

Abstract
Calcific aortic valve disease (CAVD) is the most prevalent valvular heart disease in the Western population, claiming 17000 deaths per year in the United States and affecting 25% of people older than 65 years of age. Contrary to traditional belief, CAVD is not a passive, degenerative disease but rather a dynamic disease, where initial cellular changes in the valve leaflets progress into fibrotic lesions that induce valve thickening and calcification. Advanced thickening and calcification impair valve function and lead to aortic stenosis (AS). Without intervention, progressive ventricular hypertrophy ensues, which ultimately results in heart failure and death. Currently, aortic valve replacement (AVR), surgical or transcatheter, is the only effective therapy to treat CAVD. However, these costly interventions are often delayed until the late stages of the disease. Nonetheless, 275000 are performed per year worldwide, and this is expected to triple by 2050. Given the current landscape, next-generation therapies for CAVD are needed to improve patient outcome and quality of life. Here, we first provide a background on the aortic valve (AV) and the pathobiology of CAVD as well as highlight current directions and future outlook on the development of functional 3D models of CAVD in vitro. We then consider an often-overlooked aspect contributing to CAVD: miRNA (mis)regulation. Therapeutics could potentially normalize miRNA levels in the early stages of the disease and may slow its progression or even reverse calcification. We close with a discussion of strategies that would enable the use of miRNA as a therapeutic for CAVD. This focuses on an overview of controlled delivery technologies for nucleic acid therapeutics to the valve or other target tissues.
Keywords: aortic valve, calcification, 3D model, drug delivery, microRNA (miRNA)
INTRODUCTION
The most prevalent heart valve disease in Western societies is calcific aortic valve disease (CAVD), claiming 17000 lives per year in the United States alone [1] and affecting 25% of people older than 65 years of age [2]. CAVD is a dynamic disease; initial cellular changes that begin at the base of valve leaflets manifest into fibrotic lesions that induce valve thickening, termed aortic sclerosis [3]. The disease later progresses to calcification of the valve, where large calcific masses gradually impair leaflet motion and valve function, eventually causing aortic stenosis (AS) [4]. Without intervention, progressive ventricular hypertrophy ensues, which ultimately results in heart failure and death. CAVD treatment remains challenging as it is difficult to visualize or detect calcification in the early, asymptomatic stage (Figure 1) and no pharmacological therapies have been developed to slow or halt the progression of CAVD [5]. Although lipid-lowering therapy was proposed as a potential therapy for CAVD based on identified lesions similar to those in atherosclerotic plaques, it was shown to be ineffective in a meta-analysis of randomized placebo-controlled clinical trials on 2344 patients [6]. Currently, aortic valve replacement (AVR) remains the only available clinical treatment option for AS [7]. These costly and invasive procedures are often delayed until a patient’s functional leaflet movement is severely impaired by gross calcium deposition [8]. Each year, over 275000 patients undergo surgical AVR worldwide [9] and this number is projected to triple by 2050 [9]. The emergence of transcatheter AVR technologies in 2002 created a less-invasive alternative; however, due to an increased risk for complications, this option is currently reserved for patients with severe comorbidities who are unsuitable for conventional open-heart surgery [10]. To date, there have been more than 80000 transcatheter AVRs performed worldwide [11]. As the prevalence of CAVD is expected to increase with the rising global life expectancy, CAVD becomes a growing burden that demands further understanding of the disease process and exploration of potential non-invasive (drug-based) therapies.
Figure 1. CAVD progression.
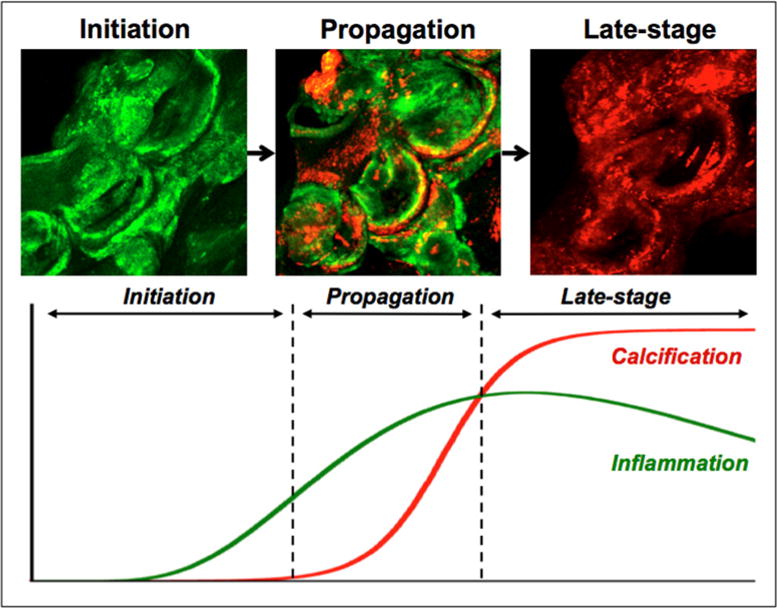
Initiation, propagation and late-stage calcification are three distinct stages of calcification in CAVD that can be identified by molecular imaging. Macrophages are labelled green and calcification is labelled red in a molecular imaging strategy to highlight the inflammation-dependent mechanism of calcification [128].
In this review, we provide a background on the aortic valve (AV) and the pathobiology of CAVD. We then highlight current directions in the community to develop functional 3D models of the AV as tools to dissect pathological processes in a controlled manner as well as forecast future directions in the development of in vitro valve models. Additionally, we consider an often-overlooked aspect of valve biology and pathology: miRNA regulation and misregulation. miRNAs present an attractive therapeutic alternative to intervene early during the disease process and potentially delay or even reverse disease processes after initiation. For miRNAs and other potential (bio)therapeutics, the controlled delivery of the molecules to the target tissues and cells of the valve is an additional challenge. In the present study, we review current directions within the drug-delivery community and suggest how they may affect the treatment of CAVD.
THE AORTIC HEART VALVE
The AV is located at the junction between the left ventricle and the aorta and ensures unidirectional blood flow into the systemic circulation from the heart. In a healthy AV, the three thin, semilunar leaflets recoil and open the pathway for blood flow during systole. When the heart relaxes during diastole, under the backpressure from the aorta, the leaflets stretch and seal the orifice, preventing regurgitation of blood into the left ventricle. Hence, an efficient opening and closing of the AV is imperative to the proper functioning of the heart. The valvular tissue microstructure, particularly the composition and orientation of the extracellular matrix (ECM), plays an important role in maintaining the mechanical and biochemical behavior of the valve. Each leaflet has a complex architecture and comprises three distinct layers: the ventricularis, the spongiosa and the fibrosa. The zona fibrosa, closest to the outflow surface, is a layer densely packed with collagen fibers that provide the tensile strength needed to bear diastolic stress and prevent leaflet prolapse. The zona spongiosa predominantly consists of glycosaminoglycans and proteoglycans and acts as a bearing surface, reducing friction between the zona fibrosa and the zona ventricularis. The zona ventricularis is an elastic layer rich in elastin fibers that allow the leaflet to extend and recoil [12]. There are two major types of valve cells that populate the AV and maintain its health: the valve endothelial cells (VECs) that coat the blood-contacting surfaces and the more abundant valve interstitial cells (VICs) that populate the three distinct layers of the leaflets. VICs are a heterogeneous and dynamic cell mixture; multiple VIC phenotypes have been identified in AV leaflets [13]. The role of VICs includes synthesizing, degrading, and repairing the ECM, which are all crucial in maintaining the tissue homoeostasis of the valve and its function.
PATHOBIOLOGY OF CAVD
Cell phenotypes contributing to the disease progression
Based on its association with aging, CAVD was traditionally assumed to be a passive, degenerative disease resulting from years of wear and tear due to mechanical stress. However, CAVD is now viewed as an active, cellular-driven disease [14]. In native healthy AVs, VICs reside primarily as quiescent fibroblast-like cells but can undergo phenotypic activation in response to injury or pathology (Figure 2). Upon exogenous pathological stimulation, VICs can differentiate into activated myofibroblast-like cells (aVICs), which undergo tissue repair and remodeling [15]. Persistent activation of VICs leads to pathological ECM remodeling and valve fibrosis [15]. These pro-fibrotic cells are characterized by increased expression of markers such as α-smooth muscle actin (α-SMA) [16]. Further, myofibroblast-like cells can differentiate into osteoblast-like cells (oVICs), which are ultimately responsible for the deposition of calcium and formation of osteogenic nodules. These pro-calcific cells are characterized by the presence of osteoblast-related proteins such as osteocalcin, osteonectin, and osteogenic transcription factors such as runt-related transcription factor 2 (Runx2) [17,18]. In addition, expression of progenitor cell markers has been identified in distinct subpopulations of VICs. Particularly, VICs positive for the progenitor cell marker ATP-binding cassette, sub-family G, member 2 (ABCG2) were found to deposit a more calcified matrix upon osteogenic induction, suggesting a possible role in the development of osteogenic VICs during valve pathology [19]. Furthermore, the level of circulating osteogenic progenitor cells in CAVD patients was significantly higher than in control patients in a study of 46 patients and 46 control subjects [20].
Figure 2. Molecular and cellular mechanisms in CAVD.
