

Abstract
The position of chromosomal neighborhoods in living cells was followed using three different methods for marking chromosomal domains occupying arbitrary locations in the nucleus; photobleaching of GFP-labeled histone H2B, local UV-marked DNA, and photobleaching of fluorescently labeled DNA. All methods revealed that global chromosomal organization can be reestablished through one cell division from mother to daughters. By simultaneously monitoring cell cycle stage in the cells in which relative chromosomal domain positions were tracked, we observed that chromosomal neighborhood organization is apparently lost in the early G1 phase of the cell cycle. However, the daughter cells eventually regain the general chromosomal organization pattern of their mothers, suggesting an active mechanism could be at play to reestablish chromosomal neighborhoods.
INTRODUCTION
Individual chromosomes are organized into discrete domains in the interphase nucleus (Cremer and Cremer, 2001; Parada and Misteli, 2002). In turn, there is order to the arrangement of chromosomal domains because the location of specific chromosome domains relative to the nuclear center and periphery correlates with gene density and chromosome size (Nagele et al., 1995; Croft et al., 1999; Tanabe et al., 2002). The organization of chromosome domains relative to each other into chromosomal neighborhoods implies that this organization can be inherited through cell division. Developments in microscopy and molecular labeling methods now allow the position of chromosomal domains to be followed through time in living cells. Thus, the question of inheritance of relative chromosomal domain position in the nucleus can be studied directly. Attempts to correlate chromosomal organization with cell cycle progression have produced conflicting conclusions about the maintenance of chromosome arrangements (Bickmore and Chubb, 2003; Gerlich and Ellenberg, 2003; Parada et al., 2003; Williams and Fisher, 2003). One study reported that relative global chromosome position is maintained through mitosis (Gerlich et al., 2003), whereas others reported that positional information is lost at metaphase and chromosomal neighborhood organization is not maintained through mitosis (Walter et al., 2003; Thomson et al., 2004).
There are several differences in the experiments of these reports that may account for their conflicting conclusions. Some features of chromosomal organization appear not to be in dispute. These include the organization of chromosomes at the metaphase plate that result in similar relative organization of chromosomal neighborhoods in the two daughter cells after division. There is also agreement on the observation that there are no large-scale reorganizations of chromosomal neighborhoods in interphase (G1-S-G2). The controversy arises when considering the inheritance of chromosomal neighborhoods from mother to daughter cells. Relatively large movements of small marked regions of chromosomes, greater than 2 μm relative to each other, have been observed in early G1 (Walter et al., 2003; Thomson et al., 2004). This movement of chromosome domains relative to each other in G1 would account for the lack of similarity in chromosome neighborhoods that were observed between mother and daughter cells. On the other hand, Gerlich et al. (2003) conclude that chromosomal neighborhoods are inherited because they observe similar patterns in mother and daughter cells.
It is crucial to determine the cell cycle stage of the individual cell being observed in order to describe the (re)arrangement of chromosomal neighborhoods throughout the cell cycle. This is of special importance when following single cells as opposed to following populations of cells where their behavior and characteristics are averaged. In the work of Gerlich et al. (2003) and Walter et al. (2003) cell cycle stages were not determined for each individual cell observed, but the kinetics of cell cycle progression were assumed to be similar for all cells observed. However, as evident in our experiments reported here, individual cells in culture can go through the cell cycle with very different kinetics. Thus, it is important to determine the cell cycle stage of the individual cell being observed in order to describe the (re)arrangement of chromosomal neighborhoods throughout the cell cycle. We have overcome this limitation by monitoring cell cycle progression through visualization of the DNA replication marker PCNA in each individual cell in which we tracked chromosomal neighborhood organization (Leonhardt et al., 2000). The organization and reorganization of chromosomal domains was followed in living cells through division such that mother and daughter cells could be unambiguously identified and compared. In addition to showing that global chromosomal organization is similar in mother and daughter cells, our data suggests that relative chromosomal domain position undergoes rearrangements in early G1 before it is reestablished. By showing that rearrangement and reestablishment of relative chromosomal domain position are part of a continuous process our results contribute to resolving the apparent contradiction among previous studies (Bickmore and Chubb, 2003; Gerlich and Ellenberg, 2003; Parada et al., 2003; Williams and Fisher, 2003).
