

Abstract
This study aimed to investigate the relationship between clinical features and prognosis of adult secondary hemophagocytic syndrome (HPS).
A retrospective analysis was conducted on the pathogenesis, clinical manifestations, laboratory examinations, treatment options, and prognosis of 47 patients with adult secondary HPS diagnosed from January 2013 to December 2015.
The average age at disease onset was (46.26 ± 18.98) years with a male:female ratio of 1:1.14. Thirteen patients died, with the highest mortality rate in patients with HPS underlying blood system malignancy (33.33%, 2/6). The mortality rate in patients with HPS underlying autoimmune disorders was the lowest (18.75%, 3/16). The Kaplan–Meier analysis indicated that signs of hemorrhage, pulmonary and nervous system involvement, serous effusion, and decrease in the blood platelet count were associated with death. The Cox regression analysis revealed that signs of hemorrhage, pulmonary involvement, serous effusion, and nervous system involvement were independent risk factors of patient death.
Adult secondary HPS has multiple etiologies and diversified clinical features. The risk of death increases in patients with signs of hemorrhage, serous effusion, pulmonary involvement, and nervous system involvement.
Keywords: clinical feature, hemophagocytic syndrome, hemorrhage, prognostic factor, serous effusion
1. Introduction
Hemophagocytic syndrome (HPS) is a clinical syndrome caused by excessive proliferation of activated lymphocytes and histiocytes, and was first reported by Risdall et al in 1979.[1] Also, immunity is ineffective, further inducing excessive inflammation of multiple organs. The main clinical features of HPS include fever, hepatosplenomegaly, liver function damage, blood cell reduction, and hemophagocytic phenomenon of tissue cells. Moreover, it is characterized by acute onset, rapid disease progression, and high mortality. Thus, early diagnosis of patients with severe HPS has great significance in improving adult HPS prognosis. In this study, clinical data of 47 patients with adult HPS were retrospectively analyzed to investigate the relationship between HPS clinical features and prognosis.
2. Methods
2.1. Ethics statements
Given the retrospective nature of the study, written consent was not obtained. However, we got the oral consent from all participants in the study by telephone contact, and patient records were anonymized and deidentified prior to analysis. Then, related data were extracted from hospital's electronic medical records. The study was reviewed and obtained the approval from Institutional Review Board of Beijing Chaoyang Hospital, Capital Medical University.
2.2. Clinical data
Data of patients with adult secondary HPS, who were clinically diagnosed and hospitalized in Beijing Chaoyang Hospital affiliated to Capital Medical University from January 2013 to December 2015, were retrospectively collected. The patients included in this study complied with HPS-2004 standards, namely, clear diagnosis of 5 of the following 8 indicators: fever: continued for more than 7 days, body temperature higher than 38.5 °C; splenomegaly (below the costal margin ≥3 cm); a decrease in blood cells (involving 2 or more lines of peripheral blood) caused by a decrease in nonbone marrow hematopoietic function; hypertriglyceridemia and/or hypofibrinogenemia; hemophagocytic cells found in bone marrow, spleen, or lymph nodes by pathology; reduction or absence of natural killer (NK) cell activity; ferritin ≥500 μg/L; and an increase in soluble interleukin (IL-2R) (sCD25).[2]
2.3. Main observed indicators
The main observed indicators were as follows: clinical features: fever, skin rash, arthritis, and clinical manifestations of blood system, respiratory system, and other systems; laboratory examinations: blood routine, liver and kidney functions, coagulation, serum ferritin, bone marrow cytology, biopsy, T cell subset analysis, NK cell activity assay, sCD25 levels, immunology, and etiology-related test results; imaging examinations: imaging of chest, abdomen, and so forth; and treatment options: glucocorticoids as a monotherapy, glucocorticoids ± etoposide ± cyclosporine A, and other combinations of chemotherapy.
2.4. Statistical methods
SPSS 19.0 statistical software (SPSS, IL) was used for data analysis. Categorical variables were expressed as frequency. Intergroup differences were assessed by the chi-square test or Fisher exact test. Continuous variables were expressed as (x ± s). Analysis of variance was used to test the intergroup homogeneity of variance. The least significant difference was used for posttest comparison of indicators with overall differences between groups; while pairwise comparison was conducted using the Kruskal–Wallis test for heterogeneity of variance. The difference had statistical significance if the P value was less than 0.05. Finally, the Kaplan–Meier method was used to compare univariate survival curves to screen risk factors for prognosis and risk factors for patients with secondary HPS. The Cox regression analysis was used for multivariate survival analysis.
