

Abstract
Background
Amiodarone (AMD) and its metabolite N-desethylamiodarone can cause some adverse effects, which include pulmonary toxicity. Some studies suggest that mitochondrial dysfunction and oxidative stress may play a role in these adverse effects. Catechin and epicatechin are recognized as important phenolic compounds with the ability to decrease oxidative stress. Therefore, the aim of this study was to evaluate the potential of catechin and epicatechin to modulate mitochondrial dysfunction and oxidative damage caused by AMD in human lung fibroblast cells (MRC-5).
Methods
Mitochondrial dysfunction was assessed through the activity of mitochondrial complex I and ATP biosynthesis. Cell viability was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Superoxide dismutase and catalase activity were measured spectrophotometrically at 480 and 240 nm, respectively. Lipid and protein oxidative levels were determined by thiobarbituric reactive substances and protein carbonyl assays, respectively. Nitric oxide (NO) levels were evaluated using the Griess reaction method.
Results
AMD was able to inhibit the activity of mitochondrial complex I and ATP biosynthesis in MRC-5 cells. Lipid and protein oxidative markers increased along with cell death, while superoxide dismutase and catalase activities and NO production decreased with AMD treatment. Both catechin and epicatechin circumvented mitochondrial dysfunction, thereby restoring the activity of mitochondrial complex I and ATP biosynthesis. Furthermore, the phenolic compounds were able to restore the imbalance in superoxide dismutase and catalase activities as well as the decrease in NO levels induced by AMD. Protein and lipid oxidative damage and cell death were reduced by catechin and epicatechin in AMD-treated cells.
Conclusions
Catechin and epicatechin reduced mitochondrial dysfunction and oxidative stress caused by AMD in MRC-5 cells.
Keywords: Arrhythmia, Cardiovascular disease, Mitochondria, Toxicity
1. Introduction
Cardiac arrhythmias are characterized by an irregular heartbeat rhythm, which could be either too slow (<60 beats/min) or too fast (>100 beats/min) [1]. Amiodarone (AMD) (Fig. 1A) is an antiarrhythmic agent widely used to treat cardiac arrhythmias, mainly atrial fibrillation [1]. Despite its pharmacological properties, AMD and its main metabolite N-desethylamiodarone (Fig. 1B) can cause some adverse effects, such as thyroid dysfunction, and hepatic and pulmonary toxicity [2], [3], [4], [5]. Pulmonary toxicity occurs in approximately 13% of the patients, who can have an associated mortality rate of 10–23% [2], [3]. The mechanism by which AMD causes human toxicity is not well understood, but some studies in mammalian cells [6], [7], [8] and an in vivo rat model [9] suggest that oxidative stress and mitochondrial dysfunction may play a role in AMD toxicity.
Fig. 1.

Chemical structure of amiodarone (AMD) and N-desethylamiodarone; (adapted from [10] and [11] respectively).
Mitochondria are recognized for their key role not only in ATP biosynthesis, but also in the maintenance of redox metabolism and apoptosis regulation, making this organelle a potential therapeutic target. Disruption of mitochondrial homeostasis is associated with an increase in reactive oxygen species (ROS), mainly in complex I (nicotinamide adenine dinucleotide/CoQ oxidoreductase) of the mitochondrial electron transport chain. In this complex, the superoxide radical (O2-·) is formed from electron escape, leading to decreased electron transport, reduced ATP biosynthesis, and increased oxidative stress [12].
Phenolic compounds are one of the most studied and effective group of bioactive compounds [13]. The flavonoids catechin (CAT) and epicatechin (EPI) (Fig. 2) are among this class of compounds [14]. It has already been demonstrated that CAT can reduce the inhibition of mitochondrial complex I induced by rotenone and N-methyl-4-phenyl-1,2,3,6-tetrahydropyridinium hydrochloride in primary rat mesencephalic cultures [15].
Fig. 2.
