

Abstract
Airway epithelial cells contribute to the inflammatory response of the lung, and their innate immune response is primarily mediated via Toll-like receptor (TLR) signaling. Cystic fibrosis (CF) airways are chronically infected with Pseudomonas aeruginosa, suggesting a modified immune response in CF. We investigated the TLR-4 expression and the inflammatory profile (IL-8 and IL-6 secretion) in CF bronchial epithelial cell line CFBE41o- and its CF transmembrane ion condcutance regulator (CFTR)-corrected counterpart grown under air–liquid interface conditions after stimulation with lipopolysaccharide (LPS) from gram-negative bacteria. In CFTR-corrected cells, IL-8 and IL-6 secretions were constitutively activated but significantly increased after LPS stimulation compared with CFBE41o-. Blocking TLR-4 by a specific antibody significantly inhibited IL-8 secretion only in CFTR-corrected cells. Transfection with specific siRNA directed against TLR-4 mRNA significantly reduced the response to LPS in both cell lines. Fluorescence-activated cell sorter analysis revealed significantly higher levels of TLR-4 surface expression in CFTR-corrected cells. In histologic lung sections of patients with CF, the TLR-4 expression in the bronchial epithelium was significantly reduced compared with healthy control subjects. In CF the loss of CFTR function appears to decrease innate immune responses, possibly by altering the expression of TLR-4 on airway epithelial cells. This may contribute to chronic bacterial infection of CF airways.
Keywords: innate immunity, cystic fibrosis transmembrane ion conductance regulator, inflammation, Toll-like receptor, interleukin-8
Cystic fibrosis (CF) is an autosomal recessive disorder caused by mutations in the gene encoding the cystic fibrosis transmembrane ion conductance regulator (CFTR) protein (1). These mutations disrupt CFTR function within epithelial cells (2). Although the defect affects ion transport in many organs in CF, the major cause of morbidity and mortality is the progressive lung disease associated with unremitting bacterial infection (3), predominantly Pseudomonas aeruginosa and Staphylococcus aureus. This persistent inflammation is dominated by neutrophils and leads to lung injury and a decrease in lung function with resulting respiratory failure (4).
Airway epithelial cells not only function as a physical barrier against inhaled pathogens, but also play an important role in innate immune responses of the host (5). Microorganisms are recognized through a variety of pattern recognition receptors (PRR) on the cell surface, mainly Toll-like receptors (TLRs), that are expressed on cells including airway epithelial cells (6, 7). TLRs comprise a highly conserved family of receptors that recognize pathogen-associated molecular patterns (PAMPs) and allow the host to detect microbial infection. TLR-4 is a receptor for lipopolysaccharide (LPS), a cell wall component of gram-negative bacteria (such as P. aeruginosa), while TLR-2 mediates responses driven by lipoproteins (e.g., lipoteichoic acid [LTA]) and peptidoglycans from gram-positive bacteria (such as S. aureus).
Binding of bacterial components to PRRs leads to recruitment and activation of kinases and results in the release of the transcription factor NF-κB, allowing it to translocate to the nucleus and mediate an increase in inflammatory cytokine gene expression (6, 8, 9). Among these cytokines IL-8 and IL-6 are members of the CXC chemokine family. IL-8 is a potent activator and chemoattractant of neutrophils. IL-6 is an important mediator of fever and activates the acute phase protein production in the liver, and further activates B cells for antibody production. Both cytokines have been shown to be expressed by epithelial cells in response to a variety of stimuli (10–13). Since there are far more epithelial cells in the airway than alveolar macrophages, it has been proposed that the epithelial cell IL-8 response serves to “amplify” the neutrophil recruitment message (14).
Studies have shown elevated IL-8 and neutrophil elastase in bronchoalveolar lavage (BAL) fluid from patients with CF even very early in life (15–19). However, CF airway epithelia fail to eradicate bacterial infection (20). Studies of immortalized cell lines give contradictory results regarding TLR mRNA and surface protein expression (6, 7) and proinflammatory cytokine production in CF (13, 21–25). Anti-inflammatory cytokines such as IL-10 are present at lower levels in CF airways (26, 27). Therefore, the hypothesis of an intrinsic dysregulated inflammatory response in CF has emerged (13). But despite the many studies investigating the distribution of TLR and the inflammatory profile in CF, it remains unclear whether the proinflammatory responses seen in CF airways are a direct result of the CFTR mutation.
To better answer these questions, we investigated the TLR-4 expression and the inflammatory profile in CF bronchial epithelial cell line CFBE41o- and its CFTR-corrected counterpart, under well-differentiating growth conditions.
MATERIALS AND METHODS
Cell Culture
CF bronchial epithelial cell line (CFBE41o-), homozygous for the ΔF508 mutation, its isogenic wild-type CFTR-complemented counterpart (corrCFBE41o-) and the (ΔF508) control plasmid cell line (dfCFBE41o-) were generated as previously described (28, 29). The human bronchial epithelial cell line (16HBE14o-) served as a non-CF control (30). For subcultivation, cells were grown in Minimum Essential Medium (MEM) with Earl's salts (Gibco, Karlsruhe, Germany), supplemented with 10% fetal calf serum (FCS; Gibco) and 1% Penicillin/Streptomycin (PAA, Pasching, Austria), on tissue culture plastic coated with a solution containing fibronectin (BD Biosciences, San Diego, CA), collagen (Cohesion Technologies, Palo Alto, CA), and BSA (Sigma-Aldrich, St. Louis, MO) at 37°C in a 5% CO2 atmosphere. All complemented CF airway epithelial cells were cultivated in medium containing 200 μg/ml Hygromycin B (Invitrogen, Carlsbad, CA). PCR and RT-PCR were used to confirm CFTR transgene expression in the complemented cells, and electrophysiologic analysis indicated restoration of cAMP-dependent Cl− transport (unpublished observation by D. C. Gruenert).
For experiments, cells were grown on 6-well Transwell inserts (Corning Inc. Life Sciences, Lowell, MA) under air–liquid interface (ALI) conditions for a minimum of 14 days to achieve polarization and differentiation (30–32), using Bronchial Epithelial Growth Medium with supplements (BEGM; Lonza, Walkersville, MD). The medium was changed both at the bottom and the top of the membrane every other day until the cells became confluent. Then, the medium was completely removed from the top and changed daily only at the bottom. ALI cultures were selected for experiments by measuring transepithelial electrical resistance (TER or Rt) with an epithelial ohmmeter (EVOM; World Precision Instruments, Sarasota, FL). Cultures were considered differentiated when Rt was more than 200–300 Ω × cm2 (33).
For immunofluorescence and experiments using siRNA, cells were grown on tissue culture plates (Greiner Bio-One, Frickenhausen, Germany) to approximately 80 to 90% confluence.
