

Abstract
Aim
Hereditary ataxias are characterized by a slowly progressive loss of gait, hand, speech, and eye coordination and cerebellar atrophy. A subset of these, including hypogonadism, are inherited as autosomal recessive traits involving coding mutations of genes involved in ubiquitination including RNF216, OTUD4, and STUB1. Cerebellar CHIPopathy (MIM 615768) is a form of autosomal recessive spinocerebellar ataxia (SCAR16) and when accompanied with hypogonadism, clinically resembles the Gordon Holmes Syndrome (GHS). A causal missense mutation in the gene that encodes the carboxy terminus of HSP-70 interacting protein (CHIP) protein was reported for the first time in 2014. CHIP−/− mice were found to phenocopy the motor deficiencies and some aspects of the hypogonadism observed in patients with STUB1 mutations. However, mechanisms responsible for these deficits are not known.
Methods
In a survey of skeletal muscle by transmission electron microscopy,
Results
CHIP−/− mice at 6 months of age were found to have morphological changes consistent with increased sarcoplasmic reticulum compartments in quadriceps muscle and gastrocnemius (toxic oligomers and tubular aggregates), but not in soleus.
Conclusion
Since CHIP has been implicated in ER stress in non-muscle cells, these findings illustrate potential parallel roles of CHIP in the muscle sarcoplasmic reticulum, a hypothesis that may be clinically relevant in a variety of common muscular and cardiac diseases.
Keywords: STUB1, muscle, ER stress
INTRODUCTION
Cerebellar CHIPopathy (MIM 615768) is a form of autosomal recessive spinocerebellar ataxia (SCAR16) that when accompanied with hypogonadism clinically resembles the Gordon Holmes Syndrome (GHS) (Holmes, 1908). This new entity was described for the first time in 2014 as a result of exome sequencing STUB1 in two patients with ataxia resembling GHS (Shi, et al., 2014). A specific mutation in the gene that encodes the carboxy terminus of HSP-70 interacting protein (CHIP) protein was found in STUB1 (c.737C>T, p.T246M). We found that the loss of CHIP function in mice (CHIP−/−) phenocopies the motor deficiencies and some aspects of the hypogonadism observed in patients with STUB1 mutations. Additionally, mice haploinsufficient for CHIP (CHIP+/−) exhibited significant physical deficits in wire hang, inverted screen, wire maneuver, and open field tasks, indicating CHIP-dependent behavioral deficits in brain and muscle (McLaughlin et al., 2012). Numerous other clinical studies identifying STUB1 mutations in subjects with cerebellar ataxia further confirmed our initial identification of a new disease (Bettencourt et al., 2015; Casarejos et al., 2014; Cordoba et al., 2014; Depondt et al., 2014; Heimdal et al., 2014; Ronnebaum et al., 2014; Shi et al., 2013; Synofzik et al., 2014). Neuroelectrophysiological examination of the two sisters affected by the CHIP T246M mutation identified a decreased amplitude motor-evoked potential in the bilateral lower limbs (Shi et al., 2014), despite normal muscle tone, power, and deep tendon reflexes of the four limbs (Shi et al., 2014). This led us to investigate the CHIP−/− musculature to identify underlying mechanisms that might be involved. The rationale for this was based on the fact that CHIP’s ubiquitin ligase activity was implicated in protein quality control and turnover of multiple proteins found ubiquitously in cells throughout the body, including skeletal muscle and sarcopenia (Altun et al., 2010). In Caenorhabditis elegans, a deletion mutant of the CHIP homolog CHN-1 in the musculature prevented motility issues in animals with mutations in dystrophin and MyoD, modeling Duchenne Muscular Dystrophy, indicating a role for CHN-1 in muscle wasting and degeneration (Nyamsuren et al., 2007). In the present study, we identify for the first time ultrastructural changes in representative CHIP−/− skeletal muscle at 6 months of age that may be related to the previously described phenotypic presentation.
MATERIALS AND METHODS
Animals
The CHIP−/− (and sibling CHIP+/+) mice were maintained on a mixed genetic background of C57BL/6 and 129SvEv and harvested at 6 months of age (Dai et al., 2003; Min et al., 2008; Willis et al., 2013). All animal husbandry and experiments were approved by the institutional care and use committee (IACUC) for animal research at the University of North Carolina at Chapel Hill.