MATERIALS AND METHODS
DNA Constructs and Cell Lines
Plasmid eGFP-PCNA was generated by inserting a cDNA encoding human PCNA into pEGFP-C1 (Clontech, Palo Alto, CA). The construct was transfected into CHO9 Chinese hamster ovary (CHO) cells and stable clones were selected using G418. The nuclear expression level of PCNA-GFP was similar to that of the endogenous PCNA protein and expression of PCNA-GFP did not interfere with cell survival nor cell cycle progression after UV damage. Time-lapse imaging of the cell line showed that the PCNA-GFP signal accumulated at sites of replication similar as previously established by immunodetection of PCNA (Leonhardt et al., 2000). Cells in early, mid- and late S phase (distinctive focal patterns) and in G1 and G2 (homogeneous PCNA distribution) phases of the cell cycle were easily distinguished. HeLa cells, stably expressing histone H2B-GFP were kindly provided by K. F. Sullivan.
Metaphase Preparations, Local UV Irradiation, and Immunofluorescence
Regular metaphase spreads were prepared following standard cytogenetic procedures (Dronkert et al., 2000). For in situ metaphase spreads, the trypsinization step was omitted. For local UV irradiation, cells were covered with a UV-blocking membrane containing 5-μm-wide pores (Millipore, Bed-ford, MA) and UV irradiated at a dose of 16 J/m2. To detect CPDs and PCNA-GFP simultaneously, cells were grown on coverslips, fixed with 2% paraformaldehyde, and permeabilized using 0.1% Triton X-100. After fixation of the cells DNA was denatured and coverslips were incubated for 1 h at 20°C with α-CPD (Mori et al., 1991) and α-GFP antibodies (rabbit polyclonal, Clontech), followed by a 1-h incubation with Alexa594-conjugated goat α-mouse and Alexa488-conjugated goat α-rabbit secondary antibodies (Molecular Probes, Eugene, OR). To detect CPDs on metaphase spreads, slides were incubated in 65% formamide for 2 min at 80°C. After washing in ice-cold 70% ethanol the slides were incubated with α-CPD and Alexa594-conjugated goat α-mouse secondary antibodies, respectively. Slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). In >1000 metaphases analyzed the local UV irradiation protocol used resulted in CPD accumulation in more than one chromosomal domain. No metaphases were observed in which only a single chromosomal domain was CPD positive.
Time-lapse Confocal Microscopy and Microinjection
Confocal images of living cells expressing GFP-tagged PCNA or histone H2B were obtained using a Zeiss LSM510 microscope (Carl Zeiss, Jena, Germany) equipped with a 25-mW Argon laser at 488 nm and a heated 63× 1.4 NA oil immersion lens. GFP fluorescence was detected using a dichroic beamsplitter (488 nm) and an additional 505–550-nm bandpass emission filter placed in front of the photo multiplier tube. Alexa546 fluorescence was detected using a 1-mW He/Ne laser at 543 nm and a dichroic beamsplitter (488/543 nm) and an additional 560–615-nm bandpass emission filter. Monitoring was done with the 488-nm Argon laser with a tube current of 6.1 A and the AOTF at 0.5% transmission for GFP and with the 543-nm He/Ne laser AOTF at 10% transmission for Alexa546-dUTP. For live cell imaging, cells were plated on coverslips, transferred to a heated chamber on the microscopic stage, and incubated at 37°C in F10-DMEM containing 10% fetal calf serum under 5% CO2 conditions throughout image acquisition.
To mark a specific pattern in histone H2B-GFP–expressing cells, the fluorescence of histone H2B-GFP was irreversibly photobleached at high laser intensity at a wavelength of 488 nm by 10 iterations (pixel dwell time of 1.76 μs) of the 488-nm Argon laser with a tube current of 6.1 A and the AOTF at 100% transmission. To irreversibly photobleach the DNA marked with fluorescent Alexa546, approximately half of the nucleus was repeatedly bleached at a wavelength of 543 nm by 25 iterations (pixel dwell time of 25.6 μs) and the AOTF at 100% transmission.
For microneedle injection of Alexa546-dUTP (Molecular Probes, Leiden, The Netherlands), PCNA-GFP–expressing cells were grown on etched grid coverslips (Bellco, Vineland, NJ) and microinjected directly into the nucleus. The concentration of the Alexa546-dUTP in the needle was 100 μM. One day after microinjection, coverslips were used in time-lapse experiments.
