

Abstract
In our efforts to address the rising incidence of hepatocellular carcinoma (HCC), we have made a commitment to the synthesis of novel molecules to combat Hep-G2 cells. A facile and highly efficient one-pot, multicomponent reaction has been successfully devised utilizing a p-toluenesulfonic acid (p-TSA)-catalyzed domino Knoevenagel/Michael/intramolecular cyclization approach for the synthesis of novel 5H-benzo[h]thiazolo[2,3-b]quinazoline and indeno[1,2-d] thiazolo[3,2-a]pyrimidine analogs bearing a bridgehead nitrogen atom. This domino protocol constructed one new ring by the concomitant formation of multiple bonds (C–C, C–N, and C=N) involving multiple steps without the use of any metal catalysts in one-pot, with all reactants effi-ciently exploited. All the newly synthesized compounds were authenticated by means of Fourier transform infrared spectroscopy, liquid chromatography–mass spectrometry, proton nuclear magnetic resonance spectroscopy, and carbon-13 nuclear magnetic resonance spectroscopy, together with elemental analysis, and their antitumor activity was evaluated in vitro on a Hep-G2 human cancer cell line by sulforhodamine B assay. Computational molecular modeling studies were carried out on cancer-related targets, including interleukin-2, interleukin-6, Caspase-3, and Caspase-8. Two compounds (4A and 6A) showed growth inhibitory activity comparable to the positive control Adriamycin, with growth inhibition of 50% <10 μg/mL. The results of the comprehensive structure–activity relationship study confirmed the assumption that two or more electronegative groups on the phenyl ring attached to the thiazolo[2,3-b]quinazoline system showed the optimum effect. The in silico simulations suggested crucial hydrogen bond and π–π stacking interactions, with a good ADMET (absorption, distribution, metabolism, excretion, and toxicity) profile and molecular dynamics, in order to explore the molecular targets of HCC which were in complete agreement with the in vitro findings. Considering their significant anticancer activity, 4A and 6A are potential drug candidates for the management of HCC.
Keywords: thiazolo[3,2-a]pyrimidine and thiazolo[2,3-b]quinazoline; hepatocellular carcinoma; domino reactions; interleukins; caspases; molecular docking; ADMET; dynamics; multi-component reactions; metal-free
Introduction
One in six human deaths globally is due to multifaceted disease, such as cancer. Liver cancer accounted for 788,000 deaths in 2015 and is the second most common cause of cancer-related deaths, worldwide. The most prevalent primary liver cancer with poor prognosis is hepatocellular carcinoma (HCC), which has potentially lethal human malignacy worldwide, particularly in Asia and Africa.1,2 In men, HCC is the fifth most commonly diagnosed cancer and the second leading cause of cancer deaths; in women, it is the seventh most frequently diagnosed cancer, and worldwide, overall, it is the sixth commonest cause of cancer-related deaths.3 In spite of continuous efforts to develop novel therapeutic strategies to treat HCC, it remains a challenge. As a consequence, therefore, increased attention has been paid, especially in the field of liver cancer therapy, to the discovery and development of safe and novel anticancer agents, together with improved cytotoxicity toward cancerous cells.4
Domino reactions involving simultaneous formation of C–C and C–heteroatom multiple bonds in a single flask are a means to achieving economical methods for the manufacture of especially unusual, fused heterocyclic, medicinally privileged scaffolds without any separation of intermediates throughout the process. Moreover, domino reactions, being attractively appealing, are frequently connected with savings in terms of energy and reaction periods, are highly convergent, have fewer environmental impacts, and show atom economy in a single-step process starting from multiple reactants.5,6
Conventional multistep chemical reactions suffer from various limitations compared with the one-pot domino technique, as they require a large number of synthetic operations, including isolation and purification of the products of each individual step. The multistep synthetic approach, therefore, has led to synthetic inefficiency, is time-consuming, or contains side reactions generating large amounts of waste.7
A key challenge in recent drug development processes is to design a rapid, versatile, and efficient synthesis that provides target molecules containing structural complexity and diversity with a choice of fascinating biologic activities.8
Thiazole-based heterocycles are central to modern chemical synthesis due to their synthetic feasibility and their incorporation into various types of therapeutically useful agents and are of paramount interest in the development of important pharmacophores in the drug discovery endeavor.9,10 Functionalized quinazoline and fused quinazolines have long been of increasing interest in the field of synthetic organic and medicinal chemistry on account of the wide range of biologic activities,11–14 including anticancer,15 anti-inflammatory,16 antituberculosis,17 anticonvulsant,18 antimalarial,19 antihypertensive,20 antidiabetic,21 and so on. Furthermore, suitably substituted thiazolo[2,3-b]quinazolines also serve as versatile building blocks in synthetic chemistry.
Nowadays, bridgehead (ring junction) nitrogen atom-containing thiazolo[3,2-a]pyrimidines command much attention as privileged scaffolds comprising a vital class of heterocyclic structures possessing exciting and varied phar-macologic activities,22–24 such as: being potential inhibitors of cyclin-dependent kinase,25 CDC25B phosphatase enzymes,26 IspF proteins,27 and YycG histidine kinase,28 antibacterial,29 analgesic, and anti-inflammatory activities,30 acting as a corticotropin-releasing factor receptor antagonist,31 hypolipidemic activity,32 antiviral including anti-HIV effects, anticancer,33,34 cardiotonic, and inotropic effects,35 acting as potent A2A adenosine receptor inverse agonists with antinociceptive activity,36 and so on.
