

Abstract
Increasing antimicrobial resistance has become a major public health crisis. New antimicrobials with novel mechanisms of action (MOA) are desperately needed. We previously developed a method, bacterial cytological profiling (BCP), which utilizes fluorescence microscopy to rapidly identify the MOA of antimicrobial compounds. BCP is based upon our discovery that cells treated with antibiotics affecting different metabolic pathways generate different cytological signatures, providing quantitative information that can be used to determine a compound’s MOA. Here, we describe a system, rapid inhibition profiling (RIP), for creating cytological profiles of new antibiotic targets for which there are currently no chemical inhibitors. RIP consists of the fast, inducible degradation of a target protein followed by BCP. We demonstrate that degrading essential proteins in the major metabolic pathways for DNA replication, transcription, fatty acid biosynthesis, and peptidoglycan biogenesis in Bacillus subtilis rapidly produces cytological profiles closely matching that of antimicrobials targeting the same pathways. Additionally, RIP and antibiotics targeting different steps in fatty acid biosynthesis can be differentiated from each other. We utilize RIP and BCP to show that the antibacterial MOA of four nonsteroidal anti-inflammatory antibiotics differs from that proposed based on in vitro data. RIP is a versatile method that will extend our knowledge of phenotypes associated with inactivating essential bacterial enzymes and thereby allow for screening for molecules that inhibit novel essential targets.
Graphical abstract

Increasing antimicrobial resistance, exacerbated by the dwindling number of new antibiotics, has become a major public health crisis. In the US alone, over 2 million people become ill and 23 thousand die due to infections caused by antibiotic resistant microbes.1 It has been estimated, based on the current trajectory, that the number of deaths worldwide could rise to 10 million per year and cumulatively cost $100 trillion in lost productivity by 2050.2 The rate of antibiotic resistance is outpacing the development of new antibiotics, the majority of which are modifications to existing scaffolds.3–5 In the past 25 years, only two new classes of antibiotics, oxazolidinones and lipopeptides, have been developed into clinically useful antibiotics.5 In order to increase the number of effective antibiotics, new chemical scaffolds that inhibit new targets are desperately needed.4,5 The recent discovery of teixobactin6 suggests that microbes remain a promising source of new scaffolds.
Antibiotics with novel mechanisms of action (MOA) need to be identified and prioritized as early as possible in the discovery process,7,8 but identifying molecules that are specifically active against new targets is notoriously challenging. Screens using purified enzymes are highly successful at identifying compounds that inhibit enzyme activity in vitro; however, most compounds identified by this process have no or only nonspecific activity against whole cells.3,4,9 In contrast, most whole cell screens are target agnostic and do not allow the selective identification of compounds that inhibit novel targets. We recently developed bacterial cytological profiling (BCP) as a rapid assay for identifying antibiotics that simultaneously identifies MOA.10 BCP utilizes high resolution fluorescent microscopy to observe cytological changes and compares cytological profiles to those from cells treated with control antibiotics to allow identification of MOA. This method has been successfully applied to understanding the MOA of antibiotics in Escherichia coli and Bacillus subtilis.10–12 It has recently been shown to identify the MOAs of compounds present in crude natural product extracts or near colonies directly on growth plates, allowing BCP to be used as a screen to identify strains and extracts with interesting bioactivities.13
The ability of BCP to identify the MOA of a compound increases as new antibiotics with novel MOAs are characterized. However, since many essential enzymes exist for which there are no known antibiotics, the cytological profiles for many potential targets are unknown. Here, we have developed a strategy for creating cytological profiles for essential bacterial proteins, allowing us to screen for inhibitors of new potential targets. We used a system in which essential proteins can be degraded with precision timing, allowing us to mimic the effect of an antibiotic targeting the protein. Then, BCP is applied to measure the cytological changes of the resulting effects of degradation. We show that the cytological profiles obtained by degrading essential proteins in a pathway mimic the profiles of antibiotics targeting the same pathway. The resulting degradation profiles can be used to identify compounds that inhibit these new targets. We have termed this method rapid inhibition profiling (RIP). RIP will allow for identification of profiles from many essential proteins in the cell, which will extend the range of MOAs we can currently identify with BCP and can be used for prioritization of compounds with novel MOAs in future antibiotic discovery programs.
RESULTS AND DISCUSSION
Establishing the RIP Degradation System
BCP works by comparing the profiles of new compounds to a library of profiles derived from reference control antibiotics. It is a fast and efficient method for screening for new antibiotics and determining the pathway they target, for determining if medicinal chemistry efforts change the MOA, and for determining if antimicrobial molecules from in vitro screens kill bacteria by the expected MOA. However, one limitation of BCP is that compounds that inhibit pathways and targets for which we lack control antibiotics will possess unique profiles that will not cluster with current MOA groups. To circumvent this limitation, we took advantage of a genetic approach that, through inducible degradation of the protein, allows the generation of profiles for many previously unexploited targets. Our degradation system is based upon a method developed by Griffith and Grossman14 in which a modified version of the E. coli ssrA tag (ssrA*) is fused to the C-terminus of a target protein. The ssrA* tag is recognized by the cognate SspB protein from E. coli (SspB*) and targeted for degradation to the native B. subtilis ClpXP protease (Figure 1A). By placing SspB* under the control of a xylose-inducible promoter (Pxyl), we can control the timing of degradation of the ssrA*-tagged protein (Figure 1A).
Figure 1.

Degradation of ssrA*-tagged proteins rapidly triggered by induction of sspB*. (A) SspB* is produced under xylose control and targets ssrA*-tagged proteins to ClpXP for degradation. (B–E). Fluorescence micrograph of B. subtilis cells containing YtsJ-GFP-ssrA* (green) with (B) no sspB* in the chromosome, (C) no induction of SspB*, (D,E) induction of SspB* for the indicated time. Membranes are stained with FM4-64 (red). Scale bar, 1 μm. (F) SspB* induced degradation of YtsJ-GFP-ssrA*. Percent fluorescence plotted versus time. Percent fluorescence was calculated as a ratio of average mean GFP intensity per cell at each time point (tx) to the same strain at 0 min (t0). Error bars indicate the standard deviation (n = 3).
