

Abstract
Epithelial–mesenchymal transition (EMT) is an essential process for morphogenesis and organ development which reversibly enables polarized epithelial cells to lose their epithelial characteristics and to acquire mesenchymal properties. It is now evident that the aberrant activation of EMT is also a critical mechanism to endow epithelial cancer cells with migratory and invasive capabilities associated with metastatic competence. This dedifferentiation program is mediated by a small cohort of pleiotropic transcription factors which orchestrate a complex array of epigenetic mechanisms for the wide-spread changes in gene expression. Here, we review major epigenetic mechanisms with an emphasis on histone modifications and discuss their implications in EMT and tumor progression. We also highlight mechanisms underlying transcription regulation concerted by various chromatin-modifying proteins and EMT-inducing transcription factors at different molecular layers. Owing to the reversible nature of epigenetic modifications, a thorough understanding of their functions in EMT will not only provide new insights into our knowledge of cancer progression and metastasis, but also facilitate the development of diagnostic and therapeutic strategies for human malignancy.
Keywords: Epithelial–mesenchymal transition, Tumor progression, Epigenetics, Histone modifications, Transcription regulation
Introduction
During embryonic gastrulation of metazoans, formation of mesoderm starts from the primitive streak of the primitive ectoderm, where a small population of polarized epithelial cells loses their tight cell–cell junctions and adhesions, undergoes dedifferentiation and eventually migrates along the extracellular space underneath the ectoderm. The process was thus defined as epithelial–mesenchymal transition (EMT) and has been observed during a variety of tissue remodeling events, including the formation of neural crest, cardiac valve, and secondary plate [1]. In addition to enabling the inter-conversions of epithelial cells to distinct cell types for tissue and organ formation during development, EMT participates in wound healing, tissue regeneration, and organ fibrosis in adulthood to generate repair-associated mesenchymal cells or fibroblasts [2, 3]. Furthermore, accumulating evidences suggests that the progression of most carcinomas is associated with the acquisition of abilities for epithelia tumor cells to escape from the primary site and invades through the basement membrane. This process recapitulates the developmental EMT program and has emerged as a critical early step for malignant progression and metastasis, the most common fatal consequence of carcinogenesis [2, 4, 5].
EMT is typically characterized by alterations in gene expression, loss of cell polarity and contacts, and gain of motility and invasiveness [6]. Certain cancer cells also acquire cancer stem cell (CSC) like properties and therapeutic resistance through this dedifferentiation program [5, 7]. The physiological activation of EMT can be triggered by extracellular signals, such as ECM, soluble growth factors TGF-β and FGF, Wnt and Notch proteins, or by intracellular cues, such as oncogenic Ras or NF-κB signaling [8, 9]. In response to ligands from nearby microenvironment, receptor-mediated signaling first activates intracellular molecules, including the Src tyrosine-kinases and the small GTPase family members. These effectors next orchestrate the changes in cytoskeletal organization and disassemble cell–cell junction complexes. A cohort of transcription factors, including two double zinc finger and homeodomain factors (ZEB1/2, the Snail family of zinc finger proteins (SNAI1/2/3), and the family of bHLH factors (TWIST1/2, E12/E47) becomes expressed and alters gene expression patterns [10, 11]. Although most EMT-inducing transcription factors (EMT-TFs) were originally implicated in embryogenesis and cell differentiation, their elevated expression has been well documented in many invasive tumors [11, 12]. These EMT-TFs function as molecular switches, convert the activated signaling pathways to transcriptional reprogramming and confer epithelial–mesenchymal plasticity.
One of the most-characterized hallmarks of EMT is the functional loss of E-cadherin, a pluripotent calcium-dependent adhesion molecule expressed in most epithelial tissues to connect adjacent epithelial cells. The loss of E-cadherin results in disaggregation of adjacent cancer cells and thus contributes to metastatic dissemination [13]. Although loss or reduction of E-cadherin expression can be occasionally caused by genetic lesions, transcriptional repression has emerged as a fundamental mechanism during EMT and tumor progression [14]. E-cadherin promoter harbors E-box elements which are directly bound by EMT-TFs for repression, such as SNAI1/2, ZEB1/2, and E47. EMT-TFs also suppress expression of other cell junction proteins, including Claudins and Desmosomes to promote EMT. Several other transcription factors, such as TWIST1, FOXC2, and TCF4 trigger EMT without binding to E-cadherin promoter. It has been shown that TWIST1 binds to SNAI2 promoter to induce its expression and epithelial gene silencing [15]. In NMuMG cells, Snail1 also induces expression of Twist1 and Ets1 which further bind to Zeb1 promoter to activate transcription [16], suggesting that different EMT-TFs also function synergistically to confer EMT. Since migrating tumor cells are believed to undergo a reverse mesenchymal-epithelial transition (MET) at the distal site to enable their colonization and metastasis, the acquisition of mesenchymal characteristics by epithelial cancer cells through EMT need not be permanent. In this scenario, the reprogramming of gene expression provides a rapid regulatory mechanism to switch epithelial–mesenchymal states during cancer progression. Given that most eukaryotic transcription factors do not have long residence times at the binding sites and turn over rapidly, a variety of epigenetic regulators and modifications are considered as critical mechanisms to integrate signals from multiple transcription factors.
Epigenetics was defined as “the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence” [17]. In eukaryotic cells, DNA is wrapped around histone octamers to form nucleosomes that are the basic repeating units of chromatin. Individual nucleosomes can pack against each other to form several higher order structures which reduce DNA availability and impede transcription. However, the chromatin structure is dynamically regulated by numerous epigenetic modifications, including chromatin remodeling, DNA methylation, histone modifications, and histone variants, which not only reflect, but also affect the transcriptional states of underlying genes [18]. These epigenetic modifications are delicately balanced in normal cells, while small changes in a given parameter can lead to major consequences and ultimately result in cellular transformation and malignant outgrowth [19]. In this review, we discuss major epigenetic mechanisms and focus on histone modifications which confer transcriptional reprogramming to regulate EMT and tumor progression.
Chromatin remodeling in EMT
ATP-dependent chromatin remodeling is one of the well-known mechanisms that permit active compaction and decompaction of DNA for gene expression regulation, DNA replication, chromosome segregation, DNA repair, and recombination. Currently, four major families of chromatin remodeling complexes have been characterized, including SWI/SNF, ISWI, CHD, and INO80. These multi-protein complexes contain a catalytic ATPase subunit which utilizes ATP hydrolysis to alter histone-DNA contacts and restructure nucleosomes [20]. They also harbor distinct non-catalytic components that are responsible for the regulation of complex targeting and specific nucleosome positioning activities to determine the gene expression programs and cell fate. The perturbation of chromatin remodeling complexes has been well documented in malignant progression [21]. Over-expression of MTA1/2, the components of the CHD family of NuRD chromatin remodeling complex, correlates with invasiveness of multiple cancers. On the contrary, MTA3 has been shown to directly inhibit transcription of SNAI1 to abrogate EMT and metastasis of breast cancer [22]. Recent cancer genome sequencing projects uncovered that ~20 % of human tumors contain mutations in at least one member of the SWI/SNF remodeling complex [23]. The catalytic subunit of SWI/SNF complex BRG1 is one of the most commonly mutated subunits across cancer types, while loss of a non-catalytic component BRM is also a contributing factor and potential marker for lung, prostate, and gastric cancers [24], suggesting that SWI/SNF complex function as a tumor suppressor. However, BRG1 can also be recruited by ZEB1 to E-cadherin promoter for transcription silencing [25]. Another SWI/SNF subunit BAF60c up-regulates WNT5a expression and activates WNT signaling to promote EMT [26], indicating that the same remodeling complex also promotes tumor progression. Recently, discrepant tumor-suppressive and oncogenic activities of Brg1 have been reported at distinct stages in a Kras-driven pancreatic cancer mouse model [27]. These findings suggest that chromatin remodeling factors possess antithetical functions that are precisely regulated during tumorigenesis and tumor progression in a stage- and/or tissue-dependent manner through different mechanisms [28].
DNA methylation in EMT
DNA methylation is a covalent modification usually occurs at the 5′-position of the cytosine ring (5mC) within CpG dinucleotides and is associated with transcriptionally repressed chromatin. It is one of the best-characterized epigenetic modifications that play pivotal roles in various physical and pathological processes [29, 30]. In mammals, about 70–80 % of CpGs are methylated by three active DNA methyltransferases (DNMTs). While DNMT1 primarily acts on hemi-methylated CpG to maintain DNA methylation patterns during cell division, DNMT3A and 3B introduce de novo DNA methylation [30]. Aberrant DNA methylation is often observed in cancers, including hypermethylation of CpG islands and hypomethylation of gene-poor regions, which leads to tumor suppressor gene silencing, genomic instability, loss of genomic imprinting, etc. [31]. The overall CpG methylation change was not detected during EMT in genome-wide profiling studies using different models. However, the DNA methylome undergoes selective CpG site methylation changes in discrete genomic regions upon induction of EMT, which are in association with transcription regulation of EMT-related genes [32, 33]. Among them, silencing of E-cadherin correlates with promoter hypermethylation in a wide range of cancer cells [34, 35]. Several EMT-TFs, including SNAI1 [36] and ZEB1 [37], are able to recruit different DNMTs to E-cadherin promoter for CpG methylation. It has also been shown that the recruitment of DNMT1 relies on TGFβ-SMAD2 signaling pathway [38], whereas DNMT3A targeting is facilitated by high-mobility group protein HMGA2 [39]. DNA methylation-associated silencing of miR-200 family members also occurs during EMT [40–42], and both DNMT1 and DNMT3A are involved in establishing DNA methylation on promoter of these key regulators of epithelial phenotypes [43, 44]. Intriguingly, recent genomic studies have identified DNMT3A mutations in 22 % of adult AML that was associated with the increased risk of relapse [45, 46], revealing a tumor suppressor function of DNMT3A in haematological malignancies [47]. Consistently, depletion of Dnmt3a in a lung cancer mouse model significantly promotes tumor growth and progression [48]. Although expression changes were observed with genes involved in angiogenesis, cell adhesion, and cell motion, whether Dnmt3a deficiency directly contributes to EMT in vivo is not clear.
It is believed that DNA methylation-mediated transcription silencing is conferred by the recruitment of various methyl-DNA-binding domain (MBD) proteins, such as MeCP2, MBD1-4, and Kaiso [49]. Through interaction with different chromatin-modifying proteins (discussed below), MBD proteins can transduce the methyl-CpG signal into a compacted chromatin environment for transcription repression [50]. It has been shown that MeCP2 and MBD1/2 bind to methylated CpG island within E-cadherin promoter in a differential manner and their binding correlates with gene silencing in various tumor cells [51]. Although different CpG methylation patterns have been proposed to directly recruit different sets of MBD proteins, other mechanisms also exist. For example, MBD2/3 is key components of NuRD chromatin remodeling complex [52] which was also co-purified with TWIST1 [53]. In pancreatic cancer cells, TWIST1 also associates with MBD1 on E-cadherin promoter for gene silencing and EMT, suggesting that MBD proteins can be recruited by this EMT-TF [54]. Moreover, promoter CpG methylation-associated E-cadherin silencing correlates with elevated MeCP2 expression in colorectal cancer tissues [55] as well as over-expression and the nuclear localization of Kaiso during progression of prostate and breast cancers [56, 57]. In addition to E-cadherin promoter, Kaiso directly binds to methylated sequence in microRNA miR-31 promoter and promotes EMT through a different pathway [56, 58]. Therefore, MBD proteins are critical effectors in the methyl-CpG-mediated epithelial gene silencing during EMT.
Although 5mC is a stable epigenetic mark, passive and active losses of DNA methylation were observed in different biological contexts. While passive DNA demethylation occurs during successive replications, active demethylation can be mediated by three ten–eleven translocation (TET1-3) proteins. Through the three consecutive oxidation reactions, these oxygenases can convert 5mC to 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) [59]. In mES cells, TET1 interacts with SIN3A co-repressor complex and significantly contributes to its recruitment and the mediated transcription repression [60]. Although SIN3A complex can be recruited by SNAI1 to E-cadherin promoter in epithelial cell lines [61], whether TET1 is involved in SNAI1-mediated gene silencing and EMT remains unknown. In several cancer cells, TET1 interacts with hypoxia inducible factors HIF-1α and HIF-2α to enhance their transactivation activities. Together with HIF-2α, TET1 functions as a transcriptional co-activator to enhance the expression of INSIG1, a master regulator of cholesterol biosynthesis and contributes to hypoxia-induced EMT [62]. Although the 5mC-to-5hC conversion was detected on the promoter, the synergistic activation of INSIG by TET1 and HIFs is independent of its DNA demethylation activity.
Consistent with the importance of DNA methylation in epithelial gene silencing and EMT, treatment with DNA methylation inhibitor 5-aza-2′-deoxycytidine (5-Aza-dC) effectively induces E-cadherin expression in a variety of cancer cell lines and restores epithelial phenotypes [63–66]. Although this approach has been applied in clinical settings to treat myelodysplastic syndrome and leukemia [67], DNA-demethylating agent also non-specifically activates the expression of many non-epithelial genes and leads to obvious side-effect toxicities. Furthermore, genome-wide DNA hypomethylation induced by Dnmt1 deficiency increases chromosome instability and thus raises tumor incidence in mice [68, 69]. Given that the reverse MET process is required for migrating cells to colonize a secondary site, inhibition of DNA methylation during the late stage of tumor progression could also promote metastasis. Thus, the utilization of DNA demethylating agents alone for anti-cancer therapy will need to be carefully evaluated.
Histone modifications
As the nucleosome core, histone octamer is composed of one H3–H4 tetramer and two separate H2A–H2B dimers. While the globular domains of histone proteins interact together and contact closely to DNA, the flexible N-terminal domains (also called histone tails) are subjected to nine different types of post-translational modifications [18]. These modifications, including acetylation, methylation, phosphorylation, ubiquitination, and their different combinations have been proposed to constitute a unique “code” to confer specific biological outcomes [70, 71]. To simplify their diverse functions in transcription regulation, histone modifications can be briefly grouped into two major categories for either transcription activation or repression, representing their associated chromatin environments. Given that a transcription reprogramming occurs during EMT, wide-spread changes in histone modifications serve as a regulatory platform to orchestrate the simultaneous repression of epithelial genes and activation of mesenchymal genes. Here, we focus on the well-studied histone acetylation and methylation, and discuss their diverse regulations and functions in this process.