3. Results
3.1. Etiology analysis on the 47 patients with secondary HPS
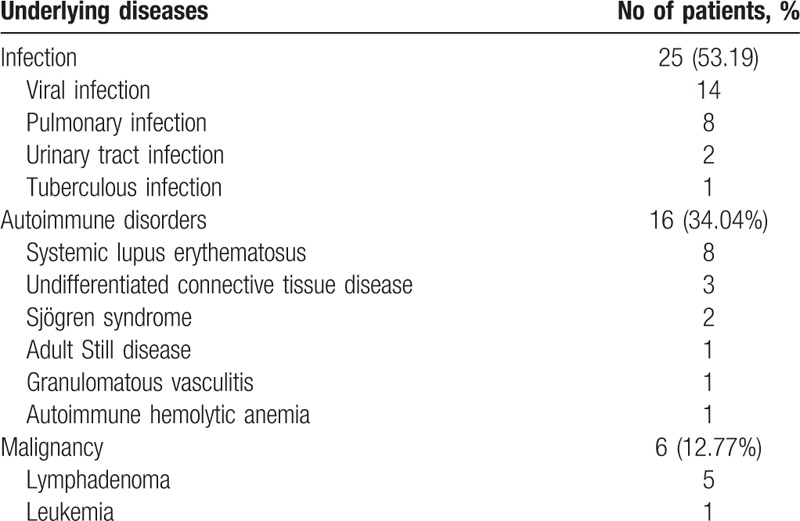
Data of 47 patients were collected in total, with 25 (53.19%) cases of HPS underlying infections, 6 (24%) patients had underlying clear Epstein–Barr (EB) viral infection, and 16 (34.04%) patients had HPS underlying autoimmune disorders. Moreover, 8 (50%) patients had systemic lupus erythematosus, and 6 (12.77%) had blood system HPS underlying malignancy. Detailed etiological classification is shown in Table 1.
Table 1.
Distribution of underlying diseases in 47 patients with hemophagocytic syndrome (HPS).
3.2. Demographic data and clinical laboratory features and prognosis
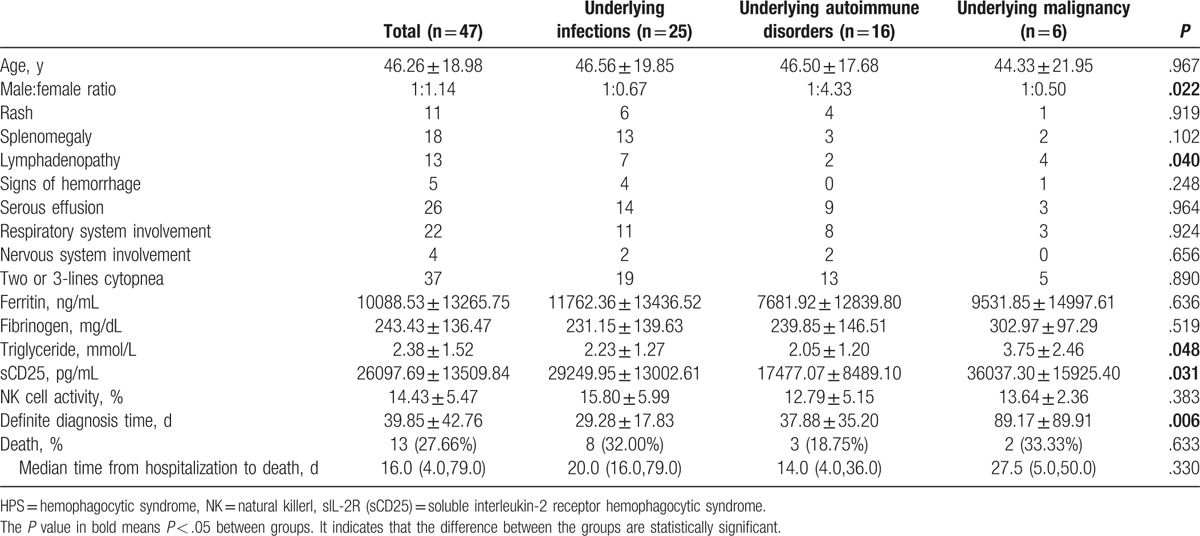
Average age at disease onset for the 47 patients was (46.26 ± 18.98) years. No statistical difference was found in ages between groups, and gender (male:female) ratio was 1:1.14. Differences in gender ratio between groups had statistical significance (P = .022), and more female patients had HPS underlying autoimmune disorders. The average diagnosis time for the 47 patients was (39.85 ± 42.76) days, while the diagnosis time was shorter for HPS underlying infection (29.28 ± 17.83 days), and longest for HPS underlying blood system malignancy (89.17 ± 89.91 days). Intergroup differences had statistical significance (P = .006) (Table 2).
Table 2.
Demographics data, clinical manifestations, and laboratory data at the onset of HPS with different causes.
Clinical features and laboratory examination results of the 47 patients included in this study were diversified: 100% patients had fever with body temperature peak: 38.5 to 41 °C, 38.30% (18/47) had splenomegaly as verified by imaging, and 25.53% (12/47) had palpable superficial lymphadenopathy. Other common clinical features included serous effusion (26/47), pulmonary involvement (24/47), signs of hemorrhage (5/47), nervous system involvement (4/47), and so forth (Table 2).