We first tested the ability of this system to degrade the abundant nonessential protein YtsJ tagged with GFP-ssrA*. SspB* was induced by the addition of xylose, and the cells were examined by fluorescence microscopy every 15 min to monitor disappearance of GFP fluorescence. Within 15 min of induction, the majority of the YtsJ-GFP-ssrA* fusion protein was degraded, and only one-third of the GFP fluorescence remained (Figure 1D,F). By 30 min, no fluorescence was detected (Figure 1E,F), demonstrating that degradation is complete and occurs rapidly. This rapid degradation during growth mirrors what we previously observed during sporulation, when we used the system for the cell-specific degradation of the SpoIIIE DNA translocase.15
In order to determine if the profiles created by degrading essential proteins matched those of antibiotic-treated cells, we constructed a reference set of strains containing ssrA*-tagged essential proteins. We then visually and quantitatively compared RIP profiles to cytological profiles of cells treated with antibiotics inhibiting these pathways. In this study, we have generated RIP profiles after degrading proteins in four major biosynthetic pathways: DNA replication, transcription, fatty acid biosynthesis, and peptidoglycan biosynthesis. Whereas no significant growth defects were observed in the absence of SspB*, the majority of strains showed large differences in the rate of growth within the first hour of SspB* induction (Figure S1), verifying that degradation occurred rapidly.
Degradation of Proteins Involved in DNA Replication and Transcription
We began our comparison with RIP of essential proteins involved in DNA replication and transcription. To target DNA replication, we tagged the beta sliding clamp of DNA polymerase (DnaN)16,17 with ssrA* (producing a DnaN-ssrA* fusion protein). Within 1 h of SspB* induction, cells were slightly elongated and contained only a single nucleoid, typically within the center of the cell and sometimes spanning the septum (Figure 2C). By 2 h, the cells had further elongated, and many were anucleate, consistent with a strong block in DNA replication (Figure 2C). The RIP profile of DnaN-ssrA* was visually similar to the profile of cells treated with ciprofloxacin for 30 min (Figure 2B), which blocks DNA replication by inactivating DNA gyrase. The only difference between RIP and ciprofloxacin was that at later time points (1 and 2 h) some cells treated with ciprofloxacin had fragmented DNA, as expected, due to the accumulation of double strand breaks18 (Figure 2B). RIP of the major transcription factor σA (SigA-ssrA*)19 resulted in elongated cells with completely decondensed DNA (Figure 2G), and the profile was similar to that of cells treated with the RNA polymerase inhibitor rifampicin (Figure 2F). Thus, the cellular effects of degrading proteins involved in DNA replication or transcription closely matched the effects of antibiotics targeting the same process.
Figure 2.
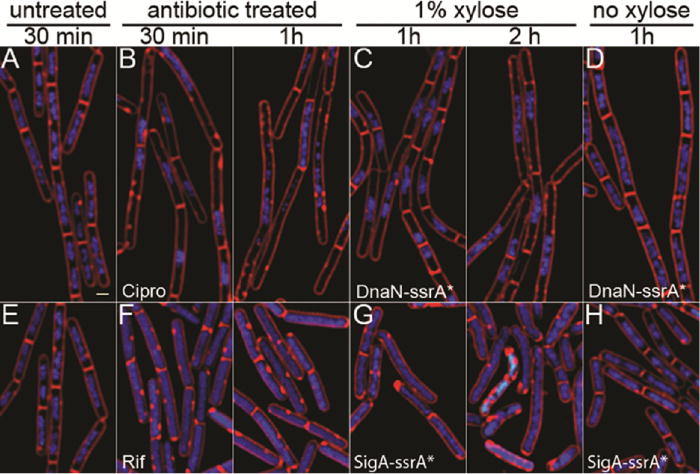
RIP of proteins involved in replication and transcription and comparison to control antibiotics. (A,E) Untreated B. subtilis (strain PY79), (B) PY79 cells treated with ciprofloxacin at 5× MIC. (C,D) Cells containing DnaN-ssrA* with (C) and without (D) induction of SspB*. (F) Cells treated with rifampicin at 5× MIC. (G-H) Cells containing σA–ssrA* with (G) and without (H) induction of SspB*. Cells are stained with FM 4-64 (red, membranes), DAPI (blue, DNA), and SYTOX Green (green, DNA). SYTOX Green is membrane impermeable and only stains cells with compromised membranes. MIC values are available in Table S2.
Degradation of Proteins Involved in Fatty Acid Biosynthesis
We next examined the fatty acid biosynthesis pathway and compared the degradation of two enzymes (AccA and FabZ) with two antibiotics (AZ105 and cerulenin) targeting the pathway (Figure 3). The acetyl-CoA carboxylase complex (ACC) catalyzes the first committed step in fatty acid biosynthesis to generate malonyl-CoA and is composed of four subunits, AccA–D.20 Malonyl-CoA is an essential precursor for both the initiation module of fatty acid biosynthesis and subsequent elongation of the fatty acid chain.21 Thus, ACC is required for both the initiation and elongation steps of fatty acid biosynthesis. We targeted the complex by degrading AccA or by inhibiting AccC with AZ105.22 We also targeted the elongation cycle by degrading FabZ or by adding cerulenin, which inhibits FabF.21
Figure 3.