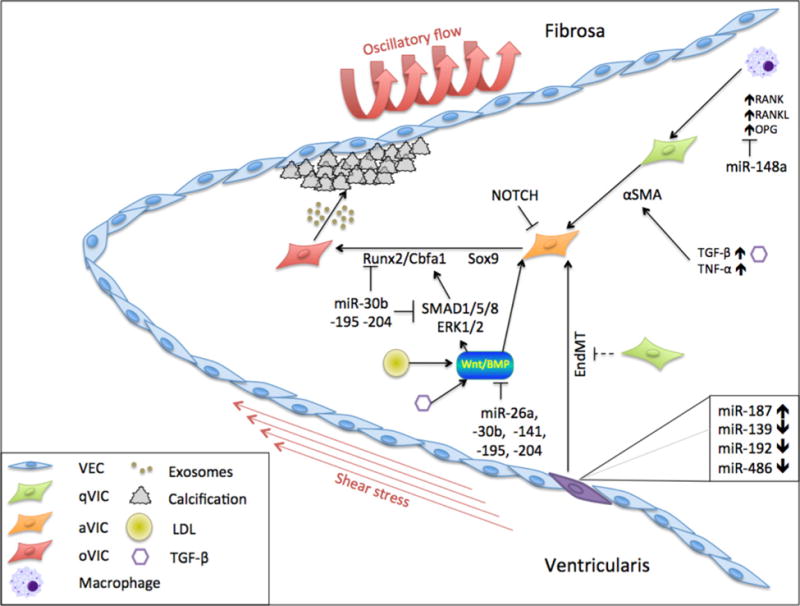
VICs differentiate from a quiescent state (qVIC) through an activated state (aVIC) into osteogenic cells (oVIC) that contribute to calcification of the fibrosa side of the AV. Mechanical stresses, EndMT, LDL, TGF-β and TNF-α affect the mechanism of disease via multiple pathways, including RANK, RANKL, OPG, α-SMA, Runx2 and BMP.
Mechanisms and signaling pathways
CAVD is multifactorial disease; many mechanisms and events contribute to the development of CAVD. In vivo studies have shown that early CAVD lesions initiate on the fibrosa side of the valve [21]. Pathological triggers such as growth factors, inflammatory cytokines, subendothelial deposits of oxidized low-density lipoproteins (LDLs), mechanical stress, and oxidative stress have been described to cause endothelial dysfunction, which in turn may initiate the activation of VICs [22–26]. In addition to VIC activation, VECs are known to have the capacity to differentiate into endothelial-derived VICs (eVICs) through endothelial-to-mesenchymal transition (EndMT) and can further differentiate into osteoblastic cells, promoting pathological remodeling in CAVD. It has been demonstrated that VIC–VEC communication is critical to maintain valve homoeostasis [27], and that disruption of VIC–VEC communication accelerates the EndMT process, advancing valvular osteogenesis [28].
At the molecular level, a number of studies have shown increased expression of various osteogenic factors and signaling pathways that may be involved in CAVD. For example, pathological ECM remodeling contributes to the release of cytokines, such as transforming growth factor β 1 (TGF-β1) and tumor necrosis factor α (TNF-α), which are potent inducers of VIC myofibroblast activation [29,30]. Notably, TGF-β1 works in synergy with its downstream effector osteoblast-cadherin through a number of downstream signaling pathways, such as extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) and the similar to mothers against decapentaplegic 2 and 3 (SMAD2/3) pathways, in the regulation of myofibroblast activation of VICs [31]. Moreover, bone morphogenetic protein 2 (BMP-2), a key inducer of VIC calcification, is thought to act through p-SMAD1/5/8 and p-ERK1/2 signaling to increase Runx2 expression [32]. On the other hand, Notch signaling has been demonstrated, through in vivo studies, as a negative regulator that represses osteoblast-like calcification pathways mediated by BMP-2 [33]. In addition, inflammation stimulates osteoblast differentiation; cytokines produced by inflammatory cells affect the expression of receptor activator of nuclear factor κ-B ligand (RANKL) and osteoprotegerin (OPG) in the RANKL/receptor activator of nuclear factor κ-B (RANK)/OPG pathway and consequently stimulate the Runx2 pathway [30]. Studies also found that excess of circulatory LDL acts through the low-density lipoprotein receptor-related protein 5 (LRP5)/Wnt signaling pathway, which stimulates the Runx2-mediated pathway and thereby induces mineralization [34]. Interestingly, studies have also suggested that mechanical stiffness promotes VIC activation through an up-regulation of phosphoinositide 3-kinase (PI3K)/Akt signaling pathway [35,36]. Although a number of pathophysiological cues have been associated with the activation of VICs leading to CAVD, knowledge on the regulation of pathologic changes in VIC phenotype is still limited due to the unavailability of adequate disease models. Understanding the mechanism involved in VIC regulation of tissue homoeostasis requires a suitable in vitro system that closely resembles the in vivo microenvironment native to the AV.
TOWARDS 3D CAVD MODELS
Understanding the cellular contribution to the progression of CAVD has proven to be challenging both in vivo and in vitro. Although in vivo animal models, such as porcine and murine models, offer complexity comparable with that in humans, these models are often based on high cholesterol diets to induce hypercholesterolaemia and subsequent atherosclerosis, which is not currently considered as a main etiology of CAVD [5]. In addition, most common 2D in vitro approaches have been shown to spontaneously activate pathological differentiation of VICs into myofibroblast-like cells. Furthermore, the contributions that knowledge of valvulogenesis could make to improve in vitro models are often overlooked [37]. Soft hydrogels have maintained VIC phenotype ex vivo [35]; however, they lack the complexity of VIC–VEC interactions and multilaminate structure in the native valve. Emerging evidence suggests that hydrogel-based 3D culture systems may provide a more tissue-like environment given their similarity to the natural ECM [38]. Recent studies have used various materials, such as hyaluronan hydrogels [39] and synthetic PEG-based hydrogels [38,40], to engineer heart valve tissue scaffolds for the study of VIC behavior. VICs cultured in materials derived from natural ECM polymers, such as collagen and fibrin, showed high viability; however, these hydrogel materials are not ideal due to their susceptibility to degradation and compaction [41]. On the other hand, methacrylated gelatin (GelMA) and methacrylated hyaluronic acid (HAMA) have successfully been used as a photocrosslinkable VIC-laden hydrogel scaffold. However, GelMA alone posed challenges to quick degradation whereas HAMA alone demonstrated limited cell adhesiveness. Recently, we utilized a hybrid GelMA–HAMA hydrogel platform to encapsulate VICs, which maintained VICs in a quiescent phenotype. These quiescent VICs further differentiated into myofibroblast-like cells upon TGF-β1 stimulation [28] and osteoblast-like cells when treated with osteogenic medium [42]. In addition, our recent study demonstrated the formation and growth of microcalcification recapitulating early disease states of native cardiovascular tissue [43]. Although these 3D hydrogel platforms provided controllable models that more closely recapitulate the in vivo environment to study the transition of VICs from a quiescent to an activated phenotype and formation of microcalcifications, they were not complete as they did not include the three distinct layers present within the native valve that contribute independently to the calcification process or the layer of endothelial cells that covers the leaflets. For future improvement, 3D-bioprinting technologies present promising approaches for engineering better representations of the native valve by incorporating all three distinct layers of the valve (Figure 3). A layer of endothelial cells can be co-cultured on top of the three layers to mimic cell–cell communication and paracrine signaling in the native valve. Moreover, the benefit of an automated system may alleviate some construct-to-construct variability and increase reproducibility as opposed to manual pipetting. This new approach could further be used to study cellular and molecular mechanisms of CAVD and serve as a drug-screening tool to aid in the development of a therapeutic treatment for CAVD. Furthermore, 3D printing opens doors to other disciplines as well, such as tissue engineering and biofabrication. Expanding the tri-layered 3D model to a geometrically accurate AV is the first step towards understanding how hemodynamic flow patterns affect the disease development when studied in a bioreactor. These efforts could lead to personalized tissue-engineered biofabricated heart valves that can be seeded with a patient’s autologous living valve cells, replacing the current prosthetic valves in AVR, bringing personalized therapy one step closer to reality. AV replacement is the current gold standard of therapy, but with the current advances of in vitro disease modeling and drug screening, an improvement through drug-based therapy is within reach and could potentially treat or even prevent CAVD.
Figure 3. 3D-bioprinted CAVD model.
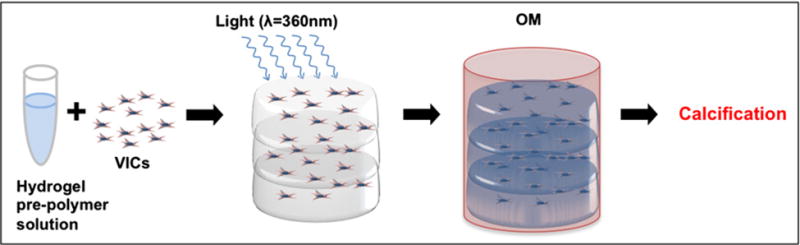
Isolated human aortic VICs are suspended in a hydrogel pre-polymer solution with photo-initiator cross-linking agent (A). Three distinct layers are bioprinted and cross-linked using UV light (B). Exposure to osteogenic medium (OM) activates the quiescent VICs to differentiate into activated VICs and osteogenic VICs (C), leading to the formation of microcalcifications, thereby mimicking CAVD progression.