Transmission electron microscopy
Tissues were perfused and fixed with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.15 mol/L sodium phosphate buffer (pH 7.4) and postfixed with 1% osmium tetroxide/0.15 mol/L sodium phosphate buffer. Samples were dehydrated with increasing concentrations of ethanol, infiltrated and embedded in Polybed 812 epoxy resin (Polysciences, Warrington, PA), and 70-nm ultrathin sections were cut with a diamond knife. Sections were mounted on 200-mesh copper grids and staining with 4% aqueous uranyl acetate and Reynolds lead citrate. Sections were observed with a LEO EM910 transmission electron microscope operating at 80 kV (LEO Electron Microscopy, Thornwood, NY) and photographed with a Gatan-Orius SC1000 CCD Digital Camera and Digital Micrograph 3.11.0 (Gatan, Pleasanton, CA).
ATPase/NADH-TR/Cytochrome C oxidase (COX) staining of fresh frozen muscle sections
Fresh frozen skeletal muscle was flash frozen, stored at −80°C, then cut cross-sectionally and stained for ATPase activity, NADH tetrazolium reductase (NADH-TR), and Cytochrome C oxidase using standard methods (Pearse, 1972;Pestronk et al., 1992; Sheehan et al., 1987).
Slide scanning and image capture
Imaging of special stained slides were obtained using Aperio Scanscope and exportd using Aperio Imagescope software (version 10.0.36.1805, Aperio Technologies, Inc., Vista, CA).
RESULTS
We surveyed soleus, quadriceps, and gastrocnemius muscle in CHIP−/− and sibling matched wild-type mice using transmission electron microscopy (TEM) for ultrastructural alterations. Compared to soleus muscle from age-matched wild-type siblings (Fig. 1A), CHIP−/− soleus muscle did not have any notable alterations in sarcomere ultrastructure (Fig. 1B). Additionally, interfibrillar mitochondria were intact, arranged in pairs (Fig. 1B), while subsarcolemmal mitochondria were intact (Fig. 1C top panels) and only rarely morphologically altered) in CHIP−/− mice (Fig. 1C lower panel, indicated by arrows). Similarly, TEM analysis of quadriceps muscle from wild-type (Fig. 2A) or CHIP−/− (Fig. 2B) found intact sarcomere morphology with no obvious defects. While the paired interfibrillar mitochondria could be found in quadriceps from both wild-type mice (Fig. 2A) and CHIP−/− mice (Fig. 2B), we observed less subsarcolemmal mitochondria in CHIP−/− quadriceps (Fig. 2C) compared to wildtype controls. Throughout the CHIP−/− quadriceps muscle, accumulation of lamellar bodies structures with the characteristic whirling pattern covering multiple sarcomere lengths (up to 15 sarcomeres) were observed (Fig. 2C, indicated by *). Analysis of gastrocnemius muscle from wild-type mice and CHIP−/− mice revealed similar intact sarcomere ultrastructure without morphological defects (Fig. 3A and 3B, respectively) and intact paired interfibrillar mitochondria (indicated by arrows in lower panels of Fig. 3A and 3B). Again in the gastrocnemius muscle from CHIP−/− mice we observed widespread multi-sarcomere accumulation of lamellar bodies (up to 15 sarcomeres long) within the expanded areas between myofibers throughout the muscle (Fig. 3C, indicated by *). These expanded areas demonstrated prominent tubular aggregates with single-walled longitudinal sections primarily in the sections observed and cut to