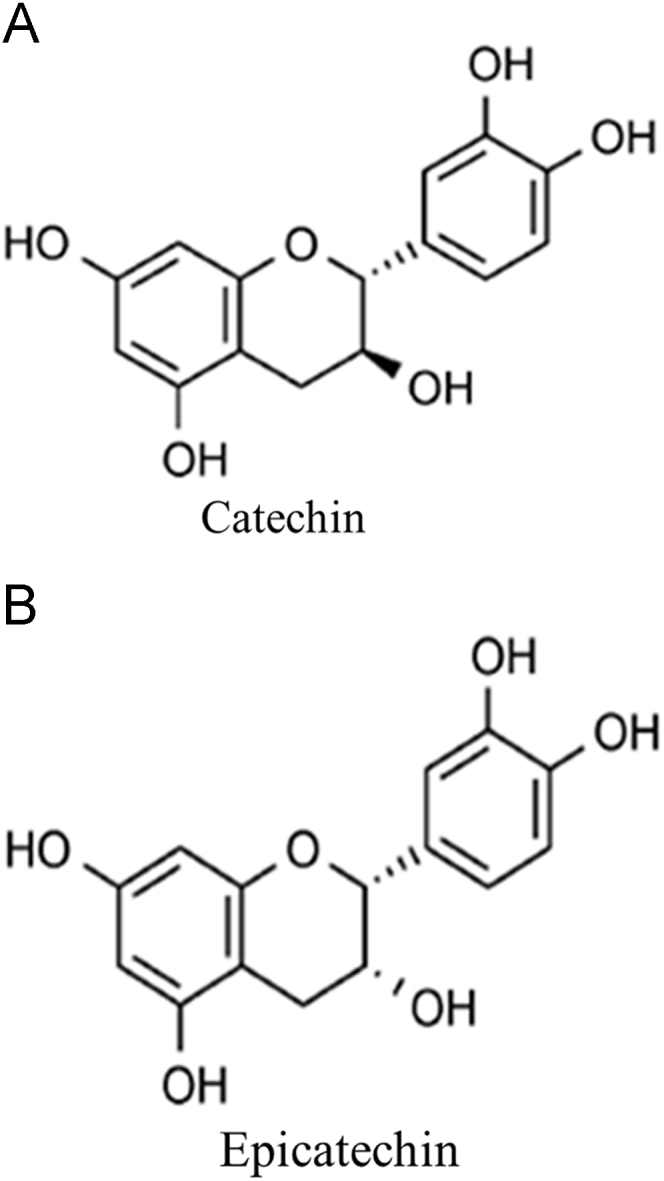
Chemical structures of catechin (CAT) and epicatechin (EPI) (adapted from [14]).
Therefore, the aim of this work was to evaluate the ability of CAT and EPI to minimize the oxidative damage and mitochondrial dysfunction induced by AMD in human lung fibroblasts (MRC-5).
2. Materials and methods
2.1. Chemicals
Amiodarone hydrochloride was obtained from Hipolabor (Brazil). Dulbecco׳s modified Eagle medium (DMEM), fetal bovine serum (FBS), trypsin-EDTA, and penicillin-streptomycin were purchased from Gibco BRL (Grand Island, NY, USA). (±)-CAT, (-)-EPI, thiobarbituric acid (TBA), trichloroacetic acid (TCA), hydrolyzed 1,1,3,3-tetramethoxypropane (TMP), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), were obtained from Sigma-Aldrich (St. Louis, MO, USA). All other reagents and solvents were obtained from Sigma (St. Louis, MO, USA).
2.2. Cell culture
MRC-5 cell line was purchased from the American Type Culture Collection (ATCC), and kept frozen in 10% (v/v) dimethyl sulfoxide. Cells were cultured in DMEM supplemented with 10% heat inactivated FBS, penicillin 100 UI/mL, and streptomycin 100 μg/mL. Prior to use in the assays, cells were incubated at 37 °C in an atmosphere of 5% CO2 with 90% humidity until they reached 80% confluence.