Fluorescent signals were analyzed using ImageJ (Wayne Rasband, National Institutes of Health, Bethesda, MD). One optical plane was taken from the time-lapse series around the mitotic division (5 cells, 18 time points). The cell of interest was segmented from the series by hand and only one cell was measured after cell division. Using the PCNA-GFP staining a mask of the nuclear area was made. This mask was used to measure the PCNA-GFP and the Alexa546-labeled DNA area (number of pixels). In all series a maximum ratio between fluorescent Alexa546-labeled DNA and PCNA-GFP was observed within 30 min after anaphase (see Figure 4D), and the time series were aligned to this point.
Figure 4.
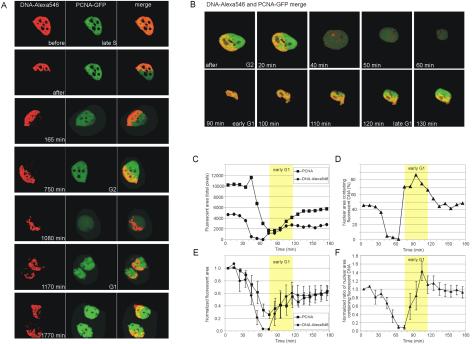
Fluorescently labeled DNA patterns are reestablished in daughter cells. (A) Simultaneous time-lapse imaging of Alexa546-labeled DNA (red) and PCNA-GFP (green). After labeling PCNA-GFP–expressing cells with Alexa546-dUTP (image before), part of the fluorescently labeled DNA, spanning about half the nucleus was photobleached (image after), leaving the PCNA-GFP signal intact, resulting in the red/green-merged image. Based on the PCNA pattern, photobleaching of the labeled DNA was done in the late S phase. Although the location of the nucleus changed, the fluorescently labeled DNA kept its original position during the late S and G2 phase in the mother cell. After cell division the fluorescent DNA pattern induced by photobleaching in the mother cell reappeared in both daughter cells. See also Supplementary Online Movie 2. (B–F) Quantitative analysis of the distribution of fluorescently labeled DNA patterns in G2 cells and redistribution after mitosis in early G1 and late G1 cells. (B) After photobleaching about half the fluorescent DNA, cells were followed through mitosis. The distinct fluorescently labeled DNA pattern that was present in the mother cell was dispersed in early G1 cells but reappeared in late G1. Note, the mother cell rounds up and detaches from the coverslip in mitosis, and in this example only one daughter is shown after mitosis. (C) The total fluorescent area of PCNA-GFP and Alexa546–labeled DNA in individual stills of the time-lapse movie of a single cell were determined. Both DNA-Alexa546 and PCNA-GFP fluorescence dropped ∼50% in the daughter cell, as the total fluorescent DNA and PCNA-GFP proteins are divided over the two cells. (D) Change in nuclear area occupied by fluorescent DNA over time. Total nuclear area is defined by PCNA-GFP fluorescence. Percent nuclear area containing fluorescent DNA is plotted as a function of time using the data shown in C. (E and F) Quantification of the normalized areas containing fluorescent DNA and PCNA-GFP during G2, mitosis, and G1. The graphs in E and F present data from five independent experiments of which one is shown in C and D, respectively. After bleaching the area containing fluorescent DNA was approximately half the size of the nuclear area, defined by PCNA-GFP fluorescence. However, in the graphs the fluorescent area for both fluorophores was set to 1 at time zero in each cell (E), which results in a ratio of 1 at time zero in the graph shown in F. Error bars, the SE of the mean.
The reestablishment of the bleached pattern in the daughter cell nuclei was independent of the orientation of the bleach border with respect to the metaphase plate (Gerlich et al., 2003). Each cell was examined using phase-contrast microscopy and DNA-Alexa546 fluorescence to determine the orientation of the metaphase plate and the orientation of boundary between the bleached and unbleached area to the spindle axis. Within this group of cells cases of perpendicular, parallel and diagonal orientation of the boundary to the spindle axis were found.