Permissive histone modifications in EMT
Histone acetylation
The actively transcribed euchromatin represents a more relaxed genome partition, where DNA is accessible to transcription machineries. These regions usually have high level of histone lysine acetylation that is catalyzed by several families of histone acetyltransferases (HATs), including PCAF, p300/CBP, TIP60, and hMOF [72]. The addition of an acetyl group to lysine residue is believed to neutralize the positive charge of histone, weaken histone-DNA, and/or nucleosome–nucleosome interactions, and thus potentiate transcription. In Wnt- and β-catenin-dependent EMT pathway, nuclear localized β-catenin binds to T-cell factor (TCF), recruits p300/CBP to assemble a transcription activation complex on target gene promoters and up-regulate their expression [73, 74]. Intriguingly, p300 and PCAF have been shown to interact with ZEB1 and acetylate this EMT-TF. The resultant acetylation dissociates ZEB1 from miR-200c/141 promoter and induces microRNA expression [75]. Furthermore, over-expression of SNAI1/2 results in an up-regulated TGFBR2 expression and increased TGF-β signaling. Although it is unknown whether SNAI1/2 directly activates transcription, increased H3K9 acetylation was detected on TGFBR2 promoter [76]. In several breast cancer cells, hMOF-catalyzed promoter H4K16 acetylation is also important to maintain expression of several EMT-related tumor suppressor genes, including TMS1, E-cadherin, and ESR1 [77]. In addition to E-boxes, E-cadherin promoter harbors four HNF3-binding sites. The hepatocyte nuclear factor HNF3 also cooperates with p300/CBP on E-cadherin promoter to activate gene expression and antagonize EMT and metastatic potential of breast cancer cells [78].
In cells, lysine acetylation is dynamically counteracted by histone deacetylases (HDACs), which restore the positive charge of the lysine, stabilize the local chromatin architecture, and thus predominantly function as transcription co-repressors. There are about 18 mammalian HDACs that are grouped into four classes. Inhibiting enzymatic activities of class I/II or class III HDACs increases H3/H4 acetylation on E-cadherin promoter and de-represses gene expression in multiple cancer cell lines further support the importance of HDACs and histone deacetylation in epithelial gene silencing and EMT [61, 79]. Similar to HATs, different HDACs often exist in large protein complexes which can be recruited to target promoters by EMT-TFs. For instance, SNAI1 has been shown to recruit HDAC1/2-containing SIN3A complex to deacetylate histones on E-cadherin promoter for gene silencing [61]. During chick neural crest EMT, SIN3A interacts with a PHD domain -containing protein PF1 which recruits the complex and SNAI2 for transcription silencing of E-cadherin and Cad6b through histone deacetylation [80]. Although the molecular details of these recruitments are not clear, blocking PF1-SIN3A interaction in triple negative breast cancer cells not only de-represses expression of E-cadherin and ESR1, but also reduces invasive morphogenesis, CSC properties, and in vivo metastasis [81]. In addition, SNAI2/SLUG binds to E-box on BRCA2 promoter and recruits HDAC1-containing CtBP complex for gene silencing [82], suggesting that it could suppress E-cadherin expression in a similar manner. ZEB1/2 could also recruit CtBP1/HDAC complex to E-cadherin promoter, as they were co-purified in a large co-repressor complex [83]. This possibility is supported by findings that ZEB1 recruits HDAC1/2 to E-cadherin promoter for repression in pancreatic cancer cells [84]. In prostate cancer cells, ZEB1 has been shown to recruit SIRT1, a class III HDAC for E-cadherin silencing, and to promote EMT and metastasis in vitro and in vivo [85]. Moreover, ZEB1-induced EMT is accompanied by repression of several other epithelial genes, including EPCAM, ST14, ESRP1, and RAB25, together with reduced H3K9 and H3K27 acetylation on their promoters. A global H3K27 deacetylation was also observed, suggesting that it is a key epigenetic event in ZEB1-induced transcriptional reprogramming [86]. In addition to EMT-TFs, HDAC1 is co-purified with TCF12, while the elevated expression of TCF12 and HDAC1 correlates with poor prognosis of gallbladder cancer patients [87], indicating that this HLH transcription factor could also recruit HDACs for epithelial gene silencing during EMT. Intriguingly, HDAC6 and SIRT1 have been shown to counteract p300-catalyzed acetylation on Cortactin and thus enhance its F-actin-binding ability to facilitate EMT and tumor progression [88, 89].
Inhibition of class I HDACs using different inhibitors, such as TSA and SAHA, displayed antigrowth activities in a variety of cancer cells accompanied by abrogated EMT and metastasis in vitro and in vivo [90–93]. So far more than 10 HDAC, small molecule inhibitors have been applied in different clinic trials [94]. Although many of them are promising in the treatment of hematological tumors, they exhibited limited effects on solid tumors, particularly on carcinomas [12]. A recent study demonstrates that mocetinostat, but not other HDAC inhibitors, specifically antagonizes ZEB1-mediated miR-203 repression in pancreatic cancer cells by restoring histone acetylation on the promoter. In cancer patients, miR-203 silencing significantly associates with recurrence after gemcitabine treatment [95]. Based on findings that HDAC1/2 is over-expressed in metastatic melanoma tissues, another report reveals that inhibition of class I HDACs sensitizes malignant melanoma cells to apoptosis following temozolomide treatment [96]. Together, these results also highlight the potential of mechanism-based combinations of selected HDAC inhibitors with standard chemotherapy in the treatment of aggressive solid tumors. Considering that persistent genes activation may require targeting of multiple epigenetic silencing machineries, HDAC inhibitors have also been utilized in combination with 5-Aza-dC as synergistic strategies [97]. In AML treatment, the combination of incorporated epigenetic inhibitor was developed [98] and led to promising results in a small cohort of patients [99]. However, recent studies uncovered that HDAC inhibitors could promote EMT in prostate and nasopharyngeal cancer cells [100, 101], indicating that the application of HDAC inhibitors in anti-cancer therapy has limitations as well.
Permissive histone methylation
Histone methylation occurs on the side chain of both lysine and arginine residues. While arginine can be mono- (me1) and di-methylated (me2, symmetrically or asymmetrically) by protein arginine methyltransferases (PRMTs), lysine can be catalyzed to mono-, di-, and tri-methylated (me3) forms by SET domain-containing histone methyltransferases (HMTases) and DOT1/DOT1L [102]. Different from acetylation, histone methylation has diverse consequences and associates with either transcription activation or repression, depending on methylation sites as well as methylation states. Among them, histone H3K4me2/3 decorates euchromatic regions particularly enhancers and promoters, while H3K36me3 associates with transcription elongation and thus marks gene bodies and 3′-ends. H3K79 methylation is also associated with transcribed genes, with H3K79me2/3 near transcription start sites and H3K79me1 spreading gene bodies [103]. A recent study reveals that the elevated DOT1L expression correlates with the metastatic potential of breast cancer [104]. This H3K79 HMTase forms a complex with c-Myc and p300 on the promoter of SNAI1 and ZEB1/2, while the catalyzed H3K79 methylation coordinates with other epigenetic marks to activate their expression to promote EMT and metastasis [105]. In a TGF-β-induced EMT model, a global increase of H3K4me3 and H3K36me3 was detected, revealing the occurrence of a genome-wide reorganization of these two permissive methylation marks [33]. Furthermore, PRMT1 has been shown to promote EMT, CSC properties, and metastasis of breast cancer cells in vitro and in vivo. In this case, PRMT1 directly catalyzes asymmetric H4R3me2 on ZEB1 promoter to activate gene expression [106].
In addition to HMTases, histone methylation is regulated by LSD and Jumoji-domain-containing families of histone demethylases (HDMs). The nuclear amine oxidase homolog LSD1 was the first identified de novo HDM which removes H3K4me1/2 via a FAD-dependent oxidative reaction [107]. Over-expression of LSD1 was detected in a variety of tumors, including colorectal, breast and prostate [108], while its role in E-cadherin gene silencing and EMT is also established. Inhibition of LSD1 in prostate cancer cells de-represses epithelial proteins, including E-cadherin and ZO1, and leads to reduced cell migration. Given that LSD1 was co-purified with CoREST, HDAC1/2 [109, 110], and SNAI1 [111], it is likely that LSD1/CoREST can be recruited by SNAI1 for H3K4me2 demethylation on E-cadherin promoter [111]. The findings that LSD1 exists in the ZEB1/2-containing CtBP1 co-repressor complex [83] suggest that it is a key player in ZEB1/2-mediated epithelial gene silencing and EMT as well. Although these results suggest an EMT-promoting role of LSD1, one study reveals that LSD1 is also an integral subunit of NuRD chromatin remodeling complex which suppresses TGF-B1 expression and negatively regulates EMT and breast cancer progression [112]. Intriguingly, TGF-β treatment up-regulates LSD1 expression in AML12 cells, which is required to confer EMT. However, the enzymatic activity of LSD1 antagonizes the same process [33]. As LSD1 can also remove the repressive H3K9 methylation after forming a complex with transcription activator AR [113], it is possible that TGFβ-induced co-activators (including AR) could interact with LSD1 and switch its substrate specificity to H3K9 [113, 114]. Therefore, LSD1 can modulate EMT through multiple mechanisms. Furthermore, over-expression of another H3K4 HDM JARID1A strongly associates with metastasis of multiple cancers. While JARID1A represses p27, but activates cyclin D1/E1 and ITGB1 expression for lung cancer cell growth and metastasis [115], it also up-regulates TNC expression to promote breast cancer cell invasion and metastasis [116]. In gastric cancer cells, TGF-β1 treatment induces expression of JARID1A which is recruited by p-SMAD3 to E-cadherin promoter for gene silencing and thus promotes malignant progression [117]. In breast cancer cells, another JARID family HDM JARID1B was also co-purified with NuRD complex, in which JARID1B and LSD1 coordinate to remove all three H3K4 methylation forms on CCL14 promoter to antagonize the chemokine pathway-mediated migration, angiogenesis, and metastasis [118]. On the contrary, JARID1B has been shown to promote EMT by suppressing the expression of miR-200s [119] or PTEN [120] through promoter H3K4 demethylation. Together, dynamic regulation of the permissive H3K4 methylation at different genomic loci through different molecular mechanisms can lead to opposite EMT outcomes.
Repressive histone modifications in EMT
Methylations on histone H3K9, H3K27, H4K20, and H4R3 (symmetric di-methylation, me2s) often associate with transcriptionally repressed genes. In mammals, H3K9 methylation is mainly introduced by G9a, GLP, SETDB1, and SUV39H1/2 [121]. G9a and GLP catalyze H3K9me1/2, while the elevated G9a expression has been correlated with poor prognosis in lung cancer patients. Mechanistically, G9a represses a cell adhesion molecule EPCAM by catalyzing H3K9me2 on the promoter to promote EMT and cancer metastasis of lung cancer cells in vitro and in vivo [122]. In breast cancer cells, G9a interacts with SNAI1 and is recruited to E-cadherin promoter for transcription silencing. Inhibiting G9a reduces promoter H3K9me2 as well as DNA methylation and further abrogates EMT and tumor metastasis [36]. The similar mechanism was also reported in head and neck squamous cell carcinoma [123], suggesting that SNAI1-G9a mediated repression could be a general mechanism during EMT. Intriguingly, SNAI1 also recruits SUV39H1 to E-cadherin promoter for H3K9me3 which is important for gene repression and EMT phenotypes in breast cancer cells [124]. Moreover, down-regulation of SETDB1 in breast cancer cells correlates with reduced EMT and CSC properties, suggesting that this HMTase has similar EMT-promoting role. However, it has been proposed that SETDB1 indirectly up-regulates STAT3 expression and induces TWIST and C-MYC to potentiate this process [125]. Alternatively, SETDB1 stabilizes ΔNp63α through protein interaction, while ΔNp63α might recruit SETDB1 to repress multiple EMT-related genes [126]. In addition, amplification of SETDB1 was observed in melanoma, lung, and liver cancers [127–129]. It represses a large set of genes, including HOX genes for melanoma progression [127] and confers outgrowth and advantages of liver cancer cells through methylating and stabilizing the oncogenic p53 mutants [128]. Surprisingly, it has also been shown that SETDB1 cooperates with SMAD2/3 to suppress EMT and metastasis of lung cancer cells [130].
As another major repressive modification, H3K27 methylation is catalyzed by EZH2 or its close homolog EZH1-containing polycomb repressive complex 2 (PRC2), which plays fundamental roles in embryonic development and stem cell differentiation [131]. Early studies uncovered that over-expression of EZH2 correlates with poor prognosis in prostate and breast cancer patients [132]. Comparing with primary tumor, PRC2/EZH2 expression is also elevated in the lymph mode metastatic breast cancer where E-cadherin expression is significantly reduced [133], indicating a promoting role of PRC2 in EMT and malignant progression. In pancreatic cancer cells, SNAI1 recruits PRC2 to E-cadherin promoter to catalyze H3K27me3 via interaction with a non-catalytic subunit SUZ12 [134]. SNAI1–PRC2 interaction can also be bridged by Ajuba LIM, PRMT5 or a mediator protein MEP50 [135, 136]. In gastric cancer cells, PRC2 is recruited through SUZ12 as well and catalyzes H3K27me3 on KLF2 and E-cadherin promoters [137]. In a TWIST1-induced EMT cell model, an H3K4me3-to-H3K27me3 switch was observed on 102 EMT marker gene promoters, including E-cadherin. The reverse H3K27me3-to-H3K4me3 switch occurred on the promoter of a set of up-regulated mesenchymal genes, including ZEB2, N-cadherin, PDGFRα, and ESRP1 [138]. Intriguingly, another set of promoters contains H3K4me3 and H3K27me3 marks simultaneously. This bivalent chromatin configuration often poises genes for subsequent activation or repression in response to specific differentiation cues [19]. The amount of genes harboring this bivalent chromatin state also increased significantly during TWIST1-induced EMT, indicating that the dedifferentiated mesenchymal cell state also associates with greater plasticity [138]. In breast cancer cells, the bivalent chromatin state was found on ZEB1 promoter to facilitate the rapid switch a CSC-like state upon different EMT signals [139].
Among five characterized H4K20 HMTases, SET8 and SUV420H1/2 are major enzymes catalyzing H4K20me1 and H4K20me2/3, respectively [140]. In breast cancer cells, SET8 interacts with TWIST1 and is targeted to E-cadherin and N-cadherin promoters for repression and activation, respectively. Although the mechanism of action is unclear, these opposite transcription regulations require the catalyzed H4K20me1 [141]. The bipolar roles of SET8 in regulating E/N-cadherin expression and EMT were also observed in prostate cancer cells, which are mediated through protein interactions with ZEB1 [142]. On the contrary, H4K20me3 plays a pivotal role in chromatin integrity, while the global loss of H4K20me3 is observed in a wide range of human cancers [143]. In breast cancer cells, loss of H4K20me3 correlates with reduced SUV420H2 expression and activation of a focal adhesion protein TNS3. Given that H4K20me3 on TNS3 promoter is necessary for gene silencing, this modification could also attenuate EMT and tumor progression through transcription regulation [144].
