

Abstract
The etiology of chronic obstructive pulmonary disease (COPD) is complex and involves an aberrant inflammatory response. Prostaglandin (PG)E2 is elevated in COPD, is a key modulator of lung fibroblast functions, and may influence COPD progression. Most studies evaluating the effects of PGE2 on lung fibroblasts have used acute exposures. The current study evaluated whether longer-term exposure would induce attenuation of PGE2 signaling as part of an autoregulatory pathway. Human fetal lung fibroblasts were pretreated with PGE2 for 24 hours, and migration and cAMP accumulation in response to acute stimulation with PGE2 were assessed. Fibroblasts from adults with and without COPD were pretreated, and migration was assessed. PGE2 pretreatment attenuated subsequent PGE2-mediated inhibition of chemotaxis and cAMP stimulation. This attenuation was predominantly due to an increase in phosphodiesterase (PDE)4-mediated degradation of cAMP rather than to decreased activation of PGE2 receptors (receptor desensitization). Albuterol- and iloprost-mediated signaling were also attenuated after PGE2 pretreatment, suggesting that activation of PDE4 was able to broadly modulate multiple cAMP-coupled pathways. Lung fibroblasts from adult control subjects pretreated with PGE2 also developed attenuation of PGE2-mediated inhibition of chemotaxis. In contrast, fibroblasts obtained from patients with COPD maintained inhibitory PGE2 signaling after PGE2 pretreatment. These data identify a PDE4-mediated attenuation of PGE2 inhibitory signaling in normal fibroblasts that appears to be altered in COPD fibroblasts. These alterations may contribute to COPD pathogenesis and could provide novel therapeutic targets.
Keywords: PGE2, desensitization, COPD, fibroblast
Cigarette smoke is the most common etiological agent associated with chronic obstructive pulmonary disease (COPD). However, the sensitivity of individuals to smoke exposure is extremely variable, and the factors accounting for this variability remain undefined. The pathogenetic mechanisms that lead from smoke exposure to disease are complex but are likely related to an aberrant inflammatory response (1). The inflammatory lipid mediator prostaglandin (PG)E2 is elevated in the lungs of patients with COPD, and the levels correlate with disease severity (2–5), which can provide a link between inflammation and altered lung structure. Lung fibroblasts are crucial in maintaining tissue structure (6). The inflammatory mediator PGE2 is a potent cAMP-dependent inhibitor of lung fibroblast repair functions, including migration (7–10). PGE2-mediated inhibition of fibroblast repair responses has been related to COPD severity and to emphysema.
Previous studies evaluating PGE2 inhibition of fibroblast repair have focused on short-term responses (7–10). Attenuation of signaling in the face of chronic exposure to stimuli is a common autoregulatory mechanism. Thus, we considered the possibility that, in normal lungs, inhibitory signaling mediated by PGE2 is transient in fibroblasts. In theory, transient inhibition of fibroblast repair would allow the initial inflammatory process to dissipate before initiation of wound healing. Conceptually, it would be counterproductive to initiate repair in the face of ongoing inflammation, a process that usually results in additional nonspecific tissue damage. We hypothesized that longer-term (24-h) PGE2 exposure would induce attenuation of PGE2-dependent inhibition of human lung fibroblast repair functions, thus permitting repair to begin after an initial delay. Persistence of this inhibition in fibroblasts obtained from the lungs of patients with COPD could contribute to altered repair after injury and altered tissue structure.
Using molecular and functional approaches, we identified a phosphodiesterase (PDE)4-dependent mechanism that mediates attenuation of PGE2 inhibitory signaling in normal lung fibroblasts and demonstrated that this process is functionally altered in lung fibroblasts obtained from patients with COPD. The inability to increase PDE4 activity and thus down-regulate PGE2-mediated inhibition of fibroblast repair functions provides a novel mechanism that may contribute to COPD pathology and serve as a novel therapeutic target.
Materials and Methods
Materials
PGE2, albuterol, and forskolin were purchased from Sigma (St. Louis, MO). EP1 agonist (ONO-DI-004), EP2 agonist (ONO-AE1-259), EP3 agonist (ONO-AE-248), EP4 agonist (ONO-AE1-329), EP1 antagonist (ONO-8713), EP3 antagonist (ONO-AE3-240), and EP4 antagonist (ONO-AE3-208) were kind gifts of ONO Pharmaceutical Co. Ltd (Osaka, Japan). EP2 antagonist (AH6809), rolipram, iloprost, and primary antibodies to the four EP receptors were purchased from Cayman Chemical (Ann Arbor, MI). Roflumilast was a kind gift from Nycomed (Zurich, Switzerland). Human fibronectin was purchased from Fisher (Hampton, NH). Primary antibodies to the four PDE4 isozymes were obtained from AbCam (Cambridge, UK). A second primary antibody to PDE4D was purchased from FabGennix (Frisco, TX). Fetal calf serum (FCS), PDE4A-C siRNA, and Lipofectamine 2000 were obtained from Invitrogen (Grand Island, NY). [3H]adenine was obtained from Perkin Elmer (Waltham, MA).
Cell Culture
Normal human fetal lung (HFL) fibroblasts were obtained from American Type Tissue Culture (Rockville, MD; CCL-153). Cells were cultured in 100-mm culture dishes (Falcon; Lincoln Park, NJ) to confluence in Dulbecco?s modified Eagle?s medium (DMEM) containing 10% FCS, 50 U/ml penicillin, 50 mg/ml streptomycin, and 0.25 mg/ml of amphotericin B (FCS media) and maintained in a humidified 5% CO2 incubator at 37°C. When confluent, cells were enzymatically detached from the dish with 0.05% trypsin in 0.53 mM EDTA and resuspended in FCS media. Cells were used between passages 13 and 21.