RESULTS AND DISCUSSION
The global distribution of chromosomes relative to each other after cell division was followed in HeLa cells expressing histone H2B-GFP with the use of local photobleaching (Kanda et al., 1998). The majority of histone H2B-GFP molecules is incorporated into chromatin and exhibits limited mobility. Therefore, patterns induced by local photobleaching in nuclei of living cells are relatively stable and can be followed in cells over time (Kimura and Cook, 2001). We followed cells with photobleached patterns through division and observed the return of the patterns in the nuclei of both daughter cells (Figure 1, A and B). Our approach differed from that of previous work with respect to the time the cells were followed through division and with respect to the size of the nuclear area that was marked and followed (Gerlich et al., 2003; Walter et al., 2003; Thomson et al., 2004). We marked global chromosome position by bleaching less than half of the nucleus and then followed the position of chromatin from interphase through mitosis to the next interphase. We observed that the photobleached patterns in chromatin of interphase nuclei were reestablished in the interphase nuclei of both daughter cells. In contrast when almost all H2B-GFP fluorescence is bleached leaving only a small region at the periphery of the nucleus, the reappearance of this pattern in daughter cells after division is not always observed (Walter et al., 2003). In addition, monitoring small chromosomal loci through binding of GFP-LacI, reveals no evidence for stable relative position through mitosis (Thomson et al., 2004). Consistent with our observations, marking large regions of the nucleus, patterns are conserved from prophase to metaphase as well as in anaphase and telophase when followed in relatively condensed chromosomes for only a short time period around mitosis (Gerlich et al., 2003).
Figure 1.
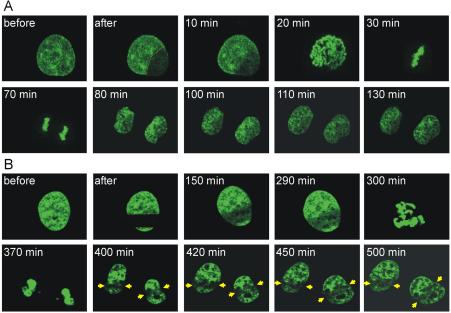
Time-lapse imaging of locally photobleached histone H2B-GFP–expressing cells. Hela cells stably expressing histone H2B-GFP were subjected to a local bleach pulse (area indicated by the dotted line) and imaged in 4D through mitosis. (A) An interphase cell, showing a typical histone H2B staining of chromatin before and after photobleaching, was followed through prophase (20 min), metaphase (30 min), anaphase (70 min), telophase (80 min), and interphase (100–130 min). The triangular shaped, photobleached pattern in the mother cell reappeared after cell division with nearly identical triangular shape in the lower right part of both daughter cells. (B) A second example of an interphase cell expressing H2B-GFP that was followed through mitosis after photobleaching. The rectangular bleached pattern (area indicated by the dotted lines) in the mother cell reappeared in two daughter cells at similar positions indicated by the arrows. Total time elapsed between local photobleaching of the mother cell in interphase until the end of imaging the two interphase daughter cells was 500 min.
The reappearance of the photobleached pattern of histone H2B-GFP in daughter nuclei indicated that global chromosomal organization is reestablished after division. There is some uncertainty in this method as newly synthesized histone H2B can exchange and redistribute, albeit to a limited extent, in the nucleus (Kimura and Cook, 2001). Therefore, we also used a completely independent method to mark the DNA of chromosomal domains by local application of UV light through nitrocellulose filters with 5-μm pores (Mone et al., 2001; Volker et al., 2001; Hoogstraten et al., 2002). For these experiments we used CHO cells in which UV-induced cyclobutyl pyrimidine dimers (CPDs) persist in DNA because they are not effectively removed by nucleotide excision repair (Hwang et al., 1998). The position of the UV-marked chromosomal domains was visualized in fixed cells by immunofluorescence with CPD-specific antibodies (Figure 2A). Metaphase chromosomes from locally irradiated cells stained for CPDs confirmed that this method marked chromosomal domains (Figure 2, B and C) in a manner analogous to irradiation of cells with a laser-UV-microbeam (Cremer et al., 1982). In addition, this technique reveals nuclear organization by marking domains on separate chromosomes that were in close proximity at the time of irradiation (for example, see Figure 2C). These cells were also stained for PCNA, a protein involved in both replicative and repair DNA synthesis. Simultaneous monitoring of CPDs and PCNA by immunofluorescence revealed that CPDs and PCNA could colocalize (Figure 2, D–F). On top of the normal PCNA staining pattern that reveals the specific stage of the cell cycle (Leonhardt et al., 2000), we observed accumulation of PCNA at the same position as the CPDs. Therefore, PCNA could be used as marker for the position of CPD-containing chromosomal domains.
Figure 2.