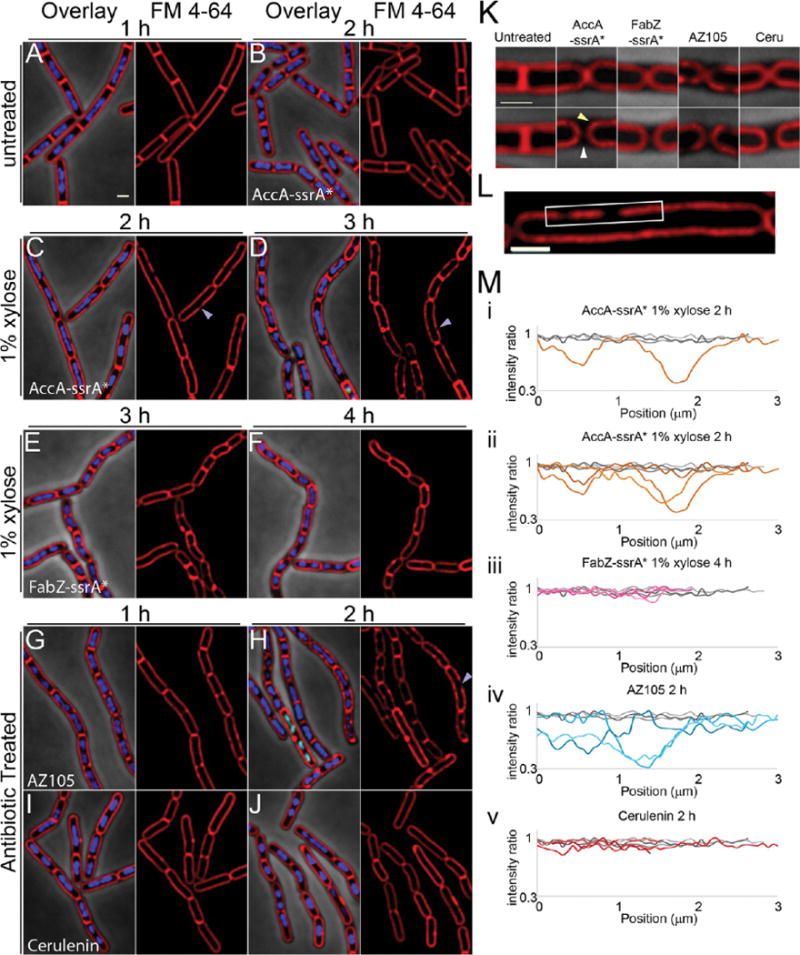
Effects of inhibiting fatty acid biosynthesis by antibiotics and RIP. (A) Untreated PY79 cells. (B–D) Cells containing AccA-ssrA* without (B) or with (C,D) induction of SspB*. Purple arrowheads point to membrane gaps. (E,F). Cells containing FabZ-ssrA* with induction of SspB*. (G,H) PY79 cells treated with AZ105 at 5× MIC. (I,J) PY79 cells treated with cerulenin at 5× MIC. (K) Expanded view of septa of untreated cells and cells after degradation of AccA-ssrA* (2h) or FabZ-ssrA* (4h), or treated with cerulenin (2h) or AZ105 (2h). The yellow arrowhead points to rounded septal membranes and the white arrowhead to the gap between contiguous cells generated after septal membrane retraction. (L) Expanded view of cell from panel C. White box indicates membrane segment where FM4–64 fluorescence intensity was measured. (M) FM4–64 fluorescence intensity ratios along selected membrane segments. Ratios were calculated based on the average intensity along the portion of the membrane measured, then corrected so the highest ratio was 1. Gray lines are untreated cells at 1 h; different colored lines indicate measurements from treated/xylose induced cells at the indicated times. (i) Single AccA-ssrA* measurement corresponding to the membrane segment boxed in white in panel L. (ii) AccA-ssrA* measurements. (iii) FabZ-ssrA* measurements. (iv) AZ105 measurements. (v) Cerulenin measurements. Cells are stained with FM 4–64 (red), DAPI (blue), and SYTOX Green (green). Error bars on the graphs represent the standard deviation of ≥3 experiments. MIC values are in Table S2, scale bars, 1 μm.
Inhibition of fatty acid biosynthesis affected the architecture of the cell membrane, as expected. Normally, B. subtilis divides by forming septa at a 90° angle relative to the long axis of the cell. During exponential growth, cells remain attached via the cell wall after division, forming chains of cells in which the two membranes of newly formed septa remain close together and are indistinguishable when viewed by staining with FM4–64 (Figure 3A,K). However, after degradation of AccA-ssrA* or FabZ-ssrA*, the two membranes of the newly formed septa retract and round up (Figure 3C–F,K, yellow arrowhead) separating from the cell wall, here visualized by phase contrast microscopy (Figure 3K, white arrowhead). This retraction of membrane from the cell wall was observed in the majority of septa (43% to 79%; n > 90) after inhibition of fatty acid biosynthesis by degradation of AccA-ssrA* or FabZ-ssrA*, or by treatment with cerulenin or AZ105 (Figure 4A). Membrane retraction from the poles also occurs during the cell lysis associated with inhibition of peptidoglycan synthesis (Figures 4A, S3). However, retraction of the membranes from the septum in the absence of cell lysis appears to be a hallmark of fatty acid biosynthesis inhibition.
Figure 4.
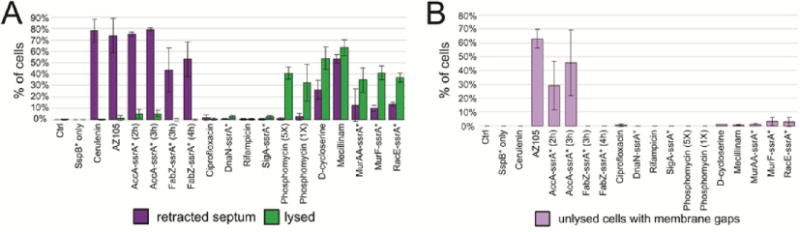
(A) Graph of percentage of cells with retracted septa and percentage of lysed cells for all antibiotics and degradation strains at 2 h (antibiotics) or selected time points (degradation strains, Table S1). Lysis is defined as visible gaps in membrane staining and permeability to SYTOX Green. (B) Graph of percentage of cells that have visible membrane gaps but are not lysed, as defined by SYTOX Green permeability.