Adult fibroblasts were obtained from patients undergoing thoracotomy for suspected lung malignancies. Patients included smokers with COPD and smokers without COPD. Normal tissue was excised from an area outside the perimeter of the mass and initiated into culture as previously described (11). Primary cell lines were frozen and shipped to the University of Nebraska Medical Center, where they were cultured as needed.
Chemotaxis Assay
Fibroblast chemotaxis towards human fibronectin (5 μg/ml) was evaluated after 24-hour pretreatment utilizing the Boyden blindwell chamber (Nucleopore, Cabin John, MD) method (12) as previously described (8). Migrated cells were counted in five high-power fields (HPF) for each well with a standard light microscope. Data for each slide were recorded onto a blank template labeled only with a corresponding identification number so as not to bias the slide evaluator. Data for chemotaxis assays with HFL cells are expressed as an average of migrated cells in all HPF's of two or three wells per condition. All experiments were repeated on at least three separate occasions.
cAMP Accumulation Assay
A modification of the Shimizu method (13) described previously (14) was used to assay cAMP accumulation. On Day 0, HFL cells were plated into 12-well culture plates (Falcon) at 50,000 cells per well in FCS. The next day (Day 1), media were replaced with fresh FCS. On Day 2, FCS media were removed and replaced with serum-free media for 30 minutes. After this serum starvation period, serum-free media were removed and then replaced with serum-free media supplemented with the appropriate compounds for pretreatment. After pretreatment, the cells were washed with 1 ml of HEPES-buffered DMEM (H-DMEM). Next, 0.5 ml of H-DMEM with 5 μCi/ml of [3H]adenine was added for 1 hour at 37°C in a non-CO2 incubator with or without experimentally specific inhibitors or antagonists. After the loading period, cells were then stimulated in 0.5 ml of H-DMEM containing various ligands for the indicated length of stimulation (5 min in most assays). Stimulation was terminated by the addition of 1.0 ml of 5% TCA containing 1 mM nonradioactive cAMP (to normalize for recovery) following media aspiration. Samples were transferred from the tissue culture plate and analyzed by column chromatography as previously described (14). Data are expressed as the percentage of [3H] ATP converted to [3H] cAMP during the stimulatory period.
Western Blot
After the pretreatment period, cells were washed twice with ice-cold PBS. Cell lysis buffer (35 mM Tris-HCl, pH 7.4, 0.4 mM EGTA, 10 mM MgCl2, 1 mM phenylmethlysulfonyl fluoride, 1:1,000 Triton, 100 mg/ml aprotinin, and 1 mg/ml leupeptin) was added to each dish and a plastic policeman used to scrape the cell layer free. Cell lysates were transferred to fresh Eppendorf tubes, sonicated, and centrifuged. Supernatants were isolated and protein concentrations quantified colormetrically with the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Samples were subjected to 10% SDS-PAGE and then transferred to an Immuno-Blot PVDF Membrane (Bio-Rad). Five percent skim milk in PBS with 0.1% Tween-20 was used as a blocking solution for 1 hour. Primary antibodies were incubated overnight at 4°C. After washing with PBS-Tween, secondary HRP-conjugated antibodies were added to the membrane for 1 hour.
Small Interfering RNA
HFL cells were plated in 100-mm dishes at an initial concentration of 6.0 × 105 cells/ dish in 10% FCS DMEM media with antibiotics. Media were replaced with SF media without antibiotics 24 hours later. The next day, Lipofectamine 2000 was diluted 1:50 in Opti-MEM. Forty-five microliters or 15 μl of 20 μM pooled siRNA (PDE4A, B, and C) was added to 0.800 ml Opti-MEM. These solutions were allowed to equilibrate for 5 minutes before combing equal amounts for a final volume of 1.6 ml. The combined solution was allowed to incubate at room temperature for 20 minutes. The volume was then expanded with Opti-MEM to 3.0 ml and a final siRNA concentration of 300 or 100 nM and incubated with the cells for 6 hours. Four milliliters of 10% FCS DMEM with antibiotics was then added and the cells were cultured for an additional 48 hours. Cells were then re-plated into 60-mm dishes for chemotaxis or 12-well plates for cAMP assays. The next day cells were pretreated with PGE2 (0.1 μM) and incubated for 24 hours. After this last incubation, cells were assessed for migration and cAMP accumulation as described above. Remaining cells were washed twice and resuspended in cold lysis buffer for confirmation of siRNA suppression by immunoblot.
Statistics
Data, unless otherwise indicated, are expressed as the mean ± SEM of at least three experiments performed separately. For mechanistic studies in HFL cells, two-way ANOVA with Bonferroni post-test was performed using GraphPad Prism version 5.04 for Windows (GraphPad Software, San Diego, CA). Student's t test was used to determine statistical differences between matched groups when appropriate (P < 0.05 was considered statistically significant). In studies utilizing adult cells, individual experiments were pooled together and the mean value for each experimental condition plotted. In addition to pooled analyses, we also calculated an index of PGE2-mediated inhibition to allow for comparisons among cells obtained from different patients. This index was calculated as follows:
where [S,S] is the total amount of migrated cells in five high-power fields in the SF pretreatment, SF chemotaxis group; [S,P] is the total migrated cells in the SF pretreatment, PGE2 chemotaxis group; [P,S] represents the total migrated cells in the PGE2 pretreatment, SF chemotaxis group; and [P,P] represents the PGE2 pretreatment, PGE2 chemotaxis group. Because the distribution of data for cells from adults is not known to be normal, nonparametric tests were used. For evaluation of studies within a group (either normal or COPD) where paired samples were available, the Wilcoxon test was used. For comparisons between groups (normal versus COPD), the unpaired, two-tailed Mann-Whitney test was used.