2.3. Cell treatments
MRC-5 cells were pre-treated with non-cytotoxic CAT and EPI concentrations of 10, 100, and 500 μM for 30 min (defined through MTT assay in previous experiments). Subsequently, cells were washed with phosphate-buffered saline (PBS) and exposed to AMD (100 µM) for 24 h to assess cell viability, oxidative damage to proteins and lipids, and NO levels. In order to analyze whether CAT and EPI could prevent mitochondrial dysfunction induced by AMD, we evaluated complex I activity and ATP biosynthesis, along with superoxide dismutase and catalase activities. For these assays, cells were treated with a low concentration of CAT and EPI (10 μM) for 30 min, and then with AMD (100 μM) for one hour. AMD time exposure was reduced in order to keep the MRC-5 cell viability at 100%.
2.4. MTT assay
To evaluate cell viability, cells at a density of 1105 were treated with phenolic compounds and AMD, and the MTT assay [16] was used. After treatment, cells were washed with PBS, exposed to 1 mg/mL per well of MTT solution, and incubated for 3 h at 37 °C. The precipitates were dissolved in 150 µL of dimethyl sulfoxide per well, and the absorbance of the resultant solution was measured with a microplate reader (Victor-X3, Perkin Elmer, Finland) at 517 nm. The results were expressed as a percentage of the control.
2.5. Oxidative stress markers
Oxidative stress assessment included the quantification of lipid and protein oxidative damage and NO production. For all assays, 1107 cells were treated with 10, 100, and 500 μM phenolic compounds and 100 μM AMD. Cells were freeze-thawed 3 times for cell lysis. The supernatants were used for all tests. Lipid oxidative damage was evaluated using the thiobarbituric acid reactive substances (TBARS) assay, according to Wills [17]. Briefly, samples containing 400 µL of cell lysate were combined with 600 µL of 15% TCA and 0.67% TBA. The mixture was heated at 100 °C for 20 min. After being cooled at 20 °C, the samples were centrifuged at 1300 g for 10 min. The supernatant fraction was isolated, and its absorbance was measured at 530 nm. TMP was used as the standard, and the results were expressed as nmol of TMP/mg of protein. Oxidative protein damage was evaluated as previously described [18]. Briefly, samples were solubilized in 2,4-dinitrophenylhydrazine (DNPH), precipitated by the addition of 20% TCA, and the absorbance was read in a spectrophotometer at 365 nm. Results were expressed as nmol DNPH/mg of protein. NO production was determined as nitrite (NO2−) formation using the Griess reaction-based method described by Green et al. [19]. Fifty microliters of cell lysate were reacted with an equal volume of Griess reagent (0.1% naphthyl ethylenediamine and 1% sulfanilamide in 5% H3PO4) for 10 min at 20 °C, and the absorbance was read at 550 nm. Sodium nitroprusside was used as the standard. The results were expressed as nmol of nitrite/mg of protein.
2.6. Mitochondrial function assessment
Cells at a density of 1107 were treated with 10 μM phenolic compounds and 100 μM AMD, washed with cold PBS, scraped, and homogenized in PBS. An assay was performed using the Complex I Enzyme Activity Microplate Assay Kit (Mitoscience, Abcam, Cambridge, MA, USA) according to the manufacturer׳s instructions. The results were expressed as percentage of the control. To verify possible alterations in ATP biosynthesis, 5104 cells/mL were treated and assayed for their ATP biosynthesis using the Cell-Titer-Glo® assay (Promega, Madison, WI) according to the manufacturer׳s instructions. The results were expressed as percentage of the control.