focus on the sarcomere structure. Skeletal muscle fiber types differ in many ways, including the number of mitochondria and metabolic capacities they have. Since our initial findings indicated that the mitochondrial compartments in CHIP−/− muscle was affected and the fact that skeletal muscle lamellar bodies (generally aggregated protein) can result from damaged mitochondria we next investigated CHIP−/− muscle fiber type in the gastrocnemius where we observed these differences in mitochondria. The mouse gastrocnemius is a mixed fiber type muscle containing both slow twitch (Type I) and fast twitch (Type II, primarily IIb) fibers. We measured fiber type composition by first staining for ATPase content, found to be higher in glycolytic (Type II) fibers. As expected, wild-type gastrocnemius muscle had a checkerboard pattern (Fig. 4A, left) with some fibers having low ATPase content (Type I) ranging to fibers with very high ATPase content (Type IIb). Conversely we stained for oxidative enzymes that are more abundant in more oxidative (Type I) fibers. The staining for cytochrome C oxidase (COX), a mitochondrial enzyme, showed a similar distribution of dark COX positive fibers and fibers with little to no COX staining in wild-type gastrocnemius muscle (Fig. 2B, left). A similar checkerboard pattern was observed in wild-type gastrocnemius muscle when we stained for oxidative enzyme NADH-tetrazolium reductase (NADH-TR), which detects a reductase present both in the mitochondria and SR, again identifying the oxidative (dark stained) and non-oxidative (light stained) fibers seen in gastrocnemius wild-type muscle (Fig. 4C, left). The staining pattern of these markers is markedly different in CHIP−/− gastrocnemius muscle. Interestingly, in gastrocnemius muscle from CHIP−/− mice, the majority of fibers had high levels of diffuse staining for ATPase (Fig. 4A, right) as well as higher levels of diffuse COX content. Strikingly, staining for NADH-TR revealed dark uniform staining in gastrocnemius from CHIP−/− mice (Fig. 4C, right) compared to the checkerboard pattern in wild-type gastrocnemius (Fig. 4C, left). Together these data suggest that the fiber composition in the gastrocnemius in affected by the loss of CHIP expression.
Figure 1.

Transmission electron micrographs of soleus muscle sarcomeres from (A) wild-type and (B) CHIP−/− mice. (C) Micrographs of CHIP−/− soleus sarcomeres with either normal (top panels) or abnormal (lower panel) subsarcolemmal mitochondrial (sm) compartments including damaged mitochondria (indicated by arrows).
Figure 2.

Transmission electron micrographs of quadriceps muscle sarcomeres from (A) wild-type and (B) CHIP−/− mice. (C) Micrographs of CHIP−/− quadriceps sarcomeres with tubular aggregates (indicated by *). Boxed region is magnified (lower right) detailing the tubular aggregate
Figure 3.
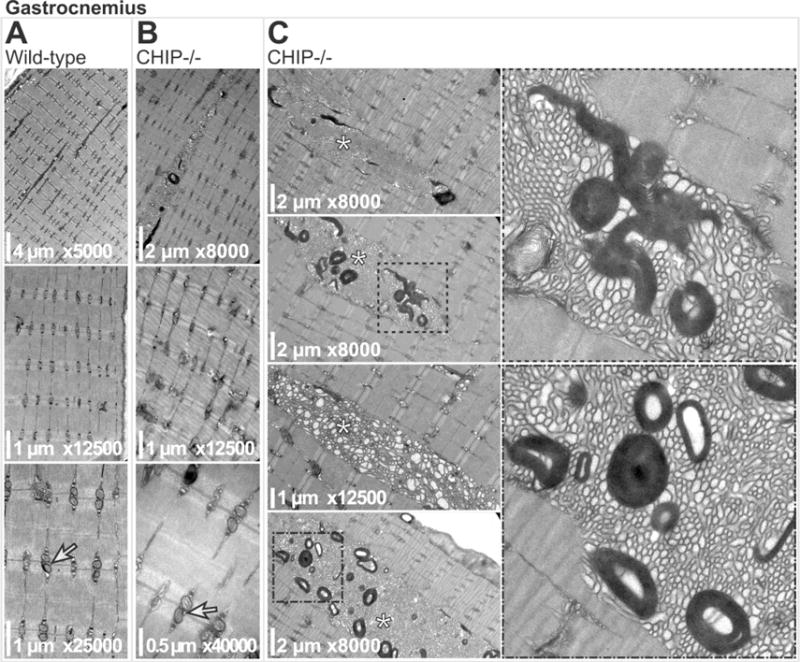
Transmission electron micrographs of gastrocnemius muscle sarcomeres from (A) wild-type and (B) CHIP−/− mice. Longitudinal doublet mitochondria are indicated (arrows in A, B, lower panels). (C) Micrographs of CHIP−/− gastrocnemius muscle sarcomeres with tubular aggregates (indicated by * in left panels). Boxed regions are magnified (right panels) highlighting the with laminar bodies
Figure 4.