Unfortunately, common valve specific drug targets required for pharmacologic approaches to treat vascular disease are not transferable to CAVD [5]. These targets could include G-protein coupled receptors, cadherin-11, or lipids, and can be targeted with small molecules, antibodies or nucleic acid therapeutics. Identifying if other pathways could be potential drug targets in CAVD and validating these approaches may be studied in appropriate 3D disease models of CAVD. In the next section, we will focus on potential drug targets and provide an overview of the candidates for therapeutic targeting, highlight modifications to improve their function and discuss potential delivery strategies.
miRNA IN AORTIC VALVE DISEASE
Biogenesis and activity of miRNA
miRNAs and miRNA regulation are often overlooked as therapeutic targets, especially in cardiac valve disease. miRNAs were first discovered by Victor Ambros and his team in 1993 [44]. They found that the lin14 gene codes not for a protein but for a pair of small RNAs, later named ‘microRNA’ (miRNA or miR). Functional mature miRNAs are single stranded, approximately 22 nucleotides long, RNAs that regulate protein expression by partial complementary binding to their target mRNA, thus inhibiting its translation. RNA polymerase II transcribes primary miRNA transcripts (pri-miRNAs) that contain one or multiple hairpin loops [45]. Drosha, a dsRNA-specific type III endoribonuclease, and DiGeorge syndrome critical region gene 8 (DGCR8) process pri-miRNAs into precursor miRNAs (pre-miRNAs) of approximately 70 nucleotides [46,47]. If correctly processed, nuclear export factor exportin 5 transports pre-miRNAs from the nucleus to the cytoplasm where Dicer, endoribonuclease III, removes the hairpin loop yielding a miRNA duplex of 18–22 bp [48,49]. When the duplex unwinds, the argonaute protein loads one strand, the guide strand or mature strand, into the RNA-induced silencing complex (RISC) [50]. This can lead to mRNA degradation or to obstruction of translation, depending on the level of complementarity between the mRNA target and the miRNA [51–53]. The other strand, the passenger strand, is often degraded although in selected cases it also serves as a functional miRNA. The complete process of strand selection is not yet fully understood. Argonaute and Dicer are two of the multiple proteins that play a role in this process [54]. Since the discovery by Ambros and his team, upwards of 1800 miRNAs have been identified in humans regulating a broad range of biological processes [55]. On account of the imperfect base pairing of miRNAs and their target mRNAs, one miRNA can affect multiple processes. Assigning the mRNA targets and biological roles to a miRNA has identified expression patterns specific to tissues, developmental processes, and disease development [56]. From these findings, miRNAs are now thought to be key regulators of pathological processes in cardiovascular diseases, including cardiovascular calcification [57]. Furthermore, circulating miRNAs can potentially function as biomarkers for early detection of heart disease [58,59] for modification of disease mechanisms, and for determining the ideal timing of surgical intervention [60]. Here, we provide an overview of studies that have identified miRNAs in CAVD and AS.
Known miRNAs in aortic valve disease
miRNAs that have been identified in cardiovascular diseases serve as regulators of multiple processes (Figure 2) [61]. Using the broad parameters “aortic | valve”, Abcam Firefly Discovery Engine identified 30 publications on 41 miRNAs related to various biological processes, including fibrosis (miR-21 [62]), inflammation (miR-125b [63]; miR-148a [64]), differentiation (miR-204 [65]), and calcification (miR-141 [66]; miR-204 [65]; miR-30b [67]; miR-214 [68]). Tables 1 and 2 provide an overview of the profiling and functional studies that have been conducted to highlight the roles of these miRNAs in their respective target pathways.
Table 1. Overview of miRNA profiling studies in CAVD.
This table provides an overview of the differentially expressed miRNAs in diseased AV tissues. AI, aortic insufficiency; TAV, tricuspid aortic valve.
| miRNA | Tissue | Finding | Method | Patients | Reference |
|---|---|---|---|---|---|
| 26a, 30b, 195 | Whole bicuspid AVs | Down-regulated | Microarray + RT-qPCR | 9 BAV patients with AS/AI | [69] |
| 30e, 32, 145, 151-3p, 152, 190, 373, 768-5p 22, 27a, 124-3-pre, 125b-1-pre, 141, 185-pre, 187, 194, 330-5p, 377, 449b, 486-3p, 551a-pre, 564, 566-pre, 575, 622, 637, 648-pre, 1202, 1282, 1469, 1908, 1972 | Bicuspid and tricuspid AVs | Up-regulated Down-regulated | Microarray + RT-qPCR | 19 BAVs 17 TAVs patients with AS | [66] |
| 30b | Calcific AV and adjacent tissue | Down-regulated | RT-qPCR | 10 patients with AS | [67] |
| 148-3p | Excised AV tissue | Down-regulated | RT-qPCR | 4 BAVs compared with healthy AV | [64] |
| 125b 374b, 602, 939 | Calcific AV and healthy AV | Up-regulated Down-regulated | Microarray + RT-qPCR | 20 patients with AS | [63] |
| 214 | Porcine AV | Up-regulated | Microarray + RT-qPCR | Ex vivo shear stress calcification model | [20] |
Table 2. Overview of functional studies of miRNAs and their roles in CAVD.
This table provides an overview of the differentially expressed miRNAs in diseased AV tissues that have been validated in functional studies. haVIC, human aortic valve interstitial cell; paVIC, porcine aortic valve interstitial cell.
| miRNA | Target (genes) | Cell | Results |
|---|---|---|---|
| 30b | RUNX2, SMAD1, CASP3, SMAD3, BMP2, NOTCH1 | haVICs | Vitiates BMP2-induced osteoblast differentiation and apoptosis through direct targeting of Runx2, Smad1 and caspase-3 [67]. Down-regulates calcification-related gene pathways [69] |
| 26a | ALPL, BMP2, SMAD1 | haVICs | Repressed several of the calcification-related genes and increased the mRNA levels of genes that may have roles inhibiting calcification (JAG2, SMAD7) [69] |
| 195 | BMP2, RUNX2, SMAD1,-3,-5 JAG2, SMAD7 | haVICs | Activates pro-calcification gene expression [69] Increases calcification repressing genes JAG2 and SMAD7 [69] |
| 148a-3p | IKBKB | haVICs | Cyclic stretch represses miR-148a-3p and activates NF-κB-dependent inflammatory signaling pathway [64] |
| 141 | BMP2 | paVICs | Blocks TGF-β-triggered BMP2 signaling [66] |
| 204 | RUNX2, BMP2 | haVICs | Negatively regulates osteogenic differentiation by repressing RUNX2 [65] |
| 214 | TGF-β1 | paVICs | Regulates TGF-β1 regulator of calcification in early stages of CAVD [68] |
As shown in Table 1, five of these studies used a miRNA candidate approach or microarray to identify the expression of miRNAs in AV leaflets from patients with AS undergoing AVR surgery. Real-time quantitative PCR (RT-qPCR) validated microarray data. First, AS patients were compared with aortic insufficiency patients, which demonstrated that in bicuspid valve samples miR-26a, -30b and -195 were down-regulated in patients with AS [69]. Second, microarray profiling identified 35 differentially expressed miRNAs between bicuspid and tricuspid AVs in patients with AS [66]. RT-qPCR validated down-regulation of miR-141. Third, a comparison of calcified and non-calcified AV tissues confirmed lower expression levels of miR-30b in the calcified valves using RT-qPCR [67]. Fourth, down-regulation of miR-148-3p was shown in bicuspid compared with tricuspid valves [64]. Fifth, microarray analysis of calcified valve tissue compared with control tissue yielded the identification of up-regulated miR-125b, and down-regulated miR-374b, -602 and -939 [63].
To understand the role these miRNAs play in AV pathophysiology, functional studies have been conducted in 2D in vitro models. Table 2 provides an overview of current knowledge of miRNAs and their target genes in fibrocalcific valve remodeling. Multiple studies demonstrated that miR-30b is functionally involved in the prevention of osteogenesis and apoptosis by direct targeting of Runx2, Smad1, and caspase-3 [67,69]. Similarly, miR-26a down-regulates multiple genes related to calcification [69], miR-141 blocks TGF-β-triggered BMP-2 signaling, and miR-204 represses Runx2 [66], consequently inhibiting osteogenic differentiation [65]. Thus, valve calcification could be caused by negative regulation of these miRs, which would abrogate various protective mechanisms. Furthermore, abnormal hemodynamics around the AV, as observed in patients with AS, aid disease progression by activating an inflammatory response through the NF-κB-dependent signaling pathway, which may be a result of miR-148a-3p repression [64]. This pathway is also activated in the aortic VECs on the fibrosa layer of the valve by shear-sensitive miR-139-3p, -187, -192, and -486-5p [70]. This inflammatory response recruits immune cells to the valve, increasing the levels of CCL4 chemokine and, as a consequence, down-regulating miR-125b in infiltrating macrophages [63], leading to calcification. Conclusively, AV disease progression is stimulated by inhibition of miRs that regulate protective mechanisms and by disturbed hemodynamics that activate an inflammatory response. Lastly, it is hypothesized that osteoblast differentiation, by repressing the activity of TGF-β type I receptor as part of the osteoblast lineage commitment program [71] and by inhibiting osteonectin expression through canonical Wnt pathway, applies to valve calcification in a similar fashion.
In addition to the classic approach of miRNA array analysis and RT-qPCR validation, a recent study identified miR-214 as a regulator of AV calcification via the TGF-β1 pathway using an ex vivo shear and calcification model with porcine aortic VECs. The porcine miRs identified in the present study are homologous with human miRs, as confirmed by miRBase. In the shear and calcification model, the cells were subjected to oscillatory shear stress for 2 days. When the fibrosa side was exposed to the oscillatory shear, calcification was triggered, whereas no calcification was observed when the ventricularis side of the valve was subjected to the same condition. In these experiments, miR-214 was significantly up-regulated by oscillatory shear in the fibrosa side compared with the ventricularis side and compared with fresh AV tissue. These results support the conclusion that in fibrosa miR-214 is regulated in a shear- and location-dependent fashion. Immunofluorescent staining of TGF-β1 revealed increased expression in the fibrosa layer in the endothelium, sub-endothelium, and in the interstitial cells upon miR-214 silencing. This strongly suggests that miR-214 serves as a key regulator of AV calcification through modulation of TGF-β1 signaling. Inhibiting miR-214, however, did not attenuate calcification initiated by exposure to oscillatory shear, which suggests that miR-214 modulates gene expression in AV disease in the early stages of AV pathogenesis rather than the late stages [68].
