

Abstract
Clathrin-mediated endocytosis in mammalian cells is critical for a variety of cellular processes including nutrient uptake and cell surface receptor down-regulation. Despite the findings that numerous endocytic accessory proteins directly or indirectly regulate actin dynamics and that actin assembly is spatially and temporally coordinated with endocytosis, direct functional evidence for a role of actin during clathrin-coated vesicle formation is lacking. Here, we take parallel biochemical and microscopic approaches to address the contribution of actin polymerization/depolymerization dynamics to clathrin-mediated endocytosis. When measured using live-cell fluorescence microscopy, disruption of the F-actin assembly and disassembly cycle with latrunculin A or jasplakinolide results in near complete cessation of all aspects of clathrin-coated structure (CCS) dynamics. Stage-specific biochemical assays and quantitative fluorescence and electron microscopic analyses establish that F-actin dynamics are required for multiple distinct stages of clathrin-coated vesicle formation, including coated pit formation, constriction, and internalization. In addition, F-actin dynamics are required for observed diverse CCS behaviors, including splitting of CCSs from larger CCSs, merging of CCSs, and lateral mobility on the cell surface. Our results demonstrate a key role for actin during clathrin-mediated endocytosis in mammalian cells.
INTRODUCTION
Clathrin-mediated endocytosis1 (CME) is critical for a variety of biological processes such as nutrient uptake, cell surface receptor down-regulation, and synaptic vesicle recycling. CME involves the spatial and temporal coordination of numerous lipid components and multiple accessory proteins (Slepnev and De Camilli, 2000). Structural proteins, including clathrin and adaptor proteins, are recruited from the cell cytosol and assemble on the plasma membrane forming a ∼200-nm clathrin-coated pit (CCP). The CCP becomes invaginated and narrows at the neck, forming a constricted coated pit which subsequently pinches off from the plasma membrane, generating a nascent clathrin-coated vesicle (CCV). Finally, the cycle is completed by disassembly of the clathrin coat, which is utilized for subsequent rounds of endocytosis.
Growing evidence has suggested a role for the actin cytoskeleton during clathrin-mediated endocytosis (Qualmann et al., 2000; Merrifield, 2004). In budding yeast, actin assembly and disassembly dynamics are tightly coupled to and essential for endocytosis (Engqvist-Goldstein and Drubin, 2003). Thus, disruption of the yeast cortical actin cytoskeleton by genetic or chemical means inhibits endocytosis. Similarly, several lines of evidence have suggested a close association between the endocytic machinery in mammalian cells and the actin cytoskeleton. First, the GTPase dynamin, a central player in CME, and several dynamin-binding partners, including cortactin, syndapin, mAbp1, intersectin, and profilin, have been shown to directly or indirectly regulate F-actin dynamics (Qualmann et al., 2000; Engqvist-Goldstein and Drubin, 2003; Merrifield, 2004). Second, Hip1R, an F-actin-binding protein, also interacts with clathrin and promotes clathrin assembly (Engqvist-Goldstein et al., 2001). Finally, it was recently shown that actin and regulators of actin polymerization, N-WASP and the Arp2/3 complex, are transiently recruited to CCSs coincident with, or shortly after, CCV detachment from the plasma membrane (Merrifield et al., 2002, 2004). Based on these and other data, F-actin has been postulated to play either a structural role in CME, controlling the localization of endocytic machinery on the plasma membrane, or a mechanical role, driving invagination, the separation of vesicles from the plasma membrane, and/or the translocation of nascent vesicles into the cytoplasm (Qualmann et al., 2000; Qualmann and Kessels, 2002; Engqvist-Goldstein and Drubin, 2003; Merrifield, 2004).
In contrast to these results, functional evidence demonstrating a direct role for actin in CME in mammalian cells is ambiguous or lacking. For example, biochemical studies have shown that chemical disruption of actin assembly/disassembly dynamics only partially inhibits endocytosis, suggesting that actin cytoskeletal dynamics are not essential for this process (Durrbach et al., 1996; Lamaze et al., 1997; Maples et al., 1997; Fujimoto et al., 2000; Lunn et al., 2000; Moskowitz et al., 2003). However, biochemical approaches used to study endocytosis measure the collective and average ability of a population of cells to internalize receptor-bound ligands. Thus, the observed partial inhibitory effects have several potential and nonexclusive interpretations. For instance, inhibition of actin dynamics may modestly reduce the rate of, but not completely block CCS internalization, suggesting that actin plays only a secondary role in CME. Alternatively, the inhibition of actin dynamics may block the internalization of a subpopulation of CCSs that are differentially sensitive to actin disruption. Finally, the uptake of all CCSs may be fully blocked, and an alternative, clathrin-independent pathway for receptor-mediated endocytosis may be induced, indicating that actin dynamics are absolutely required for CME. Microscopic studies, in contrast, have focused on the actin dynamics associated with individual CCSs. However, the behavior of these CCSs has not been analyzed after disruption of actin dynamics (Merrifield et al., 2002). Hence, the mechanistic contribution of the actin cytoskeleton to the endocytosis of CCV in mammalian cells remains uncertain.
Here, we have sought direct evidence for the functional contribution of actin dynamics to clathrin-mediated endocytosis, using parallel quantitative microscopic and biochemical approaches. We conclude that actin cytoskeletal assembly/disassembly dynamics are critical for many aspects of CCS dynamics including assembly, constriction, internalization, and lateral motility.
MATERIALS AND METHODS
Reagents
Latrunculin A (latA) and jasplakinolide were obtained from Sigma Chemical (St. Louis, MO) and resuspended in dimethyl sulfoxide (DMSO) at 10 and 1 mM concentrations, respectively. Oregon Green–labeled chicken skeletal muscle actin was prepared by covalent linkage of succinimidyl ester of Oregon Green (Molecular Probes, Eugene, OR) to lysine residues on purified skeletal muscle actin as described (Waterman-Storer, 2002). Human diferric transferrin was biotinylated as previously described (Smythe et al., 1992). All other reagents, unless otherwise noted, were obtained from Sigma.
Cell Culture
Swiss 3T3 cells (mouse fibroblasts) stably expressing clathrin light-chain DsRed (kindly provided by W. Almers, Vollum Institute, Oregon Health and Sciences University) were grown in a humidified incubator at 37°C, 5% CO2 and maintained in DMEM containing 10% fetal bovine serum (Gemini, Woodland, CA) and 400 μl/ml geneticin (Invitrogen, Carlsbad, CA).
Fluorescence Microscopy
Cells, 1.2 × 106, were plated onto poly-l-lysine (PLL; 150,000–300,000 MW 0.1 mg/ml)-coated 22 × 22-mm glass no. 1.5 coverslips (Corning, Corning, NY; n = 1.523) in a 100-mm tissue culture dish. The following day cells were viewed with an inverted microscope (Nikon TE2000U, Garden City, NY) custom modified to allow for through-the-objective multispectral total-internal reflection fluorescence microscopy and wide-field epi-fluorescence (to be described in detail elsewhere) using a 100×, 1.45 NA objective (Nikon). The wavelength and intensity of light from a 50-mW KrAr air-cooled laser (Melles Griot, Rochester, NY) were controlled with a polychromatic acousto-optical modulator, the output of which was collected by a fiberoptic to transmit it to the microscope's custom modified, dual-input epi-illuminator. Lines at 568 and 488 nm were used for total internal reflection fluorescence microscopy (TIR-FM) excitation. Wide-field epi-fluorescence (WF-EF) light was generated by a 100-W Hg arc lamp and controlled with an electronic shutter/filter wheel device containing bandpass excitation filters for 488 ± 20 and 568 ± 20 nm (Sutter Instruments, Novato, CA). Laser and arc-lamp light were both focused at the aperture plane and directed to the coverslip by a custom dual dichromatic mirror (Chroma, Brattleboro, VT). The position of the beam was adjusted with a digital readout micrometer that moved the fiber-optic coupler at its input into the illuminator. We calibrated the micrometer to enable selection of specific beam angles. We used a penetration depth of ∼100 nm of the evanescent field. Fluorescence emission was controlled with a filterwheel device (Sutter Instruments) containing narrow bandpass emission filters. Images were collected using a Hamamatsu Orca II-ERG camera (Bridgewater, NJ), operated in 14-bit mode. Control of all electronic hardware and camera acquisition was achieved with MetaMorph software (Universal Imaging, West Chester, PA). Stage temperature was controlled with a custom-modified stage incubator (20–20 Technologies, Wilmington, NC).