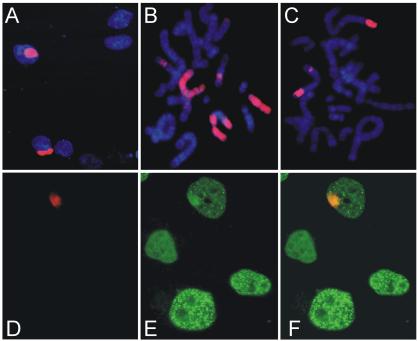
Visualization of chromosomes locally marked by UV-light in interphase nuclei of PCNA-GFP–expressing cells. (A) Local accumulation of CPDs in interphase cells. Cells were covered with a UV-blocking membrane containing 5-μm-wide pores and UV irradiated. After trypsinization and fixation, cells were spread onto microscope slides. The location of CPDs was visualized using an antibody against CPDs (red). DNA was stained with DAPI (blue). (B and C) Visualization of locally induced CPDs on metaphase chromosomes. After local UV irradiation metaphase spreads of the cells were prepared. The position of CPDs was visualized with an α-CPD antibody shown in red. (D–F-) Locally induced CPDs and PCNA colocalize. Locally induced CPDs (red) in PCNA-GFP–expressing cells were visualized by immuno-staining in D. (E) The GFP signal (green) in the same field as D. The images in D and E are merged in F. Superimposed on the PCNA pattern associated with DNA replication, PCNA accumulation at the position of the CPDs can be observed.
This information allowed us to follow CPD-marked chromosomal domains by PCNA-GFP accumulation in living cells. We performed time-lapse imaging of locally irradiated cells to follow the fate of CPD-marked chromosomal domains through cell division. Figure 3A shows an example of a cell, marked in early/mid-S phase with a local concentration of PCNA at the site of UV irradiation, that subsequently progressed through cell division (see also Supplementary Online Movie 1). Although the local PCNA accumulation at the site of CPDs disappeared in late S phase, where the prominent accumulation of PCNA is in replication foci, it was reestablished after division when the cells were in the next S phase. Importantly, this occurred in both daughter cells. We conclude that the locally induced CPDs that were in different, but neighboring, chromosomal domains return to the same position relative to each other in both daughter cells.
Figure 3.
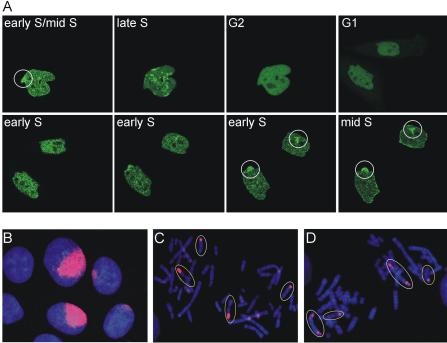
Transmission of chromosomes containing locally induced CPDs through mitosis into daughter nuclei. (A) Time-lapse imaging of PCNA-GFP in locally irradiated cells. Local UV irradiation was applied to a PCNA-GFP–expressing cell that was at the transition from early to mid-S phase. The procedure resulted in local accumulation of PCNA-GFP (indicated by the circle) on top of the standard replication associated PCNA pattern. This local PCNA-GFP accumulation in the mother cell disappeared during late S and G2. After mitosis, the local accumulation of PCNA was absent in both daughter cells in G1 and very early S phase. However, the local accumulation of PCNA-GFP, indicative of the CPD position, reappeared near the end of early S phase in both daughter cells. See also Supplementary Online Movie 1. (B) Position of CPDs in interphase cells 16 h after local UV irradiation. DNA is visualized by DAPI staining in blue and CPDs by immuno-staining in red. Two interphase cells show a large similar shaped region of CPDs, and two cells show a small similarly shaped region of CPDs. (C and D) Visualization of locally induced CPDs on metaphase chromosomes 16 h after local UV irradiation. To keep metaphases of daughter cells together, in situ metaphase spreads were made and stained with α-CPD antibodies (red). The two neighboring metaphases are from daughter cells because their metaphases revealed an identical CPD distribution on the chromosomes (indicated by white and yellow ellipses).
Similarly, when visualizing CPDs in fixed interphase cells, neighboring cells were observed with similar staining patterns (Figure 3B). The proximity of these cells suggests that they are daughters because they are too close to have resulted from irradiation through separate pores. To determine whether this was the case, we developed a protocol to make in situ metaphase spreads. If neighboring cells were daughters, then neighboring metaphase spreads should display an identical distribution of CPDs on their chromosomes. Indeed, using this protocol we detected neighboring metaphases with identical CPD distributions (Figure 3, C and D). Furthermore, the frequency of neighboring metaphases containing identical CPD patterns should increase when the time between irradiation and preparation of the metaphase spread is increased because more cells will have had the opportunity to divide. Indeed, the percentage of CPD-positive metaphases having a neighbor with an identical CPD pattern increased from 8% at 2 h to 28% at 16 h after irradiation. Thus by marking the DNA in neighboring chromosomal domains in an arbitrary region of the nucleus using UV irradiation, we show by another independent and higher resolution method that the position of chromosomal domains can reappear in a similar relative organization in daughter nuclei.
