strickland, D. K., and Gettins, P. G. (2003). Structural organization of the receptor associated protein. Biochemistry 42, 14913-14920. [DOI] [PubMed] [Google Scholar]
- Liu, J., Wang, X. B., Park, D. S., and Lisanti, M. P. (2002). Caveolin-1 expression enhances endothelial capillary tubule formation. J. Biol. Chem. 277, 10661-10668. [DOI] [PubMed] [Google Scholar]
- Liu, X., Wu, H., Byrne, M., Jeffrey, J., Krane, S., and Jaenisch, R. (1995). A targeted mutation at the known collagenase cleavage site in mouse type I collagen impairs tissue remodeling. J. Cell Biol. 130, 227-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, J. A., Kelley, D. G., and Broekelmann, T. J. (1982). Role of fibronectin in collagen deposition: Fab′ to the gelatin-binding domain of fibronectin inhibits both fibronectin and collagen organization in fibroblast extracellular matrix. J. Cell Biol. 92, 485-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown-Longo, P. J., Hanning, R., and Mosher, D. F. (1984). Binding and degradation of platelet thrombospondin by cultured fibroblasts. J. Cell Biol. 98, 22-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown-Longo, P. J., and Mosher, D. F. (1984). Mechanism of formation of disulfide-bonded multimers of plasma fibronectin with the matrix-assembly receptor of fibroblasts. J. Biol. Chem. 259, 12210-12215. [PubMed] [Google Scholar]
- McKeown-Longo, P. J., and Mosher, D. F. (1985). Interaction of the 70,000-mol-wt amino-terminal fragment of fibronectin with the matrix-assembly receptor of fibroblasts. J. Cell Biol. 100, 364-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memmo, L. M., and McKeown-Longo, P. (1998). The alphavbeta5 integrin functions as an endocytic receptor for vitronectin. J. Cell Sci. 111, 425-433. [DOI] [PubMed] [Google Scholar]
- Miekka, S. I., Ingham, K. C., and Menache, D. (1982). Rapid methods for isolation of human plasma fibronectin. Thromb. Res. 27, 1-14. [DOI] [PubMed] [Google Scholar]
- Moolenaar, W. H., Kranenburg, O., Postma, F. R., and Zondag, G. C. (1997). Lysophosphatidic acid: G-protein signalling and cellular responses. Curr. Opin. Cell Biol. 9, 168-173. [DOI] [PubMed] [Google Scholar]
- Morla, A., Zhang, Z., and Ruoslahti, E. (1994). Superfibronectin is a functionally distinct form of fibronectin. Nature 367, 193-196. [DOI] [PubMed] [Google Scholar]
- Murphy-Ullrich, J. E., and Mosher, D. F. (1987). Interactions of thrombospondin with endothelial cells: receptor-mediated binding and degradation. J. Cell Biol. 105, 1603-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutsaers, S. E., Bishop, J. E., McGrouther, G., and Laurent, G. J. (1997). Mechanisms of tissue repair: from wound healing to fibrosis. Int. J. Biochem. Cell Biol. 29, 5-17. [DOI] [PubMed] [Google Scholar]
- Nabi, I. R., and Le, P. U. (2003). Caveolae/raft-dependent endocytosis. J. Cell Biol. 161, 673-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby, A. C., and Zaltsman, A. B. (2000). Molecular mechanisms in intimal hyperplasia. J. Pathol. 190, 300-309. [DOI] [PubMed] [Google Scholar]
- Ng, T., Shima, D., Squire, A., Bastiaens, P.I.H., Gschmeissner, S., Humphries, M. J., and Parker, P. J. (1999). PKCα regulates β1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J. 18, 3909-3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, B. J. (2002). A distinct class of endosome mediates clathrin-independent endocytosis to the Golgi complex. Nat. Cell Biol. 4, 374-378. [DOI] [PubMed] [Google Scholar]