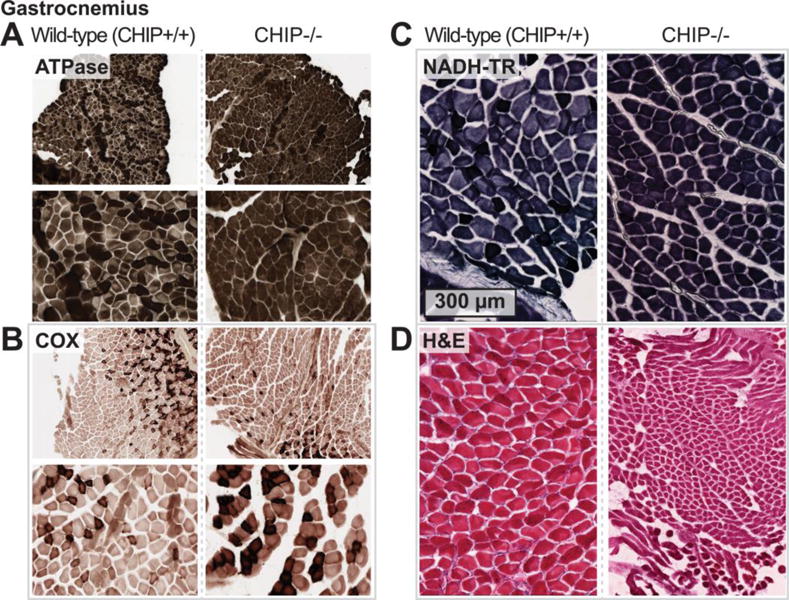
Histochemical staining wild-type and CHIP−/− gastrocnemius muscle of: (A) ATPase, (B) COX, (C) NADH-TR, and (D) (H&E).
DISCUSSION
CHIP is highly expressed in skeletal muscle, heart, pancreas, brain and placenta, with detection seen in kidney, liver, and lung (Ballinger et al., 1999; Hatakeyama et al., 2001). In a recent survey, CHIP RNA and/or Protein were present in 53 or 83 analyzed normal tissue cell types (http://www.proteinatlas.org/ENSG00000103266-STUB1/tissue). The phenotype of the CHIP−/− mouse described here (C57BL/6/129S background) is characterized by age-dependent atrophy in tissues throughout the body, including testis, thymus, gastrocnemius, quadriceps, and heart (Min et al., 2008). Significant atrophy was detected by 3 months of age in gastrocnemius and quadriceps, while significant atrophy occurred in all five organs by 12 months (JN Min, RA Whaley, NE Sharpless, P Lockyer, AL Portbury and C Patterson, 2008). These mice had an accelerated aging phenotype and altered protein quality control, indicated by significant alterations in life span (decreased), toxic oligomers (increased), proteasome activity (decreased), and indices of oxidative stress (increased) (Min et al., 2008). In the context of prior studies that identified CHIP’s role in regulating protein folding in neurodegenerative diseases (Dickey et al., 2006; Miller et al., 2005), the presence of toxic oligomers illustrates the importance of CHIP in directly regulating the pathological mechanisms involved in common neurodegenerative diseases including Alzheimer’s disease (Min et al., 2008). CHIP was initially named the carboxyl terminus of HSC70-interacting protein (CHIP) for its critical interaction with the chaperone HSC70 (Jiang et al., 2001) as well as HSP70 and HSP90 chaperones. These interactions play a role in protein triage decisions, first supporting refolding via chaperones, then targeting for degradation via its ubiquitin ligase activity if and when refolding is not possible (Connell et al., 2001). In addition to the aforementioned chaperones, CHIP interacts with multiple proteins critical to the maintenance of muscle cells, including C-Raf, Parkin ligase, DNAJB1, and RUNX2 (Ballinger et al., 1999; Dogan et al., 2008; Kharchenko et al., 1975; Li et al., 2008). The tubular aggregates described here in CHIP−/− skeletal muscle, but not wildtype controls, were previously proposed to form in aging skeletal muscle fibers, originating from the sarcoplasmic reticulum (Chevessier et al., 2005; Engel et al., 1970; Muller et al., 2001). The aggregates were hypothesized to form from the swelling of the sarcoplasmic reticulum cisternae extending into longitudinally oriented tubules (Boncompagni, et al., 2012). Multiple proteins were identified as components of the aggregates, including proteins involved in the uptake and storage of Ca2+ such as STIM1, SERCA1, Fast CSQ, Triadin 95 and 51 kDa, RYR1, Sarcolumenin (Bohm et al., 2013; Chevessier et al., 2005). Cytoskeletal markers of Spectrin, Dystrophin, and Desmin were also identified in these aggregates (Chevessier et al., 2004). In tubular aggregate myopathies, where these tubular aggregates are predominant, the swelling of the SR results from excess cytosolic Ca2+ or intraluminal Ca2+ in one study of myoblasts from a