2.7. Superoxide dismutase and catalase activities
Because mitochondrial dysfunction can lead to formation of a radical superoxide anion (O2-·) and hydrogen peroxide (H2O2) production, we also evaluated the superoxide dismutase and catalase activities. To perform these assays, 1 107 cells were treated with 10 μM phenolic compounds and 100 μM AMD, washed with PBS, scraped, and homogenized in PBS. Cells were freeze-thawed 3 times for cell lysis. Then, the supernatants were used for both enzymatic assays. Superoxide dismutase activity was measured by the inhibition of self-catalytic adrenochrome formation rate at 480 nm in a reaction medium containing 1 mmol/L adrenaline (pH 2.0) and 50 mmol/L glycine (pH 10.2) at 30 °C for 3 min as previously described [20]. Results were expressed as USod/mg of protein. One unit was defined as the amount of enzyme that inhibits the rate of adrenochrome formation by 50%. Catalase activity was measured according to the method described by Aebi [21]. The assay determined the rate of H2O2 decomposition at 30 °C for 1 min at 240 nm. Results were expressed as UCat/mg of protein. One unit was defined as the amount of enzyme that decomposes 1 nmol of H2O2 in 1 min at pH 7.4. The protein concentration was determined by the Lowry method, using bovine serum albumin as the standard [22].
2.8. Statistical analysis
All data were expressed as mean±standard derivation (SD) from at least three independent experiments. The normality of variables was evaluated by the Kolmogorov–Smirnov test. The data were analyzed by one way analysis of variance (ANOVA) followed by Duncan׳s multiple range test using statistics software package SPSS for Windows, V.21.0 (Chicago, IL, USA). Values of p < 0.05 were considered as statistically significant.
3. Results
3.1. CAT and EPI decrease the cell death and oxidative damage induced by AMD
AMD was able to induce cell death (Fig. 3), lipid and protein oxidative damage, and reduce NO production (Table 1) in MRC-5 cells. Both CAT and EPI minimized these effects at all evaluated concentrations in a dose-independent manner. Cells treated only with phenolic compounds showed neither increased oxidative damage nor changes in the NO levels (results not shown).
Fig. 3.

Viability of MRC-5 line treated with catechin (CAT) or epicatechin (EPI) for 30 min, followed by incubation with amiodarone (AMD) for 24 h. The results are expressed as the mean±SD. Different letters indicate significantly different values according to the analysis of variance (ANOVA) and Duncan post-hoc test. Statistical significance was determined at p < 0.05.
Table 1.
Thiobarbituric acid reactive substances (TBARS), protein carbonyl groups (PC), and nitric oxide (NO) levels in MRC-5 cells treated with different concentrations of catechin (CAT) or epicatechin (EPI), followed by treatment with 100 µM amiodarone (AMD).
| Treatments | TBARS (nmol of TMP/mg of protein) | PC (nmol of DNPH/mg of protein) | NO (nmol of nitrite/mg of protein) |
|---|---|---|---|
| Cell Control | 5.97±0.05a | 2.73±0.57a | 3.71±0.07a |
| AMD | 12.69±0.62f | 6.93±0.81d | 2.83±0.02d |
| CAT 10 µM + AMD | 9.60±0.39bc | 4.55±0.01c | 2.93±0.04bc |
| CAT 100 µM + AMD | 10.61±0.01d | 4.59±0.34c | 2.99±0.01b |
| CAT 500 µM + AMD | 10.48±0.85cd | 3.62±0.14b | 2.92±0.03bc |
| EPI 10 µM + AMD | 9.43±1.09b | 3.76±0.20b | 2.91±0.03bc |
| EPI 100 µM + AMD | 10.28±0.01bcd | 3.60±0.18b | 2.90±0.09cd |
| EPI 500 µM + AMD | 11.68±0.11e | 4.04±0.22bc | 2.91±0.01bc |
The results are expressed as the mean±SD. Different letters indicate significantly different values according to the analysis of variance (ANOVA) and Duncan post-hoc test. Statistical significance was determined at p< 0.05. TMP (hydrolyzed 1,1,3,3-tetramethoxypropane); DNPH (2,4-dinitrophenylhydrazine).