Results
Effect of 24-Hour PGE2 Pretreatment on PGE2-Mediated Signaling and Determination of the Specific Receptor that Mediates the Effect
Human fetal lung (HFL) cells were pretreated with or without 0.1 μM PGE2 for 24 hours after the culture protocol outlined in Materials and Methods (details are provided in the online supplement). After pretreatment, cells were trypsinized, counted, and resuspended in serum-free (SF) medium alone or in SF medium with 0.1 μM PGE2 and then assessed for migration toward human fibronectin in a Boyden blindwell chamber (Figure 1a). Control cells were susceptible to PGE2 (0.1 μM)-mediated inhibition of chemotaxis, consistent with previous results from multiple laboratories (7–10). Cells pretreated with PGE2 for 24 hours demonstrated increased baseline chemotaxis. In contrast to nonpretreated cells, the pretreated cells were refractory to PGE2-mediated inhibition of chemotaxis.
Figure 1.

Effect of 24-hour prostaglandin (PG)E2 (0.1 μM) pretreatment on acute PGE2 inhibitory signaling in human fetal lung (HFL) fibroblasts. (a) PGE2-mediated (0.1 μM) chemotaxis after 24-hour PGE2 pretreatment. (b) PGE2-stimulated cAMP accumulation in HFL fibroblasts pretreated with PGE2 (0.1 μM) for 24 hours. (c) PGE2-mediated chemotaxis after 24-hour E prostanoid receptor (EP) agonist (Ag) (1 μM) pretreatment. (d) PGE2-mediated chemotaxis after 24-hour PGE2 pretreatment in the presence of individual EP receptor antagonists (Ant). (e) PGE2-mediated cAMP accumulation after 24-hour PGE2 pretreatment in the presence of individual EP receptor antagonists. *P < 0.05; **P < 0.01. Bars are means ± SEM; n = 3 separate experiments in all panels.
PGE2 signaling occurs via four G protein–coupled receptors, two of which (E prostanoid 2 receptor [EP2r] and EP4r]) signal via elevation of cAMP. PGE2 exerts its inhibitory actions on chemotaxis via the EP2 and EP4 receptors (8, 10). Accordingly, cAMP accumulation was assessed in cells after 24-hour PGE2 pretreatment to determine if the loss of PGE2-mediated inhibition of chemotaxis after pretreatment is associated with the loss of PGE2-mediated cAMP accumulation. After 24-hour PGE2 pretreatment, PGE2 (0.1 μM) no longer stimulated cAMP accumulation as effectively as nonpretreated cells (Figure 1b), consistent with decreased cAMP accumulation as the basis for the decreased inhibition of chemotaxis.
To determine which of the four EP receptors mediates the loss of PGE2 inhibition of chemotaxis, specific EP receptor agonists were tested. Agonists were added at a fixed concentration of 1.0 μM and incubated with the cells for 24 hours, after which the cells were harvested, and chemotaxis was assessed (Figure 1c). A trend toward increased baseline chemotaxis was seen with all four receptor selective agonists. Only pretreatment with the EP2 receptor agonist resulted in loss of PGE2-mediated inhibition of chemotaxis, suggesting that the EP2 receptor is critical for the development of resistance (Figure 1c).
Similar experiments were performed with EP receptor–specific antagonists. Antagonists were added (1.0 μM) 1 hour before the addition of the PGE2 for pretreatment. After 24 hours, cells were harvested, and chemotaxis was evaluated (Figure 1d). Neither the EP1 not the EP4 receptor antagonist blocked the effect of PGE2 pretreatment. In contrast, the EP2 and EP3 receptor antagonists each blocked the loss of PGE2-mediated inhibition after pretreatment.
EP receptor antagonists were used during PGE2 pretreatment to confirm which receptors are required to mediate the loss of PGE2-mediated cAMP accumulation. Antagonists (1.0 μM) were added 1 hour before PGE2 pretreatment (24 h), after which the assay was performed using two concentrations of PGE2 (0.1 and 1.0 μM) for stimulation of cAMP accumulation (Figure 1e). PGE2 pretreatment blunted the ability of PGE2 to stimulate cAMP accumulation in the face of EP1r, EP3r, and EP4r antagonists, as expected. Only the EP2r antagonist resulted in retention of PGE2-mediated cAMP accumulation, although requiring a higher concentration of stimulating PGE2 to detect the difference, further supporting that the EP2 receptor mediates signal attenuation of cAMP accumulation and the chemotaxis response after PGE2 pretreatment.
Mechanisms Involved in the Regulation of Attenuated PGE2 Signaling after PGE2 Pretreatment
To help elucidate the precise mechanism(s) mediating decreased PGE2 signaling after PGE2 pretreatment, the time necessary to induce attenuation (see Figures E1a and E1b in the online supplement) and the time required for the effect to reverse (Figure E1c) were determined. These time frames, which are described in the online supplement in more detail, required approximately 4 hours of PGE2 treatment to induce a loss of additional inhibitory activity of PGE2. Furthermore, recovery of the ability of PGE2 to mediate cAMP accumulation after 24-hour PGE2 pretreatment required approximately 6 hours. It was determined that the loss of inhibitory PGE2 signaling was concentration dependent, with increased concentrations of PGE2 able to overcome the effect of pretreatment (Figures E2a and E2b). There was no change in total cellular protein of the EP receptors after PGE2 pretreatment (Figure E2d).
cAMP accumulation can be modulated via changes in synthesis or degradation. To determine if adenylyl cyclase (AC) activity is altered after PGE2 pretreatment, cells were pretreated with PGE2 as before, harvested, suspended in SF media with varying concentrations of forskolin (FSK) (a nonspecific AC activator), and chemotaxis was assessed (Figure 2a). Cells pretreated with PGE2 for 24 hours became resistant to FSK-mediated inhibition of chemotaxis at lower concentrations, which could be overcome by increasing the FSK concentration. Similarly, PGE2 pretreatment attenuated the ability of FSK to stimulate cAMP accumulation, which could also be overcome at higher FSK concentrations (Figure 2b). These experiments suggest that the development of resistance to PGE2-mediated inhibition of chemotaxis is due to the reduced ability of the cell to accumulate cAMP.