In addition to histone lysine methylation, symmetric di-methylation of H4R3 (H4R3me2s) has been associated with transcriptional repression. Catalyzed by PRMT5 or PRMT7, H4R3me2s prevents binding of MLL4 to nucleosomes through its PHD domain and indirectly reduces MLL4-mediated H3K4me3 [145]. PRMT5 interacts with SNAI1 through a bridging protein AJUBA, while knockdown of PRMT5 activates E-cadherin expression [135], demonstrating that PRMT5-mediated H4R3me2s is another crucial epigenetic event in SNAI1-induced repression and EMT. Consistently, PRMT5 expression reversely correlates with E-cadherin expression in a larger set of lung cell lines and has been linked to EMT and the loss of the bronchial epithelial phenotype of lung adenocarcinoma [146]. Similarly, over-expression of PRMT7 associates with EMT and breast cancer metastasis. Through a different mechanism, PRMT7 interacts with YY1 and HDAC3 and is recruited to E-cadherin promoter for transcription silencing [147].
Furthermore, these repressive methylation marks are dynamically regulated in cell. In addition to LSD1-AR complex discussed above, H3K9 methylation can be removed by several JmjC-domain-containing HDMs, including JMJD1A/B, JMJD2, PHF2/8, and KIAA1718 [108]. JMJD1A has been shown to up-regulate a long non-coding RNA MALAT1 and induce migration, invasion, and EMT in neuroblastoma cells [148]. JMJD2B is co-purified with the H3K4 HMTase MLL-2 complex, and is required for ERα-activated transcription [149]. In gastric cancer cells, JMJD2B interacts with β-catenin to activate Vimentin expression through promoter H3K9 demethylation and thus facilitates EMT and metastasis [150]. Differently, PHF8 removes H3K9 methylation on the promoter of Integrin genes and ROCK kinase to induce gene expression and promote migration and invasion of prostate cancer cells. Consistently, elevated PHF8 expression also correlates with poor prognosis of prostate cancer patients [151]. Similar to JMJD2B, a H3K27 HDM UTX interacts with another H3K4 HMTase MLL4 complex to activate expression of several oncogenes and prometastatic genes, including MMP-9/11 and SIX1, which leads to increased EMT and metastasis of breast cancer in vitro and in vivo [152]. In colon cancer cells, UTX not only demethylates H3K27me3 on the E-cadheirn promoter, but also recruits CBP for H3K27 acetylation. These epigenetic events delocalize PRC2 from the promoter and result in active transcription [153]. In breast cancer cells, UTX-MLL4 forms a complex with LSD1/HDAC1/DNMT1 on the promoter of several EMT-TFs. Through a histone demethylation independent mechanism, UTX represses SNAI1 and ZEB1/2 expression and negatively regulates EMT and CSC-like properties [154]. In mammary epithelial cells, TGF-β treatment also induces expression of JMJD3. This H3K27 HDM further activates SNAI1 expression through promoter H3K27 demethylation to facilitate EMT [155].
Many HMTases and HDMs inhibitors have been developed and characterized as well. Chaetocin is the first developed HMTase inhibitor targeting SUV39H1 without high selectivity [156]. Treatment with Chaetocin induces expression of E-cadherin and p15INK4B while reduces H3K9me3 levels on their promoters in a large panel of tumor cells, but does not affect global H3K9 methylation [157]. On the contrary, BIX01294 specifically inhibits enzymatic activity of G9a and GLP by occupying the histone peptide-binding pocket [158, 159]. BIX01294 treatment also activates E-cadherin expression and reverses EMT phenotypes in different cancer cells, accompanied by reduced H3K9me2 and increased H3K9 acetylation on the promoter [36, 123]. As BIX01294 is toxic to cells at higher concentration [159], another G9a/GLP inhibitor UNC0638 was developed with higher potency and selectivity [160]. UNC0638 treatment not only reduces H3K9me2 on G9a/GLP target gene promoters and up-regulates their expression, but also decreases H3K9me2 globally. A recent study reported that UNC0638 treatment activates E-cadherin expression in PANC-1-R cells, reverses EMT, and thus inhibits tumor growth in vivo [161]. Because of the importance of H3K27 methylation in cancer, several highly specific EZH2 inhibitors have also been developed, including GSK2816126 and EPZ-6438, which are currently in clinical trials for lymphoma and solid tumor/lymphoma [162]. However, whether they are able to reverse EMT under different clinical settings is unknown. In breast cancer and lung cancer cells, an LSD1 inhibitor Pargyline is able to reverse EMT phenotypes [111, 163]. Treatment with Parnate, another LSD1 inhibitor or blocking LSD1–SNAI1 interaction using a permeable peptide corresponding to the SNAG domain also activates E-cadherin expression and suppresses motility and invasiveness of multiple cancer cells derived from different origins in vitro and in vivo [164]. Recently, two highly specific LSD1 inhibitors, GSK2879552 and ORY-1001, have also been applied to clinical trials for the treatment of small cell lung cancer and acute leukemia [162]. Several inhibitors targeting JmjC-domain-containing HDMs have been reported as well. While JIB-04 inhibits activity of H3K4 and H4K9 HDMs and alters a subset of transcriptional pathways to attenuate lung cancer cell proliferation [165], YUKA1/2 specifically targets JARID1A with high potency and inhibits growth of different tumor cells. Although the action of mechanism is currently unknown, the ability of YUKA1 to mitigate drug tolerance to targeted therapy of breast and lung cancers highlights its clinic potential [166].
Histone modification-binding proteins in EMT
In some cases, histone modifications directly contribute to the regulation of chromatin dynamic. For example, H4K16 acetylation inhibits the folding of chromatin fiber in vitro and could thus impede the formation of chromatin compaction [167, 168]. In most cases, however, they are recognized by proteins containing distinct recognition domains. Bromodomain (BD) functions as an acetylated lysine binding motif, while two tandem BDs of BET (Bromodomain and extraterminal domain) family proteins (i.e., BRD4) recognize acetylated histones and further recruit transcription machineries to chromatin [169]. Recently, numerous studies reveal that pharmacologic inhibition of BRD4 with BET-specific BD inhibitors effectively blocks MYC expression and function in different types of cancer cells [170]. This inhibition also reduces WNT5A expression and suppresses invasion, CSC-like properties, and tumorigenicity of breast cancer cells in vitro and in vivo [171].
Given that the addition of methyl groups to lysine or arginine does not alter the charge of their side chains, histone methylation is believed to generate docking sites for binding proteins harboring specific motifs, including Chromodomain (CD), MBT, WD40 repeat, PHD finger, PWWP, Tudor, and Ankyrin repeat. These binding modules are able to distinguish methylation marks on different residues as well as different methylation states on the same residue to mediate distinct downstream functions [172]. CD-containing HP1 proteins are the first identified methyl-lysine-binding proteins which recognize methylated-H3K9 (methyl-H3K9) [173]. The conserved CD of MPP8 also functions as a specific methyl-H3K9 binding motif [174–176], while both HP1 and MPP8 have been implicated in EMT (see below). Several MBT domain family proteins can bind to mono- and di-methylated lysines, although these bindings lack sequence selectivity [177]. Four MBT-repeats domain of SFMBT1 recognize H3K4me2/3 and form a stable complex with LSD1. During TGF-β-induced EMT, SFMBT1 recruits LSD1 to E-cadherin promoter to remove H3K4me2 for gene silencing. Elevated SFMBT1 expression also correlates with several mesenchymal markers and unfavorable prognosis in breast cancer patients [178]. H3K4me2/3 is also recognized by WD40 repeat domain of WDR5 [179], and this binding lacks the lysine recognition motif in the crystal structure [180, 181]. Additionally, WDR5 is important for the assembly and activity of SET1 protein complex catalyzing H3K4me3 [179]. Under the hypoxia condition, WDR5 is induced, interacts with HDAC3, and further recruits SET1 complex to activate expression of mesenchymal genes to confer EMT [182]. Furthermore, WD40 repeat domain of EED, a key component of PRC2, recognizes H3K27me3. This binding recruits PRC2 to chromatin with pre-existing H3K27me3 to further spread the same methylation into adjacent regions [183]. Intriguingly, several chromatin-modifying enzymes also contain methyl-lysine binding motifs. DNMT3A possesses a PWWP domain which recognizes H3K36me3, and this binding facilitates nucleosomal DNA methylation [184]. The ankyrin repeats of G9a and GLP are involved in H3K9me1/2 binding which could retain G9a/GLP on chromatin for additional H3K9 methylation [185]. In addition, JMJD2A harbors a double Tudor domain which recognizes H3K4me3 and H4K20me3 [186]. Although these enzymes have been implicated in EMT, the importance of their methyl lysine binding modules still remains elusive.
In addition to inhibiting catalytic activities of chromatin-modifying enzymes, another strategy is to target specific binding proteins recognizing different histone modifications. Small molecules targeting the acetylated-histone-binding motif BD have been well characterized and the best example is the compounds selectively blocking the binding to acetylated lysine by BRD4, a well-studied BD-containing transcription co-activator [187, 188]. Given that BRD4 localizes in the super enhancer region of the key oncogene MYC, BRD4 inhibition preferentially abrogates MYC function in a variety of tumor cells [189]. It has been recently shown that the treatment of BRD4 inhibitor MS417 reduces distal metastasis of colorectal cancer in mouse xenograft models by modulating expression of several key EMT genes [190]. MS417 and another BRD4 inhibitor JQ1 also disrupt BRD4 binding to di-acetylated K73/K76 on TWIST and thus repress WNT5A expression to reverse EMT and metastasis of breast cancer in vitro and in vivo [171]. Currently, both BRD4 inhibitors are in multiple clinical trials for the treatment of different cancers. Several small molecule compounds targeting specific methylated histone-binding proteins have also been developed, including UNC1215 and UNC3866, which block the methyl-lysine binding mediated by MBT domain-containing protein L3MBTL3 [191] and CD-containing protein CBX4/7 [192], respectively. However, their function in reversing EMT and tumor progression is unknown.
Coordinated epigenetic regulations in EMT
Given that different chromatin-modifying proteins and the catalyzed modifications have been implicated in regulating the same set of genes, it is likely that they act in concert for the same pathway during EMT. One underlying mechanism is that multiple chromatin modifiers co-exist in the same protein complex to orchestrate transcription regulation [193]. Since most chromatin-modifying enzymes lack DNA-binding motifs, they are normally recruited to target promoters by different EMT-TFs. HDAC1/2, G9a/GLP and HP1 have been co-purified with ZEB1/2 in CtBP1 co-repressor complex [83, 107]. In a possible scenario, ZEB1/2 first initiates transcription repression and recruits the protein complex to E-cadherin promoter. HADC1/2 next deacetylates histones, while the primed H3K9 will be methylated by G9a/GLP. Meanwhile, LSD1 removes H3K4me1/2, and the un-methylated H3K4 could also prevent H3K9 from re-acetylation (Fig. 1a) [194, 195]. The established H3K9 methylation further recruits HP1 proteins to lock the local chromatin in a repressive state. Through protein interactions, HP1 could also recruit DNMT1 for DNA methylation [196] and turn off transcription (Fig. 1b). Although this repression pathway suggests an EMT-promoting role of HP1, its expression is down-regulated in invasive breast cancer cells. Knockdown of HP1 increases invasiveness without affecting cell growth, whereas over-expression of HP1 reduces it [197], suggesting that HP1 proteins could also attenuate EMT through mechanisms independent of E-cadherin silencing.
Fig. 1.
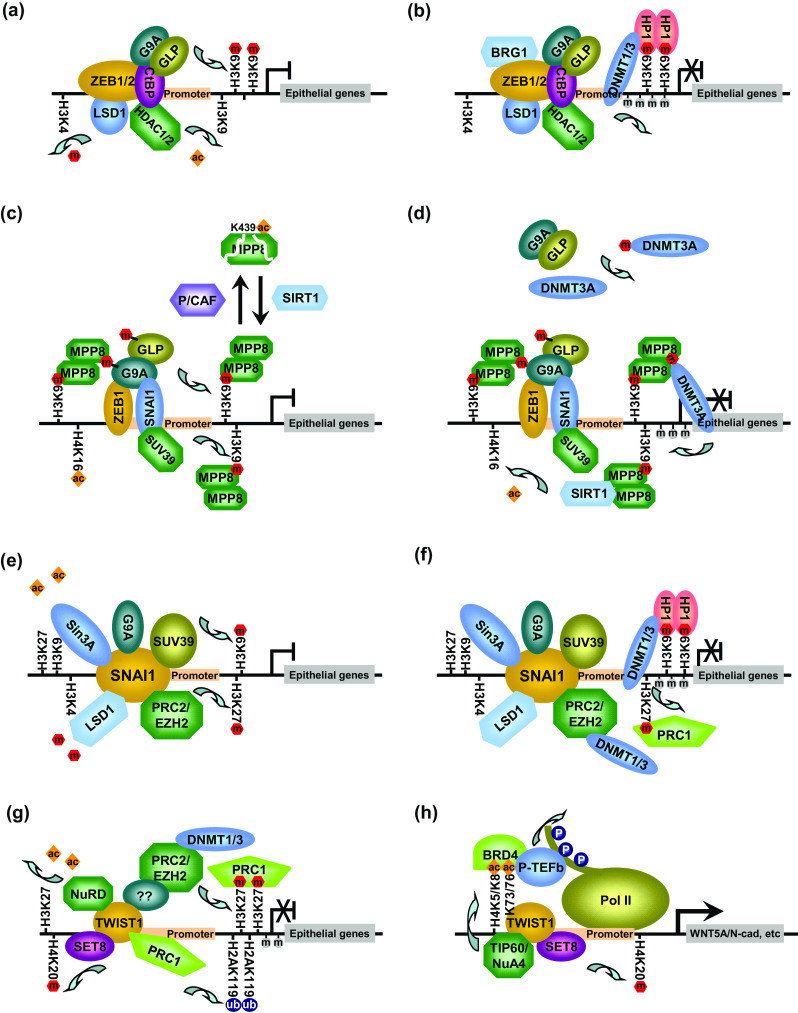
Coordinated transcription regulations mediated by different EMT-TFs and chromatin-modifying proteins. a ZEB1/2 recruits CtBP co-repressor complex, in which HDAC1/2 and LSD1 remove histone acetylation and H3K4 methylation, respectively, whereas G9a/GLP introduces H3K9 methylation. b H3K9 methylation marks are recognized by HP1 proteins which further recruit DNMTs for promoter CpG methylation. c ZEB1 and SNAI1 recruit multiple H3K9 HMTases to target promoters, while the resulting methylation and G9a/GLP self-methylation could target MPP8 for methyl-H3K9 binding. MPP8 protein stability is dynamically regulated by PCAF- and SIRT1-catalyzed MPP8–K439 acetylation and deacetylation. d G9a/GLP also methylates DNMT3A, while the resulting K47 methylation facilitates DNMT3A recruitment by MPP8. e SNAI1 recruits multiple histone-modifying enzymes to remove permissive histone acetylation and H3K4 methylation to introduce repressive H3K9 methylation and H3K27 methylations. f DNMTs are further recruited by PRC2 and HP1 proteins to catalyze promoter CpG methylation. PRC1 is also recruited by binding to methyl-H3K27 marks on the promoter. g On epithelial gene promoters, TWIST1 recruits NuRD complex, SET8, and PRC2 for histone deacetylation, H4K20 methylation, and H3K27 methylation, respectively. It also recruits PRC1 directly and indirectly to introduce H2AK119 mono-ubiquitination. h On mesenchymal gene promoters, SET8-catalyzed H4K20 methylation serves as transcription activation mark (N-cadherin promoter). TWIST1 also forms a protein complex with TIP60/NuA4 which acetylates histone H4 and TWIST1. The acetylated lysines are recognized by BRD4 which recruits P-TEFb and super elongation complex to facilitate transcription (WNT5a promoter)
Owing to the importance of H3K9 methylation in E-cadherin silencing and EMT, we have characterized MPP8 as another methyl-H3K9-binding protein. Different from HP1s, MPP8 expression is elevated in various invasive breast and lung cancer cells. Knockdown of MPP8 de-represses E-cadherin expression and reduces cell migration and invasion, indicating that it has an EMT-promoting role. MPP8 not only targets E-cadherin promoter through methyl-H3K9 binding, but also interacts with G9a/GLP and DNMT3A. Knocking-down MPP8 or disabling its methyl-H3K9 binding impairs DNMT3A binding on E-cadherin and reduces DNA methylation, but does not affect GLP and H3K9 methylation. These findings suggest a repression pathway, in which G9a/GLP methylates H3K9 on E-cadherin promoter for MPP8 targeting, while MPP8 further recruits DNMT3A for CpG methylation [175]. Furthermore, G9a/GLP methylates DNMT3A at K47 as well as their N-termini at G9a-K185 and GLP-K205, respectively [198, 199]. Intriguingly, MPP8 binding to G9a/GLP and DNMT3A is mediated through these methylated lysines. Given that the CD of MPP8 forms a dimer in structures [200, 201], MPP8 could bridge DNMT3A and G9a/GLP to assemble a repressive trimeric protein complex on the chromatin through binding to different methyl-lysines and couple H3K9 methylation and DNA methylation for epithelial genes silencing and EMT (Fig. 1c, d).