To the best of our knowledge, among the earlier reported methods devoted to the synthesis of especially 5H-benzo[h] thiazolo[2,3-b]quinazolines,37,38 only a few are described, and metal-free multicomponent domino reactions (MDRs)39,40 are still rare. In this regard, most of the reported methods have suffered from various drawbacks, for example, harsh reaction conditions, multistep synthetic routes, costly reagents/catalysts, prolonged reaction periods, tedious workups after each step, and poor availability of starting materials. There is no route for the direct construction of the indeno[1,2-d] thiazolo[3,2-a]pyrimidine moiety through domino reactions, and a literature survey revealed only one report to be available, which includes the synthesis of 5-phenylindeno[1,2-d] thiazolo[3,2-a]pyrimidine-3,6(2H,5H)-dione and 5-phenyl-2,3-dihydro indeno[1,2-d]thiazolo[3,2-a]pyrimidin-6(5H)-one in two steps.41
In order to arrive at a highly proficient and convergent synthetic strategy for the construction of these two vital structural elements, while at the same time avoiding several limitations of earlier reports, and in our venture toward the development of a modern synthetic approach, we first report herein an operationally simple and straightforward metal-free, one-pot MDR for obtaining a panel of novel 5H-benzo[h]thiazolo[2,3-b]quinazoline (1A–15A) and indeno[1,2-d]thiazolo[3,2-a]pyrimidine (1B–15B) analogs using the reactions of highly substituted α-tetralone or α-indanone with some aromatic aldehydes and distinctive 2-aminothiazoles in ethanol (EtOH) in the presence of catalytic amounts of p-toluenesulfonic acid (p-TSA; 20 mol%).7,42,43 Readily available and cheap starting materials, together with an environmentally benign and mild acidic catalyst, were employed to achieve these diversely decorated skeletons in impressive yields. They have also been synthesized by conventional two-step reactions and compared with the preferred MDRs described above.23,44–46 Additionally, all of the newly synthesized compounds were evaluated for their antitumor activity in vitro on a Hep-G2 human cancer cell line by sulforhodamine B (SRB) assay and structure–activity relationship prediction on the basis of in vitro findings. Furthermore, computational molecular modeling studies with ADMET (absorption, distribution, metabolism, excretion, and toxicity) profiling and molecular dynamic (MD) simulation were carried out on cancer-related targets, including interleukin (IL)-2, IL-6, Caspase-3, and Caspase-8 receptor sites, to identify the potential modes of action of the named compounds.
Materials and methods
General information
All of the reactions described below were carried out with commercially available chemicals of reagent grade that were used as received without further purification unless otherwise noted. Reagents were purchased from Merck and Sigma-Aldrich chemical companies. Melting points (m.p.) were determined on an m.p. apparatus and are uncorrected. Compounds were named following the International Union of Pure and Applied Chemistry rules as applied by Advanced Chemistry Development/ChemSketch. Elemental analyses were performed with a Euro EA 3000 Elemental Analyzer for C, H, N, and the results were within ±0.4% of the theoretical values. Infrared (IR) spectra were recorded on a Thermo Scientific Nicolet 6700 Fourier transform IR spectroscopy spectrometer. Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker 800 MHz NMR spectrometer (1H 800 MHz, 13C 200 MHz) (Bruker, Rheinstetten, Germany) and processed in TopSpin 2.1. Chemical shifts are expressed in parts per million (ppm) downfield from tetramethylsilane as the internal standard. Coupling constants are expressed in Hz. All of the compounds were analyzed for mass data using a liquid chromatography–mass spectrometry (LCMS)-2020 mass spectrometer (Schimadzu, Tokyo, Japan). All the compounds were dissolved in 1:1 (v/v) mixtures of acetonitrile:methanol, and 10 μL of the resulting solution was injected to acquire the data. The analysis was performed in electrospray ionization mode using a capillary column at a flow rate of 0.2 mL/min with a 50% water/methanol (1:1) mixture for 120 s. Data analysis was performed using lab solution LCMS data processing software. Reaction progress as well as the purity of the compounds were evaluated with thin layer chromatography (TLC) plates by using ethylacetate:n-hexane (3:7) as the eluent, and the developed chromatogram was visualized under ultraviolet light and iodine vapors.
General experimental procedures for characterization of the synthesized compounds
One-pot efficient synthesis of substituted 5H-benzo[h]thiazolo[2,3-b]quinazolines (1A–15A)
A mixture of substituted tetralone (1 mmol), appropriate aromatic aldehydes (1 mmol), and distinctive 2-aminothiazoles (1 mmol) in EtOH (5.0 mL) in the presence of 20 mol% p-TSA was heated under reflux for 3–4 h. The reaction mixture was poured into ice-cold water. A solid product was obtained, which was filtered, washed thoroughly with distilled water, and recrystallized from EtOH. Pure crystals were obtained (Scheme 1). The progress of the reaction was monitored by TLC on precoated silica gel-G plates using 30% ethylacetate:n-hexane as the solvent system. TLC revealed just a single spot, which proved the presence of a single product.
Scheme 1.