Cell Treatment
Twenty-four hours before the experiment, cells were switched to BEGM without supplements. Stimulation of epithelial cultures was performed with washed aliquots of living and heat-inactivated P. aeruginosa (1 × 108/ml), nonmucoid strain ATCC 27853 (kindly provided by Institute of Medical Microbiology, Marburg, Germany), and purified LPS from Salmonella enterica sv. Arizona (kindly provided by Otto Holst, Research Center Borstel, Germany) for 1 hour at 37°C. We used LPS from Salmonella enterica sv. Arizona because this was shown to have high TLR-4 activity due to its purity (unpublished observation by O. Holst). In a control experiment, cells were stimulated with LPS from P. aeruginosa (Sigma-Aldrich). Nontreated controls were processed similarly with PBS (pH 7.4) alone. As positive controls, cells were treated for 1 hour with tumor necrosis factor (TNF)-α (R&D Systems, Minneapolis, MN) at a concentration of 100 ng/ml for analysis of IL-8 and IL-6 (13). For inhibition of LPS, polymyxin B (PMX; Sigma-Aldrich), an antibiotic shown to inactivate endotoxins by binding to the lipid A portion (34), was used at a concentration of 1 μg/ml. After stimulation, cells were washed three times in PBS and incubated for 24 hours in fresh medium. Media were collected for determination of cytokine production and cells were used for mRNA or protein expression studies.
For inhibition studies, monoclonal antibody to TLR-4, clone HTA125 (Alexis, Lausen, Switzerland) was incubated with epithelial cultures at a concentration of 1 μg/ml before stimulation. After incubation for 1 hour, cells were washed with PBS and treated as described above. For experiments using siRNA, cells were seeded into 6-well tissue culture plates at a density of 2 × 105 cells per well. The day after seeding, transfection of cells with 5 nM of specific TLR-4 (Hs_TLR4_1, Hs_TLR4_7), positive control MAPK1 and negative control siRNA (Qiagen, Hilden, Germany) was performed for 48 hours using HiPerFect Transfection Reagent (Qiagen), according to the manufacturer's protocol. After incubation, cells were washed with PBS and treated as described above.
All experiments were performed in duplicates, with n = a minimum of four samples per group; for further details see figure legends.
RNA Isolation and Real-Time RT-PCR
mRNA expression of TLR-4, CD14, and MD-2 was analyzed by quantitative real-time RT-PCR. Total RNA from cell lysates was isolated using the RNeasy Mini kit (Qiagen) and DNase I digestion (Invitrogen, Karlsruhe, Germany), and cDNA was synthesized with First Strand cDNA Synthesis Kit (Fermentas, St. Leon-Rot, Germany). Generated cDNA was used as template for amplification in an iCycler (Bio-Rad, Munich, Germany), with ABsolute QPCR SYBR Green Mix (ABgene, Surrey, UK) and specific primers. Primers were generated from the respective mRNA sequences for human TLR-4, CD14, and MD-2 (European Molecular Biology Laboratory gene bank) and synthesized by Biomers (Ulm, Germany); sequences and PCR conditions available on request. ΔΔCT analysis was used to calculate expression in comparison to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and normalized to the level of CFBE cells. For qualitative analysis, PCR products were subjected to electrophoresis on a 1.5% agarose gel and visualized by ethidium bromide staining.
Fluorescence-Activated Cell Sorter Analysis
Cell surface protein expression of TLR-4 was determined by fluorescence-activated cell sorter (FACS) analysis. Briefly, cells were washed with PBS and incubated for 10 minutes with 30 mM EDTA (Sigma-Aldrich) in PBS at 37°C. Detached cells were blocked (with 10% FCS at 4°C), adjusted to 1 × 105 cells per 100 μl in PBS/5% FCS, and successively stained in darkness for 30 minutes at 4°C with fluorochrome-conjugated monoclonal antibodies for isotype control (mouse IgG1 fluorescein isothiocyanate [FITC]/mouse IgG2a phycoerythrin [PE]), epithelial cell marker CD326 (αCD326-PE), and TLR-4 (αTLR-4-FITC). Antibodies were purchased from BD Biosciences for isotype control, from Miltenyi Biotec (Bergisch Gladbach, Germany) for αCD326, and from Alexis for FITC-conjugated αTLR-4, clone HTA125, which has been characterized before (35). Analysis of 10,000 events was performed by using a FACScalibur flow cytometer (Becton Dickinson, Heidelberg, Germany) and BD CellQuest Pro software version 5.2.1. Cells were initially gated on the basis of forward and side scatter characteristics.
Immunofluorescence
For immunofluorescence analysis, cells were seeded onto 18-mm diameter coverslips (LLG, Meckenheim, Germany), in 12-well tissue culture plates, at a density of 1×105 cells per coverslip. The following day, culture medium was changed from MEM to BEGM, and cells were grown to 80 to 90% confluence. For cytoplasmic staining, cells were incubated for 30 minutes with 10 μM CellTracker Orange Fluorescent Probe (Cambrex, Walkersville, MD). Coverslips were processed by fixing the cells with 3.7% paraformaldehyde, blocking with PBS/5% BSA, and incubating with FITC-conjugated αTLR-4 (1 μg/ml) in darkness overnight at 4°C. In another experiment, fixed cells were permeabilized with PBS/0.1% Tween (Roth, Karlsruhe, Germany), processed as described above, and cellular DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI, 1.5 μg/ml; Vector Laboratories, Burlingame, CA). Coverslips were viewed using Olympus BX51 fluorescence microscope and cellF Imaging Software (Olympus, Hamburg, Germany).
Immunohistochemistry
Paraffin sections from human lungs (2 patients with CF, homozygous for the ΔF508 mutation, mean age 18.0 ± 9.9 yr; 3 donors, mean age 34.3 ± 9.9 yr) were stained by indirect immunohistochemistry. Briefly, deparaffinized and rehydrated tissue sections were blocked with 3% hydrogen peroxide in methanol followed by antigen retrieval in citrate buffer (3 × 5 min, at 450 W, 1 mM, pH 6) in a microwave oven. Immune staining for TLR-4 was done according to the standard avidin-biotin-complex (ABC) technique. Sections were blocked with 10% normal serum, incubated with primary polyconal (goat) anti–TLR-4 antibody (dilution 1:150; Alexis) for 1 hour at 37°C, and subsequently incubated with biotinylated anti-goat secondary antibody (dilution 1:100; Vector Laboratories) for 30 minutes. Signals were visualized using avidin-biotin peroxidase (Vectastain Elite kit; Vector Laboratories) with 3,3′-diamino-benzidine (DAB; Sigma, Steinheim, Germany) and counterstained with Mayer's hematoxylin. Sections were viewed using Olympus BH-2 optical microscope and cellF Imaging Software (Olympus).
Lung samples from subjects with CF undergoing lung transplantation and from lung donors without CF were a generous gift from S. von Gerlach and R. Voswinckel at the Justus-Liebig-University of Giessen. Lung sample collection was approved by the Institutional Review Board of the Justus-Liebig-University Giessen.