3.2. Mitochondrial dysfunction induced by AMD is prevented by both CAT and EPI
Mitochondrial dysfunction was evaluated through mitochondrial complex I activity and ATP biosynthesis in the treated cells. AMD was able to reduce the activity of the mitochondrial complex I by 53% (Fig. 4A) and ATP biosynthesis by 9.5% (Fig. 4B). These effects were completely prevented by 10 μM CAT or EPI addition.
Fig. 4.

Mitochondrial complex I (A), ATP biosynthesis (B), superoxide dismutase (C), and catalase (D) activities of MRC-5 cells treated with catechin (CAT) or epicatechin (EPI) and amiodarone (AMD). One USod is defined as the amount of enzyme that inhibits the rate of adrenochrome formation by 50%. One UCat is defined as the amount of enzyme that decomposes 1 mmol of H2O2 in 1 min at pH 7.4. The results are expressed as the mean±SD. Different letters indicate significantly different values according to the analysis of variance (ANOVA) and Duncan post-hoc test. Statistical significance was determined at p < 0.05.
Considering that mitochondrial dysfunction produces ROS, we evaluated the activities of antioxidant superoxide dismutase and catalase. In fact, cells treated with AMD showed a reduced activity (Fig. 4C and D) of both enzymes. CAT and EPI were able to minimize the depletion of superoxide dismutase and catalase activities induced by AMD. Treatment of the MRC-5 cells with only phenolic compounds did not modify the activity of either evaluated enzyme (results not shown).
4. Discussion
Interest in phenolic compounds has grown over the last several decades owing to the recognition of their antioxidant properties and their probable role in the prevention of a number of pathologies associated with oxidative stress [23]. Taking into account the low bioavailability of phenolic compounds [24], [25], their biological actions are more likely to be caused by their indirect effects (such as by influencing signaling systems) than their direct antioxidant effects [26]. In fact, researchers have already described how some phenolic compounds such as quercetin, resveratrol, and rutin reduced mitochondrial dysfunction induced by indomethacin in Caco-2 cells [27]. Moreover, our group demonstrated that Plinia cauliflora polyphenolic-rich extract was also able to reduce complex I inhibition and decrease the ATP biosynthesis induced by H2O2 in MRC-5 cells [28].
Although the exact mechanism of AMD toxicity has not been completely elucidated, some studies conducted in mouse liver [6], hamster lung [7], and human hepatocytes cells [8], as well as an in vivo study in a rat model [9] demonstrated that AMD could cause mitochondrial dysfunction. Therefore, our study aimed to assess whether the phenolic compounds CAT and EPI could minimize the mitochondrial dysfunction and oxidative damage induced by AMD in MRC-5 cells.
The data obtained in our work showed that AMD was able to inhibit complex I activity and ATP biosynthesis in MRC-5 cells. These effects were accompanied by an increase in lipid and protein oxidative damage and a decrease in NO levels and superoxide dismutase and catalase activities, suggesting that AMD toxicity was related to O2-· and H2O2.
Respiratory chain complex I is the most structurally and functionally complex respiratory enzyme [29], [30]. Complex I dysfunction increases O2-· production, which can be a substrate for superoxide dismutase originating H2O2, which, in turn, can be a substrate for catalase. O2-· and the H2O2 can also decrease complex I activity [31], which can feed a vicious cycle of complex I inhibition and maintain a state of cellular oxidative stress. Among the factors able to trigger the intrinsic apoptotic pathway are the oxidative stress, depolarization of the mitochondrial inner membrane, and increased release of cytochrome c [32]. Therefore, the mitochondrial dysfunction and redox imbalance induced by AMD could explain, at least, in part, MRC-5 cell death, and might be related to the lung toxicity caused by this antiarrhythmic drug. It is important to mention that these effects could be due to AMD itself and its metabolite N-desethylamiodarone. In further studies, it would be interesting to examine the effects of AMD metabolite to better understand its mechanism of action.