For imaging, coverslips of cells were preincubated for 30 min with imaging medium (DMEM [no phenol red], 10 mM HEPES, pH 7.5, 10% fetal bovine serum) containing either 5 μM latA, 1 μM jasp, or an equal volume of DMSO (0.1%) at 37°C. Coverslips were then mounted on a slide with two strips of double-sided tape spaced ∼1 cm apart. The space between the strips of tape was filled with imaging medium additionally containing either 5 μM latA or 1 μM jasp, or DMSO. The coverslip was sealed with a 1:1:1 mixture of Vaseline, lanolin, and paraffin and the slide was transferred to a prewarmed microscope stage (37°C) for imaging. One or two cells were imaged per coverslip. Pairs of WF-EFM (100–200-ms exposures) and TIR-FM (200–500-ms exposures) images were taken for 10–15 min at 2-s intervals, with WF-EFM images taken first to minimize the time between WF-EFM and TIR-FM acquisitions.
For rescue experiments, cells were imaged in a custom built perfusion chamber (Gupton and Waterman-Storer, 2005) to wash out latA during the course of imaging. After 10 min of imaging cells in the presence of 5 μM latA, cells were washed with >20 chamber volumes of DMEM (BioSource International, Camarillo, CA) +10% fetal calf serum + 10 mM HEPES, pH 7.4.
Cells were microinjected with Oregon Green G-actin (0.5 mg/ml) 4–7 h before imaging, by which time labeled actin was incorporated into the actin cytoskeleton. Cells were then imaged as above, except that culture medium was supplemented with 1.0 U oxyrase per ml (Oxyrase,) to reduce photodamage and photobleaching.
Swiss 3T3 cells stably expressing clathrin light-chain (CLC) EGFP were also analyzed for their sensitivities to drugs that disrupt the actin cytoskeletal dynamics. Similarly to the CLC-DsRed–expressing cells, we found that the CLC-EGFP dynamics were also dramatically halted when cells were treated with 5 μM latA or 1 μM jasp (unpublished data). Therefore, the effects that we see are not specific to the tag placed on the CLC.
Electron Microscopy
Cells were grown on plastic culture dishes coated with PLL overnight, treated with 5 μM latA, 1 μM jasp, or DMSO for 30 min at 37°C, and then fixed in 2.5% glutaraldehyde in 0.1 M Na cacodylate buffer (pH 7.3) for 30 min at room temperature. Samples were washed three times in 0.1 M cacodylate buffer and treated with 1% osmium tetroxide in 0.1 M cacodylate buffer for 60 min. They were then washed three times in 0.1 M cacodylate treated with 0.5% tannic acid in 0.05M cacodylate for 30 min, rinsed in 1% Na2SO4 in 0.1 M cacodylate buffer for 10 min, and then rinsed in 0.1 M cacodylate buffer. Samples were dehydrated in graded ethanols, exchanged into HPMA (2-hydroxypropyl methacrylate), embedded in LX-112 (Ladd, Burlington, VT), removed from the plastic dish, and sectioned (transversely). Sections were stained with uranyl acetate followed by Pb citrate and viewed with a Philips CM100 electron microscope (FEI, Hillsbrough, OR). For quantification of CCS morphology and surface density, cells were randomly selected and imaged at low magnification (×1900). The cell perimeter was then examined at high magnification (×18,000) to identify, classify, and count CCSs. Deeply invaginated coated pits were distinguished from curved coated pits by the inward, negative curvature at the neck of the deeply invaginated coated pits. There exists no clear morphological correlate to constricted coated pits, which are defined biochemically. The linear surface examined was measured by tracing the cell surface in the low magnification images using Metamorph software (Universal Imaging).
Image Analysis
To determine the degree of CCS internalization, CCSs were randomly identified, irrespective of size and shape, on the first frame of a 10-min time-lapse movie and analyzed to ascertain if the CCS or a portion of it was internalized. CCS internalization was detected as disappearance from TIR-FM before disappearance from WF-EFM. Internalization was also scored if a portion of a CCS separated and disappeared from TIR-FM before disappearing from WF-EFM. CCSs that disappeared simultaneously in both TIR-FM and WF-EFM were excluded from analysis. Forty to 150 CCSs were analyzed per cell. To determine the formation of CCSs, the number of new CCSs that appeared on a predefined region of the cell surface per 10-min interval was measured and presented as the fraction of CCSs that formed per unit area per 10 min relative to DMSO-treated controls. CCSs were classified as newly formed either if they appeared de novo (see Figure 1A) or if they separated from a preexisting CCS and remained on the cell surface for at least 20 s. Surface density of CCSs was determined by averaging the number of CCS in a defined region on the first frame and last frame of a time-lapse movie and expressed as the average number of CCSs/μm2 surface area.
Figure 1.

Assembled clathrin exhibits diverse behaviors. Images from time-lapse sequences showing the behavior of CLC-DsRed. TIR-FM (top panels) and WF-EFM (bottom panels). (A) Formation and internalization of CCV, Supplementary Video 1, (B) splitting and internalization of a CCV from a larger CCS, Supplementary Video 2, and (C) merging/coalescing of CCS with other CCS, Supplementary Video 3. Structures of interest are marked by arrows. Evanescent field depth ∼100 nm. Scale bar, 1 μm.
To measure the lateral mobility of CCSs, the movement of CCSs in the TIR-FM time-lapse series were manually tracked using the track points function in Metamorph software (Universal Imaging). The distance moved in consecutive frames by CCS centroids were recorded for up to 3 min. If the CCSs did not persist in the TIR-FM image for the duration of the 3 min, then they were tracked until they could no longer be detected by TIR-FM. CCSs persisting for <1 min were excluded from the analyses. To calculate the average path-length that CCS moved in a 20-s interval, the distances (μm) that the CCS traveled in consecutive 20 intervals were averaged. A total of 66–94 CCSs were analyzed from three to four cells per condition. The average rate of CCS motility was determined from CCS displacements lasting for >3 consecutive frames.
To determine colocalization of actin and clathrin, cells were fixed with 3.7% formaldehyde, stained with Alexa-488 phalloidin as previously described (Cramer et al., 2002), and mounted in phosphate-buffered saline and imaged by TIR-FM. TIR-FM images of actin and clathrin were pseudocolored and merged in Metamorph to identify by visual inspection CCSs that colocalized or partially colocalized with a local concentration of actin. The line scan function in Metamorph software was used to measure the average fluorescence intensity along a 3 pixel wide, 15 pixel long line drawn parallel to the longest clathrin, actin dimension. Data were collected from the individual actin and clathrin images. The signals were then normalized to the minimum signal along the line, and intensity was calculated as a percentage of the maximum signal along the line. CCSs and actin fluorescence were considered centered if their peak fluorescence intensities were <1 pixel (64 nm) or if their fluorescence profiles were fully overlapping. Otherwise, they were counted as spatially offset. Because the chromatic aberration of the microscope determined by measuring multispectral beads in different wavelengths was found to be less than one pixel, and because the overlap of the CLC-DsRed CCSs and the Oregon-Green–labeled actin or the Alexa-488 phalloidin were not found to be biased to one orientation (i.e., the green foci are not always to the right of the red foci, see Figures 2D and 3A), we did not correct for this aberration. Spatially offset structures were subclassified as mutually exclusive if the peaks of fluorescence intensity were 4 or more pixels (256 nm) apart as prescribed by the Rayleigh limit. Structures that did not have a clear peak were excluded from the analyses.
Figure 2.