During initial treatment, all patients showed peripheral blood abnormality. Moreover, 80.86% patients had 2- or 3-line cytopenia. The percentage of 3-line cytopenia was relatively high (50%) for patients with HPS associated with autoimmune disorders. At the peak of the disease, white blood cell count (5.02 ± 5.99) × 109/L, hemoglobin (93.60 ± 24.71) g/L, and platelet count (112.34 ± 109.77) × 109/L all obviously decreased. Serum ferritin increased to (10,088.53 ± 13,265.75) ng/mL for 97.87% of the patients; 27.27% patients showed hyperlipidemia, and 35.56% showed hypofibrinogenemia. The bone marrow cytological examination was conducted on all patients, and 36 cases (76.60%) of hemophagocytic phenomenon were found. sCD25 detection was performed on 25 patients, and its level was (26,097.69 ± 13,509.84) pg/mL. Patients with the increased sCD25 level accounted for 96% of the cases. NK cell activity was detected in 29 patients; the level was (14.43 ± 5.47)%, and reduction was found in 65.52% patients. The most commonly discovered result from the laboratory examination was hypoproteinemia (<30 g/L, 97.0%), followed by high lactate dehydrogenase acidosis (>220 U/L, 91.30%) and transaminase elevation (78.70%). The increase in the aspartate aminotransferase level was more significant compared with the alanine aminotransferase level. The average value was (80.17 ± 76.75) U/L for alanine aminotransferase and (154.86 ± 203.87) U/L for aspartate aminotransferase. The incidence of renal damage was 21.23%.
The statistical analysis of various assay indicators for patients with HPS underlying different etiologies indicated differences in blood triglyceride levels (F = 3.180, P = .048). The blood glyceride levels in patients with HPS underlying malignancy were significantly higher than those in patients with HPS underlying infections (P = .021) and HPS underlying autoimmune disorders (P = .027). Additionally, sCD25 levels between groups were also different (F = 4.071, P = .031). The sCD25 levels in patients with HPS underlying autoimmune disorders were significantly lower than those in the other 2 groups (P = .037 and .018, respectively). No significant difference was found in other assay indicators.
Furthermore, 13 cases of death were reported among 47 patients with HPS. The mortality rate was the highest for patients with HPS underlying blood system malignancy (33.33%, 2/6), and the lowest for patients with HPS underlying autoimmune disorders (18.75%, 3/16). The mortality rate was 32.00% (8/25) for patients with HPS underlying infection. The median time from hospitalization to death for the dead patients was 16.0 (4.0, 79.0) days, without any statistically significant difference between groups.
3.3. Treatment options of patients with secondary HPS
Out of the 47 cases with secondary HPS enrolled in the present study, 37 received treatment in the center during the whole process. Of the 8 who were given glucocorticoid monotherapy, 3 were given glucocorticoid pulse therapy (0.5–1.0 g/day) and the others were treated with large doses of glucocorticoids (≥1 mg/[kg·d]). A total of 24 cases received etoposide-combined glucocorticoid treatment, and glucocorticoid was given at high doses to all patients. Another 2 patients received high-dose glucocorticoid treatment combined with cyclosporine A, and 3 were given glucocorticoid treatment in combination with both etoposide and cyclosporine A.
On the basis of antiinfective treatment, most patients with HPS underlying infection received standard etoposide-combined glucocorticoid treatment. However, 3 cases did not receive this treatment because of viral infection, and they showed granulocytopenia or agranulocytosis during the early phase of the disease. All patients with HPS underlying autoimmune disorders were given above-medium doses of glucocorticoids: 6 received pulse therapy, 7 etoposide-combined treatment, 5 cyclosporine A-combined treatment, and another 4 treatment combined with other immunosuppressive agents. As for patients with HPS underlying malignancy, 2 turned to chemotherapy after being clearly diagnosed with non-Hodgkin lymphoma.
Patients with different treatment plans were divided into different groups based on whether they received etoposide-combined treatment or whether they were given large-dose glucocorticoids. The statistical analysis showed no significant difference in the death rate of different groups (P = .760 and .867).