- Ozeri, V., Tovi, A., Burstein, I., Natanson-Yaron, S., Caparon, M. G., Yamada, K. M., Akiyama, S. K., Vlodavsky, I., and Hanski, E. (1996). A two-domain mechanism for group A streptococcal adherence through protein F to the extracellular matrix. EMBO J. 15, 989-998. [PMC free article] [PubMed] [Google Scholar]
- Paddison, P. J., Caudy, A. A., Bernstein, E., Hannon, G. J., and Conklin, D. S. (2002). Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 16, 948-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, H., Le, P. U., and Nabi, I. R. (2004). Ganglioside GM1 levels are a determinant of the extent of caveolae/raft-dependent endocytosis of cholera toxin to the Golgi apparatus. J. Cell Sci. 117, 1421-1430. [DOI] [PubMed] [Google Scholar]
- Park, D. S. et al. (2002). Caveolin-1/3 double-knockout mice are viable, but lack both muscle and non-muscle caveolae, and develop a severe cardiomyopathic phenotype. Am. J. Pathol. 160, 2207-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkmans, L., and Helenius, A. (2002). Endocytosis via caveolae. Traffic 3, 311-320. [DOI] [PubMed] [Google Scholar]
- Pelkmans, L., Kartenbeck, J., and Helenius, A. (2001). Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 3, 473-483. [DOI] [PubMed] [Google Scholar]
- Pelkmans, L., Puntener, D., and Helenius, A. (2002). Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 296, 535-539. [DOI] [PubMed] [Google Scholar]
- Pereira, M., Rybarczyk, B. J., Odrljin, T. M., Hocking, D. C., Sottile, J., and Simpson-Haidaris, P. J. (2002). The incorporation of fibrinogen into extracellular matrix is dependent on active assembly of a fibronectin matrix. J. Cell Sci. 115, 609-617. [DOI] [PubMed] [Google Scholar]
- Peterson, T. E. et al. (2003). Caveolin-1 can regulate vascular smooth muscle cell fate by switching PDGF signaling from a proliferative to an apoptotic pathway. Arterioscler Thromb. Vasc. Biol. 23, 1521-1527. [DOI] [PubMed] [Google Scholar]
- Pijuan-Thompson, V., and Gladson, C. L. (1997). Ligation of integrin alpha5beta1 is required for internalization of vitronectin by integrin alphavbeta3. J. Biol. Chem. 272, 2736-2743. [DOI] [PubMed] [Google Scholar]
- Prescott, M. F., Sawyer, W. K., Von Linden-Reed, J., Jeune, M., Chou, M., Caplan, S. L., and Jeng, A. Y. (1999). Effect of matrix metalloproteinase inhibition on progression of atherosclerosis and aneurysm in LDL receptor-deficient mice overexpresing MMP-3, MMP-12, and MMP-12 and on restenosis in rats after balloon injury. Ann. NY Acad. Sci. 878, 179-190. [DOI] [PubMed] [Google Scholar]
- Raub, T. J., and Kuentzel, S. L. (1989). Kinetic and morphological evidence for endocytosis of mammalian cell integrin receptors by using an anti-fibronectin receptor beta subunit monoclonal antibody. Exp. Cell Res. 184, 407-426. [DOI] [PubMed] [Google Scholar]
- Razani, B. et al. (2001). Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 276, 38121-38138. [DOI] [PubMed] [Google Scholar]
- Rebres, R. A., McKeown-Longo, P. J., Vincent, P. A., Cho, E., and Saba, T. M. (1995). Extracellular matrix incorporation of normal and NEM-alkylated fibronectin: liver and spleen deposition. Am. J. Physiol. 269, G902-G912. [DOI] [PubMed] [Google Scholar]
- Roman, J., and McDonald, J. A. (1993). Fibulin's organization into the extracellular matrix of fetal lung fibroblasts is dependent on fibronectin matrix assembly. Am. J. Respir. Cell Mol. Biol. 8, 538-545. [DOI] [PubMed] [Google Scholar]