Moreover, the aforementioned circulating osteogenic progenitor cells are thought to play a significant role in CAVD pathogenesis. A recent study that compared AS patients with control subjects showed that these circulating osteogenic progenitor cells may be regulated by miRNAs. Specifically, miR-30c levels were higher in the AS groups compared with the control group, whereas miR-31, -106a, -148a, -204, -211, and -424 levels were lower. The study also highlighted a correlation between miR-30c expression levels and calcification in AS patients, and that the level of miR-30c and the number of circulating osteogenic progenitor cells decreased after AVR. The study concluded that differential expression of ossification-related miRNAs could be related to the differentiation into circulating osteogenic progenitor cells [20].
Recently, we have shown that extracellular vesicles play a role in cardiovascular calcification [72–74,43,75]. In addition, it was shown that these calcifying vesicles are exosomes [76] and that they may contain miRNA [77–80]. It is yet to be determined how these extracellular vesicles release their content and by which mechanisms they interact with cells and the ECM to induce formation of microcalcifications. Evidence of the presence of miRNA in calcifying extracellular vesicles gives weight to the hypothesis that these miRNAs can be potential therapeutic targets for CAVD.
miRNAs AS THERAPEUTIC TARGETS
The discovery that miRs play essential roles in a plethora of biological processes and that differential expression of miRNAs is a common contributor to cardiovascular disease, specifically to inflammatory responses and calcification, has sparked interest in targeting miRNAs with therapeutics. Expression vectors, small-molecule inhibitors, antisense oligonucleotides (ASOs) (or antagomirs or antimiRs), and miR-mimics are the four avenues that are currently explored for therapeutic intervention.
Expression vectors, or miRNA sponges, are artificial miRNA-binding sites that isolate endogenous miRNAs when overexpressed for a specific mRNA, consequently eliminating the effect the miRNAs have on their target mRNA [81]. Small molecule-based approaches function as translational regulators of the target miRNAs, rather than targeting the miRNAs themselves. Due to the high effector concentration for half-maximum response (EC50) and the unknown direct targets, the therapeutic potential of small-molecule inhibitors is constrained. AntimiRs are fully complementary to their specific target miR and, by binding to them, relieve mRNA targets from degradation or translational blockage by the specific miR. Oligonucleotides without any modification or encapsulation are prone to serum nucleases, have a low binding affinity for their target miRs, demonstrate poor pharmacodynamics/pharmacokinetics (PD/PK), and are incapable of crossing negatively charged cell membranes on account of their positive charge. The next section therefore highlights several modifications and multiple delivery strategies used to improve the therapeutic use of miR and antimiR oligonucleotides.
Chemical modifications of miRs and antimiRs
It is imperative to improve stability and efficacy of miR and antimiRs, and to reduce their degradation by serum nucleases, prior to applying these oligonucleotides as therapeutic agents. Stability can be increased by 2′-O-methyl (2′-OMe) modification, though this does not increase the resistance to serum nucleases [82]. 2′-OMe oligonucleotides can be further stabilized by replacing non-bridging oxygen atoms in the phosphate backbone with sulfur atoms, creating phosphorothioate bonds, making them less susceptible to serum nucleases that cleave phosphate bonds. Replacing all non-bridging oxygen atoms, however, often removes all binding affinity for the target miRNA [83]. Replacing a limited number of phosphodiester bonds with phosphorothioate bonds allows for more nuclease resistance without reducing the binding affinity. Injecting these phosphorothioate-modified (anti)miRs directly into the bloodstream prolonged their time in the circulation due to their higher plasma protein binding, which improved their PD/PK. This does not inhibit uptake by tissues, as the plasma protein binding affinity is still lower than the binding affinity of tissues [84].
Further chemical modification of (anti)miRNA strands leads to improved efficacy and tissue distribution in vivo. Specifically, a combination of 2′-OMe modifications, asymmetric phosphorothioate modification on the 3′ and 5′ ends, plus a 3′ cholesterol tail is now a commonly used strategy for stable miRNA modification [85,86]. At the same time, additional modifications at the 2′ sugar position were shown to improve nuclease resistance and binding affinity. These modifications are 2′-O-methoxyethyl (2′-MOE), 2′-fluoro (2′-F), and locked nucleic acid (LNA) modifications. Superior efficacy was achieved by 2′-MOE-modifications over 2′-OMe-modification [87,88]. 2′-F-modifications alone do not yield nuclease-resistant oligonucleotides, though in combination with phosphorothioate backbone modification, these oligonucleotides achieved better miRNA inhibition than any of the aforementioned modified oligonucleotides [89]. LNA modifications achieve a higher binding by tethering the 2′ oxygen via a methylene bridge to the 4′ carbon [90]. The best miRNA inhibition at a low dose with increased efficacy was achieved by combining LNA modifications in a recurring pattern of two DNA bases and one LNA base with 2′-F modifications [82]. This higher binding affinity creates room for miRNA inhibition with shorter sequences, and for oligonucleotides that only bind the seed region of their target miRNAs [91]. This evidence supports the hypothesis that one individual oligonucleotide with LNA modification can silence an entire miRNA family without inducing any off-target effects (OTEs). More recent modifications focus on changing the oligonucleotide conformation at non-nucleotide locations. N,N-diethyl-4-(4-nitronaphthalen-1-ylazo)-phenylamine (ZEN) modification at both ends of the sequence increases binding affinity and efficiency of a 2′-OMe-modified oligonucleotide, while simultaneously reducing toxicity [92]. All of these discoveries demonstrate the tremendous achievements in improving binding affinity, nuclease resistance, and efficacy of (anti)miRs. However, for in vivo applications of (anti)miRs as nucleic acid therapeutics, a delivery vehicle is often required to achieve optimal efficacy at a reasonable dose.
Delivery vehicles for miR and antimiR oligonucleotides
Despite the improvements with direct chemical modification of miRNA molecules, delivery efficiency is largely dependent on the design of delivery vehicles for in vivo applications. Limited tissue distribution and excretion occur shortly after administration when chemically modified (anti)miR oligonucleotides are introduced without a carrier, requiring a higher dosage to achieve in vivo effect, thereby increasing the chances of undesirable OTEs and increasing overall cost. Thus, the engineering of an appropriate delivery system is critical to efficient in vivo application of miR and antimiRs. The main functions of these delivery systems are protecting against nucleases to prevent premature degradation [93], avoiding recognition by the immune system, preventing non-specific interactions with other proteins and cells, preventing excretion via the liver and kidneys, exiting the circulation into the target tissue, facilitating uptake by the target cells, and releasing their content intracellularly for incorporation into the RNA processing machinery [94–100]. There are many approaches to aid in the delivery of (anti)miRs, including polymer-, lipid-, conjugation-, antibody-, microbubble-, and inorganic nanoparticle-based approaches. We elaborate on some of the most promising strategies here.
Nanoparticle/polymer-based approaches focus on the interaction between the (anti)miRs and the functional block of the polymer, allowing more flexibility while controlling nanoparticle size, yielding a nearly homogeneous solution of nanoparticles with minimal size distribution. Effective drug delivery in vivo is highly dependent on the size of the nanoparticles. Nanoparticles with a diameter between 10 and 100 nm have been shown to functionally deliver (anti)miRs, siRNA, and small molecules [101]. Common polymers used for the synthesis of this kind of nanoparticles include polyethyleneimine (PEI), poly(lactic acid) (PLA), and poly-L-lysine (PLL).
Lipid-based approaches are based on the interactions between the hydrophobic group of lipids and water molecules that lead to the formation of micelles or liposomes [102] and release their content by destroying the stability of the endosomal membrane via fusogenic lipids or pH-sensitive peptides. This creates liposomes that can be categorized as neutral liposomes, ionizable lipids, fusogenic lipids, and PEG liposomes. Neutral liposomes interact little with serum proteins, because of their lack of charge, leading to improved stability [103,104]. Depending on both internal factors (lipid composition) and external factors (e.g. type of solution, temperature), liposomes of different sizes can be created. Typically, small liposomes are 20–200 nm, large liposomes 200 nm–1 μm, and giant liposomes >1 μm [105]. Their relatively large size slows cellular uptake and can be counteracted by incorporating 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine [106]. Cationic liposomes consist of a cationic head, a connecting bond, and a hydrophobic tail, and form complexes with the (anti)miR based on their electrostatic interaction with the negatively charged (anti)miR [107]. Fusogenic lipids have the capability to fuse with the target membrane without external stimulation to release their therapeutic content into the cell, facilitating delivery of therapeutics that would otherwise not be permeable to the cell membrane [108,109]. PEG liposomes have an increased PD/PK compared with non-PEGylated liposomes due to increased water solubility, reduced enzymatic degradation, limited immunogenic responses, and lower renal clearance [110].
Conjugation-based approaches rely on direct conjugation of the cargo to the delivery material. Conjugation of siRNA to cholesterol [111] and other lipophilic molecules [112] showed the first efficacy in vivo. Other delivery materials that have been tested are antibodies, aptamers, peptides, polymers, and small molecules [113]. One example is Dynamic PolyConjugates, which are injected intravenously and target hepatocytes in the liver using N-acetylgalactosamine (GalNAc) ligands. After endocytosis, the PEG decomplexes from the membrane-disrupting polymer poly(butyl amino vinyl ether) (PBAVE) due to the increasingly acidic environment inside the endosome, exposing this polymer and allowing for endosomal escape. The bond linking the siRNA to this polymer is reduced in the cytoplasm, releasing the functional siRNA, causing RNA interference [114]. Another example of a conjugate system uses the same hepatocyte targeting ligand GalNAc conjugated with siRNA. A triantennary spacer links the 3 end of the sense strand of the siRNA to three GalNAc molecules [115,116]. Alnylam Pharmaceuticals produced a GalNAc conjugate that was administered both intravenously and subcutaneously. The latter showed greater uptake of siRNA in the liver and increased knockdown [117].