3.4. Prognosis and death predictors of patients with secondary HPS
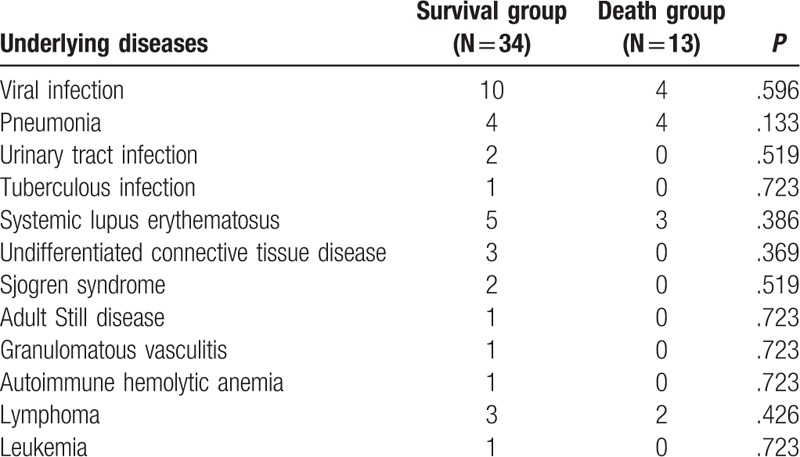
The 47 patients included in this study were divided into death group and survival group based on whether the patient survived (34 survived patients and 13 dead patients) to analyze the potential death predictors for HPS. First, the etiology of secondary HPS was compared between the death and survival groups to analyze whether the death of patient was associated with the etiology of HPS, and no significant difference was identified (Table 3).
Table 3.
The comparison of underlying diseases between hemophagocytic syndrome (HPS) patients in death and survival groups.
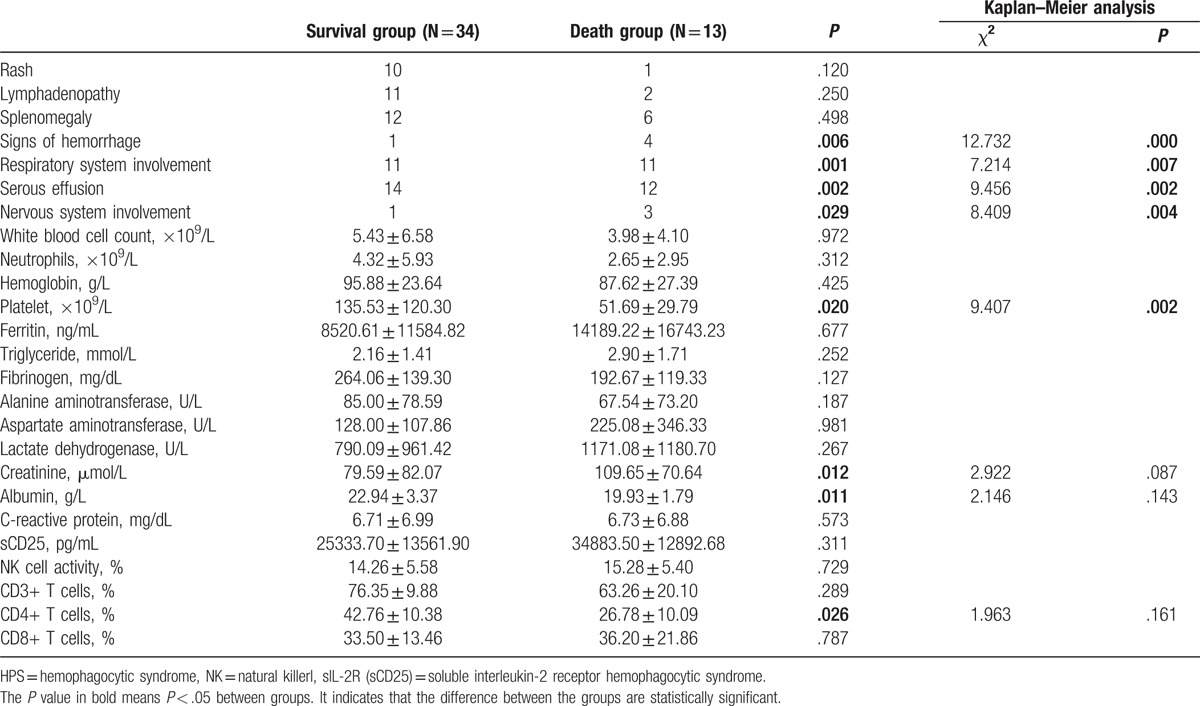
The statistical analysis of all the data about clinical features indicated significant differences in serous effusion (P = .002), signs of hemorrhage (P = .006), pulmonary involvement (P = .001), nervous system involvement (P = .029), decrease in blood platelet count (P = .02), serum creatinine elevation (P = .012), significant reduction in albumin (albumin <20 g/L) (P = .011), and decrease in the percentage of CD3 + CD4 + T cells (P = .026) between the death and survival groups. All patients with nervous system involvement (3/4) and most patients with signs of hemorrhage (3/5) were found to have a poor prognosis, but the number of cases was relatively small. However, no statistically significant difference was found between groups in white blood cell count and hemoglobin, serum ferritin level, transaminase level, hypertriglyceridemia, hypofibrinogenemia, and so forth (Table 4).
Table 4.
comparison of clinical data and predictors of death between patients with HPS in the death and survival groups.
The results from the univariate analysis by the Kaplan–Meier method indicated that patients showed signs of hemorrhage, pulmonary involvement, serous effusion, nervous system involvement, and decrease in the blood platelet count (Plt < 100 × 109/L), which were all associated with death (all P < .05).