Figure 2.
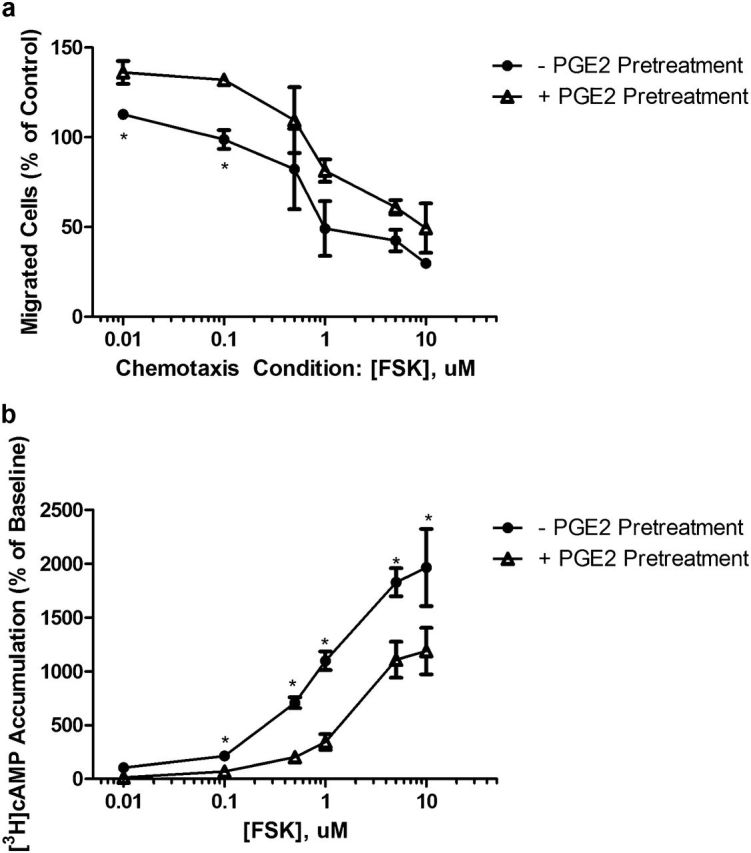
Evaluation of the mechanism mediating attenuation of acute PGE2 signaling after 24-hour PGE2 pretreatment. (a) Concentration response of forskolin (FSK)-mediated inhibition of HFL chemotaxis after 24-hour PGE2 pretreatment. (b) Concentration response of FSK-stimulated cAMP accumulation after 24-hour PGE2 pretreatment. *P < 0.05; **P < 0.01. Bars are means ± SEM; n = 3 separate experiments in all panels.
Identification of the Mechanism Mediating Attenuated PGE2 Signaling after 24-Hour PGE2 Pretreatment
To determine if PDE4 is expressed in HFL fibroblasts, whole cell lysates from HFL cells pretreated with several concentrations of PGE2 for 24 hours were subjected to SDS-PAGE and immunoblotted with primary antibodies to each of the four (PDE4A–D) isozymes (Figure 3a). PDE4A-C, but not PDE4D (two different antibodies for PDE4D were used), were detected by Western blot. There was no increase in PDE4 protein levels after 24-hour PGE2 pretreatment in any concentration of the compound. This suggests that increased total enzyme concentration in response to PGE2 pretreatment is not responsible for the development of resistance.
Figure 3.

Evaluation of the role of phosphodiesterase (PDE)4 in mediating attenuation of acute PGE2 signaling after 24-hour PGE2 pretreatment. (a) Detection of PDE4 isozyme levels after PGE2 pretreatment (24 h) in HFL cells by immunoblot. Black arrows point to a predicted band based on the documented molecular weight of specific PDE4 isozymes. (b) Effect of inhibition of PDE4 with roflumilast (rof, 1.0 μM) and rolipram (rol 1.0 μM) on PGE2-mediated inhibition of HFL chemotaxis after 24-hour PGE2 pretreatment. (c) Effect of inhibition of PDE4 with rof on PGE2-mediated cAMP accumulation after 24-hour PGE2 pretreatment. (d) Effect of inhibition of PDE4 with rol on PGE2-mediated inhibition of HFL chemotaxis after 24-hour PGE2 pretreatment. (e) Time-dependent attenuation of PGE2-mediated cAMP accumulation in HFL cells begins within 15 minutes and is complete by 90 minutes. The addition of roflumilast at 30 minutes restores cAMP accumulation. Cells pretreated with PGE2 do not accumulate cAMP until treated with roflumilast. *P < 0.05; **P < 0.01. Bars are means ± SEM; n = 3 separate experiments in all panels unless otherwise stated.
To determine if PDE4 mediates the resistance of PGE2 signaling after 24-hour PGE2 pretreatment, two specific pharmacological inhibitors of PDE4, roflumilast and rolipram, were used. After PGE2 pretreatment, HFL cells were harvested, and chemotaxis was assessed in the presence or absence of rolipram (10 μM) or roflumilast (1 μM) with or without PGE2 (0.1 μM) (Figure 3b). As before, PGE2 pretreatment resulted in a loss of PGE2-mediated inhibition of chemotaxis. Roflumilast and rolipram alone trended to reduce the increased baseline chemotaxis observed after PGE2 pretreatment. More importantly, when added with PGE2, roflumilast and rolipram restored PGE2-mediated inhibition of chemotaxis.