Both phenolic compounds CAT and EPI were able to prevent both complex I inhibition and decrease in ATP biosynthesis induced by AMD in MRC-5 cells. Consequently, the formation of O2-· and H2O2, oxidative damage, and death of MRC-5 cells were reduced. In addition, CAT and EPI minimized the reduction in NO levels induced by AMD in MRC-5 cells (Fig. 5). These results were similar for both CAT and EPI, which suggests that the chemical difference of the compounds (Fig. 2) was not related to the biological effects demonstrated by CAT and EPI. A study evaluating the ability of CAT and EPI to scavenge the O2-· and reduce the radical 2,2-diphenyl-1-picrylhydrazyl in vitro [33], also did not observe a difference in the effect of the two phenolic compounds. Additional studies using different classes of phenolic compounds would contribute to a better understanding of the relationship between the structure and biological activity of these compounds.
Fig. 5.

Effects of amiodarone (AMD), catechin (CAT), and epicatechin (EPI) in MRC-5 cells. AMD reduces NO levels and inhibits the complex I of the electron transport chain, leading to a decrease in ATP production and an increase in oxidative damage. CAT and EPI reduce these effects, thereby improving cell viability. SOD (superoxide dismutase); ETC (electron transport chain).
The mechanism by which CAT and EPI modulate the activity of complex I is not yet fully known. However, studies have already shown that CAT, resveratrol, and quercetin [34] are capable of directly or indirectly increasing proteins called sirtuins. These classes of molecules are mainly protein deacetylases involved in diverse cellular process and pathways, and they vary in cell localization and functions. Seven sirtuins have already been described in mammals, named SIRT1 to SIRT7. SIRT1 predominately localizes in the nucleus and regulates mitochondrial processes, stress response, cell proliferation, and apoptosis [35]. Furthermore, SIRT1 was found to be associated with vasodilation in rat aortic endothelial cells by increasing the activity of nitric oxide synthase [36]. SIRT3 is the major mitochondrial deacetylase, and it regulates the complex I activity, maintaining electron chain function, and therefore, ATP biosynthesis [35]. However, from the data obtained in our study, it is not possible to determine whether CAT and EPI maintain complex I activity and ATP biosynthesis, thus improving MRC-5 cell viability by directly or indirectly targeting these sirtuins. It has already been shown that EPI-rich cocoa increased the expression of SIRT1 and SIRT3 in skeletal muscle of patients with type II diabetes and heart failure [26]. Other studies should be conducted to clarify this observation and to provide perspectives for the use of sirtuins as new targets to treat AMD toxicity.
In conclusion, our data showed that the phenolic compounds CAT and EPI reduce the cytotoxicity induced by AMD in MRC-5 cells. Although extrapolation of the results of cell culture studies to human clinical situations is uncertain, this is an important finding for the possible development and application of novel therapeutic agents that can reduce the adverse effects of this arrhythmic drug.
Funding
This research was supported by a grant from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; Grant number 302885/2011-0). Mirian Salvador is the recipient of a CNPq Research Fellowship.
Conflict of Interest
All authors declare no conflict of interest related to this study.
Acknowledgments
We thank Dr. Ricardo Luiz de Almeida (INCORGS-Instituto do Coração da Serra Gaúcha) for his advice.