Determination of Cytokine Production by Enzyme-Linked Immunosorbent Assay
Concentrations of IL-8 and IL-6 in the cell culture supernatants were determined by commercially available OptEIA Sets for enzyme-linked immunosorbent assay (BD Biosciences), and normalized to the protein concentration of the lysed cells (as measured by BCA Protein Assay).
Statistical Analysis
Results are presented as mean values ± SD. Significant difference was evaluated by the unpaired Student t test with two-tailed distributions (P < 0.05). All analyses were performed by means of GraphPad Prism 4 software (San Diego, CA).
RESULTS
Inflammatory Profile in Bronchial Epithelial Cells
To determine cytokine production, epithelial cell cultures were stimulated with washed aliquots of living and heat-inactivated P. aeruginosa and LPS from Salmonella enterica sv. Arizona for 1 hour, washed three times in PBS, and incubated in fresh medium for another 24 hours. Media were collected and secreted cytokines were measured by enzyme-linked immunosorbent assay (ELISA) (Figure 1). The human bronchial epithelial cell line 16HBE14o- used as a non-CF control was also analyzed for cytokine secretion. Absolute concentrations of IL-8 and IL-6 were higher in human bronchial epithelial (HBE)- compared with all CFBE-derived cells. This is assumed to be caused by the different cell lines. Cytokine secretion of HBE cells after stimulation with living and heat-inactivated P. aeruginosa and LPS was increased by a factor of three compared with the untreated control (2,388 ± 248 pg/ml versus 806 ± 77 pg/ml).
Figure 1.

Secretion of IL-8, analyzed by enzyme-linked immunosorbent assay (ELISA), in cells from human bronchial epithelial cell line (HBE), CF bronchial epithelial cell line (CFBE), CFTR-complemented counterpart CFBE cell line (corrCFBE), and the (DF508) control plasmid cell line (dfCFBE), 24 hours after 1 hour of stimulation with living and heat-inactivated (h.i.) P. aeruginosa (P.a.) (1 × 108/ml), and LPS (1 μg/ml). TNF-α (100 ng/ml) served as positive control (n = 4). *Significantly different than control, #significantly different than CFBE and dfCFBE, with P < 0.05.
A similar response was seen in the CFTR-corrected cells (corrCFBE). These were capable of secreting more IL-8 than the CF airway epithelial cells CFBE and dfCFBE in response to living and heat-inactivated bacteria and to LPS as a bacterial component (1,198 ± 231 pg/ml versus 391 ± 53 pg/ml).
LPS-Stimulated Cytokine Secretion
Epithelial cell cultures were stimulated with purified LPS from Salmonella enterica sv. Arizona at different doses, as described above and cytokine secretions were measured by ELISA (Figures 2a and 2b). Basal concentrations of IL-8 were already increased by a factor of 2 in CFTR-corrected cells compared with CF airway epithelial cells CFBE and dfCFBE (513 ± 138 pg/ml versus 264 ± 36 pg/ml). Stimulation with LPS resulted in augmented IL-8 secretions in a dose-dependent manner in all cell lines, compared with the PBS-treated control (Figure 2a). However, in the CF airway epithelial cells, the increase was only significant at 1 μg/ml LPS, whereas in the CFTR-corrected cells, it was significant after stimulation with 0.1 and 1 μg/ml LPS (P < 0.05). For 1 μg/ml LPS, IL-8 levels increased by a factor of 2 in CFBE and dfCFBE (from 264 ± 36 pg/ml to 498 ± 78 pg/ml), while in corrCFBE the factor was 3 (from 513 ± 138 pg/ml to 1,433 ± 69 pg/ml). As a positive control, cells were incubated with TNF-α (100 ng/ml). This resulted in a significant increase of IL-8 secretion in all cells (P < 0.05).
Figure 2.
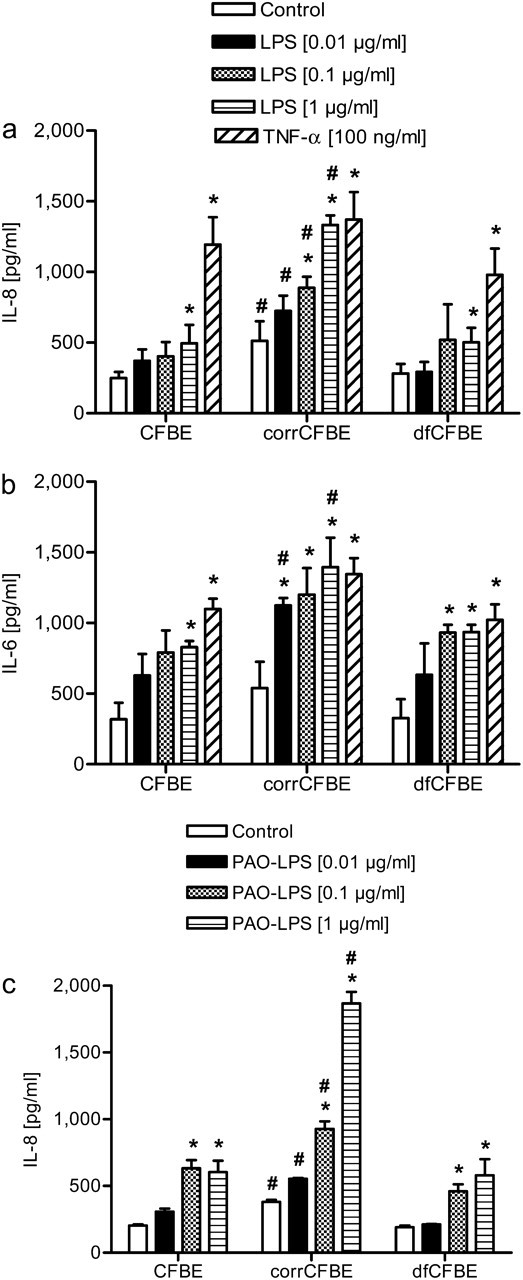
Secretion of IL-8 (a and c) and IL-6 (b), analyzed by ELISA, in CFBE, corrCFBE, and dfCFBE cells, 24 hours after 1 hour of stimulation with LPS at 0.01, 0.1, and 1 μg/ml. TNF-α (100 ng/ml) served as positive control. The cells were stimulated either with LPS from Salmonella enterica sv. Arizona (a and b) or with LPS from P. aeruginosa (c) (n = 4). *Significantly different than control, #significantly different than CFBE and dfCFBE, with P < 0.05.
Basal and LPS-stimulated concentrations of IL-6 were comparable to the results for IL-8 in all cell lines, and also showed a higher level and response to LPS in the CFTR-corrected cell line corrCFBE (Figure 2b).