Microbubbles have been used in combination with ultrasound to deliver antimiRs to the myocardium in an ischemia reperfusion mouse model [118]. The microbubbles are formed by vigor ously mixing 1,2-distearoyl-sn-glycero-3-phosphocholine, 1,2-stearoyl-3-trimethylammonnium-propane and polyoxyethylene-40-stearate in H2O, glycerol and propylene glycol, in the presence of perfluorobutane gas. The antimiRs are bound to the microbubbles via electrostatic interaction between the cationic microbubble and the anionic antimiRs, and injected intravenously. Local ultrasound waves ensured destruction of the microbubbles releasing the antimiRs at the target location, a process named ultrasound triggered microbubble destruction (UTMD). This resulted in sustained intracellular delivery of the antimiRs in cardiomyocytes without causing apoptosis and with a mild immune response.
A combinatorial approach of the aforementioned lipids results in the most effective transfection agents. Lipid nanoparticles (LNPs) (Figure 4) consist of an ionizable lipid that complexes with the siRNA, increases cellular uptake, and aids endosomal escape; a phospholipid that provides structure to the lipid bilayer; a cholesterol that provides stability to the lipid bilayer; and a lipid-anchored PEG that limits aggregations and non-specific uptake, and that lowers the degradation rate [119,120]. PEGylation of delivery vehicles reduces the non-specific interactions with serum proteins, immune cells, and non-target tissues, thereby prolonging the time until they are filtered by the liver and renal system. Most LNPs, and other delivery systems, rely on endocytosis for entering their target cells. Ionizable lipids and ligands specific for receptors on the target cells in the outer shell delivery systems aim to trigger receptors to expedite endocytosis [121]. After endocytosis, the LNPs must release their content into the cytoplasm before the endosome is degraded by lysosomes. During maturation of an endosome, its pH is lowered to 5. The pKa of common ionizable lipids is often approximately 6.5. At this pH, the nitrogen atoms on the ionizable lipid deprotonate and decomplex from the miRs and antimiRs. It is hypothesized that the ionizable lipid induces a lipid phase transition and thereby disrupts the endosomal membrane, releasing the ASOs into the intracellular environment [122]. There, the guide strand of the ASOs is loaded into the RISC machinery. Therefore, conjugation of the 5′-end of this strand must be avoided [123]. Thus, conjugation of the sequences and modifications of the backbone have to be carefully considered to establish proper strand selection by the RNA processing machinery RISC and to avoid incomplete hybridization to other, non-target mRNAs leading to OTEs [97].
Figure 4. LNP.
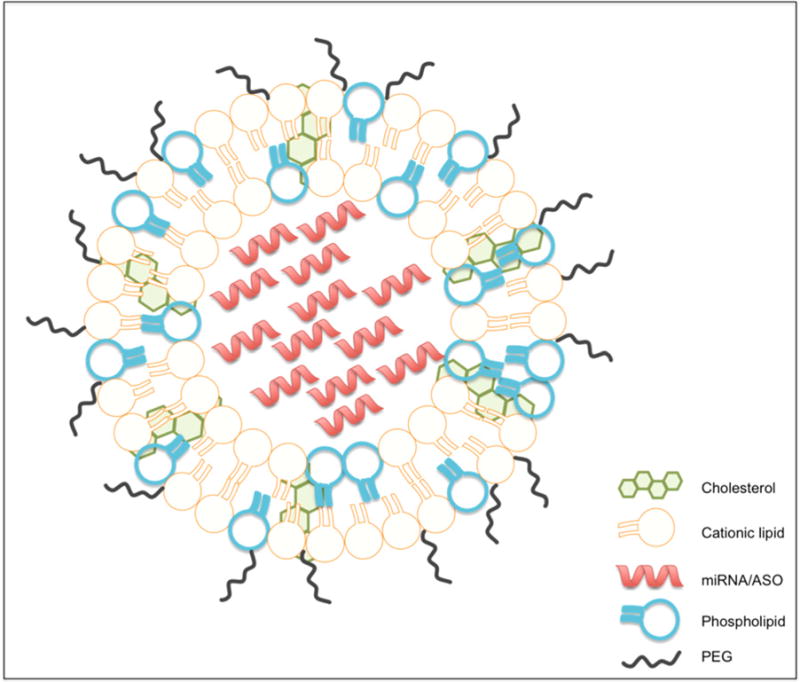
Schematic representation of a LNP made up of cationic lipids that covalently bind with the nucleic acid content, e.g. miRNA or ASO, phospholipids that provide structure to the lipid bilayer, cholesterol that provides stability of the lipid bilayer, and lipid-anchored PEG that limits aggregation and non-specific uptake.
Recently, effective delivery has been achieved with the use of gold nanoparticles as a delivery vehicle for both siRNA and miRNA. Gold nanoparticles were functionalized with siRNA, made possible by modification with thiol groups on the 5′-end of the antisense strand, and with a fusogenic peptide linked to a thiol-modified PEG for targeting. The thiol groups allow covalent bonding of the nucleic acid and the PEG–peptide complex to the gold nanoparticle. These functionalized gold nanoparticles were doped in a hydrogel and implanted in a colorectal cancer mouse model adjacent to the tumor, leading to functional in vivo silencing of Kras, an important gene in cancer progression, leading to complete remission. In further experiments, the doped hydrogels were implanted after tumor resection, preventing recurrence [124]. In a second study, miRNA was linked to gold nanoparticles and loaded in a hydrogel using the aforementioned strategy, and implanted in a breast cancer mouse model. This resulted in local, selective, and sustained delivery of the miRNA, attenuating the metastatic tendencies of the tumour [125].
FUTURE PERSPECTIVES
As illustrated, large variation between the aforementioned delivery systems exists. Many allow for efficacious drug delivery and for fine tailoring to specific needs. On the other hand, scale-up and broad application of a single nanoparticle-based delivery system remains challenging [126]. The process of mixing the individual components of these delivery systems is critical to guarantee the uniform quality of the particles. A microfluidic approach may be the answer to the scale-up of these delicate manufacturing processes [127].
Currently, nanoparticle-based drug-delivery systems mainly target tumor and the liver as these tissues are highly perfused and the endothelium is often fenestrated (not continuous), allowing for easy passage of the particles. It remains a challenge to target less accessible tissues, like the AV. This challenge can be addressed by tailor-made delivery systems, such as the injection of a reservoir of particles directly into the target tissue using injectable carriers, such as hydrogels or degradable biopolymers, or by employing strategies like UTMD.
Moreover, there is room for improving the delivery process of nucleic acid therapeutics, as the exact mechanisms remain poorly understood. Recent evidence has provided certain principles and guidelines for particle-based delivery. Currently, particles are designed to avoid renal filtration and clearance by the immune system. This limits their size to approximately 20–200 nm. Shielding particles by PEGylation has increased the circulation time and reduced serum protein binding, preventing undesirable interactions. The (anti)miRs are chemically modified to improve stability and reduce degradation by nucleases, as well as immunostimulation. Receptor-specific ligands can be included in the formulation of particles to stimulate endocytosis and improve uptake by the target cells. Acidity-dependent membrane-disrupting materials can be shielded until needed and activated after endosomal uptake to facilitate release of the content. Therefore, research efforts should focus on creative and innovative delivery platforms that can deliver particles and drugs to specific target tissues, and on elucidating the mechanisms of particle circulation, homing, uptake, and release.
Prior to making the translational step to the clinic, drug-delivery devices are validated on in vitro and in vivo disease models. Employing 3D disease models with human cells has the potential to reduce the burden on animal models. The use of human cells is beneficial, as discoveries and results based on animal studies do not always translate directly to humans. Additionally, eliminating the need for animal models greatly reduces the time between bench and bedside. Specifically, for CAVD, where in vivo models are costly and time consuming to create, a 3D in vitro disease model with human cells that mimics the native valve tri-layered structure would be a valuable tool that can aid in the design of drug-delivery devices and allow for rapid testing of the functionality of novel nucleic acid therapeutics.
An innovative and reproducible 3D-bioprinted model of human CAVD can be employed (i) to study cellular and molecular mechanisms of CAVD, and (ii) as a drug screening tool. Furthermore, it can be used to identify potential therapeutic targets for CAVD, a disease without a drug-based therapy, and it can be used as a proof of concept for drug-delivery platforms that can then be expanded to target other tissues and diseases. Specifically, the multiple approaches that aid the delivery of (anti)miRs can be tested on this 3D model, simultaneously testing the efficacy of (anti)miRs in treating CAVD by reducing calcification and testing the efficiency of the drug-delivery approach. The aforementioned approaches can be tested as is, or in combination with a delivery device, such as an implantable or an injectable hydrogel. In the clinical setting, these hydrogels would function as a local depository, allowing for a single intervention to store multiple doses of therapeutics, which are released gradually over an extended period of time.
CLINICAL PERSPECTIVES.
Employing human 3D in vitro models of CAVD for identifying the underlying mechanisms and potential targets, and for drug screening, may accelerate the discovery and validation of a drug?based therapy for CAVD. With this, patients would be able to receive treatment in an earlier stage of the disease, slowing disease progression or even reversing and curing it.