Although inhibition of HDAC1/2 also activates E-cadherin expression in multiple cancer cell lines, MPP8-mediated repression is independent of class I/II HDACs [175]. However, MPP8 interacts with a class III HDAC SIRT1 to coordinate silencing of E-cadherin and other genes [202]. In prostate and breast cancer cells, knocking-down SIRT1 or MPP8, or disabling methyl-H3K9 binding by MPP8 or MPP8–SIRT1 interaction leads to similar de-repression of E-cadherin, restoration of epithelial cell morphology, and reduction of cell migration and invasion [202]. Disabling methyl-H3K9 binding by MPP8 or MPP8–SIRT1 interaction also decreases SIRT1 localization on target promoters and increases H4K16 acetylation, suggesting that MPP8 binds to methyl-H3K9 and recruits SIRT1 for H4K16 deacetylation [202]. As H4K16 acetylation is able to de-compact chromatin directly [168], removing this modification could be critical for MPP8-mediated transcription repression. Intriguingly, SIRT1 also removes acetylation on MPP8-K439 which is catalyzed by PCAF. Given that K439 acetylation accelerates MPP8 protein degradation through the ubiquitin–proteasome pathway, SIRT1-mediated deacetylation stabilizes MPP8 as a feed-forward regulatory loop in MPP8-mediated repression pathway (Fig. 1c, d). Although SIRT1 is methylated by G9a at K622 [203], MPP8–SIRT1 interaction is independent of SIRT1–K622 methylation, revealing that MPP8 recruits SIRT1 and DNMT3A through different mechanisms. While CpG methylation prevents RNA Pol II binding on target promoters, H4K16 acetylation facilitates the release of RNA Pol II from promoter-proximal pausing and the transition into active elongation [204]. Therefore, MPP8-directed promoter DNA methylation and H4K16 deacetylation could introduce multiple barriers for transcription machineries.
Furthermore, SNAI1 associates with SIN3A and PRC2 complexes and is required for histone deacetylation and H3K27 methylation on E-cadherin promoter and gene silencing [61, 134]. A recent study also uncovered SNAI1–LSD1 interaction which not only recruits LSD1 to demethylate H3K4 on E-cadherin promoter for repression, but also increases protein stability to enhance the function of SNAI1-LSD1 co-repressor complex in EMT and metastasis of breast cancer cells [111]. Additionally, SNAI1 has been shown to recruit G9a and SUV39H1 to E-cadherin promoter through different motifs, while the catalyzed H3K9me2 and H3K9me3 are required for gene silencing [36, 124]. Inhibition of LSD1, G9a, or SUV39H1 results in similar reversed EMT phenotypes and reduced breast cancer progression and metastasis in vivo. Although these results demonstrate the role of SNAI1 in targeting different chromatin modifiers for E-cadherin silencing and EMT (Fig. 1e), whether these co-repressor protein complexes are recruited simultaneously and how they coordinate for transcription repression is currently not clear.
In addition to reduced H3K9 methylation, inhibiting G9a or SUV39H1 leads to increased H3K9 acetylation and decreased DNA methylation on E-cadherin promoter, indicating that DNA methylation plays an important role in SNAI1-mediated repression and EMT as well. Consistent with genome-wide correlation between H3K9 methylation and DNA methylation [205], G9a has been shown to interact with DNMT1 and DNMT3A/B through its N-terminal and Ankyrin-repeat domains, respectively [206, 207]. However, these protein interactions may not be the major mechanism to target DNMTs as inhibiting G9a and GLP’s enzymatic activity is sufficient to reduce DNA methylation on E-cadherin promoter [36]. The findings that HP1 proteins interact with different DNMTs [207, 208] suggest another possibility that DNMTs are recruited after HP1 binds to H3K9 methylation catalyzed by G9a/GLP and SUV39H1. SUV39H1 could also directly recruit HP1 proteins, because they form a heterodimer through their Chromo-Shadow domains (CSDs) [209]. Although MPP8 does not interact with SUV39H1, it recognizes self-methylated lysines in G9a/GLP [198]. Binding to these methyl-lysines could contribute to MPP8 promoter targeting after G9a/GLP is recruited by SNAI1 or other EMT-TFs (Fig. 1c). Owing to the higher binding affinity to methyl-H3K9 [200], MPP8 could rapidly switch to methyl-H3K9 binding on target promoters and further recruit DNMT3A through DNMT3A-K47 methylation to establish H3K9 methylation coupled DNA methylation (Fig. 1d) [175, 198]. Furthermore, PRC2 could direct promoter DNA methylation after being recruited by SNAI1 [134], because its catalytic subunit EZH2 also associates with different DNMTs [210]. Intriguingly, H3 acetylation and H3K4 methylation are able to disrupt DNMT3A’s chromatin binding through its ADD domain [211, 212]. In this case, SNAI1-recruited LSD1 and HDAC1/2 could remove these inhibitory modifications to facilitate chromatin DNA methylation by DNMT3A. Together, these findings suggest that SNAI1 not only directly recruits histone-modifying enzymes for repression, but also promotes DNA methylation through different indirect mechanisms (Fig. 1f).
TWIST1 has also been shown to associate with different chromatin-modifying enzymes. An affinity purification of Flag-TWIST identified several components of NuRD chromatin remodeling complex. Among them, TWIST directly interacts with Mi2β, MTA2, and RbAp46, and likely targets NuRD complex for histone deacetylation and chromatin remodeling on E-cadherin promoter. These epigenetic events together lead to gene silencing and promote EMT and breast cancer metastasis [53]. Under the hypoxia condition, up-regulated HIF-1α activates TWIST1 expression by directly binding to the hypoxia-response element (HRE) on the promoter and thus promotes EMT and metastatic progression [213]. In head and neck squamous cell carcinoma cells, HIF1α-induced TWIST1 binds to the E-box element in BMI1 gene, activates expression of this polycomb repressive complex 1 (PRC1) subunit, and potentiates EMT and CSC-like properties. Mechanistically, TWIST1 also interacts with BMI1 and recruits PRC1 to E-cadherin and p16INK4a promoters for H2AK119 mono-ubiquitination and gene silencing [214]. Intriguingly, knockdown of TWIST1 reduces PRC2 promoter targeting and H3K27me3, indicating that TWIST1 could also recruit PRC2. It has been shown that the PRC1 core subunits HPCs can recognize methyl-H3K27, and this H3K27me3 binding contributes to the recruitment of PRC1 [131]. Therefore, TWIST1 could also target PRC1 indirectly (Fig. 1g) and the indirect PRC1 targeting is facilitated by SNAI1 through recruitment of PRC2 [134]. Moreover, inhibition of HDACs reduces both PRC1 and PRC2 promoter localizations [214, 215], suggesting that H3K27 deacetylation is a prerequisite for PRC2-catalyzed H3K27me3 methylation. In this case, TWIST1-recruited NuRD complex [53] or other HDAC-containing complexes recruited by SNAI1 or ZEB1 [61, 83] could facilitate PRC1/2 recruitment as well. Together, these results illustrate a complex regulatory network for transcription repression which is orchestrated by different chromatin-modifying proteins and EMT-TFs.
Moreover, TWIST has been shown to associate with different sets of chromatin-modifying proteins in different cell lines. While NuRD complex was co-purified with Flag-TWIST1 from HEK-293 cells [53], a similar Flag-TWIST1 purification using HeLa-S3 cells identified E12/E47, BRD4, and NuA4 HAT complex. This finding leads to a discovery that TWIST1 is di-acetylated at K73/K76 by TIP60, the catalytic subunit of NuA4 complex which also di-acetylates H4K5/K8 [171]. Intriguingly, di-acetylated H4 and TWIST1 are recognized by BRD4 through its first and second BDs. Although mutation of both acetylation sites on TWIST1 does not affect its localization on WNT5A promoter/enhancer, it abrogates the sequential recruitment of BRD4, P-TEFb, and RNA-Pol II. These results support a model, in which TWIST1 assembles an active transcription elongation complex through the acetylated K73/K76 and BRD4 binding to activate WNT5A expression and promote EMT and tumorigenicity of breast cancer cells (Fig. 1h) [171]. In MCF7 breast cancer cells, Flag-TWIST1 was co-purified with SET8, BRCA1-associated protein BRAP, NF-κB subunit RELA, PPP2CA, and HES6 [141]. Surprisingly, SET8 and TWIST1 mediate E-cadherin repression and N-cadherin activation simultaneously to promote cell invasion and EMT. While SET8 localization on both promoters relies on TWIST1, TWIST1-mediated repression/activation also requires SET8 and the catalyzed H4K20me1, arguing that the same protein complex could contribute to opposite functions on different genomic loci (Fig. 1g, h). Although H4K20me1 has been implicated in transcription activation and repression [216, 217], the molecular mechanism underlying the bipolar contextual roles of TWIST1-SET8 is not clear. One possibility is that H4K20me1 could further coordinate different chromatin modifications or recruit distinct group of effectors to different promoters. In addition, the findings of three distinct TWIST1 protein complexes in different cell lines suggest that the diverse functions of TWIST1 could be fine-tuned by different groups of interacting proteins and different chromatin environments.
MicroRNAs and lncRNAs in EMT
In addition to various chromatins modifying proteins, the importance of several classes of non-coding RNAs (ncRNAs) in transcription regulation and EMT is now being appreciated, representing another category of epigenetic mechanisms. ncRNAs can be briefly classified into small ncRNA (<200nt) and long ncRNA (lncRNA) and contribute to a variety of cellular functions. Among them, miRNAs are the best-studied class in epithelial–mesenchymal plasticity [218]. Derived from long primary transcripts and processed through sequential cleavage steps, miRNAs function post-transcriptionally and target mRNAs for degradation, inhibition of translation, or a combination of both [219]. Dozens of miRNAs have been identified to target 3′UTR of ZEB1/2 mRNAs to suppress their expression in various cancer cells [220], while five miR-200 family members are essential regulators for differentiation and epithelial characteristics of a wide range of cell types and tissues [221–223]. During EMT, ZEB1/2 binds to miR-200s and miR-205 promoters and represses their transcription. Conversely, miR-200s and miR-205 suppress ZEB1/2 expression by directly targeting 3′-UTR of their mRNAs to maintain epithelial properties [224, 225]. Therefore, ZEB1/2 and miR-200s/205 form a reciprocal feedback regulatory loop which functions as a molecular motor for EMT, tumor progression, and metastasis [226–228]. In addition, miR-200s repress TGF-β2 and β-catenin expression to attenuate EMT [227, 229]. They also target mRNA of several chromatin-modifying enzymes, such as BMI1, SUZ12, and SIRT1, to negatively regulate EMT and CSC-like properties [230–232]. In tongue cancer cells, SNAI1/2 recruits PRC2 to suppress transcription of miR-101 which also targets 3′-UTR of EZH2 mRNA [233], representing another axis of miRNA signaling for EMT [234]. Furthermore, mRNA of SNAI1/2 and TWIST1/2 is targeted by different large groups of miRNAs, however, the reciprocal regulatory loop was not reported [220]. Intriguingly, SNAI1/2 mRNAs are directly targeted by miR34 family members in a variety of cancer cells, whereas the expression of miR34s is positively regulated by wild-type p53. These findings reveal a miR34-SNAI1/2 axis in p53 loss-of-function-induced EMT [235]. In hepatocellular carcinoma cells, p53 also up-regulates miR-200s and miR-192 family members which further target ZEB1/2 mRNAs to abrogate EMT [236]. It has been reported that oncogenic p53 mutant represses transcription of miR130b which also targets ZEB1 mRNA and inhibits EMT [237]. In addition to p53, the expression of many miRNAs is regulated by different transcription factors, including EMT-TFs. These miRNAs further target multiple EMT-related genes and contribute to EMT [220]. While MYC-activated miR-9 targets 3′-UTR of E-cadherin mRNA to facilitate EMT and metastasis of breast carcinoma cells [238], TWIST1-induced miR-424 activates N-cadherin and Fibronectin expression through unknown mechanism without affecting E-cadherin expression [239].
Moreover, expression level of miRNAs associates with various epigenetic modifications on their promoters. For example, expression of miR-200s/205 reversely correlates with promoter CpG methylation in various tumor cells [41, 42]. This DNA methylation pattern can be readily shifted during EMT or MET in vitro and in vivo [40, 240], indicating that DNA methylation is also critical to regulate miRNA expression. Consistent with this notion, a global analysis uncovered a strong inter-individual concordance between miRNA expression and epigenetic states on their promoters in human mammary epithelial cells and mammary fibroblasts. While repression of miR-200c/141 and miR-200a/200b/429 associates with promoter CpG methylation and H3K27me3, respectively, silencing of miR-205 requires both DNA methylation and H3K27me3 in fibroblasts [41]. During EMT, the promoter of miR-200a/200b/429 and miR-200c/141 is also decorated by distinct group of repressive histone modifications [41], suggesting a possibility that different chromatin-modifying enzymes and DNMTs are orchestrated with EMT-TFs to regulate transcription of these miRNAs. However, molecular details underlying these epigenetic regulations are largely unknown.