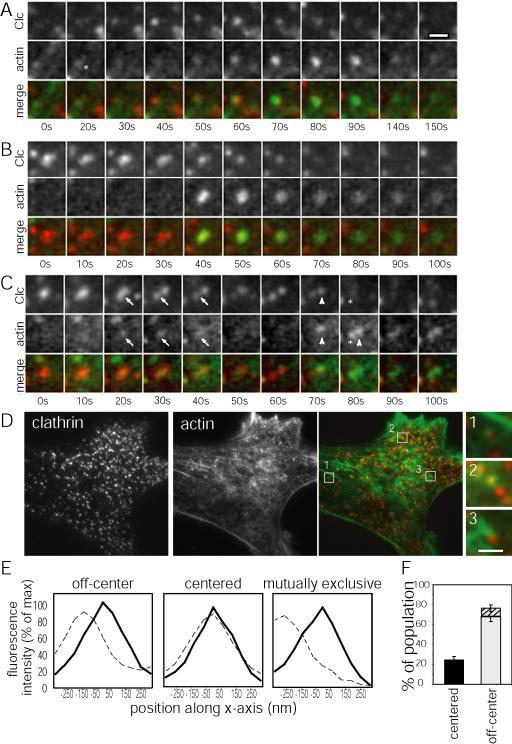
Actin and clathrin associate during endocytosis. Images from time-lapse sequences showing the behaviors of CLC-DsRed and Oregon Green actin. TIR-FM CLC-DsRed (upper panels), corresponding Oregon Green actin colocalization (middle panels), and merged images (lower panels). (A) Formation and internalization of a single CCS, Supplementary Video 4, (B) internalization of a large CCS, Supplementary Video 5, and (C) splitting and internalization of CCSs, Supplementary Video 6. Asterisk in A marks site of actin and clathrin colocalization early in CME. Actin is recruited to the site of separating CCSs (arrow in C) and then sequentially to the CCSs as they become internalized (first arrowhead, then asterisk in C). Scale bar, 1 μm. (D) TIR-FM images of fixed Swiss 3T3 CLC-DsRed cells stained with Alexa-488 phalloidin. CLC-DsRed (right), Alexa-488 phalloidin–labeled F-actin (center), merge (CLC is red, F-actin is green). Regions showing examples of (1) off-center, (2) centered, or (3) mutually exclusive CCSs are marked in the merge image and corresponding images are presented at the right. Scale bar, 5 μm. (E) Examples of average fluorescence intensity along line-scan of CLC-DsRed (solid) and F-actin (dashed) structures that are either off-center, centered, or mutually exclusive. x-axis shown in nm and centered around the peak of clathrin intensity. Measured from CCSs in fixed cells, stained with Alexa-488 phalloidin and imaged with TIR-FM. (F) Histogram showing the average percentage ± SE of the mean of CCSs and corresponding actin foci that are centered (black bar), off-center (gray bar), or off-center and mutually exclusive (hatched bar).
Figure 3.
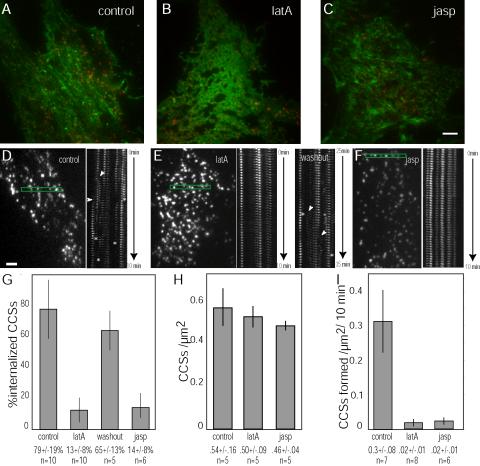
Clathrin dynamics are perturbed after disruption of actin assembly/disassembly dynamics. (A–C) Merged TIR-FM images of Oregon Green actin (green) and CLC-DsRed (red) in (A) control cells, and cells treated with (B) 5 μM latA or (C) 1 μM jasp. Scale bar is 5 μm. (D–F) Images and corresponding kymographs of rectangular regions shown in (D) control, (E) latA- and washout, and (F) jasp-treated cells. Scale bar, 2 μm and time course shown in kymograph is 10 min. Kymographs were generated by extracting the rectangular region highlighted in the micrograph of each image in the time-lapse, and assembling the regions series side-by-side in chronological order into a montage. Internalization events are marked by asterisks and formation of CCSs are marked by arrowheads. See Supplementary Video 7. (G–I) Histograms showing the effects of latA and jasp on (G) % of CCSs internalized during a 10-min internal, (H) the average surface density of CCSs, and (I) number of CCSs formed per μm2 during a 10-min interval. n = number of cells analyzed per condition. Data shown are average ± SE of the mean.
Biochemical Internalization Assays
Transferrin endocytosis assays were modified from (Smythe et al., 1992; Carter et al., 1993). Swiss 3T3-CLC-DsRed–expressing cells (n = 1.2 × 106) were plated onto PLL-coated, 12-mm circle coverslips (Fisher Scientific, Fairlawn, NJ) in 100-mm tissue culture dish (Nunc, Napierville, IL) and allowed to adhere overnight. Cells on coverslips were rinsed with SFM (DMEM + 0.2% bovine serum albumin + 10 mM HEPES, pH 7.5), transferred to four-well tissue culture dishes (Nunc; 2 dishes per time point), and incubated at 37°C with SFM containing either 5 μM latA, 1 μM jasp, or an equal concentration of DMSO as a control for 30 min. The time-course of internalization was initiated by adding SFM containing biotin-S-S-transferrin (BSST, 4 μg/ml) supplemented with drug or DMSO, and placing dishes on metal blocks in a 37°C water bath. Internalization was stopped by placing dishes on ice and washing wells three times with 0.5 ml SFM. Sequestration from avidin and cleavage of the biotin from the noninternalized transferrin by reduction of the disulfide bond was performed as previously described (Smythe et al., 1992; Carter et al., 1993), with the exception that Tris(2-carboxyethyl) phosphine hydrochloride (TCEP), a more potent and stable reducing agent, was used instead of MesNa. After the internalization of BSST, 15 mM TCEP (Pierce, Rockford, IL) in reduction buffer (50 mM HEPES, 1 mM MgCl2, 125 mM NaCl, 50 mM Tris, pH 8.0) was added to the wells and incubated at 4°C for 30 min. The reduction was quenched with iodoacetamide, and cells were solubilized in blocking buffer (1% TX-100, 0.1% SDS, 0.2% bovine serum albumin, 50 mM NaCl, 1 mM Tris, pH 7.4). Cell lysates were plated onto ELISA plates coated with anti-transferrin antibody and assayed for detectable BSST using HRP-labeled streptavidin as previously described (Smythe et al., 1992; Carter et al., 1993). Internalized BSST was expressed as the percent of total surface-bound at 4°C. The level of BSST internalization was normalized to the amount of internalization for DMSO-treated cells to account for day-to-day variation. All statistical analyses were performed using ANOVA analysis in Minitab software.
RESULTS
To study CME in real-time in living cells, we used a method pioneered by Merrifield et al. (2002) that uses nearly simultaneous total internal reflection fluorescence microscopy (TIR-FM) and wide-field epifluorescence microscopy (WF-EFM) to image CCSs in Swiss 3T3 fibroblasts stably expressing a clathrin light-chain DsRed (CLC-DsRed) fusion protein. TIR-FM enables the selective analysis of fluorescent proteins located only within a shallow evanescent excitation field (∼100 nm) that forms at the interface between the glass coverslip, upon which cells adhere, and the ventral cell cytosol (Axelrod, 2001; Steyer and Almers, 2001). This reduces the background fluorescence from deeper within the cytoplasm and improves the contrast of fluorophores at the plasma membrane. In contrast, conventional WF-EFM samples a much thicker (∼600 nm) optical section. By using near simultaneously acquired WF-EFM and TIR-FM images one can distinguish between CCV formation/internalization and CCS disassembly at the plasma membrane. Using this approach, we define CCSs as CLC-DsRed–labeled structures that exist in both WF-EFM and TIR-FM. CCV formation/internalization is signified by the selective loss of CLC-DsRed signal from the TIR-FM image, as the CCS moves out of the ∼100-nm evanescent field, followed by the subsequent loss from the WF-EFM image, as the vesicle moves further away from the plasma membrane, deeper into the cell.
Our goal was to investigate the contribution of F-actin cytoskeletal assembly and disassembly dynamics to CME directly using this microscopy-based assay. This has not previously been possible because of technical problems resulting from the fact that pharmacological disruption of the actin cytoskeleton results in detachment of cells from the substrate and prevents imaging by TIR-FM. To circumvent this problem, cells were adhered to PLL-coated coverslips. We confirmed their continued attachment after drug treatment by visualizing the plasma membrane in cells stained with the fluorescent lipophilic dye, DiOC6 (unpublished data). The frequency of CCS internalization in cells grown on PLL-coated coverslips was indistinguishable from those grown on uncoated coverslips analyzed in the microscopic assay (unpublished data), indicating that, under our conditions, the growth on PLL does not disrupt clathrin-mediated internalization.