Furthermore, validation of drug delivery platforms on a human in vitro model could expedite the translation from bench to bedside of local delivery systems.
In addition, the facile production process of the proposed delivery system allows for a broad range of applications and as miRNA therapeutics are identified for different diseases, for different target cells, tissues, or organs, similar drug delivery platforms may be applied in multiple clinical settings to treat a range of diseases.
Acknowledgments
FUNDING
This work was supported by the Netherlands CardioVascular Research Initiative (CVON: The Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Science) (to C.V., A.M. and J.P.G.S.); the Harvard Catalyst Advanced Microscopy Pilot (to C.V. and E.A.); the NIH through a Ruth L. Kirschstein National Research Service Award [grant number F32HL122009 (to M.W.T.); the National Institutes of Health (to R.L.); and the National Institutes of Health [grant numbers R01HL114805, R01HL109506 and R01HL109889 (to E.A.)]. Harvard Catalyst support is provided by The Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health Award [grant number UL1 TR001102] and financial contributions from Harvard University and its affiliated academic healthcare centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic healthcare centers, or the National Institutes of Health.
Abbreviations
- ABCG2
ATP-binding cassette, sub-family G, member 2
- AS
aortic stenosis
- ASO
antisense oligonucleotide
- AV
aortic valve
- AVR
aortic valve replacement
- BAV
bicuspid aortic valve
- BMP-2
bone morphogenetic protein 2
- CAVD
calcific aortic valve disease
- ECM
extracellular matrix
- EndMT
endothelial-to-mesenchymal transition
- ERK1/2
extracellular signal-regulated protein kinases 1 and 2
- DGCR8
DiGeorge syndrome critical region gene 8
- 2′-F
2′-fluoro
- GelMA
methacrylated gelatin
- HAMA
methacrylated hyaluronic acid
- LDL
low-density lipoprotein
- LNA
locked nucleic acid
- LNP
lipid nanoparticle
- LRP5
low-density lipoprotein receptor-related protein 5
- 2′-MOE
2′-O-methoxyethyl
- 2′-OMe
2′-O-methyl
- NF-κB
nuclear factor kappa-B
- OPG
osteoprotegerin
- OTE
off-target effects
- PD/PK
pharmacodynamics/pharmacokinetics
- PEI
polyetheleneimine
- PI3K
phosphoinositide 3-kinase
- PLA
poly(lactic acid)
- PLL
poly-L-lysine
- pre-miRNA
precursor miRNA
- pri-miRNA
primary miRNA transcript
- RANK
receptor activator of nuclear factor κ-B
- RANKL
receptor activator of nuclear factor κ-B ligand
- RISC
RNA-induced silencing complex
- RT-qPCR
real-time quantitative PCR
- Runx2
runt-related transcription factor 2
- α-SMA
α-smooth muscle actin
- SMAD
similar to mothers against decapentaplegic
- TGF-β1
transforming growth factor-β 1
- TNF-α
tumour necrosis factor-α
- UTMD
ultrasound triggered microbubble destruction
- VEC
valvular endothelial cell
- VIC
valve interstitial cell
- ZEN
N,N-diethyl-4-(4-nitronaphthalen-1-ylazo)-phenylamine
Footnotes
AUTHOR CONTRIBUTION
Casper van der Ven and Pin-Jou Wu drafted and wrote the manuscript. Mark Tibbitt, Alain van Mil, Joost Sluijter, and Robert Langer revised the manuscript. Elena Aikawa provided the concept and critically reviewed the manuscript.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després JP, Fullerton HJ, et al. Executive summary: heart disease and stroke statistics—update A report from the American Heart Association. Circulation. 2016;133:447–454. doi: 10.1161/CIR.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 2.Iung B, Vahanian A. Epidemiology of valvular heart disease in the adult. Nat Rev Cardiol. 2011;8:162–172. doi: 10.1038/nrcardio.2010.202. [DOI] [PubMed] [Google Scholar]
- 3.Rajamannan NM, Bonow RO, Rahimtoola SH. Calcific aortic stenosis: an update. Nat Rev Cardiol. 2007;4:254–262. doi: 10.1038/ncpcardio0827. [DOI] [PubMed] [Google Scholar]
- 4.Otto CM. Calcific aortic stenosis — time to look more closely at the valve. N Engl J Med. 2008;359:1395–1398. doi: 10.1056/NEJMe0807001. [DOI] [PubMed] [Google Scholar]
- 5.Hutcheson JD, Aikawa E, Merryman WD. Potential drug targets for calcific aortic valve disease. Nat Rev Cardiol. 2014;11:218–231. doi: 10.1038/nrcardio.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebo-controlled clinical trials on 2344 patients. Can J Cardiol. 2011;27:800–808. doi: 10.1016/j.cjca.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 7.Iung B, Baron G, Butchart EG, Delahaye F, Gohlke-Bärwolf C, Levang OW, Tornos P, Vanoverschelde JL, Vermeer F, Boersma E, et al. A prospective survey of patients with valvular heart disease in Europe: the Euro Heart Survey on valvular heart disease. Eur Heart J. 2003;24:1231–1243. doi: 10.1016/s0195-668x(03)00201-x. [DOI] [PubMed] [Google Scholar]
- 8.Bach DS. Prevalence and characteristics of unoperated patients with severe aortic stenosis. J Heart Valve Dis. 2011;20:284–291. [PubMed] [Google Scholar]
- 9.Takkenberg JJ, Rajamannan NM, Rosenhek R, Kumar AS, Carapetis JR, Yacoub MH, Society for Heart Valve Disease The need for a global perspective on heart valve disease epidemiology. The SHVD working group on epidemiology of heart valve disease founding statement. J Heart Valve Dis. 2008;17:135–139. [PubMed] [Google Scholar]
- 10.Lindman BR, Bonow RO, Otto CM. Current management of calcific aortic stenosis. Circ Res. 2013;113:223–237. doi: 10.1161/CIRCRESAHA.111.300084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berlin DB, Davidson MJ, Schoen FJ. The power of disruptive technological innovation: transcatheter aortic valve implantation. J Biomed Mater Res B: Appl Biomater. 2015;103:1709–1715. doi: 10.1002/jbm.b.33352. [DOI] [PubMed] [Google Scholar]
- 12.Sacks MS, Yoganathan AP. Heart valve function: a biomechanical perspective. Philos Trans R Soc B Biol Sci. 2007;362:1369–1391. doi: 10.1098/rstb.2007.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525–2532. doi: 10.1161/hc4601.099489. [DOI] [PubMed] [Google Scholar]
- 14.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O’Brien KD, et al. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group Executive summary: calcific aortic valve disease–update. Circulation. 2011;124:1783–1791. doi: 10.1161/CIRCULATIONAHA.110.006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Dis. 2004;13:841–847. [PubMed] [Google Scholar]
- 16.Taylor PM, Batten P, Brand NJ, Thomas PS, Yacoub MH. The cardiac valve interstitial cell. Int J Biochem Cell Biol. 2003;35:113–118. doi: 10.1016/s1357-2725(02)00100-0. [DOI] [PubMed] [Google Scholar]
- 17.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–2184. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohler ER, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 19.Wang H, Sridhar B, Leinwand LA, Anseth KS. Characterization of cell subpopulations expressing progenitor cell markers in porcine cardiac valves. PLoS ONE. 2013;8:e69667. doi: 10.1371/journal.pone.0069667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi K, Satoh M, Takahashi Y, Osaki T, Nasu T, Tamada M, Okabayashi H, Nakamura M, Morino Y. Dysregulation of ossification-related miRNAs in circulating osteogenic progenitor cells obtained from patients with aortic stenosis. Clin Sci (Lond Engl 1979) 2016;130:1115–1124. doi: 10.1042/CS20160094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4- and TGF-beta1-dependent pathway. Arterioscler Thromb Vasc Biol. 2009;29:254–260. doi: 10.1161/ATVBAHA.108.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arjunon S, Rathan S, Jo H, Yoganathan AP. Aortic valve: mechanical environment and mechanobiology. Ann Biomed Eng. 2013;41:1331–1346. doi: 10.1007/s10439-013-0785-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferdous Z, Jo H, Nerem RM. Strain magnitude-dependent calcific marker expression in valvular and vascular cells. Cells Tissues Organs. 2013;197:372–383. doi: 10.1159/000347007. [DOI] [PubMed] [Google Scholar]
- 24.Schoen FJ. Evolving concepts of cardiac valve dynamics. Circulation. 2008;118:1864–1880. doi: 10.1161/CIRCULATIONAHA.108.805911. [DOI] [PubMed] [Google Scholar]
- 25.Miller JD, Weiss RM, Serrano KM, Castaneda LE, Brooks RM, Zimmerman K, Heistad DD. Evidence for active regulation of pro-osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol. 2010;30:2482–2486. doi: 10.1161/ATVBAHA.110.211029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2011;108:1381–1391. doi: 10.1161/CIRCRESAHA.110.234146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gould ST, Matherly EE, Smith JN, Heistad DD, Anseth KS. The role of valvular endothelial cell paracrine signaling and matrix elasticity on valvular interstitial cell activation. Biomaterials. 2014;35:3596–3606. doi: 10.1016/j.biomaterials.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hjortnaes J, Shapero K, Goettsch C, Hutcheson JD, Keegan J, Kluin J, Mayer JE, Bischoff J, Aikawa E. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis. 2015;242:251–260. doi: 10.1016/j.atherosclerosis.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jian B, Narula N, Li Q, Mohler ER, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465. doi: 10.1016/s0003-4975(02)04312-6. [DOI] [PubMed] [Google Scholar]