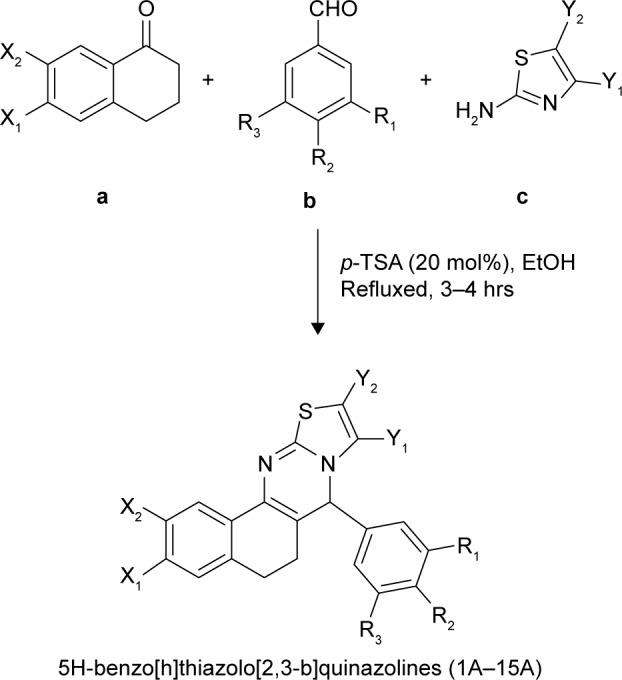
One-pot efficient synthetic route to the titled compound (1A–15A).
Note: Substituted α-tetralone (a), substituted aromatic aldehydes (b) and distinctive 2-aminothiazoles (c).
Abbreviations: EtOH, ethanol; p-TSA, p-toluenesulfonic acid.
Two-step synthesis of substituted 5H-benzo[h] thiazolo[2,3-b]quinazolines (1A–15A)
These compounds were synthesized as follows.
Step I
A mixture of substituted tetralones (1 mmol), appropriate aldehydes (1 mmol), piperidine (0.1 mmol), and glacial acetic acid (0.1 mmol) in EtOH (5.0 mL) was heated under reflux for 10–12 h. The reaction mixture was cooled to room temperature for a few minutes and then poured into ice-cold water. A solid product, substituted benzylidene-tetralones, was obtained, which was filtered, washed thoroughly with distilled water, and dried. The dry residue was recrystallized from EtOH. The progress of the reaction was monitored by TLC on precoated silica gel-G plates using 40% ethylacetate:n-hexane as the solvent system.
Step II
A solution of distinctive 2-aminothiazoles (1 mmol) and appropriate benzylidene-tetralones in glacial acetic acid (2.0 mL) was heated under reflux for 18–20 h. The solvent was evaporated under vacuum and then the residue obtained was dissolved in chloroform, washed with water, and the organic layer separated, dried, and evaporated. The resulting solid obtained was recrystallized from EtOH to yield the target compounds (1A–15A; Scheme 2).
Scheme 2.

Two-step synthetic route to the titled compound (1A–15A).
Note: Substituted α-tetralone (a), substituted aromatic aldehydes (b) and distinctive 2-aminothiazoles (c).
Abbreviation: GAA, glacial acetic acid.
7-(4-bromophenyl)-6,7-dihydro-5H-benzo[h] thiazolo[2,3-b]quinazoline (1A)
Yellow crystals; yield: 77%; m.p. 190°C–192°C; Rf=0.55 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,067.7 (C–H, aromatic), 2,944.7 (C–H), 1,665.8 (C=N), 1,595.0 (C=C), 1,295.8 (C–N), 1,068.4 (C–Br), 740.7 (C–S). 1H NMR (dimethyl sulfoxide [DMSO]-d6, 800 MHz) δ ppm: 2.95 (t, 2H, J=8 Hz), 3.07 (t, 3H, J=8 Hz), 7.40–7.44 (m, 2H), 7.50 (d, 2H, J=8 Hz), 7.59 (t, 1H, J=8 Hz), 7.67 (d, 4H, J=8 Hz), 7.97 (d, 1H, J=8 Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.12, 28.27, 39.76, 39.87, 39.97, 40.07, 40.18, 122.61, 127.54, 127.90, 129.07, 132.03, 132.43, 133.21, 134.13, 134.83, 134.91, 136.66, 143.93, 187.08. LCMS (m/z): 393.25 [M−H]−. Anal. calcd. for C20H15BrN2S: C, 60.77; H, 3.82; N, 7.09; Anal. found: C, 60.87; H, 3.83; N, 7.07.
7-(4-bromophenyl)-3-methoxy-10-methyl-6,7-dihydro-5H-benzo[h]thiazolo[2,3-b]quinazoline (2A)
Yellow crystals; yield: 82%; m.p. 200°C–202°C; Rf=0.58 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,013.1 (C–H, aromatic), 2,945.5 (C–H), 1,663.7 (C=N), 1,596.9 (C=C), 1,332.5 (C–N), 1,263.9 (C–O), 1,070.0 (C–Br), 761.9 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.92 (t, 2H, J=8 Hz), 3.03 (t, 2H, J=8 Hz), 3.55 (s, 4H), 3.86 (s, 3H), 6.93 (t, 2H, J=8 Hz), 7.48 (d, 2H, J=8 Hz), 7.62 (t, 3H, J=8 Hz), 7.94 (d, 1H, J=8 Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.16, 28.67, 39.76, 39.86, 39.97, 40.07, 40.18, 56.06, 112.81, 114.31, 122.39, 126.65, 130.44, 131.99, 132.35, 134.08, 135.08, 136.84, 146.54, 163.84, 185.75. LCMS (m/z): 437.20 [M−H]−. Anal. calcd. for C22H19BrN2OS: C, 60.14; H, 4.36; N, 6.38. Anal. found: C, 60.25; H, 4.35; N, 6.40.