A second characteristic of the RIP profile of AccA-ssrA* is the striking presence of gaps in membrane staining without compromising membrane integrity, as measured by the failure of these cells to stain with the membrane impermeable DNA stain SYTOX Green. This phenotype appeared within 2 h of degradation initiation and increased by 3 h, affecting ∼50% of cells (Figure 3C,D,J arrows, 4B). The same phenotype was observed in cells treated with AZ105, affecting ∼60% of cells after 2 h of treatment (Figure 3 H, arrows, 4B). The membrane dye FM4–64 uniformly stains the membrane of wildtype B. subtilis (Figure 3 A,K,M) and disruption of staining usually indicates lysis and increased permeability.11,23,24 The uniformity of staining can be quantified by measuring fluorescence intensity along the membrane. After normalization to the average intensity along the membrane segment, the intensity of staining for untreated cells only varied ∼10% (Figure 3L, gray lines). In contrast, after degradation of AccA-ssrA* or treatment with AZ105, some areas of the membrane have up to a 60% reduction in FM4–64 intensity. Some of the gaps in fluorescence measured over a micron in length (Figure 3Li,Lii,Liv orange and blue lines). Blocking the elongation cycle via either RIP of FabZ-ssrA* or treatment with cerulenin did not produce this phenotype (Figures 3I,J,Lv, 4B), indicating it might be specific to blocking the initiation of fatty acid biosynthesis or with simultaneously blocking inititaion and elongation.
Degradation of Proteins Involved in Peptidoglycan Biosynthesis
We next compared RIP profiles generated by degrading peptidoglycan biosynthetic enzymes to profiles for antibiotics that target various steps in peptidoglycan biosynthesis. Because degradation can only occur if the protein is cytoplasmic and accessible to SspB*, we focused on enzymes that produce cytoplasmic peptidoglycan precursors. These included MurAA, MurF, and RacE. MurAA catalyzes the first committed step in the synthesis of peptidoglycan; MurF is a ligase that adds the terminal D-ala, D-ala, generating a pentapeptide precursor, and RacE is a glutamate racemase that generates the D-glutamate required as part of the pentapeptide stem.25–27 Since disruption of the cell wall results in cell lysis (Figures 4A, S3), we performed this comparison in the presence of an osmotic stabilization agent (MSM)28 to observe changes in cell shape.
RIP of all three strains (MurAA-ssrA*, MurF-ssrA*, RacE-ssrA*) in MSM generated very similar profiles, with extensive cell width defects and minimal cell lysis after 2 h (Figure 5A–C). The profiles were remarkably similar to those produced by antibiotics (phosphomycin and D-cycloserine) targeting enzymes that synthesize peptidoglycan precursors (Figure 5D,E). For example, the RIP profile of MurAA-ssrA* was very similar to the profile of cells treated with phosphomycin (Figure 5A,D), which inhibits MurAA. In contrast, the beta-lactam antibiotic mecillinam, which targets PBP-2,29 also caused cell shape defects but still displayed substantial lysis in the presence of MSM (Figure 5F). We compared these results to the pore-forming lantibiotic nisin and the detergent triton X-100, two membrane active compounds that cause substantial cell lysis. Both resulted in permeabilization of the cells and cell lysis, but neither showed cell shape defects in the presence of MSM (Figure 5H, I), demonstrating that cell shape defects in MSM are a hallmark of inhibition of peptidoglycan biosynthesis and not a generalized indicator of cell lysis. These results show that the profiles of strains after degradation of peptidoglycan biosynthetic enzymes are similar to those of antibiotics targeting the pathway and different from those of membrane active compounds.
Figure 5.

Effects of inhibiting peptidoglycan biosynthesis by antibiotics and RIP. Cells were grown in LBMSM to facilitate visualization of cell shape defects; see Figure S3 for profiles in LB. (A) Cells containing MurAA-ssrA* without and with degradation. (B) Cells containing MurF-ssrA* without and with degradation. (C) Cells containing RacE-ssrA* without and with degradation. (D–I) Cells treated with antibiotics for either 2 h (D–F) or 30 min (G–I) with phosphomycin (D), D-cycloserine (E), mecillinam (F), nisin (H), triton (I) at 1× MIC (D, E, H), or 5× MIC (F, I) or left untreated (G). Cells are stained with FM 4–64 (red), DAPI (blue), and SYTOX Green (green). Scale bar, 1 μm. MIC values are in Table S2.
Linear Discriminant Analysis Clustering of RIP and Antibiotic Cytological Profiles
The cytological profiles of antibiotic-treated cells and RIP of the same pathways gave strikingly similar results for each pathway we tested. To quantitatively determine if the cytological profiles of the two methods are similar, we built upon the system used to classify antibiotic MOAs in BCP.10 We used CellProfiler30 to generate automated measurements of cytological parameters and linear discriminant analysis (LDA) to compile the results. Each antibiotic treatment (2 h) or protein degradation (selected time points, Table S1) was performed in triplicate, and 56 cytological parameters were measured (Tables S5 and S6). These parameters were then used for LDA, which is a supervised pattern recognition and machine learning statistical method used to find combinations of features which separate classes of objects that we have previously used to predict antibiotic susceptibility in clinical isolates of Staphylococcus aureus.31 A precategorizing control set was entered in the algorithm to set up parameter space in which the variability between categories is maximized while minimizing the variability within categories. This allows identification and weighting of the important parameters that distinguish one category from another.
We first checked to see if the antibiotics for each pathway clustered together with the expected RIP strain (Figure 6A,B), which would indicate that the two profiles were quantitatively similar. To do so, we created the dendrogram in Figure 6B by using the top three LDA values (LDA1, LDA2, LDA3, graphed in Figure 6A) that were able to group the three triplicates for each sample (RIP strains or antibiotics targeting the same pathway). The results show that in each case, the profiles for the degradation strains clustered together with the profiles for antibiotics targeting the matching pathway. For example, profiles for degradation of AccA-ssrA* (red triangles) clustered closely with profiles obtained using the fatty acid biosynthesis inhibitors AZ105 and cerulenin (red circles). Similarly, ciprofloxacin (blue circles) and DnaN-ssrA* (blue triangles) degradation profiles clustered together, indicating that the profiles generated by RIP quantitatively match the profiles of cells treated with an antibiotic that inhibits the same process.
Figure 6.