Variables were further induced in the Cox model for multivariate analysis. The results showed that signs of hemorrhage (relative risk [RR] = 0.239, 95% confidence interval [CI] 0.060–0.957, P = .043), pulmonary involvement (RR = 0.082, 95% CI 0.010–0.682, P = .021), serous effusion (RR = 0.084, 95% CI 0.010–0.719, P = .024), and nervous system involvement (RR = 0.208, 95% CI 0.045–0.965, P = .045) in patients were independent risk factors for patient death.
4. Discussion
HPS belongs to monocyte–macrophage system responsive diseases and has the characteristics of abnormal macrophage proliferation and phagocytic blood cells from the aspect of histopathology. The disease was first reported in children in 1952.[3] Risdall et al[1] first reported adult HPS in 1979. At present, HPS is thought to be a cytokine storm and multisystem inflammation that is life-threatening. It is induced by tumor necrosis factor alpha, interferon gamma, interleukin-1b, interleukin-6, sIL-2, and other inflammatory factors produced by continuously proliferating tissue cells and T lymphocytes. NK cells and cytotoxic T cell-mediated vesicle release cause cytotoxic effects; the immune system overreacts to an antigen reaction but is ineffective.[4,5]
Based on different etiologies, HPS is usually classified into 2 major categories: primary and secondary. Secondary HPS is usually further classified into 3 categories: HPS underlying infection, autoimmune disorders, and malignancy. Among the 47 patients with secondary HPS, patients with HPS caused by infectious diseases were the maximum in number, followed by patients with autoimmune disorders. Among the patients with HPS underlying infection, the most common etiology was EB viral infection. In the meanwhile, HPS underlying bacteria, fungi, and other viruses such as influenza A virus were also included in the study. A study on the Asian population showed that high levels of proinflammatory factors could be detected in patients with EB virus infection,[6] and these factors were inseparable from HPS onset. Previous studies indicated that EB virus-associated HPS onset was common in Asian countries, and its percentage was 28.74% in Japan, as reported by Ishii et al.[7] This study showed that patients with EB virus HPS underlying infections accounted for 24% of all the patients, which was close to the data reported in previous studies. In previous reports on adult HPS underlying autoimmune disorders, the main cause of disease was systemic lupus erythematosus, adult Still disease, rheumatoid arthritis, Sjögren syndrome, systemic sclerosis, and so forth.[8] The most common cause in the present study was systemic lupus erythematosus; other causes included antineutrophil cytoplasmic antibody-associated vasculitis, adult Still disease, Sjögren syndrome, and so forth, consistent with previous studies. The number of patients with HPS associated with malignancy was relatively small, with NK/T cell lymphoma as the main cause.
Clinical manifestations of adult secondary HPS were diversified and more atypical compared with those in children,[9] which might easily lead to missed diagnosis and misdiagnosis. For patients with fever as the main symptom, the diagnosis time was relatively long due to the complexity of disease itself no matter what the cause was. In particular, the difficulty in diagnosis increased because blood system malignancy and autoimmune disorders had complex disease manifestations, most of which were hard to be distinguished from HPS. From the aspect of clinical manifestations, all patients had a fever that could involve the whole body system; no obvious characteristic was reported. As for laboratory examinations, the serum ferritin level in almost all patients with adult secondary HPS increased, but no significant correlation was found with causes of the disease. The sensitivity of hypofibrinogenemia and hypertriglyceridemia, which were also included in diagnostic standards, was relatively low. However, differences were observed in laboratory examinations of patients with different causes of diseases. sCD25 in serum was one of the indicators of T lymphocyte activation, and an important immunosuppressive factor that could compete with IL-2R on the activated cell surface to bind and decrease the activity of IL-2, the level of which was positively correlated with the severity of inflammation in vivo.[10] The serum sCD25 level increased by different degrees not only for patients with HPS, but also for patients with infection, malignancy, severe trauma, rejection after organ transplantation, autoimmune disorders, and so forth.[11] Comparison of sCD25 levels for patients with secondary HPS due to different causes disclosed that sCD25 levels in patients with HPS underlying infection and blood system malignancy were significantly higher than those in patients with HPS underlying autoimmune disorders. The reason might be that most patients with autoimmune disorders included in this study were not the ones diagnosed for the first time. They were treated with corticosteroids and immunosuppressants for a long time, and thus had certain inhibition in the activation of lymphocytes. Additionally, significant differences were found in the triglyceride level that was included in HPS-2004 standards in patients with HPS underlying different causes. The triglyceride levels in patients with HPS underlying blood system malignancy were significantly higher than those in the other 2 groups. However, the result should be further analyzed because the number of patients with HPS underlying malignancy included in this study was relatively small.