References
- 1.Fu D. Cardiac arrhythmias: diagnosis, symptoms, and treatments. Cell Biochem Biophys. 2015;73:291–296. doi: 10.1007/s12013-015-0626-4. [DOI] [PubMed] [Google Scholar]
- 2.Oyama N., Yokoshiki H., Kamishima T. Detection of amiodarone-induced pulmonary toxicity in supine and prone positions. Circ J. 2005;69:466. doi: 10.1253/circj.69.466. [0] [DOI] [PubMed] [Google Scholar]
- 3.WHO - NHS Centre for the evaluation of effectiveness of health care (CeVEAS). 17th Expert Committee on the Selection and Use of Essential Medicines. Proposal for the Inclusion of Amiodarone as an Anti-arrhythmic and/or for the Treatment of Chronic Heart Failure in the Who Model List of Essential Medicines. Geneva. Available online in: 〈http://www.who.int/selection_medicines/committees/expert/17/application/amiodarone_inclusion.pdf〉. 2009, [accessed on 5.7.2012]
- 4.Ghovanloo M.R., Abdelsayed M., Ruben P.C. Effects of Amiodarone and N- desethylamiodarone on Cardiac Voltage-Gated Sodium Channels. Front Pharmacol. 2016;7:39. doi: 10.3389/fphar.2016.00039. [eCollection 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Q., Ning B., Xuan J. The role of CYP 3A4 and 1A1 in amiodarone-induced hepatocellular toxicity. Toxicol Lett. 2016;253:55–62. doi: 10.1016/j.toxlet.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fromenty B., Fisch C., Berson A. Dual effect of amiodarone on mitochondrial respiration. Initial protonophoric uncoupling effect followed by inhibition of the respiratory chain at the levels of complex I and complex II. J Pharmacol Exp Ther. 1990;255:1377–1384. [PubMed] [Google Scholar]
- 7.Bolt M.W., Card J.W., Racz W.J. Disruption of mitochondrial function and cellular ATP levels by amiodarone and N- desethylamiodarone in initiation of amiodarone-induced pulmonary cytotoxicity. J Pharmacol Exp Ther. 2001;298:1280–1289. [PubMed] [Google Scholar]
- 8.Felser A., Blum K., Lindinger P.W. Mechanisms of hepatocellular toxicity associated with dronedarone - a comparison to amiodarone. Toxicol Sci. 2013;131:480–490. doi: 10.1093/toxsci/kfs298. [DOI] [PubMed] [Google Scholar]
- 9.Serviddio G., Bellanti F., Giudetti A.M. Mitochondrial oxidative stress and respiratory chain dysfunction account for liver toxicity during amiodarone but not dronedarone administration. Free Radic Biol Med. 2011;51:2234–2242. doi: 10.1016/j.freeradbiomed.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Golli-Bennour E., Bouslimi A., Zouaoui O. Cytotoxicity effects of amiodarone on cultured cells. Exp Toxicol Pathol. 2012;64:425–430. doi: 10.1016/j.etp.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Roth F.C., Mulder J.E., Brien J.F. Cytotoxic interaction between amiodarone and desethylamiodarone in human peripheral lung epithelial cells. Chem Biol Interact. 2013;204:135–139. doi: 10.1016/j.cbi.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Cui H., Kong Y., Zhang H. Oxidative Stress, mitochondrial dysfunction, and aging. J Signal Transduct. 2012;2012:646354. doi: 10.1155/2012/646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandoval-Acuña C., Ferreira J., Speisky H. Polyphenols and mitochondria: an update on their increasingly emerging ROS-scavenging independent actions. Arch Biochem Biophys. 2014;559:75–90. doi: 10.1016/j.abb.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 14.Del Rio D., Rodrigues-Mateos A., Spencer J.P. Dietary (poly)phenolics in human health: structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid Redox Signal. 2013;18:1818–1892. doi: 10.1089/ars.2012.4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mercer L.D., Kelly B.L., Horne M.K. Dietary polyphenols protect dopamine neurons from oxidative insults and apoptosis: investigations in primary rat mesencephalic cultures. Biochem Pharmacol. 2005;69:339–345. doi: 10.1016/j.bcp.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 16.Denizot F., Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89:271–277. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- 17.Wills E.D. Mechanisms of lipid peroxide formation in animal tissues. Biochem J. 1966;99:667–676. doi: 10.1042/bj0990667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levine R.L., Garland D., Oliver C.N. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-h. [DOI] [PubMed] [Google Scholar]