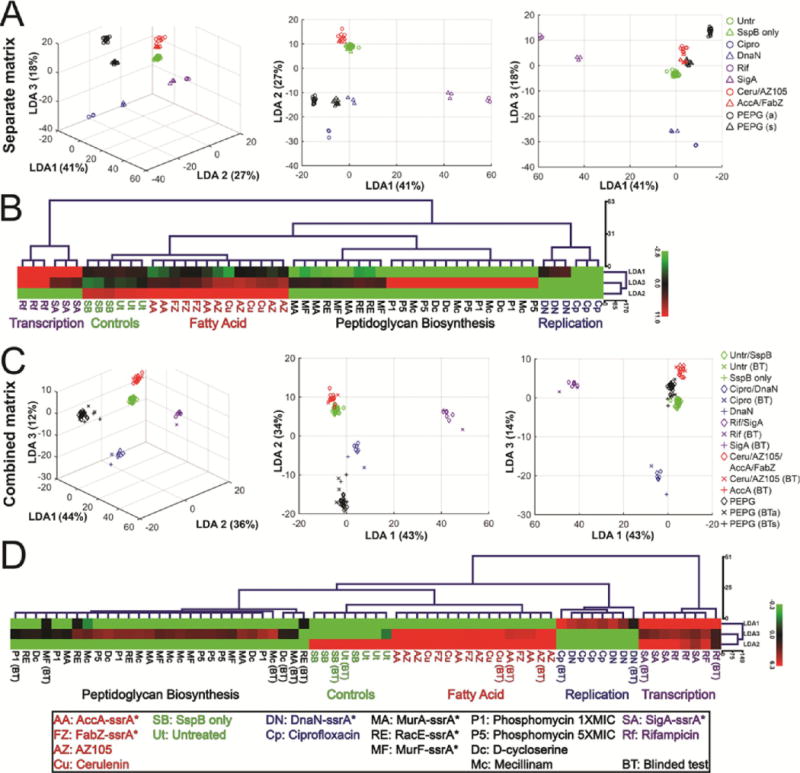
LDA and clustering analysis of profiles after antibiotic-treatment and RIP. (A) 3D and 2D scatterplots of LDA scores generated from coefficients of LDA where antibiotics (2 h) and degradation strains affecting the same pathway were defined as separate groups. Circles represent antibiotic treatments (2 h) and triangles represent degradation strains (xylose treated) at selected time points. PEPG indicates peptidoglycan biosynthesis pathway, a indicates antibiotic (D-cycloserine, phosphomycin, mecillinam), and s indicates strain with degradation (MurAA-ssrA*, MurF-ssrA*, RacE-ssrA*). (B) Euclidean distance hierarchical cluster analysis of LDA scores graphed in panel A using LDA1, LDA2, and LDA3 scores. (C) 3D and 2D scatterplots of LDA scores generated from coefficients of LDA where antibiotics (2 h) and degradation strains (selected time points) affecting the same pathway were defined as a single group (blinded test data not used to generate coefficients). Diamonds represent combined antibiotic and degradation strain data, × represents antibiotic blinded test data, + represents degradation strain blinded test data. (D) Euclidean distance hierarchical cluster analysis of LDA scores graphed in panel C using LDA1, LDA2, and LDA3 scores. Points on scatterplots and treatment names in trees are color-coded based on pathway affected: Untreated controls (green), replication (blue), transcription (purple), fatty acid biosynthesis (red), peptidoglycan (black). BT indicates blinded test data. Data used to generate graphs and trees can be found in the Supporting Information. Antibiotic concentrations are in Table S2.
We next performed a blinded-test experiment (Figure 6C,D). To do so, we first defined a new set of LDA values that were able to group the triplicates for the antibiotic-treated cells and the triplicates for the matching degradation strains (ie: DnaN and ciprofloxacin). To determine if we could properly identify the targets of unknown samples using this matrix, we performed a blinded test with both antibiotic-treated cells and degradation strains, scored them based on the combined matrix, and added the scores to both the graph and clustering analysis. Each of the blinded test samples clustered with the correct pathways, demonstrating that RIP and antibiotic data can be combined to produce robust LDA clustering to identify the target inhibited in unknown samples.
Quantitative Analysis of Membrane Active Compounds
Many antimicrobial compounds act directly on the membrane by forming pores or acting as detergent-like solubilization agents. Since these compounds quickly destroy the cell, there are typically too few remaining cells by the 2 h time point to measure. Although cells are rapidly destroyed, we found we can reliably measure 18 of the original 56 parameters after 10 min of treatment with membrane active compounds (Table S7). We treated cells with three membrane active compounds (nisin, triton X-100, and SDS) or with rifampicin as an outgroup control and collected data from triplicates (Tables S2 and S7). LDA using the 18 measurable parameters for control antibiotics after 10 min resulted in three groups (Figure 7). The largest group contains the untreated control samples as well as cells treated with antibiotics that block replication (ciprofloxacin) and peptidoglycan biogenesis (D-cycloserine, mecillinam, phosphomycin) pathways. These antibiotics have very little effect on the cytological profiles after only 10 min of exposure. The second group consists of cells treated with rifampicin, which form their own group based on the decondensation of the nucleoid, which occurs rapidly. The third group consists of the membrane active compounds nisin, triton X-100, and SDS, which immediately permeabilize the cells (Figures 8F and S3I,H). Euclidean distance hierarchical clustering of the first three LDA parameters (Figure 7B) confirms the groupings.
Figure 7.
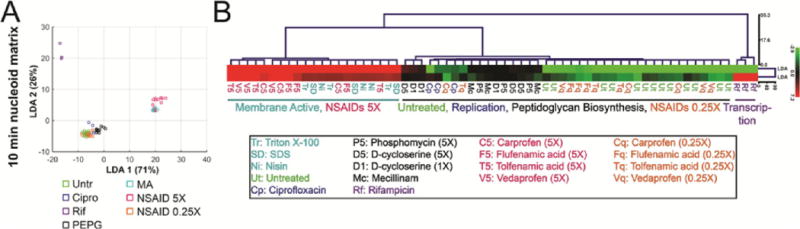
LDA and clustering analysis of cells treated with antibiotics for 10 min. (A) Scatterplot of LDA scores generated from coefficients of LDA from nucleoid measurement data after treatment with indicated antibiotics for 10 min. NSAID data were not utilized to generate the coefficients. (B) Euclidean distance hierarchical cluster analysis of LDA scores graphed in panel A using LDA1 and LDA2 scores. Points on scatterplots and treatment names in tree are color-coded based on pathway affected: controls (green), replication (blue), transcription (purple), peptidoglycan (black), membrane active (cyan), NSAIDS at 5× MIC (magenta), and NSAIDs at 0.25× MIC (orange). Data used to generate graphs and trees can be found in the Supporting Information. Antibiotic concentrations are in Table S2.