The treatment of adult secondary HPS is mostly based on the HPS-2004 protocol. However, since the clinical process of secondary HPS is different from that of primary HPS, the application of the HPS-2004 protocol should be adjusted to different types and severities of the disease, and it should be decided whether to use dexamethasone, dexamethasone + cyclosporine A, or the classical HPS-2004 protocol. This study demonstrated that for patients with infection-induced HPS, the active control of infection is the basis of HPS treatment. Patients can be first treated with glucocorticoids on the basis of antiinfective treatment. Those whose disease cannot be well controlled, the combination of etoposide is an ideal choice because this can avoid uncontrolled infection due to potent treatment at the early stage of the disease. Since most of the patients with HPS underlying autoimmune disorders have already received treatment of glucocorticoid and immunosuppressive agents before HPS diagnosis, proper adjustment made to the previous treatment program would lead to a satisfactory outcome. Some patients with HPS underlying malignancy may not tolerate the treatment of HPS and primary disease at the same time; therefore, most of them choose first to take glucocorticoids to control inflammatory factors and then to deal with the primary disease. Due to the complexity of HPS, how to determine the severity of this disease is still unclear, and therefore patient management as well as treatment should be tailored and individualized.
As for the heterogeneity in the prognosis of patients with adult HPS, the mortality rate was between 20.4% and 88% based on previous reports,[6,8,12–16] and demographic factors and follow-up time had a great impact on the results. A follow-up was conducted on 162 patients with HPS with a mortality rate 42%.[17] At the same time, previous studies showed that the mortality rate of HPS induced by autoimmune disorders was the lowest, followed by HPS underlying infections and primary HPS. HPS underlying malignancy, especially HPS underlying lymphoma, was a significantly adverse prognostic factor.[13,15,17,18] This study indicated that the overall mortality rate for patients with adult secondary HPS was 27.66%. The prognosis was the best for HPS underlying autoimmune disorders and the worst for HPS underlying blood system malignancy. The results were consistent with previous studies. However, this study did not identify a direct correlation between the etiology of HPS and death of patients. The relationship between causes of HPS and poor prognosis might be clarified if a larger sample size could be provided.
Besides causes of diseases, other commonly recognized adverse prognostic factors that have been reported included age >30 years,[19] male gender,[18] high serum ferritin levels,[15,19,20] significant decrease in the blood platelet count,[13,21] hypoalbuminemia,[15] and so forth. The 47 patients included in this study were divided into death and survival groups. The results showed that serous effusion, pulmonary involvement, signs of hemorrhage, nervous system involvement, and decrease in blood platelet were associated with death, while the 1st 4 factors were independent risk factors that could influence patient prognosis.
A large number of reports were available on serous effusion in patients with HPS; pleural, peritoneal, and pericardial effusions may also occur. A study on children with HPS revealed pleural effusion in 60% patients by imaging.[22] Seguin et al[23] conducted pulmonary high-resolution computed tomography examinations on 68 patients who were diagnosed with HPS, and 38 of them (56%) had pleural effusion. Matthias et al conducted abdominal ultrasound examinations on 9 children who were diagnosed with HPS and observed peritoneal effusion in 6 patients and pleural effusion in 2 patients,[24] indicating that the incidence of serous effusion was high in HPS. It was previously thought that HPS was mostly associated with primary diseases, but reports were rare on the relationship between serous effusion in patients with HPS and their prognosis. This study also discovered that the number of patients with serous effusion in the death group was high compared with that in the survival group, and the occurrence of serous effusion was an independent risk factor for patients with adult secondary HPS. Patients with adult secondary HPS had a poor prognosis if serous effusion occurred, which should draw the attention of researchers.
Relatively few reports were available on pulmonary involvement in HPS. The study by Seguin et al on 219 patients with HPS found pulmonary involvement at different degrees in 118 (54%) patients. Clinical manifestations of these patients had no obvious specificity; infection, pulmonary edema, and pulmonary malignancy were the most common pulmonary manifestations. Imaging manifestations included centrilobular nodules with interstitial infiltration, consolidation with unknown causes, and local ground-glass-like changes.[22] The mortality rate of patients with pulmonary involvement was obviously higher than that of patients without pulmonary involvement (52% vs 20%). However, after HPS-specific and etiotropic treatments, patients with respiratory function improvement were only 67/118 (56.7%).[23] The results from an autopsy also verified a hemophagocytic phenomenon in the lungs of patients.[25] In this study, the risk of death for patients with pulmonary involvement obviously increased, which was consistent with previous studies. Thus, clinical physicians should pay more attention to patients with pulmonary involvement.