Furthermore, stimulation with LPS from P. aeruginosa displayed a similar pattern with a significant increase of IL-8 in all cell lines at concentrations of 0.1 and 1 μg/ml LPS (Figure 2c). To test if the increase in cytokine secretion was due to the treatment with LPS, cells were incubated with LPS and polymyxin B (PMX) at the same time, because PMX is an antibiotic shown to inactivate endotoxins by binding to the lipid A portion. In all cell lines, the effect of LPS on IL-8 secretion was almost completely inhibited (data not shown).
LPS-Stimulated Cytokine Secretion Is Dependent on TLR-4 Signaling
To test if secretion of IL-8 after stimulation with LPS is induced by ligand-specific signaling via TLR-4, two sets of experiments were performed.
First, the effect of blocking TLR-4 surface receptors by incubation with a monoclonal antibody to TLR-4 (1 μg/ml) before stimulation with 1 μg/ml LPS was determined (Figure 3). In corrCFBE, a significant inhibition of signaling via TLR-4 was effective, as seen by a decrease of IL-8 secretion 24 hours after treatment with LPS (P < 0.05).
Figure 3.

Inhibition of Toll-like receptor (TLR)-4–mediated cytokine secretion by incubation with a monoclonal antibody to TLR-4. IL-8 secretion was analyzed by ELISA in CFBE, corrCFBE, and dfCFBE cells, 24 hours after 1 hour of incubation with a monoclonal antibody (Ab) to TLR-4 (1 μg/ml) and subsequent stimulation with LPS (1 μg/ml) for 1 hour (n = 4). *LPS treatment significantly different than LPS+Ab with P < 0.05.
Silencing of TLR-4 gene expression was performed in a second set of experiments. Cells were transfected with specific siRNA directed against mRNA of TLR-4 and MAPK1 and negative control siRNA for 48 hours and subsequently incubated with LPS. After another 24 hours, cells and culture media were harvested and analyzed by ELISA. The results are given as relative IL-8 compared with the negative control level because absolute values for IL-8 were lower compared with ALI experiments, due to the cell cultivation on tissue culture plastic (Figure 4). All cells treated with specific siRNA directed against TLR-4 mRNA secreted significantly less IL-8 than cells incubated either without or with negative control siRNA. Transfection efficiency was approximately 80 to 90%, as determined with negative control siRNA labeled with Alexa Fluor 488 (Qiagen) and analyzed by FACS. Efficient gene knockdown for TLR-4 was verified using real-time RT-PCR analysis, and revealed a mean reduction of mRNA expression by 60%, compared with untreated control levels. Transfection for 48 hours also resulted in a decrease of surface receptor expression by almost 21% compared with unstimulated cells, as measured by FACS. A MAPK1 siRNA served as a positive control. Western blot for MAPK1 showed reduced protein expression in cells treated with positive control siRNA (data not shown).
Figure 4.

Inhibition of TLR-4–mediated cytokine secretion by transfection of specific siRNA directed against TLR-4 mRNA. IL-8 secretion was analyzed by ELISA in CFBE, corrCFBE, and dfCFBE cells, 24 hours after 48 hours of transfection with TLR-4 and negative control siRNA, and subsequent stimulation with LPS (1 μg/ml) for 1 hour. Data are given relative to each negative control = 100% (n = 4). *Significantly different than negative control with P < 0.05.
mRNA Expression of TLR-4, CD14, and MD-2
mRNA expression of TLR-4 and adapter molecules CD14 and MD-2 under unstimulated conditions was analyzed by quantitative real-time RT-PCR, using the ΔΔCT analysis to calculate expression in comparison to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). All cell lines showed similar mRNA expression levels of the investigated genes (Figure 5a). Although there seemed to be a slight reduction of mRNA expression in corrCFBE, the differences were not significant compared with CFBE and dfCFBE.
Figure 5.

mRNA and protein expression analysis of TLR-4 in CFBE, corrCFBE, and dfCFBE cells. (a) mRNA expression of TLR-4 and adapter molecules CD14 and MD-2 under unstimulated conditions was analyzed by quantitative real-time RT-PCR, using the ΔΔCT analysis to calculate expression in comparison to GAPDH and normalized to the level of CFBE cells (n = 5). (b) FACS analysis showing surface expression of TLR-4 under basal conditions and 24 hours after 1 hour of stimulation with LPS (1 μg/ml) (n = 5) *Significantly different than CFBE and dfCFBE with P < 0.05; #significantly different than unstimulated control with P < 0.05. (c–j) Immunofluorescence analysis of CFBE (c and g), corrCFBE (d and h) and dfCFBE (e and i) cells for TLR-4, and of CFBE cells for isotype control (f and j). (c–f) Cells were incubated with CellTracker Orange Fluorescent Probe, fixed, and stained with FITC-conjugated antibody to TLR-4 (green). (g–j) Cells were fixed, permeabilized, and stained again for TLR-4, while cellular DNA was stained with DAPI (blue).
Protein Expression of TLR-4 Is Altered in Wild-Type CFTR-Corrected Cells under Basal and LPS-Stimulated Conditions
For quantification of TLR-4 surface expression, cells were analyzed by FACS (Figure 5b). The mean percentage of TLR-4–positive stained cells was significantly increased in corrCFBE, with 16.9 ± 2.3% versus 8.3 ± 1.4% in CFBE (P < 0.05). Again, the empty plasmid control dfCFBE showed results similar to those observed for CFBE. Furthermore, cell surface expression of TLR-4 after LPS stimulation was analyzed by FACS and revealed a slightly greater increase relative to control in corrCFBE, compared with CFBE and dfCFBE.
Immunofluorescence analysis for surface protein expression of TLR-4 under basal conditions confirmed that in corrCFBE, there is higher staining of the receptor at the cell surface, whereas in the CF airway epithelial cells CFBE- and dfCFBE-specific staining is low (Figures 5c–5e). In another experiment, fixed cells were permeabilized with PBS/0.1% Tween and stained again with the same antibody, which specifically recognizes human TLR-4 cell surface antigen (35). This confirmed the observation that in corrCFBE, TLR-4 was more localized at the cell surface, and permeabilizing did not change specific binding of antibody. CFBE and dfCFBE showed a strong intracellular localization of receptor components after permeabilization (Figures 5g–5i).
TLR-4 Expression in Normal and CF Lung Tissue Sections
TLR-4 expression in three normal and two CF lung tissue sections was assessed by indirect immunohistochemistry using DAB as substrate and Mayer's hematoxylin for counterstaining. In control patients, epithelial cells stained strongly positive for TLR-4, mostly in apical compartments (Figures 6a and 6b). In contrast, airways from patients with CF showed almost no or only a weak staining for TLR-4 (Figures 6c and 6d).
Figure 6.

Immunhistochemistry for TLR-4 in normal (a and b) and cystic fibrosis (CF) (c and d) lung tissue sections from two donors and two patients with CF.