Recent transcriptome studies also identified a large group of lncRNAs which are differentially transcribed in various metastatic tumors and contribute to EMT and tumor progression through diverse mechanisms [241]. Functioning as a chromatin modulator, lncRNA HOTAIR interacts with both PRC2 and LSD1 and targets these histone-modifying enzymes to repress HOX genes and many metastasis suppressor genes across genome [242]. HOTAIR expression is significantly elevated in several types of cancers and associates with metastasis and poor prognosis [243]. In contrast, lncRNA SChLAP1 impedes genome-wide chromatin targeting of SWI/SNF remodeling complex and repress its target gene expression. SChLAP1 is over-expressed in ~25 % of prostate cancers and is also critical for invasiveness and metastasis [244]. In breast cancer cells, lncRNA-ROR promotes EMT and metastasis in vitro and in vivo by antagonizing miR-205-mediated repression of ZEB2, revealing a crosstalk between lncRNA and miRNA [245]. Furthermore, ncRNA-a7 (also known as treRNA) functions as an enhancer lncRNA to facilitate transcription of its neighboring gene SNAI1 [246]. Enforced expression of ncRNA-a7 in breast cancer cells promotes EMT and metastasis, while its over-expression was detected in metastatic breast cancer tissues. Intriguingly, ncRNA-a7 also associates with a hRNP complex which binds to 3′-UTR of E-cadherin mRNA and suppresses its translation [247]. TGF-β-induced lncRNA-HIT also inhibits E-cadherin expression to promote EMT and breast cancer metastasis. However, it suppresses E-cadherin transcription via unknown mechanism [248]. Together, these examples demonstrate the critical regulatory roles of lncRNAs in control of EMT and cancer metastasis at multiple molecular layers [249].
Conclusions and perspectives
The abnormal activation of EMT, a developmental program, has been proposed to play important roles in the initiation and progression of carcinomas. This process involves a complex transcriptional reprogramming of a large set of EMT-related genes, and is triggered and regulated by different EMT-TFs. The studies within the past decade have revealed that different chromatin modifications are essential mediators of the activity of EMT-TFs and have an indispensable role in EMT and the reverse MET processes. In concert with different EMT-TFs and classical oncogenic pathways, a variety of chromatin modifications constitute a sophisticated regulatory network to coordinate the simultaneous up- and down-regulation of various mesenchymal and epithelial genes. This orchestrated action also occurs at multiple molecular layers to ensure the precise control of epithelial–mesenchymal plasticity which associates with cell motility and invasiveness that could have detrimental effects on the organism.
In spite of fast-growing information about epigenetic modifications in EMT, it will be very important to further dissect mechanisms by which different chromatin-modifying enzymes and the catalyzed modifications contribute to this dedifferentiation program. The functional importance of the non-catalytic domains of these enzymes and molecular details underlying different modification signals are recognized and transduced into biological functions are also limited. Thus, molecular elucidation will provide greater insights into the coordinated epigenetic regulation of gene expression programs during EMT. Furthermore, understanding mechanisms underlying the regulation of EMT-TFs and chromatin-modifying proteins by different EMT-inducing stimuli, ncRNAs, and alternative splicing is also important to elucidate the complex epigenetic regulatory network.
Furthermore, the biochemically reversible nature of epigenetic modifications not only provides a platform for rapid switch of a variety of epithelial and mesenchymal genes during EMT and MET, but also presents multiple therapeutic opportunities. Over the last decade, considerable progress has been achieved in the development of potent and selective small-molecule inhibitors targeting specific chromatin modifiers, and many of them are currently being evaluated in clinic studies. The application of BRD4 inhibitors also highlights the possibility to interfere specific bindings to different histone methylation marks. Comparing with inhibiting catalytic activity of chromatin-modifying enzymes, blocking specific binding event could render higher selectivity to target given epigenetic pathways in different clinic applications. Although reversing EMT is conceptually attractive for anti-cancer therapy, current understanding of EMT-induced metastasis is derived from in vitro and mouse studies. Using different mouse models, two recent studies provide evidence that EMT is not a prerequisite for metastatic dissemination, suggesting that the in vivo regulation of tumor progression and metastasis is more sophisticated. However, both studies demonstrate that EMT cells significantly contribute to chemoresistance and recurrent metastasis after standard therapy [250, 251]. Therefore, selected EMT-targeting epigenetic drugs can also be utilized to synergize the conventional chemotherapy and to overcome drug resistance. In addition, specific epigenetic inhibitors could serve as powerful tools to facilitate the functional characterization of different epigenetic pathways in EMT in vivo as well as many other important physiological and pathological processes.
Acknowledgments
This work was supported by Grant from the National Cancer Institute (R01CA172774) to J Fang. This work was also supported in part by Core Facilities at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center.
References
- 1.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–376. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 2.Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119(6):1417–1419. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 5.Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25(11):675–686. doi: 10.1016/j.tcb.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 8.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 9.Zheng H, Kang Y. Multilayer control of the EMT master regulators. Oncogene. 2014;33(14):1755–1763. doi: 10.1038/onc.2013.128. [DOI] [PubMed] [Google Scholar]
- 10.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 11.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 12.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19(11):1438–1449. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68(10):3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 14.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28(1–2):151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 15.Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011;71(1):245–254. doi: 10.1158/0008-5472.CAN-10-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dave N, Guaita-Esteruelas S, Gutarra S, Frias A, Beltran M, Peiro S, de Herreros AG. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem. 2011;286(14):12024–12032. doi: 10.1074/jbc.M110.168625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felsenfeld G. A brief history of epigenetics. Cold Spring Harb Perspect Biol. 2014 doi: 10.1101/cshperspect.a018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Ting AH, McGarvey KM, Baylin SB. The cancer epigenome-components and functional correlates. Genes Dev. 2006;20(23):3215–3231. doi: 10.1101/gad.1464906. [DOI] [PubMed] [Google Scholar]
- 20.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 21.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol Med. 2007;13(9):373–380. doi: 10.1016/j.molmed.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113(2):207–219. doi: 10.1016/S0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez-Perez A, Jene-Sanz A, Lopez-Bigas N. The mutational landscape of chromatin regulatory factors across 4623 tumor samples. Genome Biol. 2013;14(9):r106. doi: 10.1186/gb-2013-14-9-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weissman B, Knudsen KE. Hijacking the chromatin remodeling machinery: impact of SWI/SNF perturbations in cancer. Cancer Res. 2009;69(21):8223–8230. doi: 10.1158/0008-5472.CAN-09-2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanchez-Tillo E, Lazaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, Engel P, Postigo A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29(24):3490–3500. doi: 10.1038/onc.2010.102. [DOI] [PubMed] [Google Scholar]
- 26.Jordan NV, Prat A, Abell AN, Zawistowski JS, Sciaky N, Karginova OA, Zhou B, Golitz BT, Perou CM, Johnson GL. SWI/SNF chromatin-remodeling factor Smarcd3/Baf60c controls epithelial-mesenchymal transition by inducing Wnt5a signaling. Mol Cell Biol. 2013;33(15):3011–3025. doi: 10.1128/MCB.01443-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy N, Malik S, Villanueva KE, Urano A, Lu X, Von Figura G, Seeley ES, Dawson DW, Collisson EA, Hebrok M. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015;29(6):658–671. doi: 10.1101/gad.256628.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv. 2015;1(5):e1500447. doi: 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nat Struct Mol Biol. 2013;20(3):274–281. doi: 10.1038/nsmb.2518. [DOI] [PubMed] [Google Scholar]
- 30.Cedar H, Bergman Y. Programming of DNA methylation patterns. Annu Rev Biochem. 2012;81:97–117. doi: 10.1146/annurev-biochem-052610-091920. [DOI] [PubMed] [Google Scholar]
- 31.Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev. 2012;22(1):50–55. doi: 10.1016/j.gde.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 32.Carmona FJ, Davalos V, Vidal E, Gomez A, Heyn H, Hashimoto Y, Vizoso M, Martinez-Cardus A, Sayols S, Ferreira HJ, Sanchez-Mut JV, Moran S, Margeli M, Castella E, Berdasco M, Stefansson OA, Eyfjord JE, Gonzalez-Suarez E, Dopazo J, Orozco M, Gut IG, Esteller M. A comprehensive DNA methylation profile of epithelial-to-mesenchymal transition. Cancer Res. 2014;74(19):5608–5619. doi: 10.1158/0008-5472.CAN-13-3659. [DOI] [PubMed] [Google Scholar]
- 33.McDonald OG, Wu H, Timp W, Dodsi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol. 2011;18(8):867–874. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lombaerts M, van Wezel T, Philippo K, Dierssen JW, Zimmerman RM, Oosting J, van Eijk R, Eilers PH, van de Water B, Cornelisse CJ, Cleton-Jansen AM. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Brit J Cancer. 2006;94(5):661–671. doi: 10.1038/sj.bjc.6602996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reinhold WC, Reimers MA, Maunakea AK, Kim S, Lababidi S, Scherf U, Shankavaram UT, Ziegler MS, Stewart C, Kouros-Mehr H, Cui H, Dolginow D, Scudiero DA, Pommier YG, Munroe DJ, Feinberg AP, Weinstein JN. Detailed DNA methylation profiles of the E-cadherin promoter in the NCI-60 cancer cells. Mol Cancer Ther. 2007;6(2):391–403. doi: 10.1158/1535-7163.MCT-06-0609. [DOI] [PubMed] [Google Scholar]
- 36.Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, Evers BM, Zhou BP. G9a interacts with snail and is critical for snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012;122(4):1469–1486. doi: 10.1172/JCI57349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fukagawa A, Ishii H, Miyazawa K, Saitoh M. deltaEF1 associates with DNMT1 and maintains DNA methylation of the E-cadherin promoter in breast cancer cells. Cancer medicine. 2015;4(1):125–135. doi: 10.1002/cam4.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papageorgis P, Lambert AW, Ozturk S, Gao F, Pan H, Manne U, Alekseyev YO, Thiagalingam A, Abdolmaleky HM, Lenburg M, Thiagalingam S. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res. 2010;70(3):968–978. doi: 10.1158/0008-5472.CAN-09-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan EJ, Kahata K, Idas O, Thuault S, Heldin CH, Moustakas A. The high mobility group A2 protein epigenetically silences the Cdh1 gene during epithelial-to-mesenchymal transition. Nucleic Acids Res. 2015;43(1):162–178. doi: 10.1093/nar/gku1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davalos V, Moutinho C, Villanueva A, Boque R, Silva P, Carneiro F, Esteller M. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene. 2012;31(16):2062–2074. doi: 10.1038/onc.2011.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vrba L, Jensen TJ, Garbe JC, Heimark RL, Cress AE, Dickinson S, Stampfer MR, Futscher BW. Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS One. 2010;5(1):e8697. doi: 10.1371/journal.pone.0008697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wiklund ED, Bramsen JB, Hulf T, Dyrskjot L, Ramanathan R, Hansen TB, Villadsen SB, Gao S, Ostenfeld MS, Borre M, Peter ME, Orntoft TF, Kjems J, Clark SJ. Coordinated epigenetic repression of the miR-200 family and miR-205 in invasive bladder cancer. Int J Cancer. 2011;128(6):1327–1334. doi: 10.1002/ijc.25461. [DOI] [PubMed] [Google Scholar]
- 43.Ning X, Shi Z, Liu X, Zhang A, Han L, Jiang K, Kang C, Zhang Q. DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer Lett. 2015;359(2):198–205. doi: 10.1016/j.canlet.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 44.Yu Y, Wu J, Guan L, Qi L, Tang Y, Ma B, Zhan J, Wang Y, Fang W, Zhang H. Kindlin 2 promotes breast cancer invasion via epigenetic silencing of the microRNA200 gene family. Int J Cancer. 2013;133(6):1368–1379. doi: 10.1002/ijc.28151. [DOI] [PubMed] [Google Scholar]
- 45.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, Schmidt H, Zhang Q, Osborne JR, Lin L, O’Laughlin M, McMichael JF, Delehaunty KD, McGrath SD, Fulton LA, Magrini VJ, Vickery TL, Hundal J, Cook LL, Conyers JJ, Swift GW, Reed JP, Alldredge PA, Wylie T, Walker J, Kalicki J, Watson MA, Heath S, Shannon WD, Varghese N, Nagarajan R, Westervelt P, Tomasson MH, Link DC, Graubert TA, DiPersio JF, Mardis ER, Wilson RK. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, Shi JY, Zhu YM, Tang L, Zhang XW, Liang WX, Mi JQ, Song HD, Li KQ, Chen Z, Chen SJ. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43(4):309–315. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 47.Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15(3):152–165. doi: 10.1038/nrc3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao Q, Steine EJ, Barrasa MI, Hockemeyer D, Pawlak M, Fu D, Reddy S, Bell GW, Jaenisch R. Deletion of the de novo DNA methyltransferase Dnmt3a promotes lung tumor progression. Proc Natl Acad Sci USA. 2011;108(44):18061–18066. doi: 10.1073/pnas.1114946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sansom OJ, Maddison K, Clarke AR. Mechanisms of disease: methyl-binding domain proteins as potential therapeutic targets in cancer. Nat Clin Pract Oncol. 2007;4(5):305–315. doi: 10.1038/ncponc0812. [DOI] [PubMed] [Google Scholar]