Although, the relative position of chromosomal domains marked by DNA damage is reestablished in daughter cells after mitosis, the local DNA damage itself could influence nuclear location. In addition, the PCNA-GFP signal was not informative as a chromosomal domain marker at all points in the cell cycle. To follow chromosomal domain positions through the cell cycle, as well as use a method other than DNA damage for marking nuclear location, we performed the following live cell imaging experiment. Cells expressing PCNA-GFP were injected with fluorescently labeled nucleotide precursors (Zink et al., 1998). This resulted in nuclei in which both PCNA and DNA can be monitored by distinct fluorescent signals in living cells. The cell cycle stage was identified unambiguously for individual cells from the PCNA pattern. The fluorescent DNA was specifically photobleached, while leaving the PCNA-GFP signal intact, to arbitrarily mark chromosomal neighborhoods based on their position at the time of bleaching. The marked chromosomal positions were followed through mitosis by time-lapse imaging of cells (Figure 4A and Supplementary Online Movie 2). In all cases where cells could be followed through mitosis the general pattern of bleached and unbleached DNA in the daughters resembled that in their mother (Figure 4, A and B). Table 1 summarizes the cell cycle parameters for 21 individual cells. The time needed to progress through the cell cycle varied greatly for individual cells, emphasizing the necessity for a single cell marker of cell cycle stage such as PCNA. Classifying cell cycle stages by population average for the cell division cycle would have resulted in many misclassifications.
Table 1.
Cell cycle stage at the beginning and end of imaging period and duration of imaging
| Total time span of imaging (hours)
|
||||
|---|---|---|---|---|
| Cell cycle stage of the mother cell at beginning of imaging | Cell cycle stage of the daughter cell(s) at end of imaging | No. of times observed | Individual cells | Average |
| Early S | G1 | 3 | 13/20/35 | 22.7 |
| Mid S | G1 | 5 | 13/17.5/18/20/29 | 19.5 |
| Late S | G1 | 6 | 8/13/20/20/20/29 | 18.3 |
| Late S | G2 | 2 | 22.5/30 | 26.3 |
| G2 | G1 | 4 | 6/17/22.5/22.5 | 17.0 |
| G2 | MidS | 1 | 13 | 13.0 |
The table indicates the cell cycle stage of the mother cell, as determined by the PCNA pattern, in which part of the fluorescently labeled DNA was bleached and the cell cycle stage in which imaging was terminated in the daughter cell(s).
The experiment described above yielded interesting additional information on relative chromosomal domain positioning. As observed in the histone H2B-GFP photobleaching and the local UV irradiation, daughter cells recapitulated chromosomal domain position patterns marked in the mother cell. However, by following the DNA directly, we observed that there was a portion of the cell cycle, early in G1, where the pattern of bleached and unbleached chromosomal domain positions was lost. Analysis of confocal stacks confirmed that the loss of pattern was not simply due to rotation of the nuclei. To analyze the apparent loss of relative chromosomal domain position more quantitatively, we used the PCNA-GFP signal as an indicator of the total nuclear area and compared that to the portion of the nuclear area covered by Alexa546 fluorescently labeled DNA, representing the localization of the marked chromatin (Figure 4, C and D). After photobleaching the Alexa546 signal covered half of the nuclear area (Figure 4C, time 0 min), at mitosis when the cells rounded up both PCNA-GFP and DNA-Alexa546 signals were lost from view. In early G1 the cells adhered to the coverslip and coherent images of their nuclei could again be obtained. This quantification reassuringly resulted in detection of half of the original PCNA-GFP and DNA-Alexa546 fluorescence in the daughter cells (Figure 4C; compare total fluorescent area at time 0 and 180 min).