2-methoxy-7-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[h]thiazolo[2,3-b]quinazoline (3A)
Yellow crystals; yield: 83%; m.p. 214°C–216°C; Rf=0.67 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,002.4 (C–H, aromatic), 2,935.5 (C–H), 1,661 (C=N), 1,578.2 (C=C), 1,324.1 (C–N), 1,283.9, 1,248.4 (C–O), 760.5 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.88 (t, 2H, J=8 Hz), 3.13 (t, 2H, J=8 Hz), 3.72 (s, 3H), 3.83 (s, 10H), 6.86 (s, 3H), 7.18 (dd, 1H, J=8 Hz and J=8 Hz), 7.32 (d, 1H, J=8 Hz), 7.44 (d, 1H, J=8 Hz), 7.69 (s, 1H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.44, 27.54, 39.76, 39.86, 39.97, 40.07, 40.18, 55.77, 56.46, 60.59, 108.04, 110.59, 121.34, 130.34, 131.18, 134.21, 135.25, 136.32, 136.70, 138.58, 153.24, 158.65, 187. LCMS (m/z): 437.20 [M+H]+. Anal. calcd. for C24H24N2O4S: C, 66.03; H, 5.54; N, 6.42. Anal. found: C, 66.20; H, 5.52; N, 6.45.
2-methoxy-4-(2-methoxy-6,7-dihydro-5H-benzo[h] thiazolo[2,3-b]quinazolin-7-yl)phenol (4A)
Yellow crystals; yield: 82%; m.p. 218°C–220°C; Rf=0.50 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,451 (–OH), 3,020.2 (C–H, aromatic), 2,937.2 (C–H), 1,659.8 (C=N), 1,572.5 (C=C), 1,326.4 (C–N), 1,248.9 (C–O), 744 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.86 (t, 2H, J=8 Hz), 3.10 (t, 2H, J=8 Hz), 3.82 (s, 7H), 6.87 (d, 1H, J=8 Hz), 7.05 (d, 1H, J=8 Hz), 7.14–7.18 (m, 3H), 7.31 (d, 1H, J=8 Hz), 7.43 (s, 1H), 7.68 (s, 1H), 9.57 (s, 1H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.41, 39.76, 39.86, 39.97, 40.07, 55.75, 56.12, 110.61, 115.01, 115.97, 121.03, 124.23, 127, 130.22, 132.93, 134.43, 136.07, 137.28, 147.94, 148.35, 158.62, 186.89. LCMS (m/z): 391.10 [M−H]−. Anal. calcd. for C22H20N2O3S: C, 67.33; H, 5.14; N, 7.14. Anal. found: C, 67.44; H, 5.16; N, 7.13.
7-(4-bromophenyl)-3-methoxy-6,7-dihydro-5H-benzo[h]thiazolo[2,3-b]quinazoline (5A)
Yellow crystals; yield: 78%; m.p. 194°C–196°C; Rf=0.52 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,012.8 (C–H, aromatic), 2,945.7 (C–H), 1,663.2 (C=N), 1,597 (C=C), 1,332.6 (C–N), 1,263.4 (C–O), 1,070 (C–Br), 762.5 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.92 (t, 2H, J=8 Hz), 3.03 (t, 2H, J=8 Hz), 3.86 (s, 4H), 6.93–6.98 (m, 2H), 7.48 (d, 2H, J=8 Hz), 7.62 (s, 1H), 7.66 (d, 3H, J=8 Hz), 7.94 (d, 1H, J=8 Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.15, 28.67, 39.74, 39.84, 39.95, 40.05, 40.15, 56.06, 112.81, 114.31, 122.39, 126.64, 130.44, 131.99, 132.35, 134.08, 135.07, 136.84, 146.55, 163.84, 185.76. LCMS (m/z): 425.15 [M+]. Anal. calcd. for C21H17BrN2OS: C, 59.30; H, 4.03; N, 6.59. Anal. found: C, 59.19; H, 4.04; N, 6.61.
3-methoxy-7-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[h]thiazolo[2,3-b]quinazoline (6A)
Light brown crystals; yield: 86%; m.p. 214°C–216°C; Rf=0.66 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,050.6 (C–H, aromatic), 2,944.8 (C–H), 1,659 (C=N), 1,598.4 (C=C), 1,336.1 (C–N), 1,258.6 (C–O), 769.2 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.92 (t, 2H, J=8 Hz), 3.12 (t, 2H, J=8 Hz), 3.71 (s, 3H), 3.82 (s, 7H), 3.86 (s, 3H), 6.84 (s, 3H), 6.93 (s, 1H), 6.96 (s, 1H), 7.64 (s, 1H), 7.93 (d, 1H, J=8 Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.27, 28.76, 39.73, 39.84, 39.94, 40.05, 40.15, 56.04, 56.45, 60.58, 107.93, 112.79, 114.15, 126.79, 130.38, 131.34, 135.49, 135.86, 138.43, 146.46, 153.23, 163.73, 185.88. LCMS (m/z): 435.15 [M−H]−. Anal. calcd. for C24H24N2O4S: C, 66.03; H, 5.54; N, 6.42. Anal. found: C, 66.21; H, 5.56; N, 6.41.
4-(6,7-dihydro-5H-benzo[h]thiazolo[2,3-b] quinazolin-7-yl)phenol (7A)
Light brown crystals; yield: 70%; m.p. 190°C–192°C; Rf=0.45 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,204.2 (–OH), 2,951.5 (C–H, aromatic), 2,895.1 (C–H), 1,639.9 (C=N), 1,556.1 (C=C), 1,370.2 (C–N), 740.3 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.93 (t, 2H, J=8 Hz), 3.09 (t, 3H, J=8 Hz), 6.86 (d, 2H, J=8 Hz), 7.37–7.44 (m, 5H), 7.56 (t, 1H, J=8 Hz), 7.67 (s, 1H), 7.94 (d, 1H, J=8 Hz), 9.98 (s, 1H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.18, 28.29, 39.76, 39.86, 39.97, 40.07, 40.17, 116, 126.54, 127.40, 127.75, 128.88, 132.65, 132.79, 133.58, 133.73, 136.83, 143.63, 158.91, 187.06. LCMS (m/z): 331.05 [M−H]−. Anal. calcd. for C20H16N2OS: C, 72.26; H, 4.85; N, 8.43. Anal. found: C, 72.45; H, 4.87; N, 8.39.