F-actin Is Associated with Several Diverse Behaviors of CCSs
In Swiss 3T3 cells stably expressing CLC-DsRed and plated on PLL-coated coverslips, we imaged the formation of CCSs as they appeared simultaneously in TIR-FM and WF-EFM, and the internalization of CCV as they moved out of the ∼100-nm evanescent field before disappearing from the WF-EFM image (Figure 1A, Supplementary Video 1; Merrifield et al., 2002). The initial loss of the TIR-FM signal may reflect either deep invagination of the CCS or movement of the CCV after detachment away from the plasma membrane and into the cell, as previously discussed (Merrifield et al., 2002; Santini and Keen, 2002). We also observed heterogeneous behaviors for the CCSs, which ranged in size from 0.3 to 1 μm in diameter, including the splitting or separation of CCSs. CCSs that emerged from these splitting or separating CCSs either remained on the plasma membrane or were internalized as CCVs (Figure 1B, Supplementary Video 2; also see Rappoport et al., 2003; Keyel et al., 2004). We also detected the coalescence of multiple CCSs (Figure 1C, Supplementary Video 3; Keyel et al., 2004), as well as movement of CCSs at both slow (2.4 ± 0.5 μm/min, n = 16) and fast (12 ± 3 μm/min, n = 15) velocities (see also Rappoport and Simon, 2003; Rappoport et al., 2003; Keyel et al., 2004). Thus, assembled clathrin consists of a dynamic population that has numerous distinct behaviors.
In previously published studies, the recruitment of actin to CCSs was examined only in a selective subpopulation of coated structures that exhibited a stereotypical behavior (Merrifield et al., 2002). Specifically, these included individual CCSs that were smaller than 0.5 μm and did not merge with or split away from other CCSs. To extend this analysis, we microinjected cells with Oregon Green–labeled skeletal muscle actin, allowed it to incorporate into endogenous F-actin, and examined the spatial and temporal recruitment of actin to CCSs displaying a variety of heterogeneous behaviors. We first reexamined the recruitment of actin to individual, small (<0.5 μm) CCSs and observed, as previously reported (Merrifield et al., 2002), a transient increase in actin recruitment that occurred late in the maturation of the CCS as it moved out of the evanescent field (Figure 2A, Supplementary Video 4). However, we also observed other behaviors. For example, actin was recruited to larger clathrin coated clusters (Figure 2B, Supplementary Video 5) and to the cleft between separating CCSs as they moved apart and were endocytosed (Figure 2C, Supplementary Video 6). In rare instances actin associated with CCSs soon after their formation (Figure 2A). Finally, we observed that CCSs occasionally aligned along actin bundles at the ventral cell surface that likely correspond to stress fibers (unpublished data), as previously described (Bennett et al., 2001). Thus, actin and clathrin localization are spatially and temporally coordinated during a variety of heterogeneous CCS behaviors.
Interestingly, in living cells we observed that the relative distributions of CLC-DsRed structures and associated Oregon Green actin foci were not absolutely spatially correlated. To investigate and quantify the spatial relationship between clathrin and actin more carefully, we examined the relative localizations of CLC-DsRed–labeled structures and Alexa-488 phalloidin–stained F-actin in fixed and extracted cells (Figure 2D), because of lower background fluorescence in comparison to Oregon Green actin–injected cells (see Figure 3A). Although in some cases actin and clathrin signals physically overlapped and were centered (25 ± 4%, 29 of 117 CCSs from 4 cells, Figure 2, D and E, middle panel, and F, black bar), most of the CCSs associated with actin were spatially off-center from the F-actin (75 ± 4%, 88 of 117 CCSs from 4 cells, Figure 2, D and E, left panel, and F, gray bar). Of these, several were mutually exclusive from the F-actin (10% ± 4%, 10 of 88 CCSs, Figure 2, D and E, right panel, and F, hatched gray bar), with the peak of the actin fluorescence intensity falling more than 250 nm away from the peak of the CLC-DsRed intensity (see also Figure 2, A and C, merged images for examples in living cells). In living cells, overlap of the actin and clathrin signals was detected when CCSs were internalized (see Figure 2, A–C), as previously reported (Merrifield et al., 2002).
Disruption of Actin Dynamics Inhibits Internalization of CCSs
We next explored the contribution of the actin cytoskeleton to internalization and other dynamic behaviors of CCSs. CLC-DsRed dynamics were examined in cells treated with membrane permeable agents that inhibit actin assembly/disassembly dynamics, including latA, a drug that sequesters actin monomers, blocks actin polymerization, and promotes actin depolymerization (Coue et al., 1987), and jasp, a drug that binds actin filaments to stabilize them against depolymerization and promote polymerization (Bubb et al., 1994). To confirm that the drugs had the expected effects on F-actin, cells were microinjected with Oregon Green–labeled actin and treated with DMSO as a control, 5 μM latA or 1 μM jasp (Figure 3, A–C). As expected, latA-treated cells exhibited a visible disruption of the actin meshwork that appeared diffuse (Figure 3B), whereas actin remained organized in bundles in the jasp-treated cells (Figure 3C).
Kymograph analysis showed that disrupting actin assembly/disassembly dynamics with either of these drugs dramatically inhibited all CCS dynamic behaviors (Figure 3, E and F, Supplementary Video 7). Of these, we first quantified the effects on CCV internalization, which was scored when CCSs were observed to first disappear from the TIR-FM image and subsequently from the WF-EFM image. CCSs that disappeared simultaneously from both images were not counted. In control cells pretreated with DMSO, 79 ± 19% of the CCSs initially present at the onset of image acquisition were internalized within a 10-min time course (Figure 3, D and G, and white asterisks in kymographs). In contrast, levels of CCV formation in cells pretreated with either 5 μM latA (Figure 3, E and G) or 1 μM jasp (Figure 3, F and G) were significantly lower than in control cells (p < 0.001). Only 13 ± 8% or 14 ± 8% of the CCSs were internalized in 10 min in latA- and jasp-treated cells, respectively, corresponding to an 80% reduction in coated vesicle formation (Figure 3G). Importantly, the internalization defect in the presence of latA was reversible (p < 0.001) and recovered to 65 ± 13% within 15-20 min after washout of the drug (Figure 3, E and G), demonstrating that the reduction in CCV internalization in latA-treated cells was specific to drug treatment.
Disruption of Actin Dynamics Inhibits Formation of CCSs
Given our observations of actin association with CCSs and the dramatic cessation of CCS dynamics in latA- and jasptreated cells, we next examined when actin might function during CME. For example, if late stages of CME are preferentially inhibited as a result of the disruption of actin dynamics, we would expect an increase in the total number of coated structures present on the cell surface, as occurs during inhibition of endocytosis by a dominant negative dynamin mutant (Damke et al., 1994). In contrast to this hypothesis, we found that the density of CCSs on the surface of cells treated with latA or jasp was not significantly different from that of control cells (Figure 3H), suggesting that coated pit assembly and coated vesicle formation were equally affected by the disruption of actin assembly and disassembly dynamics. In control cells, CCS assembly occurred either de novo (85%) or by splitting from preexisting CCSs and remaining on the cell surface (15%). We quantified the effect of latA and jasp on the formation of new coated structures and found that CCS appearance was significantly reduced when actin dynamics were inhibited (p < 0.001). Although 0.3 ± 0.08 CCSs/μm2 were formed in control cells in a 10-min time course, only 0.02 ± 0.01 CCSs/μm2 were formed in both latA- and jasp-treated cells (Figure 3I, white arrows in kymographs), corresponding to an ∼90% reduction in CCS formation.
Actin Dynamics Are Required for Lateral Mobility of CCSs
Our analyses, and those of others (Merrifield et al., 2002), have revealed an intimate spatial and temporal relationship between actin and CCSs at multiple stages of endocytosis (Figure 2). Moreover, the kymographs in Figure 3, E and F, suggest that all aspects of CCS dynamics are halted after disruption of the actin cytoskeleton. Thus, we next quantified the effects of actin disruption on the lateral movements of CCSs moving parallel to the plane of the plasma membrane.