patient with tubular aggregate myopathy (TAM). It was also reported that excessive STIM1 oligomerization could trigger aggregate formation (Bohm et al., 2014). Previous studies have implicated CHIP in the role of ER stress, being a required ubiquitin ligase mediating IRE1/TRAF2/JNK activation (Zhu, et al., 2014) and involved in tauopathy-induced (Sakagami, et al., 2013) and beta-amyloid precursor protein (Abeta)-induced ER stress in neurodegenerative disease models (Kumar et al., 2007). Additionally, CHIP interactions with HSP70 and Parkin were linked to ER stress in Parkin-mediated Parkinson’s disease (Imai et al., 2002). The findings in the present report may illustrate previously unreported parallel roles in the muscle sarcoplasmic reticulum, which is stressed during aging related to cellular senescence. Tubular aggregates are observed in neuromuscular disorders, including occasionally in Duchenne Muscular Dystrophy and are a common finding in cases of periodic paralysis where they are often confined to type 2 fibers and in some forms of inherited myasthenia gravis (Sieb et al., 1996). Antibodies to emerin, heat shock proteins, and dysferlin were reported to localize to the tubular aggregates (Manta et al., 2004). Tubular aggregates are found in a variety of conditions and associated with multiple pathological insults. Immunohistochemical analysis of tubular aggregates identified cytoskeletal proteins and the 72 kDa heat shock protein in a case of tubular aggregate myopathy and in a case of hypokalemic periodic paralysis with tubular aggregates (Martin et al., 1991). This may indicate that heat shock proteins modulate the tertiary structure of proteins involved in the pathogenesis of tubular aggregates in muscle (Martin et al., 1991). With CHIP’s clear collaborative role with HSP70 family proteins, described above primarily in non-muscle cells, it is possible that alterations/aggregation of HSPs in CHIP−/− muscles may contribute to the buildup of toxic oligomers and tubular aggregates, to contribute to the advanced cellular senescence, a hypothesis that may be clinically relevant in a variety of common muscular and cardiac diseases (Ballinger et al., 1999; Connell et al., 2001; Dogan et al., 2008; Jiang et al., 2001; Kharchenko et al., 1975; Li et al., 2008).
Concluding points
CHIP−/− mice at 6 months of age have morphological changes consistent with increased sarcomplasmic reticulum compartments in quadriceps muscle and gastrocnemius, but not in soleus.
Lack of CHIP may contribute to the buildup of toxic oligomers and tubular aggregates, related to cellular senescence, a hypothesis that may be clinically relevant in a variety of common muscular and cardiac diseases.
Acknowledgments
We would like to thank Bob Bagnell, PhD and Vicky Madden, MS of the University of North Carolina (UNC) Microscopy Services Laboratory/Department of Pathology & Laboratory Medicine for assistance in preparing and imaging the TEM experiments. We also thank Leigh Thorne for guidance and assistance with the skeletal muscle histochemistry preparations. This work was supported by the National Institutes of Health (R01HL104129 to M.S.W., R01GM061728 to J.C.S., R37HL065619 to C.P.), an AHA Scientist Development Grant (to M.S.W.), and the Leducq Foundation Transatlantic Networks of Excellence (to M.S.W.).