- 50.McCabe MT, Brandes JC, Vertino PM. Cancer DNA methylation: molecular mechanisms and clinical implications. Clin Cancer Res. 2009;15(12):3927–3937. doi: 10.1158/1078-0432.CCR-08-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koizume S, Tachibana K, Sekiya T, Hirohashi S, Shiraishi M. Heterogeneity in the modification and involvement of chromatin components of the CpG island of the silenced human CDH1 gene in cancer cells. Nucleic Acids Res. 2002;30(21):4770–4780. doi: 10.1093/nar/gkf593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011;11(8):588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu J, Qin L, He T, Qin J, Hong J, Wong J, Liao L, Xu J. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21(2):275–289. doi: 10.1038/cr.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu J, Zhu W, Xu W, Yao W, Zhang B, Xu Y, Ji S, Liu C, Long J, Ni Q, Yu X. Up-regulation of MBD1 promotes pancreatic cancer cell epithelial-mesenchymal transition and invasion by epigenetic down-regulation of E-cadherin. Curr Mol Med. 2013;13(3):387–400. [PubMed] [Google Scholar]
- 55.Darwanto A, Kitazawa R, Maeda S, Kitazawa S. MeCP2 and promoter methylation cooperatively regulate E-cadherin gene expression in colorectal carcinoma. Cancer Sci. 2003;94(5):442–447. doi: 10.1111/j.1349-7006.2003.tb01462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jones J, Wang H, Karanam B, Theodore S, Dean-Colomb W, Welch DR, Grizzle W, Yates C. Nuclear localization of Kaiso promotes the poorly differentiated phenotype and EMT in infiltrating ductal carcinomas. Clin Exp Metastasis. 2014;31(5):497–510. doi: 10.1007/s10585-014-9644-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones J, Wang H, Zhou J, Hardy S, Turner T, Austin D, He Q, Wells A, Grizzle WE, Yates C. Nuclear Kaiso indicates aggressive prostate cancers and promotes migration and invasiveness of prostate cancer cells. Am J Pathol. 2012;181(5):1836–1846. doi: 10.1016/j.ajpath.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang H, Liu W, Black S, Turner O, Daniel JM, Dean-Colomb W, He QP, Davis M, Yates C. Kaiso, a transcriptional repressor, promotes cell migration and invasion of prostate cancer cells through regulation of miR-31 expression. Oncotarget. 2016;7(5):5677–5689. doi: 10.18632/oncotarget.6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473(7347):343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peinado H, Ballestar E, Esteller M, Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol. 2004;24(1):306–319. doi: 10.1128/MCB.24.1.306-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsai YP, Chen HF, Chen SY, Cheng WC, Wang HW, Shen ZJ, Song C, Teng SC, He C, Wu KJ. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 2014;15(12):513. doi: 10.1186/s13059-014-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Auerkari EI. Methylation of tumor suppressor genes p16(INK4a), p27(Kip1) and E-cadherin in carcinogenesis. Oral Oncol. 2006;42(1):5–13. doi: 10.1016/j.oraloncology.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 64.Corn PG, Smith BD, Ruckdeschel ES, Douglas D, Baylin SB, Herman JG. E-cadherin expression is silenced by 5′ CpG island methylation in acute leukemia. Clin Cancer Res. 2000;6(11):4243–4248. [PubMed] [Google Scholar]
- 65.Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JG. Methylation patterns of the E-cadherin 5′ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem. 2000;275(4):2727–2732. doi: 10.1074/jbc.275.4.2727. [DOI] [PubMed] [Google Scholar]
- 66.Nam JS, Ino Y, Kanai Y, Sakamoto M, Hirohashi S. 5-Aza-2′-deoxycytidine restores the E-cadherin system in E-cadherin-silenced cancer cells and reduces cancer metastasis. Clin Exp Metastasis. 2004;21(1):49–56. doi: 10.1023/B:CLIN.0000017180.19881.c1. [DOI] [PubMed] [Google Scholar]
- 67.Daskalakis M, Nguyen TT, Nguyen C, Guldberg P, Kohler G, Wijermans P, Jones PA, Lubbert M. Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment. Blood. 2002;100(8):2957–2964. doi: 10.1182/blood.V100.8.2957. [DOI] [PubMed] [Google Scholar]
- 68.Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science (New York, NY) 2003;300(5618):455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- 69.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science (New York, NY) 2003;300(5618):489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 70.Jenuwein T, Allis CD. Translating the histone code. Science (New York, NY) 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 71.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 72.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol. 2007;8(4):284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 73.Hao S, He W, Li Y, Ding H, Hou Y, Nie J, Hou FF, Kahn M, Liu Y. Targeted inhibition of beta-catenin/CBP signaling ameliorates renal interstitial fibrosis. J Am Soc Nephrol. 2011;22(9):1642–1653. doi: 10.1681/ASN.2010101079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou B, Liu Y, Kahn M, Ann DK, Han A, Wang H, Nguyen C, Flodby P, Zhong Q, Krishnaveni MS, Liebler JM, Minoo P, Crandall ED, Borok Z. Interactions between beta-catenin and transforming growth factor-beta signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP) J Biol Chem. 2012;287(10):7026–7038. doi: 10.1074/jbc.M111.276311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mizuguchi Y, Specht S, Lunz JG, 3rd, Isse K, Corbitt N, Takizawa T, Demetris AJ. Cooperation of p300 and PCAF in the control of microRNA 200c/141 transcription and epithelial characteristics. PLoS One. 2012;7(2):e32449. doi: 10.1371/journal.pone.0032449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The transcription factors Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS One. 2011;6(10):e26514. doi: 10.1371/journal.pone.0026514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kapoor-Vazirani P, Kagey JD, Powell DR, Vertino PM. Role of hMOF-dependent histone H4 lysine 16 acetylation in the maintenance of TMS1/ASC gene activity. Cancer Res. 2008;68(16):6810–6821. doi: 10.1158/0008-5472.CAN-08-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu YN, Lee WW, Wang CY, Chao TH, Chen Y, Chen JH. Regulatory mechanisms controlling human E-cadherin gene expression. Oncogene. 2005;24(56):8277–8290. doi: 10.1038/sj.onc.1208991. [DOI] [PubMed] [Google Scholar]
- 79.Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, Herman JG, Baylin SB. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2(3):e40. doi: 10.1371/journal.pgen.0020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Strobl-Mazzulla PH, Bronner ME. A PHD12-Snail2 repressive complex epigenetically mediates neural crest epithelial-to-mesenchymal transition. J Cell Biol. 2012;198(6):999–1010. doi: 10.1083/jcb.201203098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bansal N, Petrie K, Christova R, Chung CY, Leibovitch BA, Howell L, Gil V, Sbirkov Y, Lee E, Wexler J, Ariztia EV, Sharma R, Zhu J, Bernstein E, Zhou MM, Zelent A, Farias E, Waxman S. Targeting the SIN3A-PF1 interaction inhibits epithelial to mesenchymal transition and maintenance of a stem cell phenotype in triple negative breast cancer. Oncotarget. 2015;6(33):34087–34105. doi: 10.18632/oncotarget.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tripathi MK, Misra S, Khedkar SV, Hamilton N, Irvin-Wilson C, Sharan C, Sealy L, Chaudhuri G. Regulation of BRCA2 gene expression by the SLUG repressor protein in human breast cells. J Biol Chem. 2005;280(17):17163–17171. doi: 10.1074/jbc.M501375200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shi Y, Sawada J, Sui G, el Affar B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y, Shi Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422(6933):735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 84.Aghdassi A, Sendler M, Guenther A, Mayerle J, Behn CO, Heidecke CD, Friess H, Buchler M, Evert M, Lerch MM, Weiss FU. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61(3):439–448. doi: 10.1136/gutjnl-2011-300060. [DOI] [PubMed] [Google Scholar]
- 85.Byles V, Zhu L, Lovaas JD, Chmilewski LK, Wang J, Faller DV, Dai Y. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene. 2012;31(43):4619–4629. doi: 10.1038/onc.2011.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roche J, Nasarre P, Gemmill R, Baldys A, Pontis J, Korch C, Guilhot J, Ait-Si-Ali S, Drabkin H. Global decrease of histone H3K27 acetylation in ZEB1-induced epithelial to mesenchymal transition in lung cancer cells. Cancers. 2013;5(2):334–356. doi: 10.3390/cancers5020334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.He J, Shen S, Lu W, Zhou Y, Hou Y, Zhang Y, Jiang Y, Liu H, Shao Y. HDAC1 promoted migration and invasion binding with TCF12 by promoting EMT progress in gallbladder cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.8740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, Yao TP, Lane WS, Seto E. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27(2):197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Zhang M, Dong H, Yong S, Li X, Olashaw N, Kruk PA, Cheng JQ, Bai W, Chen J, Nicosia SV, Zhang X. Deacetylation of cortactin by SIRT1 promotes cell migration. Oncogene. 2009;28(3):445–460. doi: 10.1038/onc.2008.388. [DOI] [PubMed] [Google Scholar]
- 90.Shah P, Gau Y, Sabnis G. Histone deacetylase inhibitor entinostat reverses epithelial to mesenchymal transition of breast cancer cells by reversing the repression of E-cadherin. Breast Cancer Res Treat. 2014;143(1):99–111. doi: 10.1007/s10549-013-2784-7. [DOI] [PubMed] [Google Scholar]
- 91.Srivastava RK, Kurzrock R, Shankar S. MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits angiogenesis and metastasis, and reverses epithelial-mesenchymal transition in vivo. Mol Cancer Ther. 2010;9(12):3254–3266. doi: 10.1158/1535-7163.MCT-10-0582. [DOI] [PubMed] [Google Scholar]
- 92.Takai N, Desmond JC, Kumagai T, Gui D, Said JW, Whittaker S, Miyakawa I, Koeffler HP. Histone deacetylase inhibitors have a profound antigrowth activity in endometrial cancer cells. Clin Cancer Res. 2004;10(3):1141–1149. doi: 10.1158/1078-0432.CCR-03-0100. [DOI] [PubMed] [Google Scholar]
- 93.Ye Y, Xiao Y, Wang W, Yearsley K, Gao JX, Shetuni B, Barsky SH. ERalpha signaling through slug regulates E-cadherin and EMT. Oncogene. 2010;29(10):1451–1462. doi: 10.1038/onc.2009.433. [DOI] [PubMed] [Google Scholar]
- 94.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124(1):30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meidhof S, Brabletz S, Lehmann W, Preca BT, Mock K, Ruh M, Schuler J, Berthold M, Weber A, Burk U, Lubbert M, Puhr M, Culig Z, Wellner U, Keck T, Bronsert P, Kusters S, Hopt UT, Stemmler MP, Brabletz T. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med. 2015;7(6):831–847. doi: 10.15252/emmm.201404396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krumm A, Barckhausen C, Kucuk P, Tomaszowski KH, Loquai C, Fahrer J, Kramer OH, Kaina B, Roos WP. Enhanced histone deacetylase activity in malignant melanoma provokes RAD51 and FANCD2-triggered drug resistance. Cancer Res. 2016;76(10):3067–3077. doi: 10.1158/0008-5472.CAN-15-2680. [DOI] [PubMed] [Google Scholar]
- 97.Kristensen LS, Nielsen HM, Hansen LL. Epigenetics and cancer treatment. Eur J Pharmacol. 2009;625(1–3):131–142. doi: 10.1016/j.ejphar.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 98.Gilbert J, Gore SD, Herman JG, Carducci MA. The clinical application of targeting cancer through histone acetylation and hypomethylation. Clin Cancer Res. 2004;10(14):4589–4596. doi: 10.1158/1078-0432.CCR-03-0297. [DOI] [PubMed] [Google Scholar]
- 99.Gore SD, Baylin S, Sugar E, Carraway H, Miller CB, Carducci M, Grever M, Galm O, Dauses T, Karp JE, Rudek MA, Zhao M, Smith BD, Manning J, Jiemjit A, Dover G, Mays A, Zwiebel J, Murgo A, Weng LJ, Herman JG. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66(12):6361–6369. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 100.Jiang GM, Wang HS, Zhang F, Zhang KS, Liu ZC, Fang R, Wang H, Cai SH, Du J. Histone deacetylase inhibitor induction of epithelial-mesenchymal transitions via up-regulation of Snail facilitates cancer progression. Biochim Biophys Acta. 2012;1833(3):663–671. doi: 10.1016/j.bbamcr.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 101.Kong D, Ahmad A, Bao B, Li Y, Banerjee S, Sarkar FH. Histone deacetylase inhibitors induce epithelial-to-mesenchymal transition in prostate cancer cells. PLoS One. 2012;7(9):e45045. doi: 10.1371/journal.pone.0045045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 103.Butler JS, Dent SY. The role of chromatin modifiers in normal and malignant hematopoiesis. Blood. 2013;121(16):3076–3084. doi: 10.1182/blood-2012-10-451237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang L, Deng L, Chen F, Yao Y, Wu B, Wei L, Mo Q, Song Y. Inhibition of histone H3K79 methylation selectively inhibits proliferation, self-renewal and metastatic potential of breast cancer. Oncotarget. 2014;5(21):10665–10677. doi: 10.18632/oncotarget.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cho MH, Park JH, Choi HJ, Park MK, Won HY, Park YJ, Lee CH, Oh SH, Song YS, Kim HS, Oh YH, Lee JY, Kong G. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat Commun. 2015;6:7821. doi: 10.1038/ncomms8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gao Y, Zhao Y, Zhang J, Lu Y, Liu X, Geng P, Huang B, Zhang Y, Lu J. The dual function of PRMT1 in modulating epithelial-mesenchymal transition and cellular senescence in breast cancer cells through regulation of ZEB1. Sci Rep. 2016;6:19874. doi: 10.1038/srep19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 108.Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12(12):917–930. doi: 10.1038/nrd4154. [DOI] [PubMed] [Google Scholar]
- 109.Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437(7057):432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- 110.Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19(6):857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 111.Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, Evers BM, Zhou BP. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010;29(11):1803–1816. doi: 10.1038/emboj.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L, Li R, Li Y, Zhang Y, Li Q, Yi X, Shang Y. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138(4):660–672. doi: 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 113.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437(7057):436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 114.Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Lozach J, Jayes FL, Korach KS, Glass CK, Fu XD, Rosenfeld MG. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446(7138):882–887. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 115.Teng YC, Lee CF, Li YS, Chen YR, Hsiao PW, Chan MY, Lin FM, Huang HD, Chen YT, Jeng YM, Hsu CH, Yan Q, Tsai MD, Juan LJ. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 2013;73(15):4711–4721. doi: 10.1158/0008-5472.CAN-12-3165. [DOI] [PubMed] [Google Scholar]
- 116.Cao J, Liu Z, Cheung WK, Zhao M, Chen SY, Chan SW, Booth CJ, Nguyen DX, Yan Q. Histone demethylase RBP2 is critical for breast cancer progression and metastasis. Cell Rep. 2014;6(5):868–877. doi: 10.1016/j.celrep.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liang X, Zeng J, Wang L, Shen L, Ma X, Li S, Wu Y, Ma L, Ci X, Guo Q, Jia M, Shen H, Sun Y, Liu Z, Liu S, Li W, Yu H, Chen C, Jia J. Histone demethylase RBP2 promotes malignant progression of gastric cancer through TGF-beta1-(p-Smad3)-RBP2-E-cadherin-Smad3 feedback circuit. Oncotarget. 2015;6(19):17661–17674. doi: 10.18632/oncotarget.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li Q, Shi L, Gui B, Yu W, Wang J, Zhang D, Han X, Yao Z, Shang Y. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 2011;71(21):6899–6908. doi: 10.1158/0008-5472.CAN-11-1523. [DOI] [PubMed] [Google Scholar]