A number of previous studies showed that a significant decrease in the blood platelet count (<40 × 109/L) indicated poor prognosis of patients with HPS. Li et al[26] believed that the blood platelet count lower than 40 × 109/L and the fibrinogen level lower than 1.5 g/L were the factors leading to the poor prognosis of patients with HPS underlying lymphoma. The blood platelet count also indicated poor prognosis in patients with HPS caused by other reasons.[18,19,27,28] Its mechanism was not clear yet, but it was possible that the blood platelet count was more sensitive to inflammatory reactions compared with other indicators. Its decrease was a direct reflection of inflammatory cytokine storm.[12] In this study, an independent decrease in the blood platelet count might be the adverse prognostic factor for HPS (P = .05), while hypofibrinogenemia had no significant predictive function. However, this study also found that the bleeding sign was an independent risk factor for patients with adult secondary HPS. For patients with signs of hemorrhage, the blood platelet count obviously decreased, and the disease in 60% patients was complicated by hypofibrinogenemia. Thus, signs of hemorrhage could reflect the collaborative predictive function of a decrease in the blood platelet count and coagulation abnormalities.
Previously, some studies indicated that central nervous system involvement was relatively common in patients with HPS.[29–33] Some patients might only have abnormalities in the cerebrospinal fluid assay without any significant abnormal clinical manifestations, in which case they were overlooked in clinic. However, these patients with central nervous system involvement had a poorer prognosis. As a result, monitoring of clinical manifestations of the nervous system of patients was extremely important to evaluate the disease as early as possible and improve the survival rate.[34] Jovanovic et al[32] reported 30 children with HPS; 17 patients manifested central nervous system involvement, of which 10 patients died in the end and 3 of the survived patients had moderate-to-severe neurological sequelae. The central nervous system involvement was found to be an important death predictive factor. Four patients had observable nervous system damage in this study, and 3 patients died with seizures, loss of consciousness, and decrease in muscle strength for the 4 limbs as manifestations, respectively. One of the patients underwent lumbar vertebra puncture, which showed a significant increase in cerebrospinal fluid pressure (250 mm H2O) and an increase in the protein level. The analysis by Cox regression indicated that nervous system damage was an independent risk factor for death. However, further observation was needed due to the small number of cases. The reason for the relatively small number of patients with nervous system damage in this study might be that some patients had abnormalities in the assay of cerebrospinal fluid but were overlooked due to their insidious clinical manifestations. The result indicated that during clinical works, attention should be paid to whether patients with HPS had nervous system involvement. Cerebrospinal fluid examinations should be improved as soon as possible, which might help in improving the survival rate of patients. For patients with clearly diagnosed HPS, active treatment should be conducted in the case of aforementioned clinical manifestations and laboratory examination results to prevent a sharp deterioration of the disease, which could lead to poor prognosis.
5. Conclusions
This study retrospectively analyzed the clinical data of 47 patients with secondary HPS, and infection was the most common cause of adult secondary HPS. Clinical manifestations of the disease were diversified with strong heterogeneity. The death risk among the patients increased if signs of hemorrhage, serous effusion, pulmonary involvement, or nervous system involvement were observed, which should draw the attention of physicians.
Footnotes
Abbreviations: EB = Epstein–Barr, HPS = hemophagocytic syndrome, NK = natural killerl, sIL-2R (sCD25) = soluble interleukin-2 receptor.
YG and YB contributed equally to this work.
The authors have no funding and conflicts of interest to disclose.
References
- [1].Risdall RJ, McKenna RW, Nesbit ME, et al. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer 1979;44:993–1002. [DOI] [PubMed] [Google Scholar]
- [2].Henter JI, Horne A, Arico M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124–31. [DOI] [PubMed] [Google Scholar]
- [3].Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child 1952;27:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Janka G. Hemophagocytic lymphohistiocytosis: when the immune system runs amok. Klin Padiatr 2009;221:278–85. [DOI] [PubMed] [Google Scholar]
- [5].Zhang L, Zhou J, Sokol L. Hereditary and acquired hemophagocytic lymphohistiocytosis. Cancer Control 2014;21:301–12. [DOI] [PubMed] [Google Scholar]
- [6].Filipovich AH. Life-threatening hemophagocytic syndromes: current outcomes with hematopoietic stem cell transplantation. Pediatr Transplant 2005;9(Suppl 7):87–91. [DOI] [PubMed] [Google Scholar]
- [7].Ishii E, Ohga S, Imashuku S, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol 2007;86:58–65. [DOI] [PubMed] [Google Scholar]