DISCUSSION
Since CF airway epithelia fail to kill bacteria (20), there is growing evidence that a modified immune response in CF leads to chronic colonization of CF airways by P. aeruginosa. It is controversial whether excessive pulmonary inflammation is an intrinsic property of the CFTR defect or whether it is secondary to the unique environment of the CF lung.
Therefore, we wished to determine the influence of P. aeruginosa on immune responses in well-differentiating isogenic CF and CFTR-corrected human airway epithelium cell lines. We used an immortalized cell line homozygous for the deltaF508 CFTR mutation and its corrected cell line with a stable expression of the wild-type CFTR protein (29). We found that in the CF cell line (CFBE and dfCFBE), basal IL-8 secretion was reduced compared with the same cell line with the corrected CFTR defect (corrCFBE). After stimulation with LPS, IL-8 secretion increased in the CF airway epithelial cells by a factor of 2 and in the corrected cells by a factor of 3, compared with basal conditions. This suggests that the cells with the CFTR defect are able to respond to the stimulation with LPS, but the response in CFTR-corrected cells is more intense. These results are similar to the findings in two studies using the human bronchial epithelium CF cell line CFT1 (deltaF508/deltaF508), its CFTR-corrected isogenic cell line (CFT1-LCFSN), and its vector control line (CFT1-LC3), grown submerged as confluent monolayers and stimulated either with IL-1β, TNF-α, or various concentrations of LPS derived from P. aeruginosa (CFM5) (22, 25). Under basal, unstimulated conditions the CFTR-corrected cell line (CFT1-LCFSN) secreted significantly more IL-8 than the CF cell lines (CFT1 and CFT1-LC3), and heat-killed P. aeruginosa isolates caused a dose-dependent increase of IL-8 secretion by all of the cell lines, whereas purified P. aeruginosa LPS had no significant effect, suggesting a defective IL-8 secretion in the CF airway epithelial cells. Although a study using primary nasal epithelial cells (passage 1 and 2, submerged) from subjects with CF showed greater baseline NF-κB activity and higher IL-8 levels compared with cells from subjects without CF, there was a greater increase in IL-8 secretion in non-CF than in CF airway epithelial cells after stimulation with living P. aeruginosa (21). The authors concluded that CF airway epithelial cells had impairment in intranuclear NF-κB activity or were exhausted in IL-8 synthesis. In contrast, some studies showed no significant differences in IL-8 secretion at basal conditions or after stimulation with P. aeruginosa products (36, 37), while others found an increased IL-8 secretion or NF-κB activation in the CF airway epithelial cells compared with non-CF or corrected CF airway epithelial cells (23, 24, 38, 39). These results may largely differ from ours because the cells were grown submerged on plastic and are nonpolar. Here our CFBE41o- cells were grown as differentiating ALI cultures where they form tight junctions, maintain cell polarity, and express high levels of CFTR (complemented cells) (29, 30, 40, 41).
Further experiments suggested a TLR-4–dependent induction of proinflammatory cytokines. Although mRNA expression levels of TLR-4 were comparable in all cell lines, cell surface expression of TLR-4 was significantly reduced in CF airway epithelial cells compared with the CFTR-corrected cells. In two studies with CF and normal control cell lines and cells in primary culture, protein expression of TLRs was readily visualized along basolateral aspects using confocal laser scanning microscopy, but not quantified (6, 7). However, Hauber and coworkers were able to show a significantly reduced TLR-4 expression in the airway epithelium of patients with CF (n = 7) compared with control subjects (n = 6) using bronchial biopsies (42), which we confirmed in histologic lung sections from the bronchial epithelium of patients with CF (n = 2) compared with healthy control subjects (n = 3). Our in vitro and in vivo findings of a reduced TLR-4 surface expression in the CF bronchial epithelium might explain the diminished IL-8 response that results from a decreased binding to and activation of TLR-4. Consistent with this the LPS induced IL-8 secretion was significantly inhibited by incubation with a monoclonal antibody to TLR-4 only in corrCFBE cells. This suggests that in the CF airway epithelial cells, TLR-4 localized at the cell surface plays a very limited role in immune responses to LPS. However, the localization and activation of TLR-4 remains controversial. Some studies have shown that the receptor is functionally present at the cell surface (6, 43), whereas other studies demonstrated an exclusively intracellular localization and activation process of TLR-4 requiring internalization of LPS (44, 45). The latter studies focused on intestinal epithelial cells, tracheobronchial epithelial cell line BEAS-2B, and carcinoma alveolar epithelial cell line A549. Therefore, different adaptations to diverse cellular environments might explain the observed variability between the cell lines. Our immunofluorescence images suggest that in CF airway epithelial cells, TLR-4 is mainly localized intracellularly, while after CFTR correction it is abundantly expressed on the cell surface. Inhibition of TLR-4 mRNA expression by transfection of specific siRNA not only resulted in decreased surface receptor expression by almost 21% compared with unstimulated cells, but also blocked the induction of IL-8 secretion in all cell lines. This supports the hypothesis that intracellular binding of internalized LPS to TLR-4 in CF airway epithelial cells may be absent or delayed. However, the compromised TLR-4 signaling pathway does not explain reduced IL-8 secretions in CF airway epithelial cells under unstimulated conditions. We speculate that the regulation of basal cytokine secretion is a more complex matter that might be influenced by the cellular environment and is not dependent on one immune receptor.
A possible explanation for the observed intracellular localization and consequently reduced surface expression of TLR-4 in CF airway epithelial cells might be that in CF, mutant versions of the CFTR are recognized as abnormal and remain incompletely processed in the endoplasmic reticulum (ER), where they are subsequently degraded (46–48). Almost 100% of the deltaF508 CFTR variants undergo rapid degradation before exiting from the ER. The molecular components of this stringently applied quality control system may also affect other membrane proteins that together with CFTR are substrates for proteasomal degradation during maturation within the ER (49–51). One of the affected membrane proteins could be TLR-4, and this would lead to less expression on the surface of CF airway epithelial cells, whereas in CFTR-corrected cells, a less activated cytosolic proteasome may lead to restored surface expression. However, our immunofluorescence images showed an intracellular localization of TLR-4 in CF airway epithelial cells. Furthermore, preliminary studies using a proteasome inhibitor did not lead to increased surface expression of TLR-4 in CF airway epithelial cells, whereas inhibiting protein glycosylation with tunicamycin resulted in a decrease of surface expression only in HBE and CFTR-corrected cells (unpublished observation by our group). It has been shown that the CFTR defect leads to alterations of post-translational modifications, such as glycosylation, sulphation, and sialylation of proteins affecting secreted and cell surface molecules as well as their cellular localization (52, 53). For example, sialylation of proteins and lipids such as the glycolipid GM1 was reduced in CF epithelial cells, with an increase of asialoGM1 on the cell surface (54). TLR-4 also undergoes glycosylation (43), which could be compromised in CF. In addition, a functional receptor requires the presence of adapter molecules CD14 and MD-2 (55, 56), and membrane organization in lipid raft domains is dependent on factors such as lysophospholipids (57). Although mRNA expression levels of CD14 and MD-2 were not different between the cell lines, protein levels of several factors could be affected in CF airway epithelial cells. Preliminary studies of co-staining for TLR-4 and the lipid raft marker GM1 (58) indicated co-localization on the cell surface of CFTR-corrected cells. However, the results for CFBE and dfCFBE did not support the hypothesis of an intracellular localization of TLR-4 due to GM1 surface reduction or mislocalization (see the online supplement).