7-(4-chlorophenyl)-2-methoxy-9-methyl-6,7-dihydro-5H-benzo[h]thiazolo[2,3-b]quinazoline (8A)
Yellow crystals; yield: 65%; m.p. 196°C–198°C; Rf=0.40 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,067.6 (C–H, aromatic), 2,941.2 (C–H), 1,660.7 (C=N), 1,589.1 (C=C), 1,322.7 (C–N), 1,251.5 (C–O), 1,031.5 (C–Cl), 743.9 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.88 (t, 4H, J=8 Hz), 3.04 (t, 4H, J=8 Hz), 3.36 (s, 3H), 7.19 (dd, 1H, J=16 Hz and J=16 Hz), 7.32 (d, 1H, J=16 Hz), 7.45 (d, 1H, J=8 Hz), 7.53–7.58 (m, 4H), 7.69 (s, 1H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.30, 27.46, 39.78, 39.88, 39.99, 40.09, 40.19, 55.79, 110.61, 121.49, 129.10, 130.43, 132.19, 133.83, 134.06, 134.59, 134.83, 136.39, 136.56, 158.68, 186.92. LCMS (m/z): 397.20 [M+2]. Anal. calcd. for C22H19ClN2OS: C, 66.91; H, 4.85; N, 7.09. Anal. found: C, 66.79; H, 4.87; N, 7.12.
7-(4-chlorophenyl)-3-methoxy-9-methyl-6,7-dihydro-5H-benzo[h]thiazolo[2,3-b]quinazoline (9A)
Yellow crystals; yield: 68%; m.p. 196°C–198°C; Rf=0.42 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,050.8 (C–H, aromatic), 2,943.9 (C–H), 1,660.5 (C=N), 1,587.9 (C=C), 1,336.3 (C–N), 1,274.7 (C–O), 1,030.8 (C–Cl), 760.5 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.92 (t, 4H, J=8 Hz), 3.04 (t, 4H, J=8 Hz), 3.43 (s, 3H), 6.93–6.97 (m, 2H), 7.52 (dd, 4H, J=24 Hz and J=24 Hz), 7.64 (s, 1H), 7.94 (d, 1H, J=8 Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.14, 28.68, 39.78, 39.88, 39.99, 40.09, 40.19, 56.06, 112.81, 114.30, 126.67, 129.06, 130.43, 132.10, 133.65, 134.01, 136.78, 146.53, 163.84, 185.73. LCMS (m/z): 395.25 [M+]. Anal. calcd. for C22H19ClN2OS: C, 66.91; H, 4.85; N, 7.09. Anal. found: C, 66.75; H, 4.86; N, 7.11.
7-(4-chlorophenyl)-9-methyl-6,7-dihydro-5H-benzo[h]thiazolo[2,3-b]quinazoline (10A)
Yellow crystals; yield: 64%; m.p. 188°C–190°C; Rf=0.44 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,067.7 (C–H, aromatic), 2,944.7 (C–H), 1,668.1 (C=N), 1,596.4 (C=C), 1,317.4 (C–N), 1,027.7 (C–Cl), 743 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.95 (t, 4H, J=8 Hz), 3.07 (t, 4H, J=8 Hz), 7.39–7.44 (m, 2H), 7.54–7.61 (m, 5H), 7.70 (s, 1H), 7.97 (s, 1H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.11, 28.28, 39.78, 39.88, 39.99, 40.09, 40.20, 127.53, 127.89, 129.11, 132.20, 133.23, 133.85, 134.12, 134.58, 134.76, 136.61, 143.92, 187.07. LCMS (m/z): 365.20 [M+]. Anal. calcd. for C21H17ClN2S: C, 69.12; H, 4.70; N, 7.68. Anal. found: C, 69.25; H, 4.71; N, 7.67.
7-(4-chlorophenyl)-2-methoxy-6,7-dihydro-5H-benzo[h]thiazolo[2,3-b]quinazoline (11A)
Yellow crystals; yield: 70%; m.p. 194°C–196°C; Rf=0.49 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,067.8 (C–H, aromatic), 2,940.6 (C–H), 1,660.6 (C=N), 1,589.3 (C=C), 1,323.0 (C–N), 1,252.2 (C–O), 1,031.6 (C–Cl), 744.8 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.88 (t, 2H, J=8 Hz), 3.04 (t, 2H, J=8 Hz), 3.82 (s, 4H), 7.20 (d, 1H, J=8 Hz), 7.33 (d, 1H, J=8 Hz), 7.45 (s, 1H), 7.53–7.58 (m, 5H), 7.69 (s, 1H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.30, 27.46, 39.78, 39.88, 39.99, 40.09, 40.19, 55.79, 110.62, 121.50, 129.10, 130.44, 132.19, 133.83, 134.07, 134.83, 136.56, 158.69, 186.98. LCMS (m/z): 381.25 [M+]. Anal. calcd. for C21H17ClN2OS: C, 66.22; H, 4.50; N, 7.35. Anal. found: C, 66.32; H, 4.52; N, 7.33.