Although CCSs are not highly motile, they are not static (Gaidarov et al., 1999; Merrifield et al., 2002; Rappoport and Simon, 2003; Rappoport et al., 2003; Keyel et al., 2004). Instead, CCSs appear to fluctuate locally, and a subset transition between stationary and mobile periods. In a 20-s time interval, the average path-length moved by CCSs in control cells was 0.32 ± 0.05 μm and with an average velocity during movement of 2.4 ± 0.5 μm/min (Figure 4, A and C), consistent with previous reports (Figure 4A, Keyel et al., 2004; Rappoport et al., 2003). These movements are much slower than rates reported for microtubule-based kinesin or dynein driven motility (30–60 μm/min; Vale et al., 1985; Vallee et al., 1988) and are consistent with motilities mediated by actin polymerization (1.2–12 μm/min; Theriot et al., 1992; Loisel et al., 1999). Indeed, we have observed actin recruitment behind motile CCSs that is reminiscent of actin comet tails seen behind motile Listeria monocytogenes (Figure 4B, Supplementary Video 8). On disruption of the actin cytoskeleton, the average CCS path-length in cells treated with latA or jasp was dramatically reduced to 0.13 ± 0.04 or 0.08 ± 0.09 μm of displacement per 20 s, respectively, significantly lower than the average path lengths measured in control cells (Figure 4C, p < 0.001). Furthermore, as lateral motility was required for CCS splitting and coalescence, these behaviors were also specifically inhibited by disruption of actin assembly/disassembly dynamics.
Figure 4.

Lateral motility of CCSs requires a dynamic actin cytoskeleton. (A) TIR-FM images of CCSs and paths that structures followed in a 20-s interval in control, latA-, and jasp-treated cells. (B) Actin recruitment behind a motile CCS, indicated by arrow. See Supplementary Video 8. Scale bar, 1 μm. (C) Box plots showing the range of average path length that CCSs moved in 20-s intervals in control, latA-, and jasp-treated cells. Outliers shown as circles. Averages ± SE of the mean are shown below box plots. n = 63–93 CCS from three to four cells per condition.
Actin Dynamics Participate in the Formation of Constricted Coated Pits
Biochemical analyses of the role of actin during endocytosis in mammalian cells have yielded inconsistent and/or ambiguous results (Durrbach et al., 1996; Lamaze et al., 1997; Maples et al., 1997; Fujimoto et al., 2000; Lunn et al., 2000; Moskowitz et al., 2003). Previous studies have used both a variety of cell types as well as distinct assays to monitor the effect of disrupting actin dynamics on endocytosis, preventing a direct comparison between the different assays. In an effort to reconcile these observations, we biochemically measured the effect of disrupting actin dynamics on CME using the same conditions and cells that we used for microscopy (i.e., assay was performed on cells bound to PLL-coated coverslips, see Materials and Methods). We used a stage specific assay that quantitatively measures the internalization of transferrin, which is biotinylated through a cleavable disulfide bond (BSST), and relies on the principle that at different stages of clathrin-coated pit maturation, the BSST becomes protected from exogenously added agents of different sizes (Smythe et al., 1992; Carter et al., 1993; Figure 5A). Thus, when receptor-bound BSST is either sequestered in constricted coated pits or internalized into coated vesicles, it acquires inaccessibility to exogenously added avidin, a bulky molecule that will bind to and mask the BSST exposed on the cell surface. In contrast, BSST that is retained in constricted coated pits remains accessible to a small-molecular-weight, membrane-impermeant reducing agent, TCEP (Tris (2-carboxyethyl) phosphine hydrochloride), that can cleave the biotin and render the transferrin undetectable. Thus, BSST becomes inaccessible to TCEP only after it has been internalized into sealed coated vesicles. Cells treated with latA or jasp were 40–45% less efficient than control cells in their ability to sequester BSST from avidin (Figure 5B, black symbols) and exhibited a corresponding 20 ± 10% increase in the levels of cell surface–associated transferrin. These data indicate that disruption of actin assembly/disassembly dynamics inhibits uptake of transferrin into constricted coated pits and/or coated vesicles without an equivalent block of recycling pathways, consistent with previous results (Lamaze et al., 1997; Fujimoto et al., 2000).
Figure 5.
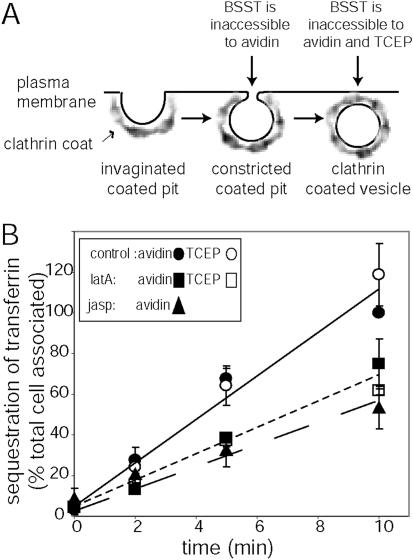
Transferrin internalization is partially inhibited at an intermediate stage of CCV formation after disruption of F-actin cytoskeletal dynamics. (A) Schematic describing the stage specific biochemical assay used. Biotinylated transferrin (BSST) is sequestered from avidin in constricted CCPs and from avidin and TCEP in sealed coated vesicles. (B) Time-course of transferrin sequestration from either avidin (black symbols) or TCEP (white symbols) in control (•, ○), latA-(▪, □), or jasp (▴)-treated cells. Data are expressed as the percent of the total cell surface associated BSST (average ± SD of the mean). Data from at least three independent experiments.
If actin assembly functions exclusively during late events leading to the scission of CCPs, as suggested by the late burst of actin recruitment visualized by microscopy, then we would predict that after disruption of actin dynamics, the detectable BSST might be trapped at the cell surface in constricted coated pits. This scenario would also be consistent with the inhibition of CCS internalization (i.e., departure from the cell surface) observed by TIR-FM. To determine if inhibition of actin assembly/disassembly dynamics results in the accumulation of constricted coated pits, we measured the sequestration of BSST from TCEP. Interestingly, inhibition of actin dynamics with latA had indistinguishable effects on the avidin and TCEP signals (Figure 5B, avidin, black symbols and TCEP, white symbols), indicating that in the absence of actin dynamics, constricted coated pits do not accumulate and the defect in BSST uptake derives from a defect in the formation of constricted coated pits and coated vesicles.
Deeply Invaginated CCSs Accumulate after Disruption of Actin Assembly/Disassembly Dynamics
To further investigate the stage at which actin assembly/disassembly dynamics contribute to CME, we quantified the effects of disruption of actin dynamics on the ultrastructural morphology of CCSs in CLC-DsRed–expressing Swiss 3T3 cells grown on PLL-coated dishes using EM. CCSs at all stages were found in control, latA-, or jasp-treated cells, including shallow CCSs, curved CCSs, deeply invaginated CCSs, and CCVs (see for example, Figure 6), as previously reported (Lamaze et al., 1997). However, in contrast to control cells, a significant fraction of the deeply invaginated CCS in latA- and jasp-treated cells formed clusters (33 and 15%, respectively; see Figure 6A), where multiple discrete areas of coats associated with a large invagination. Although the density of coated pits did not significantly differ (p = 0.1) between control (120 ± 30 CCSs/mm) and latA-(170 ± 40 CCSs/mm), or jasp-treated cells (120 ± 30 CCSs/mm), the abundance of CCS morphologies was different (Figure 6B). Disruption of actin assembly/disassembly dynamics resulted in a shift of CCSs from shallow CCSs to more deeply invaginated CCSs (Figure 6B). This suggests that inhibition of actin dynamics does not block CCS invagination, but blocks at a late stage of CME. Constricted coated pits, which are defined biochemically (see Figure 5), do not have an identified morphological correlate, and cannot be distinguished morphologically from deeply invaginated structures (Schmid and Carter, 1990; Sever et al., 2000). In addition, fewer CCVs were found in latA- and jasp-treated cells than in control cells, as expected. Those CCVs that were found were primarily located at the top, nonadherent side of the cells (78% of control CCVs were at the top, Figure 6B). After perturbation of actin dynamics, CCVs were only rarely detected at the bottom of the cells, suggesting that membrane at the top of the cells is more active in the formation of CCVs. These data suggest that invagination is not blocked after disruption of actin assembly/disassembly dynamics and is consistent with the interpretation that actin dynamics are important for a late stage of CME (i.e., constriction and possibly detachment).