Figure 8.
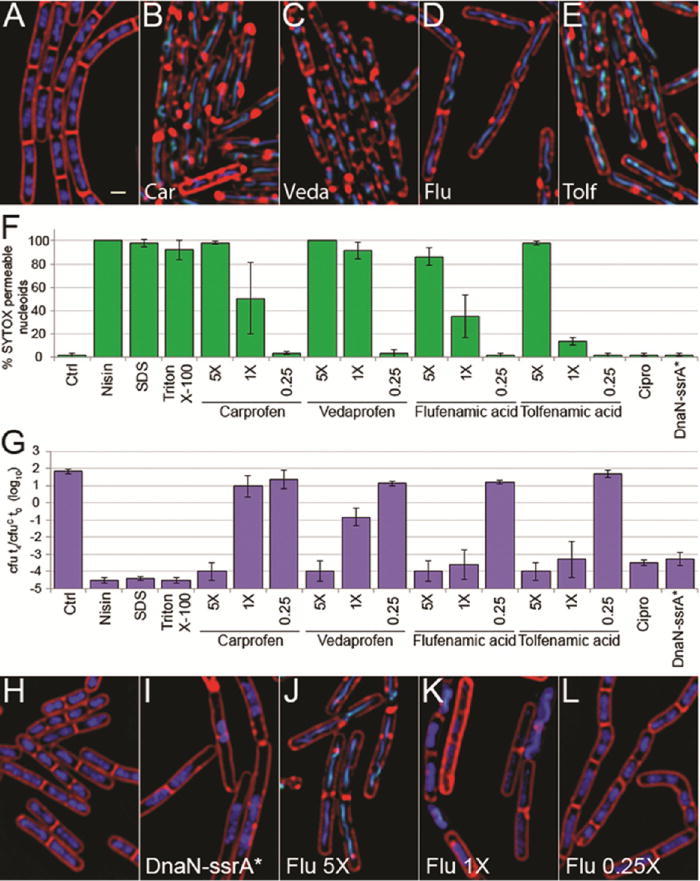
Effect of NSAIDs on the morphology and viability of B. subtilis. (A–E) Fluorescence micrographs of PY79 cells treated for 30 min with 5× MIC of NSAIDs carprofen (B), vedaprofen (C), flufenamic acid (D), tolfenamic acid (E), or untreated (A). (F) Permeability of cells treated with NSAIDs and membrane active compounds as measured by SYTOX permeability as described in the methods. Error bars represent the standard deviation of ≥3 experiments. (G) Effect on cell viability of treatment with NSAIDs and membrane active compounds. Cell viability is shown as the ratio of colony-forming units (cfu) at 4 h (t4) to the cfu at the initial time of treatment (t0) for the control (cfuC) in log10 units, where 0 indicates no change in viability. Error bars represent the standard deviation of ≥3 experiments. (H–L) Fluorescence micrographs of indicated concentrations of flufenamic acid as compared to cells after induction of DnaN-ssrA* degradation for 2 h (I). Cells were treated for 2 h with flufenamic acid at 5× (J), 1× (K), and 0.25× (L) the MIC or not treated (H). Cells are stained with FM 4–64 (red), DAPI (blue), and SYTOX Green (green). Scale bar, 1 μm. Antibiotic concentrations are shown in Table S2.
MOA Determination for Nonsteroidal Anti-Inflammatory Antibiotics
We next wanted to use our quantitation to determine the MOA for an antibiotic with an unconfirmed target. DnaN is essential for DNA replication and is an attractive potential new target for which few known inhibitors exist. A recent study found nonsteroidal anti-inflammatory antibiotics (NSAIDs) bind DnaN in vitro with IC50s ranging from 200 to 1500 μM in the top five candidates, and with most MIC values in the 300 μM range in B. subtilis and between 2500 and >5000 in E. coli.17 We picked the four NSAIDs with the lowest B. subtilis MICs and performed BCP with the compounds to determine if inhibition of DnaN is the likely MOA by comparing the profile to that obtained after RIP of DnaN. We profiled each compound over a range of concentrations from 0.25× up to 5× the MIC. All four resulted in identical profiles at 5× the MIC and look strikingly similar to the membrane active compound nisin (Figures 8B–E and S3H,I). The addition of NSAIDs to cells at 5× the MIC results in permeabilization of 90–100% of the cells within 10 min, similar to nisin, triton X-100, and SDS (Figure 8F). Since NSAIDs are clearly membrane active at 5× the MIC, we examined lower concentrations to see if we could observe evidence for an effect on DNA replication, as we would expect if they are inhibiting DnaN. As the concentrations decreased, CFUs mL−1 increased (Figure 8G), but we did not observe any effect on DNA replication in vivo. Instead, we found a clear dose response between the percentage of cells permeabilized within 10 min and NSAID concentration (Figure 8F,J–L). NSAID cytological profiles were visually identical to those of membrane active compounds, and when quantitated using LDA, for cells treated at 5× the MIC, all NSAIDs tested clustered with membrane active compounds, an effect that is lost at 0.25× the MIC where the compounds no longer kill (Figure 7). Our results show that NSAIDs kill B. subtilis by permeabilizing the membrane and not through DnaN inhibition.