Altered processing and localization of TLR-4 might have considerable consequences for the innate immunity. A limited response by the CF bronchial airway epithelium at the time of initial bacterial colonization, caused by a reduced bacterial recognition, may compromise the local immune response and delay neutrophil chemotaxis and migration across the epithelium. This delayed neutrophil activation favors bacterial growth and biofilm formation, and neutrophil degradation increases sputum tenacity and impairs mucociliary clearance due to the release of large amounts of DNA (59–65).
Because the impaired CFTR function increases asialoGM1 receptors, this may also lead to increased adherence of P. aeruginosa, S. aureus, and Haemophilus influenzae to CF airway epithelial cells and reduced clearance from the airways (54, 66–68). Furthermore, the CFTR protein itself may act as a pattern recognition receptor (PRR) that extracts P. aeruginosa LPS from the outer membrane into epithelial cells and causes an increase of proinflammatory cytokines (69). Due to the lack of CFTR protein in apical membranes of CF airway epithelial cells, a further mechanism of recognizing and clearing gram-negative bacteria immediately after the first contact to the bronchial epithelium is also compromised. All this might explain the survival of P. aeruginosa in the CF airway at the time of an initial infection leading to a hyperinflammatory response and chronic bacterial infection clinically observed in patients with CF (15, 16, 26, 70, 71).
Taken together, we have shown that TLR-4 is produced in the CF airway epithelial cells, though its cell surface expression is reduced, which is possibly related to a diminished translocation to the cell surface. This is associated with a reduced inflammatory response to LPS from gram-negative bacteria and may predispose the CF lung to bacterial colonization and chronic infection. After correction of the CFTR defect TLR-4 is effectively expressed on the airway epithelial cell surface.
Additional material
Supplementary data supplied by authors.
Acknowledgments
The authors thank I. Schaffrath for technical assistance, and S. von Gerlach and R. Voswinckel at the Justus-Liebig-University of Giessen for the generous gift of the lung samples.
This study was supported by Christiane Herzog Stiftung, Germany.
This study was published as an abstract in Toronto at the International Conference of the American Thoracic Society (ATS) 2008.
Originally Published in Press as DOI: 10.1165/rcmb.2008-0408OC on June 5, 2009
Conflict of Interest Statement: M.O.H. received consultancy fees from Bayer for $1,001–$5,000, a grant from Boehringer Ingelheim for $50,001–$100,000, and Christiane Herzog Stiftung for $50,001–$100,000. D.C.G. has received grants from NIH for over $100,000 and from PACFI for $10,001–$50,000. He has received royalties from UCSF for $10,001–$50,000 and consultancy fees from CFF for less than $1,000. G.J. has received a grant from Christiane Herzog Stiftung, Germany for $50,001–$100,000. B.K.R. has received consultancy fees from RegeneRx Biopharmaceuticals, Inc. for $1,000–$5,000 and Pfizer Global Research & Development for $1,000–$5,000. He also served on advisory boards for Pharmaxis for $1,000–$5,000 and Schering-Plough Pharmaceuticals for $1,000–$5,000. He has received grants from Trudell Medical International for $10,000–$50,000, GlaxoSmithKline for over $100,000, Bayer Schering Pharma for $50,001–$100,000, and Reckitt Benckiser Group plc for $50,001–$100,000. None of the other authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989;245:1059–1065. [DOI] [PubMed] [Google Scholar]
- 2.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993;73:1251–1254. [DOI] [PubMed] [Google Scholar]
- 3.Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med 1996;154:1229–1256. [DOI] [PubMed] [Google Scholar]
- 4.Heijerman H. Infection and inflammation in cystic fibrosis: a short review. J Cyst Fibros 2005;4:3–5. [DOI] [PubMed] [Google Scholar]
- 5.Machen TE. Innate immune response in CF airway epithelia: hyperinflammatory? Am J Physiol Cell Physiol 2006;291:C218–C230. [DOI] [PubMed] [Google Scholar]
- 6.Greene CM, Carroll TP, Smith SG, Taggart CC, Devaney J, Griffin S, O'Neill SJ, McElvaney NG. TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J Immunol 2005;174:1638–1646. [DOI] [PubMed] [Google Scholar]
- 7.Muir A, Soong G, Sokol S, Reddy B, Gomez MI, Van HA, Prince A. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am J Respir Cell Mol Biol 2004;30:777–783. [DOI] [PubMed] [Google Scholar]
- 8.Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of Toll-like receptor-mediated immune responses. Nat Rev Immunol 2005;5:446–458. [DOI] [PubMed] [Google Scholar]
- 9.O'Neill LA. How Toll-like receptors signal: what we know and what we don't know. Curr Opin Immunol 2006;18:3–9. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura H, Yoshimura K, McElvaney NG, Crystal RG. Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in a human bronchial epithelial cell line. J Clin Invest 1992;89:1478–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kube D, Sontich U, Fletcher D, Davis PB. Proinflammatory cytokine responses to P. aeruginosa infection in human airway epithelial cell lines. Am J Physiol Lung Cell Mol Physiol 2001;280:L493–L502. [DOI] [PubMed] [Google Scholar]
- 12.DiMango E, Zar HJ, Bryan R, Prince A. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J Clin Invest 1995;96:2204–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stecenko AA, King G, Torii K, Breyer RM, Dworski R, Blackwell TS, Christman JW, Brigham KL. Dysregulated cytokine production in human cystic fibrosis bronchial epithelial cells. Inflammation 2001;25:145–155. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura H, Yoshimura K, Jaffe HA, Crystal RG. Interleukin-8 gene expression in human bronchial epithelial cells. J Biol Chem 1991;266:19611–19617. [PubMed] [Google Scholar]
- 15.Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med 1995;151:1075–1082. [DOI] [PubMed] [Google Scholar]
- 16.Armstrong DS, Grimwood K, Carlin JB, Carzino R, Gutierrez JP, Hull J, Olinsky A, Phelan EM, Robertson CF, Phelan PD. Lower airway inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med 1997;156:1197–1204. [DOI] [PubMed] [Google Scholar]