- 30.Kaden JJ, Dempfle CE, Grobholz R, Fischer CS, Vocke DC, Kiliç R, Sarıkoç A, Piñol R, Hagl S, Lang S, et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc Pathol. 2005;14:80–87. doi: 10.1016/j.carpath.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Leinwand LA, Anseth KS. Roles of transforming growth factor-β1 and OB-cadherin in porcine cardiac valve myofibroblast differentiation. FASEB J. 2014;28:4551–4562. doi: 10.1096/fj.14-254623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X, Meng X, Su X, Mauchley DC, Ao L, Cleveland JC, Jr, Fullerton DA. Bone morphogenic protein 2 induces Runx2 and osteopontin expression in human aortic valve interstitial cells: role of Smad1 and extracellular signal-regulated kinase 1/2. J Thorac Cardiovasc Surg. 2009;138:1008–1015. doi: 10.1016/j.jtcvs.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 33.Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. 2009;47:828–834. doi: 10.1016/j.yjmcc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rajamannan NM, Subramaniam M, Caira F, Stock SR, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation. 2005;112(Suppl 9):I-229–I-234. doi: 10.1161/01.CIRCULATIONAHA.104.524306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang H, Tibbitt MW, Langer SJ, Leinwand LA, Anseth KS. Hydrogels preserve native phenotypes of valvular fibroblasts through an elasticity-regulated PI3K/AKT pathway. Proc Natl Acad Sci USA. 2013;110:19336–19341. doi: 10.1073/pnas.1306369110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kloxin AM, Benton JA, Anseth KS. In situ elasticity modulation with dynamic substrates to direct cell phenotype. Biomaterials. 2010;31:1–8. doi: 10.1016/j.biomaterials.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riem Vis PW, Kluin J, Sluijter JP, van Herwerden LA, Bouten CV. Environmental regulation of valvulogenesis: implications for tissue engineering. Eur J Cardiothorac Surg. 2011;39:8–17. doi: 10.1016/j.ejcts.2010.05.032. [DOI] [PubMed] [Google Scholar]
- 38.Mabry KM, Lawrence RL, Anseth KS. Dynamic stiffening of poly(ethylene glycol)-based hydrogels to direct valvular interstitial cell phenotype in a three-dimensional environment. Biomaterials. 2015;49:47–56. doi: 10.1016/j.biomaterials.2015.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puperi DS, O’Connell RW, Punske ZE, Wu Y, West JL, Grande-Allen KJ. Hyaluronan hydrogels for a biomimetic spongiosa layer of tissue engineered heart valve scaffolds. Biomacromolecules. 2016;17:1766–1775. doi: 10.1021/acs.biomac.6b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puperi DS, Balaoing LR, O’Connell RW, West JL, Grande-Allen KJ. 3-Dimensional spatially organized PEG-based hydrogels for an aortic valve co-culture model. Biomaterials. 2015;67:354–364. doi: 10.1016/j.biomaterials.2015.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-β implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004;95:253–260. doi: 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- 42.Hjortnaes J, Goettsch C, Hutcheson JD, Camci-Unal G, Lax L, Scherer K, Body S, Schoen FJ, Kluin J, Khademhosseini A, et al. Simulation of early calcific aortic valve disease in a 3D platform: a role for myofibroblast differentiation. J Mol Cell Cardiol. 2016;94:13–20. doi: 10.1016/j.yjmcc.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, Yabusaki K, Faits T, Bouten C, Franck G, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. 2016;15:335–343. doi: 10.1038/nmat4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 45.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 46.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 47.Gregory RI, Yan K, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. The microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 48.Lund E, Güttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 49.Hutvágner G, McLachlan J, Pasquinelli AE, Bálint É, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 50.Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 51.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 52.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meijer HA, Smith EM, Bushell M. Regulation of miRNA strand selection: follow the leader? Biochem Soc Trans. 2014;42:1135–1140. doi: 10.1042/BST20140142. [DOI] [PubMed] [Google Scholar]
- 55.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34(Suppl 1):D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goettsch C, Hutcheson JD, Aikawa E. MicroRNA in cardiovascular calcification: focus on targets and extracellular vesicle delivery mechanisms. Circ Res. 2013;112:1073–1084. doi: 10.1161/CIRCRESAHA.113.300937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oerlemans MI, Mosterd A, Dekker MS, de Vrey EA, van Mil A, Pasterkamp G, Doevendans PA, Hoes AW, Sluijter JPG. Early assessment of acute coronary syndromes in the emergency department: the potential diagnostic value of circulating microRNAs. EMBO Mol Med. 2012;4:1176–1185. doi: 10.1002/emmm.201201749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deddens JC, Colijn JM, Oerlemans MI, Pasterkamp G, Chamuleau SA, Doevendans PA, Sluijter JPG. Circulating microRNAs as novel biomarkers for the early diagnosis of acute coronary syndrome. J Cardiovasc Transl Res. 2013;6:884–898. doi: 10.1007/s12265-013-9493-9. [DOI] [PubMed] [Google Scholar]
- 60.Romaine SP, Tomaszewski M, Condorelli G, Samani NJ. MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart. 2015;101:921–928. doi: 10.1136/heartjnl-2013-305402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sluijter JPG. MicroRNAs in cardiovascular regenerative medicine: directing tissue repair and cellular differentiation. Int Sch Res Not. 2013;2013:e593517. [Google Scholar]
- 62.Villar AV, Garćıa R, Merino D, Llano M, Cobo M, Montalvo C, Martin-Durán R, Hurlé MA, Nistal JF. Myocardial and circulating levels of microRNA-21 reflect left ventricular fibrosis in aortic stenosis patients. Int J Cardiol. 2013;167:2875–2881. doi: 10.1016/j.ijcard.2012.07.021. [DOI] [PubMed] [Google Scholar]
- 63.Ohukainen P, Syväranta S, Näpänkangas J, Rajamäki K, Taskinen P, Peltonen T, Helske-Suihko S, Kovanen PT, Ruskoaho H, Rysä J. MicroRNA-125b and chemokine CCL4 expression are associated with calcific aortic valve disease. Ann Med. 2015;47:423–429. doi: 10.3109/07853890.2015.1059955. [DOI] [PubMed] [Google Scholar]
- 64.Patel V, Carrion K, Hollands A, Hinton A, Gallegos T, Dyo J, Sasik R, Leire E, Hardiman G, Mohamed SA, et al. The stretch responsive microRNA miR-148a-3p is a novel repressor of IKBKB, NF-B signaling, and inflammatory gene expression in human aortic valve cells. FASEB J. 2015;29:1859–1868. doi: 10.1096/fj.14-257808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Chen S, Deng C, Li F, Wang Y, Hu X, Shi F, Dong N. MicroRNA-204 targets Runx2 to attenuate BMP-2-induced osteoblast differentiation of human aortic valve interstitial cells. J Cardiovasc Pharmacol. 2015;66:63–71. doi: 10.1097/FJC.0000000000000244. [DOI] [PubMed] [Google Scholar]
- 66.Yanagawa B, Lovren F, Pan Y, Garg V, Quan A, Tang G, Singh KK, Shukla PC, Kalra NP, Peterson MD, et al. miRNA-141 is a novel regulator of BMP-2–mediated calcification in aortic stenosis. J Thorac Cardiovasc Surg. 2012;144:256–262. doi: 10.1016/j.jtcvs.2011.10.097. [DOI] [PubMed] [Google Scholar]
- 67.Zhang M, Liu X, Zhang X, Song Z, Han L, He Y, Xu Z. MicroRNA-30b is a multifunctional regulator of aortic valve interstitial cells. J Thorac Cardiovasc Surg. 2014;147:1073–1080. doi: 10.1016/j.jtcvs.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 68.Rathan S, Ankeny CJ, Arjunon S, Ferdous Z, Kumar S, Esmerats JF, Heath JM, Nerem RM, Yoganathan AP, Jo H. Identification of side- and shear-dependent microRNAs regulating porcine aortic valve pathogenesis. Sci Rep. 2016;6 doi: 10.1038/srep25397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nigam V, Sievers HH, Jensen BC, Sier HA, Simpson PC, Srivastava D, Mohamed SA. Altered micrornas in bicuspid aortic valve: a comparison between stenotic and insufficient valves. J Heart Valve Dis. 2010;19:459–465. [PMC free article] [PubMed] [Google Scholar]
- 70.Holliday CJ, Ankeny RF, Jo H, Nerem RM. Discovery of shear- and side-specific mRNAs and miRNAs in human aortic valvular endothelial cells. Am J Physiol Heart Circ Physiol. 2011;301:H856–H867. doi: 10.1152/ajpheart.00117.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Z, Hassan MQ, Volinia S, van Wijnen AJ, Stein JL, Croce CM, Lian JB, Stein GS. A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc Natl Acad Sci USA. 2008;105:13906–13911. doi: 10.1073/pnas.0804438105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E. Macrophage-Derived Matrix Vesicles: An Alternative Novel Mechanism for Microcalcification in Atherosclerotic Plaques. Circ Res. 2013;113:72–77. doi: 10.1161/CIRCRESAHA.113.301036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.New SE, Aikawa E. Role of extracellular vesicles in de novo mineralization: an additional novel mechanism of cardiovascular calcification. Arterioscler Thromb Vasc Biol. 2013;33:1753–1758. doi: 10.1161/ATVBAHA.112.300128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krohn JB, Hutcheson JD, Martínez-Martínez E, Aikawa E. Extracellular vesicles in cardiovascular calcification: expanding current paradigms. J Physiol. 2016;594:2895–2903. doi: 10.1113/JP271338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, Nykjaer A, Kjolby M, Rogers M, Michel T, Shibasaki M, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest. 2016;126:1323–1336. doi: 10.1172/JCI80851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RTM, Alvarez-Hernandez D, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. 2015;116:1312–1323. doi: 10.1161/CIRCRESAHA.116.305012. [DOI] [PubMed] [Google Scholar]