Non-standard abbreviations
- DNAJB1
DnaJ (Hsp40) Homolog, Subfamily B, Member 1
- CHIP
carboxyl terminus of Hsc70 interacting protein
- STUB1
STIP1 Homology And U-Box Containing Protein 1
- HSPA1A
heat shock 70 kDa protein 1
- HSPA4
heat shock 70 kDa protein 4
- HSPA8
heat shock protein 70 kDa 8 (aka heat shock cognate 71 kDa protein or Hsc70 or HSP73)
- MuRF1
muscle ring finger-1
- MyoD
transcription factor regulating myocyte differentiation
- OTUD4
OUT domain-containing protein 4
- RNF216
ring finger protein 216
- RUNX2
runt-related transcription factor 2
- SCAR16
Spinocerebellar ataxia, autosomal recessive 16
- ATPase
adenosine triphosphatase- an enzyme that converts ATP to ADP
- COX
cytochrome C oxidase
- NADH-TR
NADH-tetrazolium reductase
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Compliance with Ethical Standards
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
References
- Altun M, Besche HC, Overkleeft HS, Piccirillo R, Edelmann MJ, Kessler BM, Goldberg AL, Ulfhake B. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J Biol Chem. 2010;285:39597–39608. doi: 10.1074/jbc.M110.129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19:4535–4545. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt C, de Yebenes JG, Lopez-Sendon JL, Shomroni O, Zhang X, Qian SB, Bakker IM, Heetveld S, Ros R, Quintans B, Sobrido MJ, Bevova MR, Jain S, Bugiani M, Heutink P, Rizzu P. Clinical and Neuropathological Features of Spastic Ataxia in a Spanish Family with Novel Compound Heterozygous Mutations in STUB 1. Cerebellum. 2015;14:378–381. doi: 10.1007/s12311-014-0643-7. [DOI] [PubMed] [Google Scholar]
- Bird TD. Hereditary Ataxia Overview. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews(R) 2003. [Google Scholar]
- Bohm J, Chevessier F, Koch C, Peche GA, Mora M, Morandi L, Pasanisi B, Moroni I, Tasca G, Fattori F, Ricci E, Penisson-Besnier I, Nadaj-Pakleza A, Fardeau M, Joshi PR, Deschauer M, Romero NB, Eymard B, Laporte J. Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIM1. J Med Genet. 2014;51:824–833. doi: 10.1136/jmedgenet-2014-102623. [DOI] [PubMed] [Google Scholar]
- Bohm J, Chevessier F, Maues De Paula A, Koch C, Attarian S, Feger C, Hantai D, Laforet P, Ghorab K, Vallat JM, Fardeau M, Figarella-Branger D, Pouget J, Romero NB, Koch M, Ebel C, Levy N, Krahn M, Eymard B, Bartoli M, Laporte J. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am J Hum Genet. 2013;92:271–278. doi: 10.1016/j.ajhg.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompagni S, Protasi F, Franzini-Armstrong C. Sequential stages in the age-dependent gradual formation and accumulation of tubular aggregates in fast twitch muscle fibers: SERCA and calsequestrin involvement. Age (Dordr) 2012;34:27–41. doi: 10.1007/s11357-011-9211-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarejos MJ, Perucho J, Lopez-Sendon JL, Garcia de Yebenes J, Bettencourt C, Gomez A, Ruiz C, Heutink P, Rizzu P, Mena MA. Trehalose improves human fibroblast deficits in a new CHIP-mutation related ataxia. PLoS One. 2014;9:e106931. doi: 10.1371/journal.pone.0106931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevessier F, Bauche-Godard S, Leroy JP, Koenig J, Paturneau-Jouas M, Eymard B, Hantai D, Verdiere-Sahuque M. The origin of tubular aggregates in human myopathies. J Pathol. 2005;207:313–323. doi: 10.1002/path.1832. [DOI] [PubMed] [Google Scholar]
- Chevessier F, Marty I, Paturneau-Jouas M, Hantai D, Verdiere-Sahuque M. Tubular aggregates are from whole sarcoplasmic reticulum origin: alterations in calcium binding protein expression in mouse skeletal muscle during aging. Neuromuscul Disord. 2004;14:208–216. doi: 10.1016/j.nmd.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- Cordoba M, Rodriguez-Quiroga S, Gatto EM, Alurralde A, Kauffman MA. Ataxia plus myoclonus in a 23-year-old patient due to STUB1 mutations. Neurology. 2014;83:287–288. doi: 10.1212/WNL.0000000000000600. [DOI] [PubMed] [Google Scholar]
- Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V, Li HH, Madamanchi N, Xu W, Neckers L, Cyr D, Patterson C. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003;22:5446–5458. doi: 10.1093/emboj/cdg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depondt C, Donatello S, Simonis N, Rai M, van Heurck R, Abramowicz M, D’Hooghe M, Pandolfo M. Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations. Neurology. 2014;82:1749–1750. doi: 10.1212/WNL.0000000000000416. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, Zehr C, West G, Cao S, Clark AM, Caldwell GA, Caldwell KA, Eckman C, Patterson C, Hutton M, Petrucelli L. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci. 2006;26:6985–6996. doi: 10.1523/JNEUROSCI.0746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan T, Harms GS, Hekman M, Karreman C, Oberoi TK, Alnemri ES, Rapp UR, Rajalingam K. X-linked and cellular IAPs modulate the stability of C-RAF kinase and cell motility. Nat Cell Biol. 2008;10:1447–1455. doi: 10.1038/ncb1804. [DOI] [PubMed] [Google Scholar]
- Engel WK, Bishop DW, Cunningham GG. Tubular aggregates in type II muscle fibers: ultrastructural and histochemical correlation. J Ultrastruct Res. 1970;31:507–525. doi: 10.1016/s0022-5320(70)90166-8. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Yada M, Matsumoto M, Ishida N, Nakayama KI. U box proteins as a new family of ubiquitin-protein ligases. J Biol Chem. 2001;276:33111–33120. doi: 10.1074/jbc.M102755200. [DOI] [PubMed] [Google Scholar]
- Heimdal K, Sanchez-Guixe M, Aukrust I, Bollerslev J, Bruland O, Jablonski GE, Erichsen AK, Gude E, Koht JA, Erdal S, Fiskerstrand T, Haukanes BI, Boman H, Bjorkhaug L, Tallaksen CM, Knappskog PM, Johansson S. STUB1 mutations in autosomal recessive ataxias - evidence for mutation-specific clinical heterogeneity. Orphanet J Rare Dis. 2014;9:146. doi: 10.1186/s13023-014-0146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes G. A form of famlial degeneration of the cerebellum. Brain. 1908;30:466–489. [Google Scholar]
- Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI, Takahashi R. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol Cell. 2002;10:55–67. doi: 10.1016/s1097-2765(02)00583-x. [DOI] [PubMed] [Google Scholar]
- Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Hohfeld J, Patterson C. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem. 2001;276:42938–42944. doi: 10.1074/jbc.M101968200. [DOI] [PubMed] [Google Scholar]
- Kharchenko AD, Burgasova VA, Ermolenko RI. Indicators of the activity of sympathetic-adrenal system at different stages of development of diabetes mellitus. Pediatriia. 1975:34–36. [PubMed] [Google Scholar]
- Kumar P, Ambasta RK, Veereshwarayya V, Rosen KM, Kosik KS, Band H, Mestril R, Patterson C, Querfurth HW. CHIP and HSPs interact with beta-APP in a proteasome-dependent manner and influence Abeta metabolism. Hum Mol Genet. 2007;16:848–864. doi: 10.1093/hmg/ddm030. [DOI] [PubMed] [Google Scholar]
- Li X, Huang M, Zheng H, Wang Y, Ren F, Shang Y, Zhai Y, Irwin DM, Shi Y, Chen D, Chang Z. CHIP promotes Runx2 degradation and negatively regulates osteoblast differentiation. J Cell Biol. 2008;181:959–972. doi: 10.1083/jcb.200711044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manta P, Terzis G, Papadimitriou C, Kontou C, Vassilopoulos D. Emerin expression in tubular aggregates. Acta Neuropathol. 2004;107:546–552. doi: 10.1007/s00401-004-0851-1. [DOI] [PubMed] [Google Scholar]
- Martin JE, Mather K, Swash M, Gray AB. Expression of heat shock protein epitopes in tubular aggregates. Muscle Nerve. 1991;14:219–225. doi: 10.1002/mus.880140304. [DOI] [PubMed] [Google Scholar]