To determine if the roflumilast and rolipram effect was associated with restoration of cAMP accumulation, analogous experiments were performed assessing cAMP accumulation. Roflumilast completely restored the ability of PGE2 to stimulate cAMP accumulation (Figure 3c). Rolipram, a less potent PDE4 inhibitor, only partially restored PGE2-mediated cAMP accumulation (Figure 3d). Together, these experiments provide evidence that the loss of PGE2-mediated cAMP accumulation and the subsequent loss of inhibition by PGE2 are mediated by increased cAMP hydrolysis by PDE4.
To confirm the role of PDE4 in mediating the loss of PGE2-mediated cAMP accumulation in HFL cells, an alternative approach was used. After 24-hour pretreatment, PGE2 was added to stimulate cAMP accumulation for varying amounts of time (Figure 3e). In nonpretreated cells, cAMP accumulated and peaked within roughly 15 minutes, after which a gradual decline in cAMP accumulation occurred, returning to baseline after 90 minutes. Cells pretreated with PGE2 for 24 hours lost this transient increase in cAMP accumulation in response to acute PGE2 and do not accumulate cAMP. After the 30-minute time point, roflumilast (final concentration, 1.0 μM) was directly added to the media containing the stimulating PGE2, and cAMP accumulation was measured at 60 and 90 minutes after the initial PGE2 challenge. The addition of roflumilast restored cAMP accumulation in SF pretreated cells. The addition of roflumilast to cells pretreated with PGE2, which previously were unable to accumulate cAMP, restored the ability of PGE2 to stimulate cAMP accumulation. This suggests PDE4 activity increases relatively rapidly and confirms the concept that PGE2 signaling is being regulated downstream of the EP receptors.
To further confirm the role of PDE4 in the development of resistance after PGE2 pretreatment and to address concerns with the nonspecific effects inherent in all pharmacological inhibitors, we used the alternate strategy of inhibiting PDE4 using siRNA for the three isozymes detectable by immunoblot. We determined that cells transfected with pooled PDE4A-C siRNA, which resulted in detectable down-regulation of PDE4A and PDE4C only (Figure E3a), before pretreatment with PGE2 remained sensitive to further inhibitory PGE2 signaling (Figures E3b and E3c). Through individual transfection experiments, it was determined that the PDE4A and PDE4C work in redundant fashion to mediate the loss of inhibitory PGE2 signaling after PGE2 pretreatment (Figures E4a–E4e).
Specificity of the cAMP Attenuation Pathway
cAMP accumulation is stimulated by several distinct ligand-receptor transduction pathways. To determine if the PDE4-mediated resistance after PGE2 pretreatment is specific for PGE2 or generalized for other cAMP-coupled signaling pathways, iloprost (Ilo), a specific agonist for the IP receptor, and albuterol (Alb), an agonist of the β2 adrenergic receptor, were used. After PGE2 pretreatment, the ability of Ilo and Alb to inhibit HFL fibroblast chemotaxis was attenuated (Figure 4a). After PGE2 pretreatment, inhibition of PDE4 with roflumilast was able to restore the ability of Ilo and Alb to mediate inhibition of chemotaxis in the presence of PGE2 (Figure 4a). Similarly, PGE2 pretreatment blocked the ability of FSK, Ilo, and Alb to mediate cAMP accumulation (Figure 4b). Inhibition of PDE4 with roflumilast restored the ability of these compounds to mediate cAMP accumulation in PGE2-pretreated cells. These data suggest that activation of PDE4 by PGE2 pretreatment is able to modulate cellular cAMP accumulation in response to several pathways that induce cAMP production.
Figure 4.

Effect of 24-hour PGE2 pretreatment on iloprost and albuterol signaling in HFL cells. (a) Effect of iloprost (Ilo, 0.1 μM) and albuterol (Alb, 0.1 μM) on fibroblast chemotaxis after PGE2 pretreatment in the presence or absence of PDE4 inhibition with Rof (1.0 μM). (b) Effect of Ilo (0.1 μM) and Alb (0.1 μM) on fibroblast cAMP accumulation after PGE2 pretreatment in the presence or absence of PDE4 inhibition with Rof (1.0 μM). *P < 0.05; **P < 0.01. Bars are means ± SEM; n = 3 separate experiments in all panels.
Effect of PGE2 Pretreatment on Normal Adult and COPD Lung Fibroblast Chemotaxis
Altered repair processes have been reported by our laboratory and others in fibroblasts obtained from patients with COPD. To determine if deficient repair in COPD might result, in part, from a lack of PGE2-induced attenuation of inhibitory signaling, chemotaxis was assessed in fibroblasts cultured from the lungs of normal subjects and patients with COPD. To compare strains, an index of PGE2-mediated inhibition was determined. As with HFL cells, treatment of fibroblasts from control subjects with PGE2 inhibited chemotaxis, and this inhibition was blocked by pretreatment of the cells with PGE2 for 24 hours (Figure 5a, top panel). In contrast, cells cultured from the lungs of patients with COPD exhibited no loss of inhibition when the cells were pretreated with PGE2 (Figure 5a, bottom panel). That is, the COPD cells failed to demonstrate resistance to long-term PGE2 exposure. Alternatively, we have presented these data in the form of a PGE2 inhibition index (Figures E5a and E5b). This presentation better displays intersubject data. The change in inhibition indexes before and after PGE2 pretreatment between control and COPD cells was statistically significant (P < 0.01) (Figure E5b).
Figure 5.

Effect of PGE2 pretreatment (24 h) on PGE2-mediated inhibition of chemotaxis in lung fibroblasts obtained from patients with chronic obstructive pulmonary disease (COPD) and healthy control subjects. (a-1) PGE2-mediated chemotaxis of pooled control (n = 8) fibroblast strains pretreated in the presence or absence PGE2 for 24 hours. (a-2) PGE2-mediated chemotaxis of pooled COPD (n = 12) fibroblast strains pretreated in the presence or absence PGE2 for 24 hours. (b) Schema depicting cAMP saturation of PDE4 enzymes after increased agonist stimulation. (c) Schema of global activity of PGE2 activated PDE4 enzymes on multiple cAMP-coupled pathways. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05. Bars are means ± SEM. AC = adenylyl cyclase; B2AR = β2 adrenergic receptor; Gs = Gαs G protein; IPr = prostacyclin receptor.