Figure 6.

Clathrin-coated pit morphology is altered after disruption of actin cytoskeletal dynamics. (A) Electron micrographs of representative classifications of CCSs. Image containing clustered CCS (cl, electron-dense coats marked by asterisks) also contains curved CCS (cv). Scale bar, 250 nm. (B) Histogram showing the percentage of CCSs in each morphology classification (shown schematically below bars, white-shallow, light gray-curved, dark gray-deeply invaginated, black-vesicle). CCSs found to be located at top of cells (solid bar), CCSs located at bottom of cells (hatched bars). n = number of CCSs counted per condition.
DISCUSSION
We have taken parallel, quantitative approaches using fluorescence microscopy, EM, and biochemical methods to address the role of F-actin assembly/disassembly dynamics during CME in mammalian cells. Actin dynamics are critical for slow lateral movements and numerous distinct behaviors of CCSs. Importantly, we establish that actin polymerization/depolymerization dynamics are required for the vast majority of clathrin-mediated endocytic events. In addition, we have identified multiple actin-dependent stages of CME including new CCS formation, existing CCS constriction, and CCV internalization.
The Timing of the Contribution of Actin Dynamics during CME
Actin polymerization/depolymerization dynamics contribute to many events during the formation of a CCV (Figure 7). Disruption of actin assembly/disassembly dynamics inhibited the formation of new CCSs both as a consequence of reduced de novo assembly and reduced CCS splitting from preexisting CCSs. It has been proposed that the actin cytoskeleton functions as a scaffold for the recruitment of accessory proteins to the plasma membrane (Qualmann et al., 2000), thereby establishing “endocytic hot spots” that define sites for the formation of new coated pits (Gaidarov et al., 1999) or otherwise spatially restrict CCS assembly (Ehrlich et al., 2004). Thus, the inhibition of CCS formation we observed after disruption of actin cytoskeletal dynamics may reflect an inability to organize new endocytic hot spots. Alternatively, the effect may be indirect; by disrupting internalization and/or the lateral mobility of existing CCSs, new sites for coat assembly on the plasma membrane might be masked.
Figure 7.

Model showing the requirement for F-actin dynamics in CME. F-actin assembly/disassembly dynamics are required for (1) CCS assembly, (2) CCS motility, (3) CCS constriction, and possibly (4) scission to form a CCV and (5) translocation of the nascent vesicle away from the plasma membrane. CCS invagination proceeds in the absence of actin dynamics. Actin assembly may proceed from the edge of the CCS and assemble inwards, facilitating CCS constriction, scission, and translocation.
Later events in CCV formation also required actin assembly/disassembly dynamics (Figure 7). Fluorescence microscopic assays revealed a dramatic inhibition of CCS internalization in latA- and jasp-treated cells, whereas stage-specific biochemical assays showed that actin disruption blocked the formation of constricted CCPs, consistent with earlier findings (Lamaze et al., 1997). Electron microscopic analyses showing an accumulation of deeply invaginated CCPs after inhibition of actin dynamics are also consistent with fluorescence microscopic and biochemical results. A role for actin assembly/disassembly dynamics in endocytic CCV formation is consistent with results from the budding yeast S. cerevisiae, where disruption of actin dynamics also inhibits endocytosis (Ayscough, 2000; Engqvist-Goldstein and Drubin, 2003; Kaksonen et al., 2003), suggesting a conserved role for actin cytoskeletal dynamics in endocytosis. Our data establish a direct role for actin dynamics in CCS constriction. However, because blocking earlier events in vesicle formation would prevent the accumulation of downstream structures, we cannot exclude the possibility that actin polymerization could also contribute to membrane scission or facilitate the propulsion of nascent vesicles away from the plasma membrane (Figure 7). In fact, actin tails have been seen associated with endocytically derived vesicles (Ma et al., 1998; Merrifield et al., 1999; Rozelle et al., 2000; Taunton et al., 2000; Frischknecht and Way, 2001). Interestingly, a study on the forces exerted by actin polymerization on artificial lipid vesicles moving through cytoplasmic extract showed that the greatest forces on the vesicle were compressive forces, orthogonal to the direction of motility (Giardini et al., 2003; Merrifield, 2004). Such actin generated forces may be exerted at CCSs to promote constriction, fission, and/or translocation (Figure 7).
Coordination of Actin Dynamics at CCSs
The recruitment of F-actin to CCSs was highly coordinated with CME, as was previously observed (Merrifield et al., 2002). Although in rare instances this association occurs early in the formation of the CCS, in the majority of events, actin begins to accumulate just before initiation of the loss of TIR-FM CLC-DsRed signal, indicating CCS invagination or translocation away from the plasma membrane during or after fission (Merrifield et al., 2002; Santini and Keen, 2002). However, because the fraction of deeply invaginated CCSs was increased after the inhibition of actin dynamics, it is more likely that actin assembly/disassembly dynamics participate in a step after invagination. The observed recruitment of actin to CCSs may reflect the accumulation of preformed actin filaments, stabilization of monomeric actin, or localized actin polymerization. We favor the latter because N-WASP and the Arp2/3 complex, molecules that stimulate actin polymerization, have also been shown to transiently localize with CCSs (Merrifield et al., 2004). In addition, actin polymerization has been detected at sites of endocytosis in budding yeast and in mammalian cells under perturbed conditions (Kaksonen et al., 2003; Engqvist-Goldstein et al., 2004).
We found that peaks of actin and clathrin fluorescence were spatially offset and sometimes mutually exclusive. On the basis of these observations, we propose that during CCS assembly, actin and clathrin do not occupy the same space on the cell cortex. This is consistent with EM analyses showing the mutually exclusive distributions of actin and clathrin (Fujimoto et al., 2000; Engqvist-Goldstein et al., 2001). We speculate that the spatially offset actin and clathrin reflects the path of actin assembly at the CCS. Actin may be nucleated at the edge of the CCS and subsequently be assembled and swept inward toward the center of the CCS as the invaginating CCS is displaced from the plasma membrane (Figure 7).
Actin Dynamics and the Early Endocytic Pathway
Disrupting actin dynamics caused an 80% inhibition in the internalization of CCVs measured microscopically. Interestingly, the percentage of CCSs that were internalized in cells where actin assembly/disassembly was disrupted (13%) corresponds strongly with the percentage of CCSs previously shown to be internalized without a corresponding recruitment of actin (20%; Merrifield et al., 2002). In contrast, when measured biochemically, BSST internalization was only inhibited by 40–45%. What might account for these quantitative differences? If we assume that 10–20% of the transferrin endocytosis measured biochemically occurs through a bulk internalization pathway independent of clathrin and dynamin (Conner and Schmid, 2003), 35–50% of the transferrin internalization measured biochemically after the disruption of actin dynamics may still occur through a clathrin-mediated pathway. This is still a modestly larger fraction than the ∼20% residual internalization of CCSs measured in the microscopy-based assay and leaves ∼15–30% of biochemically internalized transferrin unaccounted for. There are several possible explanations for this discrepancy that are not mutually exclusive. One possibility is that the residual transferrin uptake may result from differences in the actin requirements for internalization at the adherent bottom of the cell that is analyzed exclusively using TIR-FM compared with those at the nonadherent top of the cell, additionally sampled biochemically. Remarkably, we found that the majority of CCVs detected by EM were located at the top of the cell. Alternatively, transferrin receptors might be more highly concentrated in the fraction of CCVs that are able to detach in the latA- or jasp-treated cells than those in control cells. Consistent with this hypothesis, we and others have found that the levels of surface-associated transferrin receptor increases after the inhibition of actin assembly/disassembly dynamics (see also Lamaze et al., 1997; Fujimoto et al., 2000; Moskowitz et al., 2003). Moreover, it has been reported that the fraction of transferrin receptors found in CCPs in cells with disrupted actin dynamics was the same as in CCPs in control cells, suggesting that the number of receptors per coated pit is higher (Lamaze et al., 1997). Finally, it is possible that an alternative clathrin- and actin-independent pathway of endocytosis may be used in the internalization of transferrin or that a compensatory clathrin- and actin-independent pathway for transferrin uptake is induced upon drug treatment. In such a case, although CCVs do not form, CCSs may still function as sites of receptor concentration. Also consistent with the existence of independent clathrin- and actin-mediated pathways is the observation that disruption of clathrin and actin dynamics have additive effects on the reduction in transferrin endocytosis (Moskowitz et al., 2003).