7-(4-chlorophenyl)-6,7-dihydro-5H-benzo[h] thiazolo[2,3-b]quinazoline (12A)
Yellow crystals; yield: 61%; m.p. 184°C–186°C; Rf=0.43 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,069.4 (C–H, aromatic), 2,944.7 (C–H), 1,668.1 (C=N), 1,596.4 (C=C), 1,317.4 (C–N), 1,020.1 (C–Cl), 742.7 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.97 (t, 2H, J=8 Hz), 3.09 (t, 3H, J=8 Hz), 7.40–7.44 (m, 2H), 7.54–7.61 (m, 6H), 7.70 (s, 1H), 7.98 (d, 1H, J=8 Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.11, 28.28, 39.78, 39.88, 39.99, 40.09, 40.19, 127.53, 127.89, 129.06, 129.10, 132.20, 133.23, 133.85, 134.11, 134.58, 134.76, 136.60, 143.92, 187.07. LCMS (m/z): 353.0 [M+2]. Anal. calcd. for C20H15ClN2S: C, 68.46; H, 4.31; N, 7.98. Anal. found: C, 68.53; H, 4.34; N, 7.95.
7-(4-chlorophenyl)-2-methoxy-10-methyl-6,7-dihydro-5H-benzo[h]thiazolo[2,3-b]quinazoline (13A)
Yellow crystals; yield: 74%; m.p. 198°C–200°C; Rf=0.46 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,067.9 (C–H, aromatic), 2,940.4 (C–H), 1,660.7 (C=N), 1,591 (C=C), 1,324.7 (C–N), 1,256.8 (C–O), 1,033.4 (C–Cl), 748.3 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.88 (t, 4H, J=8 Hz), 3.04 (t, 4H, J=8 Hz), 3.47 (s, 3H), 7.19 (d, 1H, J=8 Hz), 7.32 (d, 1H, J=8 Hz), 7.45 (s, 1H), 7.53–7.58 (m, 4H), 7.68 (s, 1H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.30, 27.46, 39.78, 39.88, 39.99, 40.09, 40.19, 55.78, 110.61, 121.48, 129.09, 130.42, 132.18, 133.83, 134.06, 134.59, 134.82, 136.38, 136.54, 158.67, 186.91. LCMS (m/z): 393.25 [M−H]−. Anal. calcd. for C22H19ClN2OS: C, 66.91; H, 4.85; N, 7.09. Anal. found: C, 66.83; H, 4.87; N, 7.10.
7-(4-chlorophenyl)-3-methoxy-10-methyl-6,7-dihydro-5H-benzo[h]thiazolo[2,3-b]quinazoline (14A)
Light brown crystals; yield: 76%; m.p. 198°C–200°C; Rf=0.48 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,051.0 (C–H, aromatic), 2,938.8 (C–H), 1,653.8 (C=N), 1,602.6 (C=C), 1,339 (C–N), 1,266.3 (C–O), 1,091.8 (C–Cl), 764.1 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.94 (t, 3H, J=8 Hz), 3.06 (t, 4H, J=8 Hz), 3.86 (s, 4H), 6.93–6.98 (m, 2H), 7.53–7.56 (m, 4H), 7.64 (s, 1H), 7.95 (d, 1H, J=8 Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.14, 28.68, 39.78, 39.88, 39.99, 40.09, 40.19, 56.06, 112.81, 114.30, 126.67, 129.07, 130.43, 132.11, 133.65, 134.01, 134.74, 136.79, 146.54, 163.84, 185.74. LCMS (m/z): 393.25 [M−H]−. Anal. calcd. for C22H19ClN2OS: C, 66.91; H, 4.85; N, 7.09. Anal. found: C, 66.79; H, 4.86; N, 7.07.
7-(4-chlorophenyl)-10-methyl-6,7-dihydro-5H-benzo[h]thiazolo[2,3-b]quinazoline (15A)
Yellow crystals; yield: 79%; m.p. 186°C–188°C; Rf=0.56 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,069.7 (C–H, aromatic), 2,944.8 (C–H), 1,668.1 (C=N), 1,596.1 (C=C), 1,317.3 (C–N), 1,089.4 (C–Cl), 742.3 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 2.97 (t, 4H, J=8 Hz), 3.09 (t, 4H, J=8 Hz), 7.40–7.44 (m, 2H), 7.55–7.61 (m, 5H), 7.70 (s, 1H), 7.98 (d, 1H, J=8 Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 27.11, 28.29, 39.79, 39.89, 39.99, 40.09, 40.20, 127.54, 127.90, 129.07, 129.11, 132.21, 133.24, 133.85, 134.13, 134.59, 134.76, 136.61, 143.93, 187.08. LCMS (m/z): 367.30 [M+2]. Anal. calcd. for C21H17ClN2S: C, 69.12; H, 4.70; N, 7.68. Anal. found: C, 69.23; H, 4.72; N, 7.66.
One-pot efficient synthesis of substituted indeno[1,2-d]thiazolo[3,2-a]pyrimidines (1B–15B)
A mixture of substituted α-indanone (1 mmol), appropriate aromatic aldehydes (1 mmol), and distinctive 2-aminothiazoles (1 mmol) in EtOH (5.0 mL) in the presence of 20 mol% p-TSA was heated under reflux for 3–4 h. The reaction mixture was then poured into ice-cold water. A solid product was obtained, which was filtered, washed thoroughly with distilled water, and recrystallized from EtOH. Pure crystals were obtained (Scheme 3). The progress of the reaction was monitored by TLC on precoated silica gel-G plates using 30% ethylacetate:n-hexane as the solvent system. TLC revealed just a single spot, which proved the presence of a single product.
Scheme 3.