Discussion
COPD, a chronic disease characterized by irreversible and progressive loss of lung function, is now the third leading cause of death in the United States (15). An abnormal inflammatory response is a well recognized feature of COPD, but the mechanisms that underlie this response have not been fully delineated. In addition, COPD is characterized by tissue disruption that includes damage and inadequate repair. The current study demonstrates that resistance to PGE2-mediated inhibition of fibroblast repair in response to prolonged PGE2 exposure occurs in normal cells and represents a normal physiologic mechanism of feedback regulation. This regulation appears to be absent in lung fibroblasts from patients with COPD and illustrates a novel pathogenetic mechanism for COPD. The attenuation of this PGE2 response in normal fibroblasts is due to enhanced degradation of cAMP by PDE4 rather than to receptor desensitization.
Regulation of the sensitivity of autocrine, paracrine, and endocrine signaling pathways is a critical step in the modulation of hormone signal strength and integration. Many studies investigating PGE2-dependent signaling have been performed in cells engineered to overexpress receptors of interest, and these studies have determined that the EP2 receptor does not undergo receptor desensitization (16–19). This approach is useful in determining receptor dynamics on the cell surface but ignores the complex molecular machinery that can be unique to each specific cell type. The current data document the development of resistance to PGE2-mediated inhibition of chemotaxis after pretreatment with PGE2 in HFL fibroblasts, a normal, genetically intact, and widely used lung fibroblast cell strain (20–23). Our approach is more likely to yield results applicable to normal fibroblast physiology rather than approaches that rely on genetically altered cell systems.
The attenuation of PGE2-dependent signaling after PGE2 pretreatment was due to reduced cAMP accumulation, which developed over several hours. Receptor protein levels remained unchanged, and the resistance could be overcome by increasing PGE2 levels, consistent with intracellular regulation rather than classical receptor desensitization. PDE enzymes are able to modulate cyclic nucleotide-dependent signaling (24). HFL cells express PDE4 enzymes (PDE4A, -B, and- C) (Figure 3a) detectable by immunoblot. Inhibition of these enzymes pharmacologically or by siRNA restored PGE2-mediated cAMP accumulation and PGE2-mediated inhibition of chemotaxis after PGE2 pretreatment. This suggests that increased PDE4 activity is the mechanism for the development of resistance to PGE2 signaling, likely by the PDE4A and PDE4C isozymes, specifically. Using two separate anti-PDE4D antibodies, we were unable to detect PDE4D protein. This does not, however, rule out the potential role for PDE4D in regulating cAMP dynamics in lung fibroblasts. PGE2 pretreatment did not change PDE4 isozyme protein expression, suggesting increased activity without increased enzyme levels.
Resistance to PGE2-mediated inhibition of chemotaxis (Figure E2a) and cAMP accumulation (Figure E2b) after PGE2 pretreatment could be overcome by increasing the concentration of stimulating ligand likely due to increased activation of AC. This suggests that as the amount of ligand-stimulated cAMP within the cells increases, the PDE4 enzymes eventually become saturated (Figure 5c). Excess cAMP then accumulates and leads to inhibition of fibroblast function.
PGE2 pretreatment also led to resistance to the two other cAMP-coupled receptor signaling pathways that were tested, the β2 adrenergic and IP receptor agonists. Similarly, this effect was sensitive to PDE4 inhibition and suggests that PGE2-mediated activation of PDE4 activity is able to broadly mediate decreased cAMP accumulation within the cell, independent of the receptor that activates adenylyl cyclase (Figure 5d). We used this approach to confirm our primary observation. The pathophysiological implications of receptor cross regulation between EP receptors and other cAMP coupled receptors before and after chronic PGE2 exposure is of interest.
PGE2 pretreatment induced resistance to further PGE2-mediated inhibition in fibroblasts derived from control subjects, indicating that this effect is not limited to the HFL cell system. In contrast, development of resistance did not occur in fibroblasts obtained from patients with COPD (Figure 5a; Figures E5a and E5b). These adult cells were derived from smokers undergoing surgery for suspected malignancy. Because they were of similar age, smoking history, and final cancer diagnosis but differed in the diagnosis of COPD, the differences in PGE2-induced resistance to PGE2-mediated inhibition are not a function of age, smoking history, or malignancy and are most likely related to their COPD status. Whether cells from patients with COPD who differed clinically from these individuals would behave similarly remains to be determined.
Differences between normal and COPD fibroblasts have been observed for a variety of fibroblast repair functions, including decreased proliferation (11), decreased migration (25), decreased contraction of 3D collagen gels (25), increased senescence (26, 27), and increased sensitivity to the toxic effects of cigarette smoke extract (28). Decreased development of PGE2 resistance would render COPD fibroblasts more sensitive to PGE2 inhibition of repair and could further potentiate these functional differences in vivo. Abnormal inflammation is thought to be such a fundamental feature of COPD that it is included in the definition of the condition (29). Considering that PGE2 is a well known inflammatory mediator that is a potent inhibitor of fibroblast-mediated repair in vitro, the current study provides a mechanistic basis for how part of the “abnormal” inflammation present in COPD may result in altered tissue homeostasis and resultant pathological airway remodeling.