For 5 of 21 cells imaged through G1 there were time points in early G1 when the DNA-Alexa546 pattern was lost. DNA-Alexa546 fluorescence was no longer limited to half of the nucleus but often coincided almost completely with the PCNA signal representing the complete nuclear area (Figure 4B, 90- and 100-min image and corresponding time points in Figure 4D). In the remaining 16 cells this early stage of G1 was missed because of the 10-min intervals between image acquisitions. This mixing of PCNA and DNA-Alexa546 fluorescence was observed in all five examples where at least one daughter cell was followed throughout the very early portion of G1. In addition, the apparent loss of organization of the DNA-Alexa546 pattern is unlikely to be due to changes in nuclear size and shape in early G1. At the time points where the DNA-Alexa546 fluorescence was no longer limited to half of the nucleus, the cells were clearly no longer in mitosis. They were reattached to the coverslip and their nuclei had again flattened, as evident by detection of PCNA fluorescence in an area about half as large as in the mother cell (see Figure 4B, 110 min and recovery of PCNA fluorescent area by 100–120 min in Figure 4, C and E). The measurements presented in Figure 4 were taken from a 1.5-μm-thick confocal slice closest to the coverslip. To confirm that the loss of the DNA-Alexa546 pattern occurs in the whole nucleus and was not due to a change in orientation presenting a different portion of the nucleus in the confocal slice analyzed, we performed the same analysis on the second and third confocal slices of one set of mother and two daughter cells. Here again in this segment of the nucleus, the pattern of DNA-Alexa546 was lost in both daughters in early G1 (relative fluorescence profile the same as in Figure 4F). We estimate that the confocal slices analyzed represented more than 70% of the nuclear volume in which there was no apparent pattern to the DNA-Alexa546 fluorescence, suggesting that relative chromosomal domain positions were lost at these time points. However, the DNA-Alexa546 fluorescent pattern, localized to half of the nucleus, was reestablished at later times (Figure 4, E and F). This qualitative and quantitative analysis indicates that there was a period early in G1 when the chromosomes were uncondensed but not yet organized into the same or similar neighborhoods as in the mother nucleus.
We have shown by three independent DNA marking methods that the relative organization of chromosomal domain position is similar between mother and daughter cells. In addition, there was a period early in G1 where this organization was apparently lost. This loss of organization was not due to a specific orientation of the bleached boundary relative to the spindle axis as the cells analyzed included examples of all relative orientations, parallel, perpendicular, and diagonal. Chromosome decondensation in G1 could account for the loss of relative chromosomal domain organization. If this event is not highly coordinated among chromosomes and occurs asynchronously, organization maintained at metaphase would be disturbed. The purpose of maintenance of relative chromosomal domain position and the mechanism for its reestablishment after cell division remain intriguing. The simplest reason to organize chromosomes into domains in the nucleus is to prevent them from becoming entangled. Establishing chromosomal domains into neighborhoods may not be a deterministic process but could result from the physical properties of chromosomes, the time of their separation, their rate of decondensation, and perhaps their differential interaction with components of the reforming nuclear envelope. We followed the general pattern of chromosomal domain organization through one cell division. Though the daughter nuclei resembled their mothers, the similarity was not absolute. Thus, the organization of chromosomal neighborhoods changed somewhat through cell divisions. If different chromosomal neighborhood organization is functionally important, this variation would allow additional epigenetic mechanisms to affect cell differentiation during development.
Supplementary Material
Acknowledgments
We thank Jan Ellenberg for discussion and comments on the manuscript. HeLa cells, stably expressing histone H2B-GFP, were kindly provided by K. F. Sullivan. This work was supported by grants from the Netherlands Organization for Scientific Research, the Dutch Cancer Society, and the European Union.
Article published online ahead of print in MBC in Press on December 1, 2004 (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-10-0876).