We have begun to explore these possibilities by imaging transferrin receptor-EGFP (TfnR-GFP) dynamics using TIR-FM. In preliminary studies, we find that TfnR-GFP is concentrated in CCSs in both control and latA-treated cells (see also Lamaze et al., 1997), and we can detect the disappearance of TfnR-GFP fluorescence independent of a loss of CLC-DsRed fluorescence in latA-treated cells (unpublished data). However, insufficient contrast obtained in WF-EFM images of TfnR-GFP precludes our ability to unambiguously measure internalization events. Thus, although this suggests that alternative, clathrin-independent pathways of transferrin internalization may be used after disruption of actin assembly/disassembly dynamics, new assays for the unambiguous detection of internalization of fluorescently labeled cargo molecules will be needed to rule out other possible interpretations.
CCS Lateral Motility and Actin Dynamics
Actin assembly/disassembly dynamics are required for the lateral mobility of CCSs on the plasma membrane. Actin comet tails, like those observed behind motile bacteria (Pantaloni et al., 2001) or vesicles (Ma et al., 1998; Merrifield et al., 1999; Rozelle et al., 2000; Taunton et al., 2000; Frischknecht and Way, 2001) could be detected on motile CCSs, and disruption of actin dynamics blocked CCS lateral movements, including coalescence into larger CCSs and splitting into smaller CCSs. The large CLC-DsRed–labeled structures could represent clusters containing multiple closely packed CCSs not resolvable by light microscopy, or complex structures of clathrin lattices interspersed with invaginated CCSs, both of which have been observed by EM on the ventral surfaces of substrate-attached cells (Heuser and Anderson, 1989; Fujimoto et al., 2000; Rappoport et al., 2004). Although it is not clear how these lateral movements contribute to endocytosis, such motility may be involved in the scanning of the cell surface for cargo or in the communication between signaling receptors that reside in different CCSs. Indeed, others have shown that agonist-induced clustering of coated pits was inhibited by the disruption of actin assembly/disassembly dynamics (Santini et al., 2002). In addition, the lateral mobility of endocytic components in budding yeast is abrogated after disruption of actin dynamics (Kaksonen et al., 2003). The molecular composition of these laterally mobile CCSs, in particular their content of cargo adaptor molecules, may be distinct from CCSs undergoing internalization (Rappoport et al., 2003; Keyel et al., 2004).
It was previously reported that disruption of actin dynamics has a stimulatory effect on the average range of CCS lateral movement (Gaidarov et al., 1999). In contrast, we find that disruption of actin dynamics blocks lateral movement. This difference may reflect the different experimental procedures used, specifically, the adhesion of cells to the coverslip using PLL and imaging of CCS dynamics using TIR-FM. We are only able to observe the previously reported increase in the trajectory of CCSs upon latA treatment when cells are not grown on PLL and imaged using WF-EFM (unpublished data). Therefore, we suggest that the increase in trajectory may be either a cell-specific difference in the sensitivity of CCSs to disruption of the actin cytoskeletal dynamics or a consequence of increased movement of the plasma membrane after disruption of actin dynamics.
In summary, our results clearly demonstrate that dynamic actin assembly and disassembly is required for multiple stages of clathrin-mediated endocytosis in mammalian cells including the formation of new CCSs, CCS constriction, CCV internalization, and the lateral movement of CCSs.
Supplementary Material
Acknowledgments
We thank Wolf Almers (Vollum Institute, Oregon Health and Sciences University) for Swiss 3T3 stably expressing CLC-DsRed, Malcolm Wood (TSRI EM core facility) for help with EM and preparing the sections for EM, Mike Adams and Bill Shin from the Waterman lab for technical help with microscopy, and Iain Cheeseman and members of the Schmid lab for comments on the manuscript. This work was supported by National Institutes of Health (NIH) grants MH61345 to S.L.S. and GM67230 to C.W.S. D.Y. was supported by NIH training grant 5T32HL07695 and ACS Fellowship PF-04-105-01-CSM. This is The Scripps Research Institute Manuscript No. 16897-CB
Article published online ahead of print in MBC in Press on December 15, 2004 (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-09-0774).
Abbreviations used: CME, clathrin-mediated endocytosis; CCP, clathrin-coated pit; CCS, clathrin-coated structure; CCV, clathrincoated vesicle; TIR-FM, total internal reflection fluorescence microscopy; WF-EFM, wide-field epi-fluorescence microscopy; CLC-DsRed, clathrin light-chain DsRed; Pll, poly-l-lysine; latA, latrunculin A; jasp, jasplakinolide.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Ayscough, K. R. (2000). Endocytosis and the development of cell polarity in yeast require a dynamic F-actin cytoskeleton. Curr. Biol. 10, 1587-1590. [DOI] [PubMed] [Google Scholar]
- Axelrod, D. (2001). Total internal reflection fluorescence microscopy in cell biology. Traffic 2, 764-774. [DOI] [PubMed] [Google Scholar]
- Bennett, E. M., Chen, C. Y., Engqvist-Goldstein, A. E., Drubin, D. G., and Brodsky, F. M. (2001). Clathrin hub expression dissociates the actin-binding protein Hip1R from coated pits and disrupts their alignment with the actin cytoskeleton. Traffic 2, 851-858. [DOI] [PubMed] [Google Scholar]
- Bubb, M. R., Senderowicz, A. M., Sausville, E. A., Duncan, K. L., and Korn, E. D. (1994). Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 269, 14869-14871. [PubMed] [Google Scholar]
- Carter, L. L., Redelmeier, T. E., Woollenweber, L. A., and Schmid, S. L. (1993). Multiple GTP-binding proteins participate in clathrin-coated vesicle-mediated endocytosis. J. Cell Biol. 120, 37-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner, S. D., and Schmid, S. L. (2003). Regulated portals of entry into the cell. Nature 422, 37-44. [DOI] [PubMed] [Google Scholar]
- Coue, M., Brenner, S. L., Spector, I., and Korn, E. D. (1987). Inhibition of actin polymerization by latrunculin A. FEBS Lett. 213, 316-318. [DOI] [PubMed] [Google Scholar]
- Cramer, L. P., Briggs, L. J., and Dawe, H. R. (2002). Use of fluorescently labelled deoxyribonuclease I to spatially measure G-actin levels in migrating and non-migrating cells. Cell Motil. Cytoskelet. 51, 27-38. [DOI] [PubMed] [Google Scholar]
- Damke, H., Baba, T., Warnock, D. E., and Schmid, S. L. (1994). Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 127, 915-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrbach, A., Louvard, D., and Coudrier, E. (1996). Actin filaments facilitate two steps of endocytosis. J. Cell Sci. 109(Pt 2), 457-465. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein, A. E., Zhang, C. X., Carreno, S., Barroso, C., Heuser, J. E., and Drubin, D. G. (2004). RNAi-mediated Hip1R silencing results in stable association between the endocytic machinery and the actin assembly machinery. Mol. Biol. Cell 15, 1666-1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engqvist-Goldstein, A. E., and Drubin, D. G. (2003). Actin assembly and endocytosis: from yeast to mammals. Annu. Rev. Cell Dev. Biol. 19, 287-332. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein, A. E., Warren, R. A., Kessels, M. M., Keen, J. H., Heuser, J., and Drubin, D. G. (2001). The actin-binding protein Hip1R associates with clathrin during early stages of endocytosis and promotes clathrin assembly in vitro. J. Cell Biol. 154, 1209-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich, M., Boll, W., Van Oijen, A., Hariharan, R., Chandran, K., Nibert, M. L., and Kirchhausen, T. (2004). Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell 118, 591-605. [DOI] [PubMed] [Google Scholar]
- Frischknecht, F., and Way, M. (2001). Surfing pathogens and the lessons learned for actin polymerization. Trends Cell Biol. 11, 30-38. [DOI] [PubMed] [Google Scholar]
- Fujimoto, L. M., Roth, R., Heuser, J. E., and Schmid, S. L. (2000). Actin assembly plays a variable, but not obligatory role in receptor-mediated endocytosis in mammalian cells. Traffic 1, 161-171. [DOI] [PubMed] [Google Scholar]
- Gaidarov, I., Santini, F., Warren, R. A., and Keen, J. H. (1999). Spatial control of coated-pit dynamics in living cells. Nat. Cell Biol. 1, 1-7. [DOI] [PubMed] [Google Scholar]
- Giardini, P. A., Fletcher, D. A., and Theriot, J. A. (2003). Compression forces generated by actin comet tails on lipid vesicles. Proc. Natl. Acad. Sci. USA 100, 6493-6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton, S. L., and Waterman-Storer, C. M. (2005). Live-cell fluorescent speckle microscopy (FSM) of actin cytoskeletal dynamics and their perturbation by drug perfusion. In: Cell Biology: A Laboratory Handbook, 3rd ed., ed J. Celis and J. V. Small, New York: Elsevier (in press).