- Rotundo, R. F., Vincent, P. A., McKeown-Longo, P. J., Blumenstock, F. A., and Saba, T. M. (1999). Hepatic fibronectin matrix turnover in rats: involvement of the asialoglycoprotein receptor. Am. J. Physiol. 277, G1189-G1199. [DOI] [PubMed] [Google Scholar]
- Salicioni, A. M., Mizelle, K. S., Loukinova, E., Mikhailenko, I., Strickland, D. K., and Gonias, S. L. (2002). The low density lipoprotein receptor-related protein mediates fibronectin catabolism and inhibits fibronectin accumulation on cell surfaces. J. Biol. Chem. 277, 16160-16166. [DOI] [PubMed] [Google Scholar]
- Sarkissian, M., and Lafyatis, R. (1998). Transforming growth factor-beta and platelet derived growth factor regulation of fibrillar fibronectin matrix formation by synovial fibroblasts. J. Rheumatol. 25, 613-622. [PubMed] [Google Scholar]
- Sasaki, T., Wiedemann, H., Matzner, M., Chu, M.-L., and Timpl, R. (1996). Expression of fibulin-2 by fibroblasts and deposition with fibronectin into a fibrillar matrix. J. Cell Sci. 109, 2895-2904. [DOI] [PubMed] [Google Scholar]
- Schmid, S. L., McNiven, M. A., and De Camilli, P. (1998). Dynamin and its partners: a progress report. Curr. Opin. Cell Biol. 10, 504-512. [DOI] [PubMed] [Google Scholar]
- Schubert, W., Frank, P. G., Razani, B., Park, D. S., Chow, C. W., and Lisanti, M. P. (2001). Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J. Biol. Chem. 276, 48619-48622. [DOI] [PubMed] [Google Scholar]
- Sechler, J. L., and Schwarzbauer, J. E. (1998). Control of cell cycle progression by fibronectin matrix architecture. J. Biol. Chem 273, 25533-25536. [DOI] [PubMed] [Google Scholar]
- Smart, E. J., and Anderson, R. G. (2002). Alterations in membrane cholesterol that affect structure and function of caveolae. Methods Enzymol. 353, 131-139. [DOI] [PubMed] [Google Scholar]
- Sommers, C. E., and Mosher, D. F. (1993). PKC modulation of fibronectin matrix assembly. J. Biol. Chem. 268, 22277-22280. [PubMed] [Google Scholar]
- Sottile, J., and Hocking, D. C. (2002). Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol. Biol. Cell 13, 3546-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile, J., Hocking, D. C., and Langenbach, K. J. (2000). Fibronectin polymerization stimulates cell growth by RGD-dependent and -independent mechanisms. J. Cell Sci. 113, 4287-4299. [DOI] [PubMed] [Google Scholar]
- Sottile, J., Hocking, D. C., and Swiatek, P. (1998). Fibronectin matrix assembly enhances adhesion-dependent cell growth. J. Cell Sci. 111, 2933-2943. [DOI] [PubMed] [Google Scholar]
- Sottile, J., and Mosher, D. F. (1997). N-terminal type I modules required for fibronectin binding to fibroblasts and to fibronectin's III1 module. Biochem. J. 323, 51-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile, J., and Wiley, S. (1994). Assembly of amino-terminal fibronectin dimers into the extracellular matrix. J. Biol. Chem. 269, 17192-17198. [PubMed] [Google Scholar]
- Streuli, C. (1999). Extracellular matrix remodelling and cellular differentiation. Curr. Opin. Cell Biol. 11, 634-640. [DOI] [PubMed] [Google Scholar]
- Tchougounova, E., Forsberg, E., Angelborg, G., Kjellen, L., and Pejler, G. (2000). Altered processing of fibronectin in mice lacking heparin. A role for heparin-dependent mast cell chymase in fibronectin degradation. J. Biol. Chem. 276, 3772-3777. [DOI] [PubMed] [Google Scholar]
- Tomasini-Johansson, B. R., Kaugman, N. R., Ensenberger, M. G., Ozeri, V., Hanski, E., and Mosher, D. F. (2001). Peptide from adhesin F1 inhibits fibronectin matrix assembly. J. Biol. Chem. 276, 23430-23439. [DOI] [PubMed] [Google Scholar]