- 119.Enkhbaatar Z, Terashima M, Oktyabri D, Tange S, Ishimura A, Yano S, Suzuki T. KDM5B histone demethylase controls epithelial-mesenchymal transition of cancer cells by regulating the expression of the microRNA-200 family. Cell Cycle (Georgetown, Tex) 2013;12(13):2100–2112. doi: 10.4161/cc.25142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tang B, Qi G, Tang F, Yuan S, Wang Z, Liang X, Li B, Yu S, Liu J, Huang Q, Wei Y, Zhai R, Lei B, Yu H, Jiao X, He S. JARID1B promotes metastasis and epithelial-mesenchymal transition via PTEN/AKT signaling in hepatocellular carcinoma cells. Oncotarget. 2015;6(14):12723–12739. doi: 10.18632/oncotarget.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mozzetta C, Boyarchuk E, Pontis J, Ait-Si-Ali S. Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat Rev Mol Cell Biol. 2015;16(8):499–513. doi: 10.1038/nrm4029. [DOI] [PubMed] [Google Scholar]
- 122.Chen MW, Hua KT, Kao HJ, Chi CC, Wei LH, Johansson G, Shiah SG, Chen PS, Jeng YM, Cheng TY, Lai TC, Chang JS, Jan YH, Chien MH, Yang CJ, Huang MS, Hsiao M, Kuo ML. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010;70(20):7830–7840. doi: 10.1158/0008-5472.CAN-10-0833. [DOI] [PubMed] [Google Scholar]
- 123.Liu S, Ye D, Guo W, Yu W, He Y, Hu J, Wang Y, Zhang L, Liao Y, Song H, Zhong S, Xu D, Yin H, Sun B, Wang X, Liu J, Wu Y, Zhou BP, Zhang Z, Deng J. G9a is essential for EMT-mediated metastasis and maintenance of cancer stem cell-like characters in head and neck squamous cell carcinoma. Oncotarget. 2015;6(9):6887–6901. doi: 10.18632/oncotarget.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dong C, Wu Y, Wang Y, Wang C, Kang T, Rychahou PG, Chi YI, Evers BM, Zhou BP. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene. 2013;32(11):1351–1362. doi: 10.1038/onc.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang H, Cai K, Wang J, Wang X, Cheng K, Shi F, Jiang L, Zhang Y, Dou J. MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells (Dayton, Ohio) 2014;32(11):2858–2868. doi: 10.1002/stem.1795. [DOI] [PubMed] [Google Scholar]
- 126.Regina C, Compagnone M, Peschiaroli A, Lena A, Annicchiarico-Petruzzelli M, Piro MC, Melino G, Candi E. Setdb1, a novel interactor of DeltaNp63, is involved in breast tumorigenesis. Oncotarget. 2016 doi: 10.18632/oncotarget.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferre F, Bourque C, Burke CJ, Turner L, Uong A, Johnson LA, Beroukhim R, Mermel CH, Loda M, Ait-Si-Ali S, Garraway LA, Young RA, Zon LI. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471(7339):513–517. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fei Q, Shang K, Zhang J, Chuai S, Kong D, Zhou T, Fu S, Liang Y, Li C, Chen Z, Zhao Y, Yu Z, Huang Z, Hu M, Ying H, Chen Z, Zhang Y, Xing F, Zhu J, Xu H, Zhao K, Lu C, Atadja P, Xiao ZX, Li E, Shou J. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat Commun. 2015;6:8651. doi: 10.1038/ncomms9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rodriguez-Paredes M, Martinez de Paz A, Simo-Riudalbas L, Sayols S, Moutinho C, Moran S, Villanueva A, Vazquez-Cedeira M, Lazo PA, Carneiro F, Moura CS, Vieira J, Teixeira MR, Esteller M. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene. 2014;33(21):2807–2813. doi: 10.1038/onc.2013.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wu PC, Lu JW, Yang JY, Lin IH, Ou DL, Lin YH, Chou KH, Huang WF, Wang WP, Huang YL, Hsu C, Lin LI, Lin YM, Shen CK, Tzeng TY. H3K9 histone methyltransferase, KMT1E/SETDB1, cooperates with the SMAD2/3 pathway to suppress lung cancer metastasis. Cancer Res. 2014;74(24):7333–7343. doi: 10.1158/0008-5472.CAN-13-3572. [DOI] [PubMed] [Google Scholar]
- 131.Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol. 2013;20(10):1147–1155. doi: 10.1038/nsmb.2669. [DOI] [PubMed] [Google Scholar]
- 132.Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yu H, Simons DL, Segall I, Carcamo-Cavazos V, Schwartz EJ, Yan N, Zuckerman NS, Dirbas FM, Johnson DL, Holmes SP, Lee PP. PRC2/EED-EZH2 complex is up-regulated in breast cancer lymph node metastasis compared to primary tumor and correlates with tumor proliferation in situ. PLoS One. 2012;7(12):e51239. doi: 10.1371/journal.pone.0051239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Herranz N, Pasini D, Diaz VM, Franci C, Gutierrez A, Dave N, Escriva M, Hernandez-Munoz I, Di Croce L, Helin K, Garcia de Herreros A, Peiro S. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol. 2008;28(15):4772–4781. doi: 10.1128/MCB.00323-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hou Z, Peng H, Ayyanathan K, Yan KP, Langer EM, Longmore GD, Rauscher FJ., 3rd The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol Cell Biol. 2008;28(10):3198–3207. doi: 10.1128/MCB.01435-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Langer EM, Feng Y, Zhaoyuan H, Rauscher FJ, 3rd, Kroll KL, Longmore GD. Ajuba LIM proteins are snail/slug corepressors required for neural crest development in Xenopus. Dev Cell. 2008;14(3):424–436. doi: 10.1016/j.devcel.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Xia R, Jin FY, Lu K, Wan L, Xie M, Xu TP, De W, Wang ZX. SUZ12 promotes gastric cancer cell proliferation and metastasis by regulating KLF2 and E-cadherin. Tumour Biol. 2015;36(7):5341–5351. doi: 10.1007/s13277-015-3195-7. [DOI] [PubMed] [Google Scholar]
- 138.Malouf GG, Taube JH, Lu Y, Roysarkar T, Panjarian S, Estecio MR, Jelinek J, Yamazaki J, Raynal NJ, Long H, Tahara T, Tinnirello A, Ramachandran P, Zhang XY, Liang S, Mani SA, Issa JP. Architecture of epigenetic reprogramming following Twist1-mediated epithelial-mesenchymal transition. Genome Biol. 2013;14(12):R144. doi: 10.1186/gb-2013-14-12-r144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D’Alessio AC, Young RA, Weinberg RA. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013;154(1):61–74. doi: 10.1016/j.cell.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yang H, Mizzen CA. The multiple facets of histone H4-lysine 20 methylation. Biochem Cell Biol. 2009;87(1):151–161. doi: 10.1139/O08-131. [DOI] [PubMed] [Google Scholar]
- 141.Yang F, Sun L, Li Q, Han X, Lei L, Zhang H, Shang Y. SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012;31(1):110–123. doi: 10.1038/emboj.2011.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hou L, Li Q, Yu Y, Li M, Zhang D. SET8 induces epithelialmesenchymal transition and enhances prostate cancer cell metastasis by cooperating with ZEB1. Mol Med Rep. 2016;13(2):1681–1688. doi: 10.3892/mmr.2015.4733. [DOI] [PubMed] [Google Scholar]
- 143.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37(4):391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 144.Shinchi Y, Hieda M, Nishioka Y, Matsumoto A, Yokoyama Y, Kimura H, Matsuura S, Matsuura N. SUV420H2 suppresses breast cancer cell invasion through down regulation of the SH2 domain-containing focal adhesion protein tensin-3. Exp Cell Res. 2015;334(1):90–99. doi: 10.1016/j.yexcr.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 145.Dhar SS, Lee SH, Kan PY, Voigt P, Ma L, Shi X, Reinberg D, Lee MG. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012;26(24):2749–2762. doi: 10.1101/gad.203356.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ibrahim R, Matsubara D, Osman W, Morikawa T, Goto A, Morita S, Ishikawa S, Aburatani H, Takai D, Nakajima J, Fukayama M, Niki T, Murakami Y. Expression of PRMT5 in lung adenocarcinoma and its significance in epithelial-mesenchymal transition. Hum Pathol. 2014;45(7):1397–1405. doi: 10.1016/j.humpath.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 147.Yao R, Jiang H, Ma Y, Wang L, Wang L, Du J, Hou P, Gao Y, Zhao L, Wang G, Zhang Y, Liu DX, Huang B, Lu J. PRMT7 induces epithelial-to-mesenchymal transition and promotes metastasis in breast cancer. Cancer Res. 2014;74(19):5656–5667. doi: 10.1158/0008-5472.CAN-14-0800. [DOI] [PubMed] [Google Scholar]
- 148.Tee AE, Ling D, Nelson C, Atmadibrata B, Dinger ME, Xu N, Mizukami T, Liu PY, Liu B, Cheung B, Pasquier E, Haber M, Norris MD, Suzuki T, Marshall GM, Liu T. The histone demethylase JMJD1A induces cell migration and invasion by up-regulating the expression of the long noncoding RNA MALAT1. Oncotarget. 2014;5(7):1793–1804. doi: 10.18632/oncotarget.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shi L, Sun L, Li Q, Liang J, Yu W, Yi X, Yang X, Li Y, Han X, Zhang Y, Xuan C, Yao Z, Shang Y. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc Natl Acad Sci USA. 2011;108(18):7541–7546. doi: 10.1073/pnas.1017374108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhao L, Li W, Zang W, Liu Z, Xu X, Yu H, Yang Q, Jia J. JMJD2B promotes epithelial-mesenchymal transition by cooperating with beta-catenin and enhances gastric cancer metastasis. Clin Cancer Res. 2013;19(23):6419–6429. doi: 10.1158/1078-0432.CCR-13-0254. [DOI] [PubMed] [Google Scholar]
- 151.Bjorkman M, Ostling P, Harma V, Virtanen J, Mpindi JP, Rantala J, Mirtti T, Vesterinen T, Lundin M, Sankila A, Rannikko A, Kaivanto E, Kohonen P, Kallioniemi O, Nees M. Systematic knockdown of epigenetic enzymes identifies a novel histone demethylase PHF8 overexpressed in prostate cancer with an impact on cell proliferation, migration and invasion. Oncogene. 2012;31(29):3444–3456. doi: 10.1038/onc.2011.512. [DOI] [PubMed] [Google Scholar]
- 152.Kim JH, Sharma A, Dhar SS, Lee SH, Gu B, Chan CH, Lin HK, Lee MG. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014;74(6):1705–1717. doi: 10.1158/0008-5472.CAN-13-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zha L, Cao Q, Cui X, Li F, Liang H, Xue B, Shi H. Epigenetic regulation of E-cadherin expression by the histone demethylase UTX in colon cancer cells. Med Oncol. 2016;33(3):21. doi: 10.1007/s12032-016-0734-z. [DOI] [PubMed] [Google Scholar]
- 154.Choi HJ, Park JH, Park M, Won HY, Joo HS, Lee CH, Lee JY, Kong G. UTX inhibits EMT-induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD1 and HDAC1. EMBO Rep. 2015;16(10):1288–1298. doi: 10.15252/embr.201540244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ramadoss S, Chen X, Wang CY. Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J Biol Chem. 2012;287(53):44508–44517. doi: 10.1074/jbc.M112.424903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat Chem Biol. 2005;1(3):143–145. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]
- 157.Lakshmikuttyamma A, Scott SA, DeCoteau JF, Geyer CR. Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 inhibition. Oncogene. 2010;29(4):576–588. doi: 10.1038/onc.2009.361. [DOI] [PubMed] [Google Scholar]
- 158.Chang Y, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, Snyder JP, Bedford MT, Cheng X. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat Struct Mol Biol. 2009;16(3):312–317. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25(3):473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 160.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, Dong A, Tempel W, Wang SC, Chen X, Chau I, Mangano TJ, Huang XP, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7(8):566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pan MR, Hsu MC, Chen LT, Hung WC. G9a orchestrates PCL3 and KDM7A to promote histone H3K27 methylation. Sci Rep. 2015;5:18709. doi: 10.1038/srep18709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Finley A, Copeland RA. Small molecule control of chromatin remodeling. Chem Biol. 2014;21(9):1196–1210. doi: 10.1016/j.chembiol.2014.07.024. [DOI] [PubMed] [Google Scholar]
- 163.Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, Song Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS One. 2012;7(4):e35065. doi: 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ferrari-Amorotti G, Fragliasso V, Esteki R, Prudente Z, Soliera AR, Cattelani S, Manzotti G, Grisendi G, Dominici M, Pieraccioli M, Raschella G, Chiodoni C, Colombo MP, Calabretta B. Inhibiting interactions of lysine demethylase LSD1 with snail/slug blocks cancer cell invasion. Cancer Res. 2013;73(1):235–245. doi: 10.1158/0008-5472.CAN-12-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, Ruiz J, Bruick R, Pena-Llopis S, Xu J, Babinski DJ, Frantz DE, Brekken RA, Quinn AM, Simeonov A, Easmon J, Martinez ED. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. 2013;4:2035. doi: 10.1038/ncomms3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gale M, Sayegh J, Cao J, Norcia M, Gareiss P, Hoyer D, Merkel JS, Yan Q. Screen-identified selective inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance. Oncotarget. 2016 doi: 10.18632/oncotarget.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Robinson PJ, An W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J Mol Biol. 2008;381(4):816–825. doi: 10.1016/j.jmb.2008.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science (New York, NY) 2006;311(5762):844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 169.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, Arrowsmith CH, Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149(1):214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54(5):728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, Rusinova E, Zhang G, Wang C, Zhu H, Yao J, Zeng YX, Evers BM, Zhou MM, Zhou BP. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25(2):210–225. doi: 10.1016/j.ccr.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wagner T, Robaa D, Sippl W, Jung M. Mind the methyl: methyllysine binding proteins in epigenetic regulation. Chem Med Chem. 2014;9(3):466–483. doi: 10.1002/cmdc.201300422. [DOI] [PubMed] [Google Scholar]
- 173.Maison C, Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol. 2004;5(4):296–304. doi: 10.1038/nrm1355. [DOI] [PubMed] [Google Scholar]
- 174.Bua DJ, Kuo AJ, Cheung P, Liu CL, Migliori V, Espejo A, Casadio F, Bassi C, Amati B, Bedford MT, Guccione E, Gozani O. Epigenome microarray platform for proteome-wide dissection of chromatin-signaling networks. PLoS One. 2009;4(8):e6789. doi: 10.1371/journal.pone.0006789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kokura K, Sun L, Bedford MT, Fang J. Methyl-H3K9-binding protein MPP8 mediates E-cadherin gene silencing and promotes tumour cell motility and invasion. EMBO J. 2010;29(21):3673–3687. doi: 10.1038/emboj.2010.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Quinn AM, Bedford MT, Espejo A, Spannhoff A, Austin CP, Oppermann U, Simeonov A. A homogeneous method for investigation of methylation-dependent protein-protein interactions in epigenetics. Nucleic Acids Res. 2010;38(2):e11. doi: 10.1093/nar/gkp899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nady N, Krichevsky L, Zhong N, Duan S, Tempel W, Amaya MF, Ravichandran M, Arrowsmith CH. Histone recognition by human malignant brain tumor domains. J Mol Biol. 2012;423(5):702–718. doi: 10.1016/j.jmb.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 178.Tang M, Shen H, Jin Y, Lin T, Cai Q, Pinard MA, Biswas S, Tran Q, Li G, Shenoy AK, Tongdee E, Lin S, Gu Y, Law BK, Zhou L, McKenna R, Wu L, Lu J. The malignant brain tumor (MBT) domain protein SFMBT1 is an integral histone reader subunit of the LSD1 demethylase complex for chromatin association and epithelial-to-mesenchymal transition. J Biol Chem. 2013;288(38):27680–27691. doi: 10.1074/jbc.M113.482349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell. 2005;121(6):859–872. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 180.Couture JF, Collazo E, Trievel RC. Molecular recognition of histone H3 by the WD40 protein WDR5. Nat Struct Mol Biol. 2006;13(8):698–703. doi: 10.1038/nsmb1116. [DOI] [PubMed] [Google Scholar]