- 19.Green L.C., Tannenbaum S.R., Goldman P. Nitrate synthesis in the germ free and conventional rat. Science. 1981;212:56–58. doi: 10.1126/science.6451927. [DOI] [PubMed] [Google Scholar]
- 20.Bannister J.V., Calabrese L. Assays for Superoxide dismutase. Methods Biochem Anal. 1987;32:279–312. doi: 10.1002/9780470110539.ch5. [DOI] [PubMed] [Google Scholar]
- 21.Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–126. doi: 10.1016/s0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- 22.Lowry O.H., Rosebrough N.J., Farr A.L. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 23.Li A.N., Li S., Zhang Y.J. Resources and biological activities of natural polyphenols. Nutrients. 2014;6:6020–6047. doi: 10.3390/nu6126020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crozier A., Jaganath I.B., Clifford M.N. Dietary phenolics: chemistry, bioavailability and effects on health. Nat Prod Rep. 2009;26:965–1096. doi: 10.1039/b802662a. [DOI] [PubMed] [Google Scholar]
- 25.Wiese S., Esatbeyoglu T., Winterhalter P. Comparative biokinetics and metabolism of pure monomeric, dimeric, and polymeric flavan-3-ols: a randomized cross-over study in humans. Mol Nutr Food Res. 2015;59:610–621. doi: 10.1002/mnfr.201400422. [DOI] [PubMed] [Google Scholar]
- 26.Ramirez-Sanchez I., Taub P.R., Ciaraldi T.P. (−)-Epicatechin rich cocoa mediated modulation of oxidative stress regulators in skeletal muscle of heart failure and type 2 diabetes patients. Int J Cardiol. 2013;168:3982–3990. doi: 10.1016/j.ijcard.2013.06.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carrasco-Pozo C., Mizgier M.L., Speisky H. Differential Protective effects of quercetin, resveratrol, rutin and epigallocatechin gallate against mitochondrial dysfunction induced by indomethacin in Caco-2 cells. Chem Biol Interact. 2012;195:199–205. doi: 10.1016/j.cbi.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Calloni C., Dall Agnol R., Martínez L.R. Jaboticaba (Plinia trunciflora (O. Berg) Kausel) fruit reduces oxidative stress in human fibroblasts cells (MRC-5) Food Res Int. 2015;70:15–22. [Google Scholar]
- 29.Fendel U., Tocilescu M.A., Kerscher S. Exploring the inhibitor binding pocket of respiratory complex I. Biochim Biophys Acta. 2008;1777:660–665. doi: 10.1016/j.bbabio.2008.04.033. [DOI] [PubMed] [Google Scholar]
- 30.Verkhovskaya M., Bloch D.A. Energy-converting respiratory Complex I: on the way to the molecular mechanism of the proton pump. Int J Biochem Cell Biol. 2013;45:491–511. doi: 10.1016/j.biocel.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 31.Brown G.C., Borutaite V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim Biophys Acta. 2004;1658:44–49. doi: 10.1016/j.bbabio.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 32.Cosentino K., García-Sáez A.J. Mitochondrial alterations in apoptosis. Chem Phys Lipids. 2014;18:62–75. doi: 10.1016/j.chemphyslip.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Nanjo F., Mori M., Goto K. Radical scavenging activity of tea catechins and their related compounds. Biosci Biotechnol Biochem. 1999;63:1621–1623. doi: 10.1271/bbb.63.1621. [DOI] [PubMed] [Google Scholar]
- 34.Chung S., Yao H., Caito S., Arunachalam G., Rahman I. Regulation of SIRT1 in cellular functions: role of polyphenols. Arch Biochem Biophys. 2010;501:79–90. doi: 10.1016/j.abb.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dang W. The controversial world of sirtuins. Drug Discov Today Technol. 2014;12:9–17. doi: 10.1016/j.ddtec.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mattagajasingh I., Kim C.S., Naqvi A. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]