Overall, we have demonstrated that RIP can be used to generate cytological profiles for essential proteins for which no current antibiotics exist. We can use these novel profiles to specifically screen for antibiotics that inhibit these new targets. Since many available antibiotics inhibit the activity of specific essential proteins, we envision that this approach will be broadly applicable to the discovery of new antimicrobial molecules. However, there are some limitations that are important to consider. First, our current system is not suitable for proteins in which the C-terminus is inaccessible to the cytoplasmic protease or the C-terminal fusions are nonfunctional. Some such proteins might be able to be degraded and profiled by adopting N-terminal degradation systems.32 Second, some antibiotics can have more than one target, which might yield complex cytological profiles not matching any individual RIP profile. This limitation can by circumvented by titrating the antibiotic concentration, since typically a lower concentration is required to inhibit primary targets than secondary targets.33 We also have evidence that simultaneous degradation of proteins from different pathways produces hybrid profiles, which might help to tease apart individual targets of multitarget antimicrobials. Third, some antibiotics might act by modifying the function of the target, rather than by inhibiting a specific essential pathway, which might produce unique cytological profiles that cannot by identified by RIP. To account for these cases, it is essential to keep expanding our antibiotic-based BCP library to identify profiles that cannot be obtained by RIP. Together, RIP and antibiotic-based BCP hold great promise to accelerate the identification of the MOA of new antimicrobial molecules. By extending RIP to more essential proteins, we will be able to create a collection of profiles for many unexploited cellular targets allowing the identification of molecules that inhibit these proteins in vivo.
METHODS
Strains and Culture Conditions
The strains used in this study are listed in Table S1 and are all derivatives of B. subtilis PY79. All cultures were grown in LB (unless otherwise indicated) at 37 °C in a roller. Cultures in exponential phase (OD600 0.2–0.5) were diluted down to OD600 0.002 and grown to early exponential phase (OD600∼0.15), then treated with antibiotics, xylose (1%), or a solvent in a final volume of 1 mL (microscopy, viable cell counts) or 1.5 mL (growth curves). Cultures containing FabZ-ssrA* were grown for 45 min (OD600∼0.01) before xylose was added instead of waiting for OD600∼0.15. Cultures grown in LBMSM were handled identically to LB cultures. LBMSM was made by mixing 2× magnesium-sucrose-maleic acid (MSM) pH 7 (40 mM MgCl2, 1 M sucrose, and 40 mM maleic acid) 1:1 with 2× LB. Antibiotic stock and treatment concentrations are listed in Table S2.
Fluorescence Microscopy
Cells were cultured for fluorescence microscopy as described above and treated in a final volume of 1 mL at the concentrations indicated in Table S2. Samples were collected for imaging at 10 min, 30 min, 1 h, and 2 h for antibiotic treated cultures or every hour for 3 h for degradation strains. For degradation strains, xylose (1% final) or a matching volume of water was added. A total of 6 μL of cells was added to 1.5 μL of dye mix (30 μg mL−1 FM 4–64, 5 μg mL−1 DAPI, and 2.5 μM SYTOX Green in 1× Tbase) and transferred to an agarose pad (20% LB, 1% agarose) for visualization. Cultures grown in LBMSM were treated as described above, except agarose pads had a final concentration of 1×MSM in addition to the 20% LB. Cells were visualized on an Applied Precision DV Elite optical sectioning microscope equipped with a Photometrics Cool-SNAP-HQ2 camera. Pictures were deconvolved using SoftWoRx v5.5.1 (Applied Precision). The median focal plane is shown. The phase contrast, FM 4–64, and DAPI images were adjusted for best visualization, whereas the SYTOX Green intensities were normalized between all images (LBMSM images were normalized on a different scale than LB only images); thus they reflect relative intensities of SYTOX Green between the images. Exposure times for DAPI and SYTOX Green images were kept constant throughout all experiments.
Generation of Cell Morphology Measurements
All z-stack images were processed into tifs and medial focal planes determined. Tifs were created in Fiji and adjusted in Photoshop (CS6) to facilitate later identification of objects during thresholding. Images were then analyzed using CellProfiler v. 2.1.1.30 Cells were initially identified using the FM 4–64 images, and the objects were expanded using the phase image as a guide to obtain final cell objects. Nucleoids were identified separately and then associated with corresponding cells. Additional description is available in the Supporting Information.
Supplementary Material
Acknowledgments
We would like to thank AstraZeneca, A. Miller and T. Keating for providing AZ105, and A. Miller for helpful comments on the manuscript. We would also like to thank the people at the CellProfiler forum for troubleshooting assistance. These studies were supported by the National Institute of Health (R01-AI113295 and R01-GM57045). J.L.-G. was supported by an EMBO Long Term fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b01050.