- 17.Meyer KC, Sharma A. Regional variability of lung inflammation in cystic fibrosis. Am J Respir Crit Care Med 1997;156:1536–1540. [DOI] [PubMed] [Google Scholar]
- 18.Gutierrez JP, Grimwood K, Armstrong DS, Carlin JB, Carzino R, Olinsky A, Robertson CF, Phelan PD. Interlobar differences in bronchoalveolar lavage fluid from children with cystic fibrosis. Eur Respir J 2001;17:281–286. [DOI] [PubMed] [Google Scholar]
- 19.Noah TL, Black HR, Cheng PW, Wood RE, Leigh MW. Nasal and bronchoalveolar lavage fluid cytokines in early cystic fibrosis. J Infect Dis 1997;175:638–647. [DOI] [PubMed] [Google Scholar]
- 20.Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 1996;85:229–236. [Published erratum appears in Cell 1996;87:Following 355.] [DOI] [PubMed] [Google Scholar]
- 21.Carrabino S, Carpani D, Livraghi A, Di CM, Costantini D, Copreni E, Colombo C, Conese M. Dysregulated interleukin-8 secretion and NF-kappaB activity in human cystic fibrosis nasal epithelial cells. J Cyst Fibros 2006;5:113–119. [DOI] [PubMed] [Google Scholar]
- 22.Massengale AR, Quinn F Jr, Yankaskas J, Weissman D, McClellan WT, Cuff C, Aronoff SC. Reduced interleukin-8 production by cystic fibrosis airway epithelial cells. Am J Respir Cell Mol Biol 1999;20:1073–1080. [DOI] [PubMed] [Google Scholar]
- 23.Scheid P, Kempster L, Griesenbach U, Davies JC, Dewar A, Weber PP, Colledge WH, Evans MJ, Geddes DM, Alton EW. Inflammation in cystic fibrosis airways: relationship to increased bacterial adherence. Eur Respir J 2001;17:27–35. [DOI] [PubMed] [Google Scholar]
- 24.Aldallal N, McNaughton EE, Manzel LJ, Richards AM, Zabner J, Ferkol TW, Look DC. Inflammatory response in airway epithelial cells isolated from patients with cystic fibrosis. Am J Respir Crit Care Med 2002;166:1248–1256. [DOI] [PubMed] [Google Scholar]
- 25.Black HR, Yankaskas JR, Johnson LG, Noah TL. Interleukin-8 production by cystic fibrosis nasal epithelial cells after tumor necrosis factor-alpha and respiratory syncytial virus stimulation. Am J Respir Cell Mol Biol 1998;19:210–215. [DOI] [PubMed] [Google Scholar]
- 26.Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, Berger M. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med 1995;152:2111–2118. [DOI] [PubMed] [Google Scholar]
- 27.Bonfield TL, Konstan MW, Burfeind P, Panuska JR, Hilliard JB, Berger M. Normal bronchial epithelial cells constitutively produce the anti-inflammatory cytokine interleukin-10, which is downregulated in cystic fibrosis. Am J Respir Cell Mol Biol 1995;13:257–261. [DOI] [PubMed] [Google Scholar]
- 28.Cozens AL, Yezzi MJ, Chin L, Simon EM, Finkbeiner WE, Wagner JA, Gruenert DC. Characterization of immortal cystic fibrosis tracheobronchial gland epithelial cells. Proc Natl Acad Sci USA 1992;89:5171–5175. [Published erratum appears in Proc Natl Acad Sci USA 1992;89:7849.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shuto T, Furuta T, Oba M, Xu H, Li JD, Cheung J, Gruenert DC, Uehara A, Suico MA, Okiyoneda T, et al. Promoter hypomethylation of Toll-like receptor-2 gene is associated with increased proinflammatory response toward bacterial peptidoglycan in cystic fibrosis bronchial epithelial cells. FASEB J 2006;20:782–784. [DOI] [PubMed] [Google Scholar]
- 30.Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol 1994;10:38–47. [DOI] [PubMed] [Google Scholar]
- 31.Yamaya M, Finkbeiner WE, Chun SY, Widdicombe JH. Differentiated structure and function of cultures from human tracheal epithelium. Am J Physiol 1992;262:L713–L724. [DOI] [PubMed] [Google Scholar]
- 32.Gray TE, Guzman K, Davis CW, Abdullah LH, Nettesheim P. Mucociliary differentiation of serially passaged normal human tracheobronchial epithelial cells. Am J Respir Cell Mol Biol 1996;14:104–112. [DOI] [PubMed] [Google Scholar]
- 33.Gruenert DC, Willems M, Cassiman JJ, Frizzell RA. Established cell lines used in cystic fibrosis research. J Cyst Fibros 2004;3:191–196. [DOI] [PubMed] [Google Scholar]
- 34.Morrison DC, Jacobs DM. Binding of polymyxin B to the lipid A portion of bacterial lipopolysaccharides. Immunochemistry 1976;13:813–818. [DOI] [PubMed] [Google Scholar]
- 35.Jiang Q, Akashi S, Miyake K, Petty HR. Lipopolysaccharide induces physical proximity between CD14 and Toll-like receptor 4 (TLR4) prior to nuclear translocation of NF-kappa B. J Immunol 2000;165:3541–3544. [DOI] [PubMed] [Google Scholar]
- 36.Pizurki L, Morris MA, Chanson M, Solomon M, Pavirani A, Bouchardy I, Suter S. Cystic fibrosis transmembrane conductance regulator does not affect neutrophil migration across cystic fibrosis airway epithelial monolayers. Am J Pathol 2000;156:1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bedard M, McClure CD, Schiller NL, Francoeur C, Cantin A, Denis M. Release of interleukin-8, interleukin-6, and colony-stimulating factors by upper airway epithelial cells: implications for cystic fibrosis. Am J Respir Cell Mol Biol 1993;9:455–462. [DOI] [PubMed] [Google Scholar]
- 38.DiMango E, Ratner AJ, Bryan R, Tabibi S, Prince A. Activation of NF-kappaB by adherent Pseudomonas aeruginosa in normal and cystic fibrosis respiratory epithelial cells. J Clin Invest 1998;101:2598–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joseph T, Look D, Ferkol T. NF-kappaB activation and sustained IL-8 gene expression in primary cultures of cystic fibrosis airway epithelial cells stimulated with Pseudomonas aeruginosa Am J Physiol Lung Cell Mol Physiol 2005;288:L471–L479. [DOI] [PubMed] [Google Scholar]
- 40.Shuto T, Furuta T, Cheung J, Gruenert DC, Ohira Y, Shimasaki S, Suico MA, Sato K, Kai H. Increased responsiveness to TLR2 and TLR4 ligands during dimethylsulfoxide-induced neutrophil-like differentiation of HL-60 myeloid leukemia cells. Leuk Res 2007;31:1721–1728. [DOI] [PubMed] [Google Scholar]
- 41.Furuta T, Shuto T, Shimasaki S, Ohira Y, Suico MA, Gruenert DC, Kai H. DNA demethylation-dependent enhancement of Toll-like receptor-2 gene expression in cystic fibrosis epithelial cells involves SP1-activated transcription. BMC Mol Biol 2008;9:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hauber HP, Tulic MK, Tsicopoulos A, Wallaert B, Olivenstein R, Daigneault P, Hamid Q. Toll-like receptors 4 and 2 expression in the bronchial mucosa of patients with cystic fibrosis. Can Respir J 2005;12:13–18. [DOI] [PubMed] [Google Scholar]