- 77.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mittelbrunn M, Gutiérrez-Vázquez C, Villarroya-Beltri C, González S, Sánchez-Cabo F, González MÁ, Bernad A, Sanchez-Madrid F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. 2011;2:282. doi: 10.1038/ncomms1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012;119:756–766. doi: 10.1182/blood-2011-02-338004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li J, Zhang Y, Liu Y, Dai X, Li W, Cai X, Baty CJ, Gibson GA, Erdos G, Wang Z, et al. Microvesicle-mediated transfer of microRNA-150 from monocytes to endothelial cells promotes angiogenesis. J Biol Chem. 2013;288:23586–23596. doi: 10.1074/jbc.M113.489302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lennox KA, Behlke MA. A direct comparison of anti-microRNA oligonucleotide potency. Pharm Res. 2010;27:1788–1799. doi: 10.1007/s11095-010-0156-0. [DOI] [PubMed] [Google Scholar]
- 83.Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with “antagomirs. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 84.Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2009;5:381–391. doi: 10.1517/17425250902877680. [DOI] [PubMed] [Google Scholar]
- 85.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 86.Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, Teruya-Feldstein J, Bell GW, Weinberg RA. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28:341–347. doi: 10.1038/nbt.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Manoharan M. 2′-carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochim Biophys Acta BBA - Gene Struct Expr. 1999;1489:117–130. doi: 10.1016/s0167-4781(99)00138-4. [DOI] [PubMed] [Google Scholar]
- 88.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 89.Davis S, Lollo B, Freier S, Esau C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 2006;34:2294–2304. doi: 10.1093/nar/gkl183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Singh SK, Koshkin AA, Wengel J, Nielsen P. LNA (locked nucleic acids): synthesis and high-affinity nucleic acid recognition. Chem Commun. 1998;4:455–456. [Google Scholar]
- 91.Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. 2011;43:371–378. doi: 10.1038/ng.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lennox KA, Owczarzy R, Thomas DM, Walder JA, Behlke MA. Improved performance of anti-miRNA oligonucleotides using a novel non-nucleotide modifier. Mol Ther Nucleic Acids. 2013;2:e117. doi: 10.1038/mtna.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Larson SD, Jackson LN, Chen LA, Rychahou PG, Evers BM. Effectiveness of siRNA uptake in target tissues by various delivery methods. Surgery. 2007;142:262–269. doi: 10.1016/j.surg.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 97.Kanasty RL, Whitehead KA, Vegas AJ, Anderson DG. Action and reaction: the biological response to siRNA and its delivery vehicles. Mol Ther. 2012;20:513–524. doi: 10.1038/mt.2011.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schroeder A, Levins CG, Cortez C, Langer R, Anderson DG. Lipid-based nanotherapeutics for siRNA delivery. J Intern Med. 2010;267:9–21. doi: 10.1111/j.1365-2796.2009.02189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 100.van Mil A, Doevendans PA, Sluijter JP. The potential of modulating small RNA activity in vivo. Mini Rev Med Chem. 2009;9:235–248. doi: 10.2174/138955709787316029. [DOI] [PubMed] [Google Scholar]
- 101.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 102.Malam Y, Loizidou M, Seifalian AM. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009;30:592–599. doi: 10.1016/j.tips.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 103.Zhao Z, Zhuang S, Qi XR. Comparative study of the in vitro and in vivo characteristics of cationic and neutral liposomes. Int J Nanomedicine. 2011;6:3087–3098. doi: 10.2147/IJN.S25399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Castaing M, Loiseau A, Mulliert G. Multidrug resistance modulator interactions with neutral and anionic liposomes: membrane binding affinity and membrane perturbing activity. J Pharm Pharmacol. 2005;57:547–554. doi: 10.1211/0022357055911. [DOI] [PubMed] [Google Scholar]
- 105.Jesorka A, Orwar O. Liposomes: technologies and analytical applications. Annu Rev Anal Chem (Palo Alto Calif) 2008;1:801–832. doi: 10.1146/annurev.anchem.1.031207.112747. [DOI] [PubMed] [Google Scholar]
- 106.Jubeli E, Raju L, Khalique NA, Bilchuk N, Zegel C, Chen A, Lou HH, Øpstad CL, Zeeshan M, Sliwka HR, et al. Polyene-based cationic lipids as visually traceable siRNA transfer reagents. Eur J Pharm Biopharm. 2015;89:280–289. doi: 10.1016/j.ejpb.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 107.Dar GH, Gopal V, Rao NM. Systemic delivery of stable siRNA-encapsulating lipid vesicles: optimization, biodistribution, and tumor suppression. Mol Pharm. 2015;12:610–620. doi: 10.1021/mp500677x. [DOI] [PubMed] [Google Scholar]
- 107.Bailey AL, Cullis PR. Liposome fusion. In: Epand R, editor. Current topics in Membranes. Academic Press; San Diego: 1997. pp. 359–373. [Google Scholar]
- 109.Bailey AL, Cullis PR. Modulation of membrane fusion by asymmetric transbilayer distributions of amino lipids. Biochemistry (Mosc) 1994;33:12573–12580. doi: 10.1021/bi00208a007. [DOI] [PubMed] [Google Scholar]
- 110.Milla P, Dosio F, Cattel L. PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr Drug Metab. 2012;13:105–119. doi: 10.2174/138920012798356934. [DOI] [PubMed] [Google Scholar]
- 111.Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 112.Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol. 2007;25:1149–1157. doi: 10.1038/nbt1339. [DOI] [PubMed] [Google Scholar]
- 113.Jeong JH, Mok H, Oh YK, Park TG. siRNA conjugate delivery systems. Bioconjugate Chem. 2009;20:5–14. doi: 10.1021/bc800278e. [DOI] [PubMed] [Google Scholar]
- 114.Rozema DB, Ekena K, Lewis DL, Loomis AG, Wolff JA. Endosomolysis by masking of a membrane-active agent (EMMA) for cytoplasmic release of macromolecules. Bioconjugate Chem. 2003;14:51–57. doi: 10.1021/bc0255945. [DOI] [PubMed] [Google Scholar]
- 115.Biessen EA, Beuting DM, Roelen HC, van de Marel GA, van Boom JH, van Berkel TJ. Synthesis of cluster galactosides with high affinity for the hepatic asialoglycoprotein receptor. J Med Chem. 1995;38:1538–1546. doi: 10.1021/jm00009a014. [DOI] [PubMed] [Google Scholar]
- 116.Rensen PC, van Leeuwen SH, Sliedregt LA, van Berkel TJ, Biessen EA. Design and synthesis of novel N-acetylgalactosamine-terminated glycolipids for targeting of lipoproteins to the hepatic asialoglycoprotein receptor. J Med Chem. 2004;47:5798–5808. doi: 10.1021/jm049481d. [DOI] [PubMed] [Google Scholar]
- 117.Kallanthottathil R. Conjugation strategies for in vivo siRNA delivery. 2012 http://www.alnylam.com/web/wp-content/uploads/2012/11/ALNY-OTS-Conjugate-Oct2012.pdf.
- 118.Kwekkeboom RF, Sluijter JP, van Middelaar BJ, Metz CH, Brans MA, Kamp O, Paulus WJ, Musters RJP. Increased local delivery of antagomir therapeutics to the rodent myocardium using ultrasound and microbubbles. J Control Release. 2016;222:18–31. doi: 10.1016/j.jconrel.2015.11.020. [DOI] [PubMed] [Google Scholar]
- 119.Dong Y, Love KT, Dorkin JR, Sirirungruang S, Zhang Y, Chen D, Bogorad RL, Yin H, Chen Y, Vegas AJ, et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc Natl Acad Sci USA. 2014;111:3955–3960. doi: 10.1073/pnas.1322937111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR, Qin J, Cantley W, Qin LL, Racie T, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci USA. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yu B, Zhao X, Lee LJ, Lee RJ. Targeted delivery systems for oligonucleotide therapeutics. AAPS J. 2009;11:195–203. doi: 10.1208/s12248-009-9096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 123.Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 124.Conde J, Oliva N, Zhang Y, Artzi N. Local triple-combination therapy results in tumour regression and prevents recurrence in a colon cancer model. Nat Mater. 2016;15:1128–1138. doi: 10.1038/nmat4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gilam A, Conde J, Weissglas-Volkov D, Oliva N, Friedman E, Artzi N, Shomron N. Local microRNA delivery targets palladin and prevents metastatic breast cancer. Nat Commun. 2016;7 doi: 10.1038/ncomms12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ragelle H, Danhier F, Préat V, Langer R, Anderson DG. Nanoparticle-based drug delivery systems: a commercial and regulatory outlook as the field matures. Expert Opin Drug Deliv. 2016 doi: 10.1080/17425247.2016.1244187. in the press. [DOI] [PubMed] [Google Scholar]
- 127.Gindy ME, Leone AM, Cunningham JJ. Challenges in the pharmaceutical development of lipid-based short interfering ribonucleic acid therapeutics. Expert Opin Drug Deliv. 2012;9:171–182. doi: 10.1517/17425247.2012.642363. [DOI] [PubMed] [Google Scholar]
- 128.Aikawa E, Otto CM. Look more closely at the valve imaging calcific aortic valve disease. Circulation. 2012;125:9–11. doi: 10.1161/CIRCULATIONAHA.111.073452. [DOI] [PubMed] [Google Scholar]