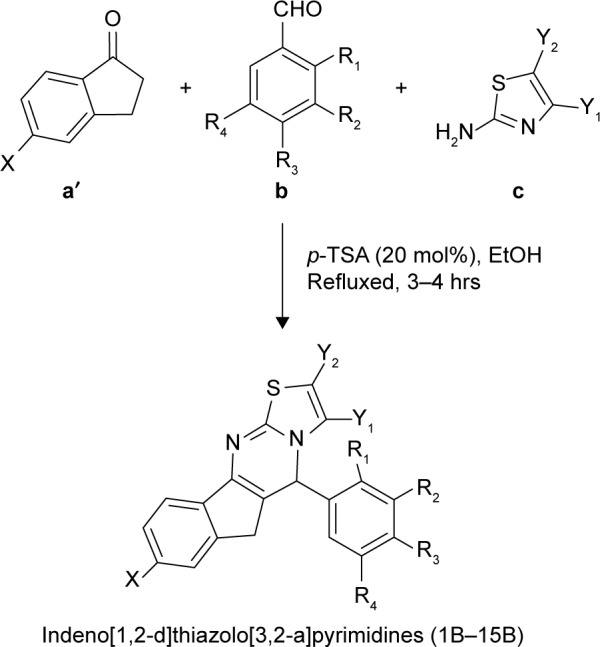
One-pot efficient synthetic route to the titled compound (1B–15B).
Note: Substituted α-indanone (a′), substituted aromatic aldehydes (b) and distinctive 2-aminothiazoles (c).
Abbreviations: EtOH, ethanol; p-TSA, p-toluenesulfonic acid.
Two-step synthesis of substituted indeno[1,2-d] thiazolo[3,2-a]pyrimidines (1B–15B)
These compounds were synthesized as follows.
Step I
A mixture of substituted indanones (1 mmol), appropriate aldehydes (1 mmol), piperidine (0.1 mmol), and glacial acetic acid (0.1 mmol) in EtOH (5.0 mL) was heated under reflux for 10–12 h. The reaction mixture was cooled to room temperature for a few minutes and then poured into ice-cold water. A solid product, substituted benzylidene-indanones, was obtained, which was filtered, washed thoroughly with distilled water, and dried. The dry residue was recrystallized from EtOH. The progress of the reaction was monitored by TLC on precoated silica gel-G plates using 40% ethylacetate:n-hexane as the solvent system.
Step II
A solution of distinctive 2-aminothiazoles (1 mmol) and appropriate benzylidene-indanones in glacial acetic acid (2.0 mL) was heated under reflux for 18–20 h. The solvent was evaporated under vacuum and then the residue obtained was dissolved in chloroform, washed with water, and the organic layer separated, dried, and evaporated. The resulting solid obtained was recrystallized from EtOH to yield the target compounds (1B–15B; Scheme 4).
Scheme 4.

Two-step synthetic route to the titled compound (1B–15B).
Note: Substituted α-indanone (a′), substituted aromatic aldehydes (b) and distinctive 2-aminothiazoles (c).
Abbreviation: GAA, glacial acetic acid.
5-(4-bromophenyl)-5,6-dihydroindeno[1,2-d] thiazolo[3,2-a]pyrimidine (1B)
Yellow crystals; yield: 75%; m.p. 186°C–188°C; Rf=0.54 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,023.4 (C–H, aromatic), 2,928.9 (C–H), 1,693.2 (C=N), 1,620.8 (C=C), 1,325.1 (C–N), 733.9 (C–S), 1,070.9 (C–Br). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 4.14 (s, 3H), 7.50–7.54 (m, 2H), 7.70–7.82 (m, 8H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.32, 39.76, 39.86, 39.97, 40.07, 40.17, 123.83, 124.19, 127.21, 128.28, 132.01, 132.47, 133.11, 134.61, 135.59, 136.38, 137.56, 150.54, 193.80. LCMS (m/z): 381.25 [M+]. Anal. calcd. for C19H13BrN2S: C, 59.85; H, 3.44; N, 7.35. Anal. found: C, 59.97; H, 3.42; N, 7.34.
5-(2-chlorophenyl)-5,6-dihydroindeno[1,2-d] thiazolo[3,2-a]pyrimidine (2B)
Yellow crystals; yield: 66%; m.p. 178°C–180°C; Rf=0.41 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,064.6 (C–H, aromatic), 2,922.5 (C–H), 1,697.1 (C=N), 1,621.3 (C=C), 1,326.1 (C–N), 740.4 (C–S), 1,037.4 (C–Cl). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 4.14 (s, 3H), 7.49–7.53 (m, 4H), 7.63 (d, 1H, 1H, J=8 Hz), 7.68 (d, 1H, J=8 Hz), 7.74 (t, 1H, J=8 Hz), 7.83 (s, 1H), 7.98 (d, 1H, J=8 Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 31.84, 39.76, 39.86, 39.97, 40.07, 40.17, 124.32, 127.24, 128.33, 130.56, 130.97, 131.70, 132.96, 135.39, 135.76, 137.43, 138.14, 150.80, 193.62. LCMS (m/z): 339.20 [M+2]. Anal. calcd. for C19H13ClN2S: C, 67.75; H, 3.89; N, 8.32. Anal. found: C, 67.65; H, 3.91; N, 8.30.