- [8].Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology (Oxford) 2008;47:1686–91. [DOI] [PubMed] [Google Scholar]
- [9].Zhang Z, Wang J, Ji B, et al. Clinical presentation of hemophagocytic lymphohistiocytosis in adults is less typical than in children. Clinics (Sao Paulo) 2016;71:205–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Matera G, Puccio R, Giancotti A, et al. Impact of interleukin-10, soluble CD25 and interferon-gamma on the prognosis and early diagnosis of bacteremic systemic inflammatory response syndrome: a prospective observational study. Crit Care 2013;17:R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Goudy K, Aydin D, Barzaghi F, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol 2013;146:248–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li F, Yang Y, Jin F, et al. Clinical characteristics and prognostic factors of adult hemophagocytic syndrome patients: a retrospective study of increasing awareness of a disease from a single-center in China. Orphanet J Rare Dis 2015;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Arca M, Fardet L, Galicier L, et al. Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: impact of triggering disease and early treatment with etoposide. Br J Haematol 2015;168:63–8. [DOI] [PubMed] [Google Scholar]
- [14].Aulagnon F, Lapidus N, Canet E, et al. Acute kidney injury in adults with hemophagocytic lymphohistiocytosis. Am J Kidney Dis 2015;65:851–9. [DOI] [PubMed] [Google Scholar]
- [15].Parikh SA, Kapoor P, Letendre L, et al. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc 2014;89:484–92. [DOI] [PubMed] [Google Scholar]
- [16].Takahashi N, Chubachi A, Kume M, et al. A clinical analysis of 52 adult patients with hemophagocytic syndrome: the prognostic significance of the underlying diseases. Int J Hematol 2001;74:209–13. [DOI] [PubMed] [Google Scholar]
- [17].Riviere S, Galicier L, Coppo P, et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med 2014;127:1118–25. [DOI] [PubMed] [Google Scholar]
- [18].Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore) 2014;93:100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kaito K, Kobayashi M, Katayama T, et al. Prognostic factors of hemophagocytic syndrome in adults: analysis of 34 cases. Eur J Haematol 1997;59:247–53. [DOI] [PubMed] [Google Scholar]
- [20].Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol 2015;90:220–4. [DOI] [PubMed] [Google Scholar]
- [21].Buyse S, Teixeira L, Galicier L, et al. Critical care management of patients with hemophagocytic lymphohistiocytosis. Intensive Care Med 2010;36:1695–702. [DOI] [PubMed] [Google Scholar]
- [22].Fitzgerald NE, MacClain KL. Imaging characteristics of hemophagocytic lymphohistiocytosis. Pediatr Radiol 2003;33:392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Seguin A, Galicier L, Boutboul D, et al. Pulmonary involvement in patients with hemophagocytic lymphohistiocytosis. Chest 2016;149:1294–301. [DOI] [PubMed] [Google Scholar]
- [24].Schmidt MH, Sung L, Shuckett BM. Hemophagocytic lymphohistiocytosis in children: abdominal US findings within 1 week of presentation. Radiology 2004;230:685–9. [DOI] [PubMed] [Google Scholar]
- [25].Ost A, Nilsson-Ardnor S, Henter JI. Autopsy findings in 27 children with haemophagocytic lymphohistiocytosis. Histopathology 1998;32:310–6. [DOI] [PubMed] [Google Scholar]
- [26].Li F, Li P, Zhang R, et al. Identification of clinical features of lymphoma-associated hemophagocytic syndrome (LAHS): an analysis of 69 patients with hemophagocytic syndrome from a single-center in central region of China. Med Oncol 2014;31:902. [DOI] [PubMed] [Google Scholar]
- [27].Trottestam H, Berglof E, Horne A, et al. Risk factors for early death in children with haemophagocytic lymphohistiocytosis. Acta Paediatr 2012;101:313–8. [DOI] [PubMed] [Google Scholar]
- [28].Dao AT, Luong VT, Nguyen TT, et al. Risk factors for early fatal outcomes among children with hemophagocytic lymphohistiocytosis (HLH): a single-institution case-series in Vietnam. Pediatr Hematol Oncol 2014;31:271–81. [DOI] [PubMed] [Google Scholar]
- [29].Goo HW, Weon YC. A spectrum of neuroradiological findings in children with haemophagocytic lymphohistiocytosis. Pediatr Radiol 2007;37:1110–7. [DOI] [PubMed] [Google Scholar]
- [30].Shinoda J, Murase S, Takenaka K, et al. Isolated central nervous system hemophagocytic lymphohistiocytosis: case report. Neurosurgery 2005;56:187. [PubMed] [Google Scholar]
- [31].Rooms L, Fitzgerald N, McClain KL. Hemophagocytic lymphohistiocytosis masquerading as child abuse: presentation of three cases and review of central nervous system findings in hemophagocytic lymphohistiocytosis. Pediatrics 2003;111:e636–40. [DOI] [PubMed] [Google Scholar]
- [32].Jovanovic A, Kuzmanovic M, Kravljanac R, et al. Central nervous system involvement in hemophagocytic lymphohistiocytosis: a single-center experience. Pediatr Neurol 2014;50:233–7. [DOI] [PubMed] [Google Scholar]
- [33].Gratton SM, Powell TR, Theeler BJ, et al. Neurological involvement and characterization in acquired hemophagocytic lymphohistiocytosis in adulthood. J Neurol Sci 2015;357:136–42. [DOI] [PubMed] [Google Scholar]
- [34].Kim MM, Yum MS, Choi HW, et al. Central nervous system (CNS) involvement is a critical prognostic factor for hemophagocytic lymphohistiocytosis. Korean J Hematol 2012;47:273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]