- Heuser, J. E., and Anderson, R. G. (1989). Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J. Cell Biol. 108, 389-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen, M., Sun, Y., Drubin, D. G. (2003). A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell 115, 475-487. [DOI] [PubMed] [Google Scholar]
- Keyel, P. A., Watkins, S. C., and Traub, L. M. (2004). Endocytic adaptor molecules reveal an endosomal population of clathrin by total internal reflection fluorescence microscopy. J. Biol. Chem. 279, 13190-13204. [DOI] [PubMed] [Google Scholar]
- Lamaze, C., Fujimoto, L. M., Yin, H. L., and Schmid, S. L. (1997). The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J. Biol. Chem. 272, 20332-20335. [DOI] [PubMed] [Google Scholar]
- Loisel, T. P., Boujemaa, R., Pantaloni, D., and Carlier, M. F. (1999). Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature 401, 613-616. [DOI] [PubMed] [Google Scholar]
- Lunn, J. A., Wong, H., Rozengurt, E., and Walsh, J. H. (2000). Requirement of cortical actin organization for bombesin, endothelin, and epidermal growth factor receptor internalization. Am. J. Physiol. Cell Physiol. 279, C2019-C2027. [DOI] [PubMed] [Google Scholar]
- Ma, L., Cantley, L. C., Janmey, P. A., and Kirschner, M. W. (1998). Corequirement of specific phosphoinositides and small GTP-binding protein Cdc42 in inducing actin assembly in Xenopus egg extracts. J. Cell Biol. 140, 1125-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maples, C. J., Ruiz, W. G., and Apodaca, G. (1997). Both microtubules and actin filaments are required for efficient postendocytotic traffic of the polymeric immunoglobulin receptor in polarized Madin-Darby canine kidney cells. J. Biol. Chem. 272, 6741-6751. [DOI] [PubMed] [Google Scholar]
- Merrifield, C. J. (2004). Seeing is believing: imaging actin dynamics at single sites of endocytosis. Trends Cell Biol. 14, 352-358. [DOI] [PubMed] [Google Scholar]
- Merrifield, C. J., Feldman, M. E., Wan, L., and Almers, W. (2002). Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol. 4, 691-698. [DOI] [PubMed] [Google Scholar]
- Merrifield, C. J., Moss, S. E., Ballestrem, C., Imhof, B. A., Giese, G., Wunderlich, I., and Almers, W. (1999). Endocytic vesicles move at the tips of actin tails in cultured mast cells. Nat. Cell Biol. 1, 72-74. [DOI] [PubMed] [Google Scholar]
- Merrifield, C. J., Qualmann, B., Kessels, M. M., and Almers, W. (2004). Neural Wiskott Aldrich Syndrome Protein (N-WASP) and the Arp2/3 complex are recruited to sites of clathrin-mediated endocytosis in cultured fibroblasts. Eur. J. Cell Biol. 83, 13-18. [DOI] [PubMed] [Google Scholar]
- Moskowitz, H. S., Heuser, J., McGraw, T. E., and Ryan, T. A. (2003). Targeted chemical disruption of clathrin function in living cells. Mol. Biol. Cell 14, 4437-4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni, D., Le Clainche, C., and Carlier, M. F. (2001). Mechanism of actin-based motility. Science 292, 1502-1506. [DOI] [PubMed] [Google Scholar]
- Qualmann, B., and Kessels, M. M. (2002). Endocytosis and the cytoskeleton. Int. Rev. Cytol. 220, 93-144. [DOI] [PubMed] [Google Scholar]
- Qualmann, B., Kessels, M. M., and Kelly, R. B. (2000). Molecular links between endocytosis and the actin cytoskeleton. J. Cell Biol. 150, F111-F116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappoport, J., Simon, S., and Benmerah, A. (2004). Understanding living clathrin-coated pits. Traffic 5, 327-337. [DOI] [PubMed] [Google Scholar]
- Rappoport, J. Z., and Simon, S. M. (2003). Real-time analysis of clathrin-mediated endocytosis during cell migration. J. Cell Sci. 116, 847-855. [DOI] [PubMed] [Google Scholar]
- Rappoport, J. Z., Taha, B. W., Lemeer, S., Benmerah, A., and Simon, S. M. (2003). The AP-2 complex is excluded from the dynamic population of plasma membrane-associated clathrin. J. Biol. Chem. 278, 47357-47360. [DOI] [PubMed] [Google Scholar]
- Rozelle, A. L., Machesky, L. M., Yamamoto, M., Driessens, M. H., Insall, R. H., Roth, M. G., Luby-Phelps, K., Marriott, G., Hall, A., and Yin, H. L. (2000). Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr. Biol. 10, 311-320. [DOI] [PubMed] [Google Scholar]
- Santini, F., Gaidarov, I., and Keen, J. H. (2002). G protein-coupled receptor/arrestin3 modulation of the endocytic machinery. J. Cell Biol. 156, 665-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini, F., and Keen, J. H. (2002). A glimpse of coated vesicle creation? Well almost! Nat. Cell Biol. 4, E230-E232. [DOI] [PubMed] [Google Scholar]
- Schmid, S. L., and Carter, L. (1990). ATP is required for receptor-mediated endocytosis in intact cells. J. Cell Biol. 111, 2307-2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever, S., Damke, H., and Schmid, S. L. (2000). Dynamin:GTP control the formation of constricted coated pits, the rate limiting step in clathrin-mediated endocytosis. J. Cell Biol. 150, 1137-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepnev, V. I., and De Camilli, P. (2000). Accessory factors in clathrin-dependent synaptic vesicle endocytosis. Nat. Rev. Neurosci. 1, 161-172. [DOI] [PubMed] [Google Scholar]
- Smythe, E., Carter, L. L., and Schmid, S. L. (1992). Cytosol- and clathrin-dependent stimulation of endocytosis in vitro by purified adaptors. J. Cell Biol. 119, 1163-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyer, J. A., and Almers, W. (2001). A real-time view of life within 100 nm of the plasma membrane. Nat. Rev. Mol. Cell. Biol. 2, 268-275. [DOI] [PubMed] [Google Scholar]
- Taunton, J., Rowning, B. A., Coughlin, M. L., Wu, M., Moon, R. T., Mitchison, T. J., and Larabell, C. A. (2000). Actin-dependent propulsion of endosomes and lysosomes by recruitment of N-WASP. J. Cell Biol. 148, 519-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot, J. A., Mitchison, T. J., Tilney, L. G., and Portnoy, D. A. (1992). The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature 357, 257-260. [DOI] [PubMed] [Google Scholar]
- Vale, R. D., Reese, T. S., and Sheetz, M. P. (1985). Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42, 39-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee, R. B., Wall, J. S., Paschal, B. M., and Shpetner, H. S. (1988). Microtubule-associated protein 1C from brain is a two-headed cytosolic dynein. Nature 332, 561-563. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer, C. M. (2002). Fluorescent speckle microscopy (FSM) of microtubules and actin in living cells. In: Current Protocols in Cell Biology, ed. J. S. Bonifacino, M. Dasso, J. B. Harford, J. Lippincott-Schwartz, and K. M. Yamada, New York: John Wiley, Unit 4.10. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.