- 43.Armstrong L, Medford AR, Uppington KM, Robertson J, Witherden IR, Tetley TD, Millar AB. Expression of functional Toll-like receptor-2 and -4 on alveolar epithelial cells. Am J Respir Cell Mol Biol 2004;31:241–245. [DOI] [PubMed] [Google Scholar]
- 44.Guillot L, Medjane S, Le-Barillec K, Balloy V, Danel C, Chignard M, Si-Tahar M. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem 2004;279:2712–2718. [DOI] [PubMed] [Google Scholar]
- 45.Hornef MW, Frisan T, Vandewalle A, Normark S, Richter-Dahlfors A. Toll-like receptor 4 resides in the golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J Exp Med 2002;195:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990;63:827–834. [DOI] [PubMed] [Google Scholar]
- 47.Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 1995;83:121–127. [DOI] [PubMed] [Google Scholar]
- 48.Kopito RR. Biosynthesis and degradation of CFTR. Physiol Rev 1999;79:S167–S173. [DOI] [PubMed] [Google Scholar]
- 49.Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 1995;83:129–135. [DOI] [PubMed] [Google Scholar]
- 50.Howell GJ, Holloway ZG, Cobbold C, Monaco AP, Ponnambalam S. Cell biology of membrane trafficking in human disease. Int Rev Cytol 2006;252:1–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jarosch E, Lenk U, Sommer T. Endoplasmic reticulum-associated protein degradation. Int Rev Cytol 2003;223:39–81. [DOI] [PubMed] [Google Scholar]
- 52.Barasch J, Kiss B, Prince A, Saiman L, Gruenert D, Al Awqati Q. Defective acidification of intracellular organelles in cystic fibrosis. Nature 1991;352:70–73. [DOI] [PubMed] [Google Scholar]
- 53.Cheng PW, Boat TF, Cranfill K, Yankaskas JR, Boucher RC. Increased sulfation of glycoconjugates by cultured nasal epithelial cells from patients with cystic fibrosis. J Clin Invest 1989;84:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saiman L, Prince A. Pseudomonas aeruginosa pili bind to asialoGM1 which is increased on the surface of cystic fibrosis epithelial cells. J Clin Invest 1993;92:1875–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohnishi T, Muroi M, Tanamoto K. MD-2 is necessary for the Toll-like receptor 4 protein to undergo glycosylation essential for its translocation to the cell surface. Clin Diagn Lab Immunol 2003;10:405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.da Silva CJ, Soldau K, Christen U, Tobias PS, Ulevitch RJ. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex. transfer from CD14 to TLR4 and MD-2. J Biol Chem 2001;276:21129–21135. [DOI] [PubMed] [Google Scholar]
- 57.Jackson SK, Abate W, Parton J, Jones S, Harwood JL. Lysophospholipid metabolism facilitates Toll-like receptor 4 membrane translocation to regulate the inflammatory response. J Leukoc Biol 2008;84:86–92. [DOI] [PubMed] [Google Scholar]
- 58.Moreno-Altamirano MM, Aguilar-Carmona I, Sanchez-Garcia FJ. Expression of GM1, a marker of lipid rafts, defines two subsets of human monocytes with differential endocytic capacity and lipopolysaccharide responsiveness. Immunology 2007;120:536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Henke MO, Renner A, Huber RM, Seeds MC, Rubin BK. MUC5AC and MUC5B mucins are decreased in cystic fibrosis airway secretions. Am J Respir Cell Mol Biol 2004;31:86–89. [DOI] [PubMed] [Google Scholar]
- 60.Henke MO, John G, Germann M, Lindemann H, Rubin BK. MUC5AC and MUC5B mucins increase in cystic fibrosis airway secretions during pulmonary exacerbation. Am J Respir Crit Care Med 2007;175:816–821. [DOI] [PubMed] [Google Scholar]
- 61.Matthews LW, Spector S, Lemm J, Potter JL. Studies on pulmonary secretions: I. The overall chemical composition of pulmonary secretions from patients with cystic fibrosis, bronchiectasis, and laryngectomy. Am Rev Respir Dis 1963;88:199–204. [DOI] [PubMed] [Google Scholar]
- 62.Chernick WS, Barbero GJ. Composition of tracheobronchial secretions in cystic fibrosis of the pancreas and bronchiectasis. Pediatrics 1959;24:739–745. [PubMed] [Google Scholar]
- 63.Potter JL, Spector S, Matthews LW, Lemm J. Studies on pulmonary secretions: 3. The nucleic acids in whole pulmonary secretions from patients with cystic fibrosis, bronchiectasis, and laryngectomy. Am Rev Respir Dis 1969;99:909–916. [DOI] [PubMed] [Google Scholar]
- 64.Picot R, Das I, Reid L. Pus, deoxyribonucleic acid, and sputum viscosity. Thorax 1978;33:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lethem MI, James SL, Marriott C, Burke JF. The origin of DNA associated with mucus glycoproteins in cystic fibrosis sputum. Eur Respir J 1990;3:19–23. [PubMed] [Google Scholar]
- 66.de Bentzmann S, Roger P, Dupuit F, Bajolet-Laudinat O, Fuchey C, Plotkowski MC, Puchelle E. Asialo GM1 is a receptor for Pseudomonas aeruginosa adherence to regenerating respiratory epithelial cells. Infect Immun 1996;64:1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Imundo L, Barasch J, Prince A, Al Awqati Q. Cystic fibrosis epithelial cells have a receptor for pathogenic bacteria on their apical surface. Proc Natl Acad Sci USA 1995;92:3019–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saiman L, Cacalano G, Gruenert D, Prince A. Comparison of adherence of Pseudomonas aeruginosa to respiratory epithelial cells from cystic fibrosis patients and healthy subjects. Infect Immun 1992;60:2808–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schroeder TH, Lee MM, Yacono PW, Cannon CL, Gerceker AA, Golan DE, Pier GB. CFTR is a pattern recognition molecule that extracts Pseudomonas aeruginosa LPS from the outer membrane into epithelial cells and activates NF-kappa B translocation. Proc Natl Acad Sci USA 2002;99:6907–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Armstrong DS, Grimwood K, Carzino R, Carlin JB, Olinsky A, Phelan PD. Lower respiratory infection and inflammation in infants with newly diagnosed cystic fibrosis. BMJ 1995;310:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Armstrong DS, Hook SM, Jamsen KM, Nixon GM, Carzino R, Carlin JB, Robertson CF, Grimwood K. Lower airway inflammation in infants with cystic fibrosis detected by newborn screening. Pediatr Pulmonol 2005;40:500–510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.