- Towbin, H. T., Staehlin, T., and Grodon, J. (1979). Electrophoretic transfer of protein from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76, 4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upla, P., Marjomaki, V., Kankaanpaa, P., Ivaska, J., Hyypia, T., Van Der Goot, F. G., and Heino, J. (2004). Clustering induces a lateral redistribution of alpha 2 beta 1 integrin from membrane rafts to caveolae and subsequent PKC-dependent internalization. Mol. Biol. Cell 15, 625-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nhieu, G. T., Krukonis, E. S., Reszka, A. A., Horwitz, A. F., and Isberg, R. R. (1996). Mutations in the cytoplasmic domain of the integrin beta1 chain indicate a role for endocytosis factors in bacterial internalization. J. Biol. Chem. 271, 7665-7672. [DOI] [PubMed] [Google Scholar]
- Velling, T., Risteli, J., Wennerberg, K., Mosher, D. F., and Johansson, S. (2002). Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins alpha 11beta 1 and alpha 2beta 1. J. Biol. Chem. 277, 37377-37381. [DOI] [PubMed] [Google Scholar]
- Vu, T. H., Shipley, J. M., Bergers, G., Berger, J. E., Helms, J.A., Hanahan, D., Shapiro, S. D., Senior, R. M., and Werb, Z. (1998). MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 93, 411-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienke, D., MacFadyen, J. R., and Isacke, C.M. (2003). Identification and characterization of the endocytic transmembrane glycoprotein Endo180 as a novel collagen receptor. Mol. Biol. Cell 14, 3592-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciechowski, K., Chang, C. H., and Hocking, D. C. (2004). Expression, production, and characterization of full-length vitronectin in Escherichia coli. Protein Expr. Purif. 36, 131-138. [DOI] [PubMed] [Google Scholar]
- Wu, C., Keightley, S. Y., Leung-Hagesteijn, C., Radeva, G., Coppolino, M., Goicoechea, S., McDonald, J. A., and Dedhar, S. (1998). Integrin-linked protein kinase regulates fibronectin matrix assembly, E-cadherin expression and tumorigenicity. J. Biol. Chem. 273, 528-536. [DOI] [PubMed] [Google Scholar]
- Yanagishita, M., and Hascall, V. C. (1984). Metabolism of proteoglycans in rat ovarian glanulosa cell culture. Multiple intracellular degradative pathways and the effect of chloroquine. J. Biol. Chem. 259, 10270-10283. [PubMed] [Google Scholar]
- Yatohgo, T., Izumi, M., Kashiwagi, H., and Hayashi, M. (1988). Novel purification of vitronectin from human plasma by heparin affinity. Cell Struct. Funct. 13, 281-292. [DOI] [PubMed] [Google Scholar]
- Yong, L. C. (1997). The mast cell: origin, morphology, distribution, and function. Exp. Toxicol. Pathol. 49, 409-424. [DOI] [PubMed] [Google Scholar]
- Zeisberg, M., Strutz, F., and Muller, G. A. (2000). Role of fibroblast activation in inducing interstitial fibrosis. J. Nephrol. 13(Suppl 3), S111-S120. [PubMed] [Google Scholar]
- Zhang, Q., Magnusson, M. K., and Mosher, D. F. (1997). Lysophosphatidic acid and microtuble-destabilizing agents stimulate fibronectin matrix assembly through rho-dependent actin stress fiber formation and cell contraction. Mol. Biol. Cell 8, 1415-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, C., Chrzanowska-Wodnicka, M., Brown, J., Shaub, A., Belkin, A. M., and Burridge, K. (1998). Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J. Cell Biol. 141, 539-551. [DOI] [PMC free article] [PubMed] [Google Scholar]