- McLaughlin B, Buendia MA, Saborido TP, Palubinsky AM, Stankowski JN, Stanwood GD. Haploinsufficiency of the E3 ubiquitin ligase C-terminus of heat shock cognate 70 interacting protein (CHIP) produces specific behavioral impairments. PLoS One. 2012;7:e36340. doi: 10.1371/journal.pone.0036340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ, Davidson BL, Rebagliati MR, Paulson HL. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci. 2005;25:9152–9161. doi: 10.1523/JNEUROSCI.3001-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JN, Whaley RA, Sharpless NE, Lockyer P, Portbury AL, Patterson C. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol Cell Biol. 2008;28:4018–4025. doi: 10.1128/MCB.00296-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HD, Vielhaber S, Brunn A, Schroder JM. Dominantly inherited myopathy with novel tubular aggregates containing 1–21 tubulofilamentous structures. Acta Neuropathol. 2001;102:27–35. doi: 10.1007/s004010000342. [DOI] [PubMed] [Google Scholar]
- Nyamsuren O, Faggionato D, Loch W, Schulze E, Baumeister R. A mutation in CHN-1/CHIP suppresses muscle degeneration in Caenorhabditis elegans. Dev Biol. 2007;312:193–202. doi: 10.1016/j.ydbio.2007.09.033. [DOI] [PubMed] [Google Scholar]
- Pearse AGE. HISTOCHEMISTRY -THEORETICAL and APPLIED. Harcourt Brace/Churchill Livingstone; 1972. [Google Scholar]
- Pestronk GJ, Kaiser KK, Brooke MH. ATPase stain in muscle histochemistry. Muscle Nerve. 1992;15:258. [PubMed] [Google Scholar]
- Ronnebaum SM, Patterson C, Schisler JC. Emerging evidence of coding mutations in the ubiquitin–proteasome system associated with cerebellar ataxias. Hum Genome Var. 2014;1 doi: 10.1038/hgv.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakagami Y, Kudo T, Tanimukai H, Kanayama D, Omi T, Horiguchi K, Okochi M, Imaizumi K, Takeda M. Involvement of endoplasmic reticulum stress in tauopathy. Biochem Biophys Res Commun. 2013;430:500–504. doi: 10.1016/j.bbrc.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Sheehan DC, Hrapchak BB. Theory and practice of histotechnology. Battelle Memorial institute; 1987. [Google Scholar]
- Shi CH, Schisler JC, Rubel CE, Tan S, Song B, McDonough H, Xu L, Portbury AL, Mao CY, True C, Wang RH, Wang QZ, Sun SL, Seminara SB, Patterson C, Xu YM. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet. 2014;23:1013–1024. doi: 10.1093/hmg/ddt497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Wang J, Li JD, Ren H, Guan W, He M, Yan W, Zhou Y, Hu Z, Zhang J, Xiao J, Su Z, Dai M, Wang J, Jiang H, Guo J, Zhou Y, Zhang F, Li N, Du J, Xu Q, Hu Y, Pan Q, Shen L, Wang G, Xia K, Zhang Z, Tang B. Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS One. 2013;8:e81884. doi: 10.1371/journal.pone.0081884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieb JP, Tolksdorf K, Dengler R, Jerusalem F. An autosomal-recessive congenital myasthenic syndrome with tubular aggregates in a Libyan family. Neuromuscul Disord. 1996;6:115–119. doi: 10.1016/0960-8966(95)00034-8. [DOI] [PubMed] [Google Scholar]
- Synofzik M, Schule R, Schulze M, Gburek-Augustat J, Schweizer R, Schirmacher A, Krageloh-Mann I, Gonzalez M, Young P, Zuchner S, Schols L, Bauer P. Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts. Orphanet J Rare Dis. 2014;9:57. doi: 10.1186/1750-1172-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis MS, Min JN, Wang S, McDonough H, Lockyer P, Wadosky KM, Patterson C. Carboxyl terminus of Hsp70-interacting protein (CHIP) is required to modulate cardiac hypertrophy and attenuate autophagy during exercise. Cell Biochem Funct. 2013;31:724–735. doi: 10.1002/cbf.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zhang J, Sun H, Jiang C, Dong Y, Shan Q, Su S, Xie Y, Xu N, Lou X, Liu S. Ubiquitination of inositol-requiring enzyme 1 (IRE1) by the E3 ligase CHIP mediates the IRE1/TRAF2/JNK pathway. J Biol Chem. 2014;289:30567–30577. doi: 10.1074/jbc.M114.562868. [DOI] [PMC free article] [PubMed] [Google Scholar]