The lungs of patients with COPD have increased levels of many inflammatory mediators, including PGE2 (2–5), and the level of PGE2 in the lung correlates with disease severity. Fibroblasts are a significant source of PGE2 in vivo, and fibroblasts derived from the lungs of patients with COPD produce increased levels of PGE2 in vitro (25). COPD lungs are chronically inflamed. With this inflammation, there are increased levels of inflammatory mediators, including IL-1β and TNF-α, which also result in PGE2 release from fibroblasts. Thus, the inflammatory milieu may also directly contribute to increased PGE2 release from fibroblasts and may result in autocrine regulation of fibroblast function. PGE2 inhibits many repair functions of fibroblasts. Thus, chronically elevated levels of PGE2 in the COPD lung combined with a lack of normal attenuation mechanisms could result in chronic inhibition of fibroblast-mediated repair. The resulting deficiency of fibroblast-mediated tissue repair in the presence of chronic inflammation and ongoing tissue damage may result in emphysema. Within this context, it is possible that emphysema development requires two “hits.” The first “hit” is uncontrolled chronic inflammation in response to noxious stimuli (cigarette smoke), which results in increased PGE2 lung burden. In individuals not susceptible to the effects of cigarette smoke, disease does not occur. This may be explained by the ability of resident lung fibroblasts to become resistant to the inhibitory actions of PGE2 and subsequently to mediate effective repair in the face of ongoing tissue destruction. The second “hit” represents a change in fibroblast phenotype that results in a deficiency in the cell’s ability to modulate these chronic PGE2 signals. These fibroblasts would be chronically inhibited by elevated levels of PGE2 in the COPD lung and rendered unable to mediate normal repair responses, possibly resulting in emphysema.
Alterations in PGE2-mediated signaling may contribute to other chronic lung diseases. In idiopathic pulmonary fibrosis, a disease process that is in many ways opposite to those occurring in emphysema, decreased levels of PGE2 have been reported (30–33). These low or absent levels of PGE2 are thought to contribute to uncontrolled fibroblast activity that results in fibrosis. Manipulation of PGE2 signaling may thus be useful in multiple diseases.
In summary, we present evidence for an autoregulatory feedback mechanism that provides resistance to inhibitory PGE2 signaling in normal fibroblasts, which is mediated by activation of PDE4 enzymes and not regulated at the level of the receptor. PGE2-mediated induction of this mechanism, which occurs through the EP2 receptor, modulates multiple cAMP-coupled pathways related to airway repair. We present data that this mechanism of signal attenuation is altered in COPD cells. However, the role of PDE4 in mediating this attenuation in adult fibroblasts remains unclear. Deficient PGE2 signal attenuation in COPD fibroblasts is a novel pathogenic mechanism for COPD and provides a mechanistic basis for the abnormal inflammatory response that characterizes COPD and a link between inflammation and deficient repair.
Additional material
Supplementary data supplied by authors.
Acknowledgments
The authors thank Mrs. Lillian Richards for excellent secretarial support, Nycomed and Dr. Hermon Tenor for the kind gift of roflumilast, and ONO Pharmaceuticals for the kind gift of EP receptor agonists and antagonist.
Footnotes
This work was supported by the Kuhl Respiratory Research Award, University of Nebraska Graduate Studies Fellowship, National Institutes of Health grant 1 R01 HL064088, and the Larson Endowment, University of Nebraska Medical Center.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2012-0057OC on October 4, 2012
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Yao H, Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl Pharmacol 2011;254:72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Y, Chen P, Hanaoka M, Droma Y, Kubo K. Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology 2008;13:1014–1021. [DOI] [PubMed] [Google Scholar]
- 3.Montuschi P, Kharitonov SA, Ciabattoni G, Barnes PJ. Exhaled leukotrienes and prostaglandins in COPD. Thorax 2003;58:585–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verhoeven GT, Garrelds IM, Hoogsteden HC, Zijlstra FJ. Effects of fluticasone propionate inhalation on levels of arachidonic acid metabolites in patients with chronic obstructive pulmonary disease. Mediators Inflamm 2001;10:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y, Chen P. [Expressions of cyclooxygenase-2 and matrix metalloproteinase-2 and airflow obstruction in patients with chronic obstructive pulmonary disease]. Zhonghua Jie He He Hu Xi Za Zhi 2005;28:324–327. [PubMed] [Google Scholar]
- 6.Horowitz JC, Martinez FJ, Thannickal VJ. Mesenchymal cell fate and phenotypes in the pathogenesis of emphysema. COPD 2009;6:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kohyama T, Ertl RF, Valenti V, Spurzem J, Kawamoto M, Nakamura Y, Veys T, Allegra L, Romberger D, Rennard SI. Prostaglandin E(2) inhibits fibroblast chemotaxis. Am J Physiol Lung Cell Mol Physiol 2001;281:L1257–L1263. [DOI] [PubMed] [Google Scholar]
- 8.Li YJ, Wang XQ, Sato T, Kanaji N, Nakanishi M, Kim M, Michalski J, Nelson AJ, Sun JH, Farid M, et al. Prostaglandin E inhibits human lung fibroblast chemotaxis through disparate actions on different E-prostanoid receptors. Am J Respir Cell Mol Biol 2011;44:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Togo S, Liu X, Wang X, Sugiura H, Kamio K, Kawasaki S, Kobayashi T, Ertl RF, Ahn Y, Holz O, et al. PDE4 inhibitors roflumilast and rolipram augment PGE2 inhibition of TGF-{beta}1-stimulated fibroblasts. Am J Physiol Lung Cell Mol Physiol 2009;296:L959–L969. [DOI] [PubMed] [Google Scholar]