Supporting Methods, Figures S1–S3 (PDF)
Tables S1–S10 (XLSX)
Notes
The authors declare the following competing financial interest(s): K.P. and J.P. have an equity interest in Linnaeus Bioscience Incorporated and receive consulting income from the company. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
References
- 1.Antibiotic Resistance Threats in the United States, 2013. Centers for Disease Control and Prevention; Atlanta, GA: 2013. [Google Scholar]
- 2.O’Neill J. Review on Antimicrobial Resistance. London: 2014. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. [Google Scholar]
- 3.Chopra I. The 2012 Garrod lecture: discovery of antibacterial drugs in the 21st century. J Antimicrob Chemother. 2013;68:496–505. doi: 10.1093/jac/dks436. [DOI] [PubMed] [Google Scholar]
- 4.Fair RJ, Tor Y. Antibiotics and bacterial resistance in the 21st century. Perspect Med Chem. 2014;6:25–64. doi: 10.4137/PMC.S14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabtu N, Enoch DA, Brown NM. Antibiotic resistance: what, why, where, when and how? Br Med Bull. 2015 doi: 10.1093/bmb/ldv041. ldv041. [DOI] [PubMed] [Google Scholar]
- 6.Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schaberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, Lewis K. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Neill J. Review on Antimicrobial Resistance. London: 2015. Securing New Drugs for Future Generations: The Pipeline of Antibiotics. [Google Scholar]
- 8.Fernandes P. The global challenge of new classes of antibacterial agents: an industry perspective. Curr Opin Pharmacol. 2015;24:7–11. doi: 10.1016/j.coph.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 9.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discovery. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 10.Nonejuie P, Burkart M, Pogliano K, Pogliano J. Bacterial cytological profiling rapidly identifies the cellular pathways targeted by antibacterial molecules. Proc Natl Acad Sci U S A. 2013;110:16169–16174. doi: 10.1073/pnas.1311066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamsa A, Liu WT, Dorrestein PC, Pogliano K. The Bacillus subtilis cannibalism toxin SDP collapses the proton motive force and induces autolysis. Mol Microbiol. 2012;84:486–500. doi: 10.1111/j.1365-2958.2012.08038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu WT, Lamsa A, Wong WR, Boudreau PD, Kersten R, Peng Y, Moree WJ, Duggan BM, Moore BS, Gerwick WH, Linington RG, Pogliano K, Dorrestein PC. MS/MS-based networking and peptidogenomics guided genome mining revealed the stenothricin gene cluster in Streptomyces roseosporus. J Antibiot. 2014;67:99–104. doi: 10.1038/ja.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nonejuie P, Trial RM, Newton GL, Lamsa A, Ranmali Perera V, Aguilar J, Liu WT, Dorrestein PC, Pogliano J, Pogliano K. Application of bacterial cytological profiling to crude natural product extracts reveals the antibacterial arsenal of Bacillus subtilis. J Antibiot. 2015 doi: 10.1038/ja.2015.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffith KL, Grossman AD. Inducible protein degradation in Bacillus subtilis using heterologous peptide tags and adaptor proteins to target substrates to the protease ClpXP. Mol Microbiol. 2008;70:1012–1025. doi: 10.1111/j.1365-2958.2008.06467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin JY, Lopez-Garrido J, Lee SH, Diaz-Celis C, Fleming T, Bustamante C, Pogliano K. Visualization and functional dissection of coaxial paired SpoIIIE channels across the sporulation septum. eLife. 2015;4:e06474. doi: 10.7554/eLife.06474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kong XP, Onrust R, O’Donnell M, Kuriyan J. Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell. 1992;69:425–437. doi: 10.1016/0092-8674(92)90445-i. [DOI] [PubMed] [Google Scholar]
- 17.Yin Z, Wang Y, Whittell LR, Jergic S, Liu M, Harry E, Dixon NE, Kelso MJ, Beck JL, Oakley AJ. DNA replication is the target for the antibacterial effects of nonsteroidal anti-inflammatory drugs. Chem Biol. 2014;21:481–487. doi: 10.1016/j.chembiol.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Mitscher LA. Bacterial topoisomerase inhibitors: quinolone and pyridone antibacterial agents. Chem Rev. 2005;105:559–592. doi: 10.1021/cr030101q. [DOI] [PubMed] [Google Scholar]
- 19.Haldenwang WG. The sigma factors of Bacillus subtilis. Microbiol Rev. 1995;59:1–30. doi: 10.1128/mr.59.1.1-30.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cronan JE, Jr, Waldrop GL. Multi-subunit acetyl-CoA carboxylases. Prog Lipid Res. 2002;41:407–435. doi: 10.1016/s0163-7827(02)00007-3. [DOI] [PubMed] [Google Scholar]
- 21.Parsons JB, Rock CO. Bacterial lipids: metabolism and membrane homeostasis. Prog Lipid Res. 2013;52:249–276. doi: 10.1016/j.plipres.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA. ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discovery. 2015;14:529–542. doi: 10.1038/nrd4572. [DOI] [PubMed] [Google Scholar]
- 23.Pogliano J, Osborne N, Sharp MD, Abanes-De Mello A, Perez A, Sun YL, Pogliano K. A vital stain for studying membrane dynamics in bacteria: a novel mechanism controlling septation during Bacillus subtilis sporulation. Mol Microbiol. 1999;31:1149–1159. doi: 10.1046/j.1365-2958.1999.01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pogliano J, Pogliano N, Silverman JA. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J Bacteriol. 2012;194:4494–4504. doi: 10.1128/JB.00011-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kimura K, Tran LS, Itoh Y. Roles and regulation of the glutamate racemase isogenes, racE and yrpC, in Bacillus subtilis. Microbiology. 2004;150:2911–2920. doi: 10.1099/mic.0.27045-0. [DOI] [PubMed] [Google Scholar]
- 26.El Zoeiby A, Sanschagrin F, Levesque RC. Structure and function of the Mur enzymes: development of novel inhibitors. Mol Microbiol. 2003;47:1–12. doi: 10.1046/j.1365-2958.2003.03289.x. [DOI] [PubMed] [Google Scholar]
- 27.Gautam A, Rishi P, Tewari R. UDP-N-acetylglucosamine enolpyruvyl transferase as a potential target for antibacterial chemotherapy: recent developments. Appl Microbiol Biotechnol. 2011;92:211–225. doi: 10.1007/s00253-011-3512-z. [DOI] [PubMed] [Google Scholar]
- 28.Leaver M, Dominguez-Cuevas P, Coxhead JM, Daniel RA, Errington J. Life without a wall or division machine in Bacillus subtilis. Nature. 2009;457:849–853. doi: 10.1038/nature07742. [DOI] [PubMed] [Google Scholar]
- 29.Spratt BG. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc Natl Acad Sci U S A. 1975;72:2999–3003. doi: 10.1073/pnas.72.8.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quach DT, Sakoulas G, Nizet V, Pogliano J, Pogliano K. Bacterial Cytological Profiling (BCP) as a Rapid and Accurate Antimicrobial Susceptibility Testing Method for Staphylococcus aureus. EBioMedicine. 2016;4:95–103. doi: 10.1016/j.ebiom.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sekar K, Gentile AM, Bostick JW, Tyo KE. N-Terminal-Based Targeted, Inducible Protein Degradation in Escherichia coli. PLoS One. 2016;11:e0149746. doi: 10.1371/journal.pone.0149746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silver LL. Multi-targeting by monotherapeutic antibacterials. Nat Rev Drug Discovery. 2007;6:41–55. doi: 10.1038/nrd2202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.