- 181.Ruthenburg AJ, Wang W, Graybosch DM, Li H, Allis CD, Patel DJ, Verdine GL. Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat Struct Mol Biol. 2006;13(8):704–712. doi: 10.1038/nsmb1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, Teng SC, Wu KJ. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell. 2011;43(5):811–822. doi: 10.1016/j.molcel.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 183.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D, Gamblin SJ. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461(7265):762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem. 2010;285(34):26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Collins RE, Northrop JP, Horton JR, Lee DY, Zhang X, Stallcup MR, Cheng X. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat Struct Mol Biol. 2008;15(3):245–250. doi: 10.1038/nsmb.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science (New York, NY) 2006;312(5774):748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 187.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hu Y, Zhou J, Ye F, Xiong H, Peng L, Zheng Z, Xu F, Cui M, Wei C, Wang X, Wang Z, Zhu H, Lee P, Zhou M, Jiang B, Zhang DY. BRD4 inhibitor inhibits colorectal cancer growth and metastasis. Int J Mol Sci. 2015;16(1):1928–1948. doi: 10.3390/ijms16011928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.James LI, Barsyte-Lovejoy D, Zhong N, Krichevsky L, Korboukh VK, Herold JM, MacNevin CJ, Norris JL, Sagum CA, Tempel W, Marcon E, Guo H, Gao C, Huang XP, Duan S, Emili A, Greenblatt JF, Kireev DB, Jin J, Janzen WP, Brown PJ, Bedford MT, Arrowsmith CH, Frye SV. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat Chem Biol. 2013;9(3):184–191. doi: 10.1038/nchembio.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Stuckey JI, Dickson BM, Cheng N, Liu Y, Norris JL, Cholensky SH, Tempel W, Qin S, Huber KG, Sagum C, Black K, Li F, Huang XP, Roth BL, Baughman BM, Senisterra G, Pattenden SG, Vedadi M, Brown PJ, Bedford MT, Min J, Arrowsmith CH, James LI, Frye SV. A cellular chemical probe targeting the chromodomains of Polycomb repressive complex 1. Nat Chem Biol. 2016;12(3):180–187. doi: 10.1038/nchembio.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Suganuma T, Workman JL. Crosstalk among Histone Modifications. Cell. 2008;135(4):604–607. doi: 10.1016/j.cell.2008.10.036. [DOI] [PubMed] [Google Scholar]
- 194.Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P, Reinberg D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16(4):479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wang H, Cao R, Xia L, Erdjument-Bromage H, Borchers C, Tempst P, Zhang Y. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol Cell. 2001;8(6):1207–1217. doi: 10.1016/S1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- 196.Smallwood A, Esteve PO, Pradhan S, Carey M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007;21(10):1169–1178. doi: 10.1101/gad.1536807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Norwood LE, Moss TJ, Margaryan NV, Cook SL, Wright L, Seftor EA, Hendrix MJ, Kirschmann DA, Wallrath LL. A requirement for dimerization of HP1Hsalpha in suppression of breast cancer invasion. J Biol Chem. 2006;281(27):18668–18676. doi: 10.1074/jbc.M512454200. [DOI] [PubMed] [Google Scholar]
- 198.Chang Y, Sun L, Kokura K, Horton JR, Fukuda M, Espejo A, Izumi V, Koomen JM, Bedford MT, Zhang X, Shinkai Y, Fang J, Cheng X. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat Commun. 2011;2:533. doi: 10.1038/ncomms1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sampath SC, Marazzi I, Yap KL, Sampath SC, Krutchinsky AN, Mecklenbrauker I, Viale A, Rudensky E, Zhou MM, Chait BT, Tarakhovsky A. Methylation of a histone mimic within the histone methyltransferase G9a regulates protein complex assembly. Mol Cell. 2007;27(4):596–608. doi: 10.1016/j.molcel.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 200.Chang Y, Horton JR, Bedford MT, Zhang X, Cheng X. Structural insights for MPP8 chromodomain interaction with histone H3 lysine 9: potential effect of phosphorylation on methyl-lysine binding. J Mol Biol. 2011;408(5):807–814. doi: 10.1016/j.jmb.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Li J, Li Z, Ruan J, Xu C, Tong Y, Pan PW, Tempel W, Crombet L, Min J, Zang J. Structural basis for specific binding of human MPP8 chromodomain to histone H3 methylated at lysine 9. PLoS One. 2011;6(10):e25104. doi: 10.1371/journal.pone.0025104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sun L, Kokura K, Izumi V, Koomen JM, Seto E, Chen J, Fang J. MPP8 and SIRT1 crosstalk in E-cadherin gene silencing and epithelial-mesenchymal transition. EMBO Rep. 2015;16(6):689–699. doi: 10.15252/embr.201439792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Moore KE, Carlson SM, Camp ND, Cheung P, James RG, Chua KF, Wolf-Yadlin A, Gozani O. A general molecular affinity strategy for global detection and proteomic analysis of lysine methylation. Mol Cell. 2013;50(3):444–456. doi: 10.1016/j.molcel.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kapoor-Vazirani P, Kagey JD, Vertino PM. SUV420H2-mediated H4K20 trimethylation enforces RNA polymerase II promoter-proximal pausing by blocking hMOF-dependent H4K16 acetylation. Mol Cell Biol. 2011;31(8):1594–1609. doi: 10.1128/MCB.00524-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Cheng X, Blumenthal RM. Coordinated chromatin control: structural and functional linkage of DNA and histone methylation. Biochemistry. 2010;49(14):2999–3008. doi: 10.1021/bi100213t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Epsztejn-Litman S, Feldman N, Abu-Remaileh M, Shufaro Y, Gerson A, Ueda J, Deplus R, Fuks F, Shinkai Y, Cedar H, Bergman Y. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol. 2008;15(11):1176–1183. doi: 10.1038/nsmb.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20(22):3089–3103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Fuks F. DNA methylation and histone modifications: teaming up to silence genes. Curr Opin Genet Dev. 2005;15(5):490–495. doi: 10.1016/j.gde.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 209.Schotta G, Ebert A, Krauss V, Fischer A, Hoffmann J, Rea S, Jenuwein T, Dorn R, Reuter G. Central role of Drosophila SU(VAR)3-9 in histone H3-K9 methylation and heterochromatic gene silencing. EMBO J. 2002;21(5):1121–1131. doi: 10.1093/emboj/21.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F. The polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439(7078):871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 211.Otani J, Nankumo T, Arita K, Inamoto S, Ariyoshi M, Shirakawa M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009;10(11):1235–1241. doi: 10.1038/embor.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I, Rathert P, Brandt O, Reinhardt R, Fischle W, Jeltsch A. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010;38(13):4246–4253. doi: 10.1093/nar/gkq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10(3):295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 214.Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, Chang SY, Lee OK, Wu KJ. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010;12(10):982–992. doi: 10.1038/ncb2099. [DOI] [PubMed] [Google Scholar]
- 215.Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, Mehra R, Laxman B, Cao X, Yu J, Kleer CG, Varambally S, Chinnaiyan AM. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008;27(58):7274–7284. doi: 10.1038/onc.2008.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kalakonda N, Fischle W, Boccuni P, Gurvich N, Hoya-Arias R, Zhao X, Miyata Y, Macgrogan D, Zhang J, Sims JK, Rice JC, Nimer SD. Histone H4 lysine 20 monomethylation promotes transcriptional repression by L3MBTL1. Oncogene. 2008;27(31):4293–4304. doi: 10.1038/onc.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Li Z, Nie F, Wang S, Li L. Histone H4 Lys 20 monomethylation by histone methylase SET8 mediates Wnt target gene activation. Proc Natl Acad Sci USA. 2011;108(8):3116–3123. doi: 10.1073/pnas.1009353108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10(2):126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 220.Abba ML, Patil N, Leupold JH, Allgayer H. MicroRNA Regulation of epithelial to mesenchymal transition. J Clin Med. 2016 doi: 10.3390/jcm5010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop—a motor of cellular plasticity in development and cancer? EMBO Rep. 2010;11(9):670–677. doi: 10.1038/embor.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Bracken CP, Gregory PA, Khew-Goodall Y, Goodall GJ. The role of microRNAs in metastasis and epithelial-mesenchymal transition. Cell Mol Life Sci. 2009;66(10):1682–1699. doi: 10.1007/s00018-009-8750-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Gregory PA, Bracken CP, Bert AG, Goodall GJ. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle (Georgetown, Tex) 2008;7(20):3112–3118. doi: 10.4161/cc.7.20.6851. [DOI] [PubMed] [Google Scholar]
- 224.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283(22):14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22(7):894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68(19):7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 227.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9(6):582–589. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 229.Xia H, Ng SS, Jiang S, Cheung WK, Sze J, Bian XW, Kung HF, Lin MC. miR-200a-mediated downregulation of ZEB2 and CTNNB1 differentially inhibits nasopharyngeal carcinoma cell growth, migration and invasion. Biochem Biophys Res Commun. 2010;391(1):535–541. doi: 10.1016/j.bbrc.2009.11.093. [DOI] [PubMed] [Google Scholar]
- 230.Eades G, Yao Y, Yang M, Zhang Y, Chumsri S, Zhou Q. miR-200a regulates SIRT1 expression and epithelial to mesenchymal transition (EMT)-like transformation in mammary epithelial cells. J Biol Chem. 2011;286(29):25992–26002. doi: 10.1074/jbc.M111.229401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010;39(5):761–772. doi: 10.1016/j.molcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, Dirbas FM, Somlo G, Pera RA, Lao K, Clarke MF. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138(3):592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, Brenner JC, Yu J, Kim JH, Han B, Tan P, Kumar-Sinha C, Lonigro RJ, Palanisamy N, Maher CA, Chinnaiyan AM. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science (New York, NY) 2008;322(5908):1695–1699. doi: 10.1126/science.1165395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Zheng M, Jiang YP, Chen W, Li KD, Liu X, Gao SY, Feng H, Wang SS, Jiang J, Ma XR, Cen X, Tang YJ, Chen Y, Lin YF, Tang YL, Liang XH. Snail and Slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget. 2015;6(9):6797–6810. doi: 10.18632/oncotarget.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, Cha SY, Ryu JK, Yoon D, Fearon ER, Rowe RG, Lee S, Maher CA, Weiss SJ, Yook JI. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011;195(3):417–433. doi: 10.1083/jcb.201103097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, Pineau P, Marchio A, Palatini J, Suh SS, Alder H, Liu CG, Dejean A, Croce CM. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208(5):875–883. doi: 10.1084/jem.20110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Dong P, Karaayvaz M, Jia N, Kaneuchi M, Hamada J, Watari H, Sudo S, Ju J, Sakuragi N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. 2013;32(27):3286–3295. doi: 10.1038/onc.2012.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, Weinberg RA. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12(3):247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Drasin DJ, Guarnieri AL, Neelakantan D, Kim J, Cabrera JH, Wang CA, Zaberezhnyy V, Gasparini P, Cascione L, Huebner K, Tan AC, Ford HL. TWIST1-Induced miR-424 Reversibly Drives Mesenchymal Programming while Inhibiting Tumor Initiation. Cancer Res. 2015;75(9):1908–1921. doi: 10.1158/0008-5472.CAN-14-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Neves R, Scheel C, Weinhold S, Honisch E, Iwaniuk KM, Trompeter HI, Niederacher D, Wernet P, Santourlidis S, Uhrberg M. Role of DNA methylation in miR-200c/141 cluster silencing in invasive breast cancer cells. BMC Res Notes. 2010;3:219. doi: 10.1186/1756-0500-3-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016 doi: 10.1007/s00018-016-2174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science (New York, NY) 2010;329(5992):689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Prensner JR, Iyer MK, Sahu A, Asangani IA, Cao Q, Patel L, Vergara IA, Davicioni E, Erho N, Ghadessi M, Jenkins RB, Triche TJ, Malik R, Bedenis R, McGregor N, Ma T, Chen W, Han S, Jing X, Cao X, Wang X, Chandler B, Yan W, Siddiqui J, Kunju LP, Dhanasekaran SM, Pienta KJ, Feng FY, Chinnaiyan AM. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat Genet. 2013;45(11):1392–1398. doi: 10.1038/ng.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Hou P, Zhao Y, Li Z, Yao R, Ma M, Gao Y, Zhao L, Zhang Y, Huang B, Lu J. LincRNA-ROR induces epithelial-to-mesenchymal transition and contributes to breast cancer tumorigenesis and metastasis. Cell Death Dis. 2014;5:e1287. doi: 10.1038/cddis.2014.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143(1):46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Gumireddy K, Li A, Yan J, Setoyama T, Johannes GJ, Orom UA, Tchou J, Liu Q, Zhang L, Speicher DW, Calin GA, Huang Q. Identification of a long non-coding RNA-associated RNP complex regulating metastasis at the translational step. EMBO J. 2013;32(20):2672–2684. doi: 10.1038/emboj.2013.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Richards EJ, Zhang G, Li ZP, Permuth-Wey J, Challa S, Li Y, Kong W, Dan S, Bui MM, Coppola D, Mao WM, Sellers TA, Cheng JQ. Long non-coding RNAs (LncRNA) regulated by transforming growth factor (TGF) beta: LncRNA-hit-mediated TGFbeta-induced epithelial to mesenchymal transition in mammary epithelia. J Biol Chem. 2015;290(11):6857–6867. doi: 10.1074/jbc.M114.610915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Dhamija S, Diederichs S. From junk to master regulators of invasion: lncRNA functions in migration. EMT and metastasis. Int J Cancer. 2016;139(2):269–280. doi: 10.1002/ijc.30039. [DOI] [PubMed] [Google Scholar]
- 250.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, Schwabe RF, Vahdat LT, Altorki NK, Mittal V, Gao D. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–476. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]