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Bickmore, W. A., and Chubb, J. R. (2003). Chromosome position: now, where was I? Curr. Biol. 13, R357-R359. [DOI] [PubMed] [Google Scholar]
- Cremer, T., and Cremer, C. (2001). Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2, 292-301. [DOI] [PubMed] [Google Scholar]
- Cremer, T., Cremer, C., Baumann, H., Luedtke, E. K., Sperling, K., Teuber, V., and Zorn, C. (1982). Rabl's model of the interphase chromosome arrangement tested in Chinese hamster cells by premature chromosome condensation and laser-UV-microbeam experiments. Hum. Genet. 60, 46-56. [DOI] [PubMed] [Google Scholar]
- Croft, J. A., Bridger, J. M., Boyle, S., Perry, P., Teague, P., and Bickmore, W. A. (1999). Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145, 1119-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronkert, M. L., Beverloo, H. B., Johnson, R. D., Hoeijmakers, J. H., Jasin, M., and Kanaar, R. (2000). Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol. Cell. Biol. 20, 3147-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlich, D., Beaudouin, J., Kalbfuss, B., Daigle, N., Eils, R., and Ellenberg, J. (2003). Global chromosome positions are transmitted through mitosis in mammalian cells. Cell 112, 751-764. [DOI] [PubMed] [Google Scholar]
- Gerlich, D., and Ellenberg, J. (2003). Dynamics of chromosome positioning during the cell cycle. Curr. Opin. Cell Biol. 15, 664-671. [DOI] [PubMed] [Google Scholar]
- Hoogstraten, D., Nigg, A. L., Heath, H., Mullenders, L. H., van Driel, R., Hoeijmakers, J. H., Vermeulen, W., and Houtsmuller, A. B. (2002). Rapid switching of TFIIH between RNA polymerase I and II transcription and DNA repair in vivo. Mol. Cell 10, 1163-1174. [DOI] [PubMed] [Google Scholar]
- Hwang, B. J., Toering, S., Francke, U., and Chu, G. (1998). p48 Activates a UV-damaged-DNA binding factor and is defective in xeroderma pigmentosum group E cells that lack binding activity. Mol. Cell. Biol. 18, 4391-4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda, T., Sullivan, K. F., and Wahl, G. M. (1998). Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8, 377-385. [DOI] [PubMed] [Google Scholar]
- Kimura, H., and Cook, P. R. (2001). Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153, 1341-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt, H., Rahn, H. P., Weinzierl, P., Sporbert, A., Cremer, T., Zink, D., and Cardoso, M. C. (2000). Dynamics of DNA replication factories in living cells. J. Cell Biol. 149, 271-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mone, M. J., Volker, M., Nikaido, O., Mullenders, L. H., van Zeeland, A. A., Verschure, P. J., Manders, E. M., and van Driel, R. (2001). Local UV-induced DNA damage in cell nuclei results in local transcription inhibition. EMBO Rep. 2, 1013-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, T., Nakane, M., Hattori, T., Matsunaga, T., Ihara, M., and Nikaido, O. (1991). Simultaneous establishment of monoclonal antibodies specific for either cyclobutane pyrimidine dimer or (6–4)photoproduct from the same mouse immunized with ultraviolet-irradiated DNA. Photochem. Photobiol. 54, 225-232. [DOI] [PubMed] [Google Scholar]
- Nagele, R., Freeman, T., McMorrow, L., and Lee, H. Y. (1995). Precise spatial positioning of chromosomes during prometaphase: evidence for chromosomal order. Science 270, 1831-1835. [DOI] [PubMed] [Google Scholar]
- Parada, L., and Misteli, T. (2002). Chromosome positioning in the interphase nucleus. Trends Cell Biol. 12, 425-432. [DOI] [PubMed] [Google Scholar]
- Parada, L. A., Roix, J. J., and Misteli, T. (2003). An uncertainty principle in chromosome positioning. Trends Cell Biol. 13, 393-396. [DOI] [PubMed] [Google Scholar]
- Tanabe, H., Muller, S., Neusser, M., von Hase, J., Calcagno, E., Cremer, M., Solovei, I., Cremer, C., and Cremer, T. (2002). Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. Proc. Natl. Acad. Sci. USA 99, 4424-4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, I., Gilchrist, S., Bickmore, W. A., and Chubb, J. R. (2004). The radial positioning of chromatin is not inherited through mitosis but is established de novo in early G1. Curr. Biol. 14, 166-172. [DOI] [PubMed] [Google Scholar]
- Volker, M., Mone, M. J., Karmakar, P., van Hoffen, A., Schul, W., Vermeulen, W., Hoeijmakers, J. H., van Driel, R., van Zeeland, A. A., and Mullenders, L. H. (2001). Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 8, 213-224. [DOI] [PubMed] [Google Scholar]
- Walter, J., Schermelleh, L., Cremer, M., Tashiro, S., and Cremer, T. (2003). Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J. Cell Biol. 160, 685-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, R. R., and Fisher, A. G. (2003). Chromosomes, positions please! Nat. Cell Biol. 5, 388-390. [DOI] [PubMed] [Google Scholar]
- Zink, D., Cremer, T., Saffrich, R., Fischer, R., Trendelenburg, M. F., Ansorge, W., and Stelzer, E. H. (1998). Structure and dynamics of human interphase chromosome territories in vivo. Hum. Genet. 102, 241-251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.