- 10.White ES, Atrasz RG, Dickie EG, Aronoff DM, Stambolic V, Mak TW, Moore BB, Peters-Golden M. Prostaglandin E(2) inhibits fibroblast migration by E-prostanoid 2 receptor-mediated increase in PTEN activity. Am J Respir Cell Mol Biol 2005;32:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holz O, Zuhlke I, Jaksztat E, Muller KC, Welker L, Nakashima M, Diemel KD, Branscheid D, Magnussen H, Jorres RA. Lung fibroblasts from patients with emphysema show a reduced proliferation rate in culture. Eur Respir J 2004;24:575–579. [DOI] [PubMed] [Google Scholar]
- 12.Bitterman PB, Wewers MD, Rennard SI, Adelberg S, Crystal RG. Modulation of alveolar macrophage-driven fibroblast proliferation by alternative macrophage mediators. J Clin Invest 1986;77:700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimizu H, Daly JW, Creveling CR. A radioisotopic method for measuring the formation of adenosine 3′,5′-cyclic monophosphate in incubated slices of brain. J Neurochem 1969;16:1609–1619. [DOI] [PubMed] [Google Scholar]
- 14.Kassel KM, Wyatt TA, Panettieri RA, Toews ML. Inhibition of human airway smooth muscle cell proliferation by beta 2-adrenergic receptors and cAMP is PKA independent: evidence for EPAC involvement. Am J Physiol Lung Cell Mol Physiol 2008;294:L131–L138. [DOI] [PubMed] [Google Scholar]
- 15.Miniño AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008 Natl Vital Stat Rep 2011;59:1–126. [PubMed] [Google Scholar]
- 16.Wilson RJ, Rhodes SA, Wood RL, Shield VJ, Noel LS, Gray DW, Giles H. Functional pharmacology of human prostanoid EP2 and EP4 receptors. Eur J Pharmacol 2004;501:49–58. [DOI] [PubMed] [Google Scholar]
- 17.Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol 2001;41:661–690. [DOI] [PubMed] [Google Scholar]
- 18.Honda A, Sugimoto Y, Namba T, Watabe A, Irie A, Negishi M, Narumiya S, Ichikawa A. Cloning and expression of a cDNA for mouse prostaglandin E receptor EP2 subtype. J Biol Chem 1993;268:7759–7762. [PubMed] [Google Scholar]
- 19.Regan JW, Bailey TJ, Pepperl DJ, Pierce KL, Bogardus AM, Donello JE, Fairbairn CE, Kedzie KM, Woodward DF, Gil DW. Cloning of a novel human prostaglandin receptor with characteristics of the pharmacologically defined EP2 subtype. Mol Pharmacol 1994;46:213–220. [PubMed] [Google Scholar]
- 20.Chen YJ, Wang T, Liu DS, Xu D, Chen L, An J, Wen FQ. Effect of budesonide on TGF-beta1-enhanced VEGF production by lung fibroblasts. Cell Biol Int 2010;34:777–782. [DOI] [PubMed] [Google Scholar]
- 21.Petecchia L, Sabatini F, Usai C, Carnevali S, Ognibene M, Vanni C, Eva A, Fabbri LM, Rossi GA, Ricciardolo FL. Mechanisms of bradykinin-induced contraction in human fetal lung fibroblasts. Eur Respir J 2010;36:655–664. [DOI] [PubMed] [Google Scholar]
- 22.Kohyama T, Yamauchi Y, Takizawa H, Kamitani S, Kawasaki S, Nagase T. Histamine stimulates human lung fibroblast migration. Mol Cell Biochem 2010;337:77–81. [DOI] [PubMed] [Google Scholar]
- 23.Yang Z, Zhu X, Guo C, Sun K. Stimulation of 11beta-HSD1 expression by IL-1beta via a C/EBP binding site in human fetal lung fibroblasts. Endocrine 2009;36:404–411. [DOI] [PubMed] [Google Scholar]
- 24.Antoniu SA. New therapeutic options in the management of COPD: focus on roflumilast. Int J Chron Obstruct Pulmon Dis 2011;6:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Togo S, Holz O, Liu X, Sugiura H, Kamio K, Wang X, Kawasaki S, Ahn Y, Fredriksson K, Skold CM, et al. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am J Respir Crit Care Med 2008;178:248–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nyunoya T, Monick MM, Klingelhutz A, Yarovinsky TO, Cagley JR, Hunninghake GW. Cigarette smoke induces cellular senescence. Am J Respir Cell Mol Biol 2006;35:681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller KC, Paasch K, Feindt B, Welker L, Watz H, Weise M, Schmid RA, Nakashima M, Branscheid D, Magnussen H, et al. In contrast to lung fibroblasts: no signs of senescence in skin fibroblasts of patients with emphysema. Exp Gerontol 2008;43:623–628. [DOI] [PubMed] [Google Scholar]
- 28.Nobukuni S, Watanabe K, Inoue J, Wen FQ, Tamaru N, Yoshida M. Cigarette smoke inhibits the growth of lung fibroblasts from patients with pulmonary emphysema. Respirology 2002;7:217–223. [DOI] [PubMed] [Google Scholar]
- 29.Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A, Barnes PJ, Fabbri LM, Martinez FJ, Nishimura M, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. GOLD Executive Summary. Am J Respir Crit Care Med (In press) [DOI] [PubMed]
- 30.Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest 1995;95:1861–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borok Z, Gillissen A, Buhl R, Hoyt RF, Hubbard RC, Ozaki T, Rennard SI, Crystal RG. Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am Rev Respir Dis 1991;144:1080–1084. [DOI] [PubMed] [Google Scholar]
- 32.Moore BB, Ballinger MN, White ES, Green ME, Herrygers AB, Wilke CA, Toews GB, Peters-Golden M. Bleomycin-induced E prostanoid receptor changes alter fibroblast responses to prostaglandin E2. J Immunol 2005;174:5644–5649. [DOI] [PubMed] [Google Scholar]
- 33.Huang SK, Fisher AS, Scruggs AM, White ES, Hogaboam CM, Richardson BC, Peters-Golden M. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am J Pathol 2010;177:2245–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.