5-(4-bromophenyl)-2-methyl-5,6-dihydroindeno [1,2-d]thiazolo[3,2-a]pyrimidine (3B)
Yellow crystals; Yield: 80%; m.p. 192°C–194°C; Rf=0.57 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,024.1 (C–H, aromatic), 2,926.7 (C–H), 1,693.3 (C=N), 1,621.2 (C=C), 1,325.3 (C–N), 733.5 (C–S), 1,070.4 (C–Br). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 4.14 (s, 6H), 7.50–7.54 (m, 2H), 7.69–7.78 (m, 6H), 7.81 (d, 1H, J=8Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 34.53, 41.97, 42.08, 42.18, 42.29, 42.39, 126.04, 126.40, 129.42, 130.49, 134.23, 134.67, 135.31, 136.82, 137.79, 138.59, 139.77, 152.75, 196. LCMS (m/z): 396.95 [M+H]+. Anal. calcd. for C20H15BrN2S: C, 60.77; H, 3.82; N, 7.09. Anal. found: C, 60.85; H, 3.81; N, 7.08.
5-(4-bromophenyl)-8-methoxy-3-methyl-5,6-dihydroindeno[1,2-d]thiazolo[3,2-a]pyrimidine (4B)
Yellow crystals; yield: 69%; m.p. 198°C–200°C; Rf=0.48 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,018.7 (C–H, aromatic), 2,903.2 (C–H), 1,687.0 (C=N), 1,622.9 (C=C), 1,335.3 (C–N), 1,288.7 (C–O), 763.9 (C–S), 1,071.3 (C–Br). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 3.91 (s, 5H), 4.08 (s, 4H), 7.05 (d, 1H, J=8 Hz), 7.20 (s, 1H), 7.44 (s, 1H), 7.71–7.75 (m, 5H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.39, 39.76, 39.86, 39.97, 40.07, 40.18, 56.32, 110.65, 115.99, 123.49, 126.06, 130.67, 130.88, 132.42, 132.89, 134.77, 136.98, 153.47, 165.53, 191.97. LCMS (m/z): 425.20 [M+]. Anal. calcd. for C21H17BrN2OS: C, 59.30; H, 4.03; N, 6.59. Anal. found: C, 59.20; H, 4.04; N, 6.61.
5-(4-bromophenyl)-3-methyl-5,6-dihydroindeno [1,2-d]thiazolo[3,2-a]pyrimidine (5B)
Yellow crystals; yield: 78%; m.p. 192°C–194°C; Rf=0.54 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,025.1 (C–H, aromatic), 2,928.1 (C–H), 1,693.1 (C=N), 1,622.1 (C=C), 1,324.7 (C–N), 732.9 (C–S), 1,070.1 (C–Br). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 4.14 (s, 6H), 7.51–7.54 (m, 2H), 7.70–7.77 (m, 6H), 7.82 (d, 1H, J=8 Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.32, 39.76, 39.86, 39.97, 40.07, 40.18, 123.83, 124.19, 127.21, 128.28, 132.01, 132.46, 133.10, 134.61, 135.59, 136.38, 137.56, 150.54, 193.79. LCMS (m/z): 395.00 [M+]. Anal. calcd. for C20H15BrN2S: C, 60.77; H, 3.82; N, 7.09. Anal. found: C, 60.89; H, 3.80; N, 7.11.
5-(4-bromophenyl)-8-methoxy-5,6-dihydroindeno[1,2-d]thiazolo[3,2-a]pyrimidine (6B)
Yellow crystals; yield: 75%; m.p. 188°C–190°C; Rf=0.61 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,018.4 (C–H, aromatic), 2,902.6 (C–H), 1,686.9 (C=N), 1,622.6 (C=C), 1,335.0 (C–N), 1,288.6 (C–O), 763.9 (C–S), 1,071.1 (C–Br). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 3.91 (s, 3H), 4.08 (s, 3H), 7.06 (d, 1H, J=8 Hz), 7.20 (s, 1H), 7.44 (s, 1H), 7.72–7.75 (m, 6H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.39, 39.76, 39.86, 39.97, 40.07, 40.18, 56.32, 110.65, 115.99, 123.49, 126.06, 130.67, 130.88, 132.42, 132.89, 134.77, 136.98, 153.47, 165.53, 192.08. LCMS (m/z): 413.25 [M+2]. Anal. calcd. for C20H15BrN2OS: C, 58.40; H, 3.68; N, 6.81. Anal. found: C, 58.31; H, 3.70; N, 6.83.
5-(4-bromophenyl)-8-methoxy-2-methyl-5,6-dihydroindeno[1,2-d]thiazolo[3,2-a]pyrimidine (7B)
Yellow crystals; yield: 77%; m.p. 198°C–200°C; Rf=0.47 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,018.2 (C–H, aromatic), 2,902.5 (C–H), 1,687.1 (C=N), 1,622.7 (C=C), 1,335.7 (C–N), 1,288.7, 1,246.9 (C–O), 763.5 (C–S), 1,071.9 (C–Br). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 3.90 (s, 5H), 4.07 (s, 4H), 7.04 (dd, 1H, J=8 Hz and J=8 Hz), 7.19 (d, 1H, J=8 Hz), 7.44 (s, 1H), 7.70–7.75 (m, 5H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.39, 39.76, 39.86, 39.97, 40.07, 40.18, 56.31, 110.64, 115.98, 123.49, 126.05, 130.66, 130.88, 132.41, 132.89, 134.77, 136.97, 153.46, 165.52, 191.96. LCMS (m/z): 425.00 [M+]. Anal. calcd. for C21H17BrN2OS: C, 59.30; H, 4.03; N, 6.59. Anal. found: C, 59.18; H, 4.04; N, 6.60.
5-(3,4,5-trimethoxyphenyl)-5,6-dihydroindeno[1,2-d] thiazolo[3,2-a]pyrimidine (8B)
Yellow crystals; yield: 72%; m.p. 206°C–208°C; Rf=0.52 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,046.1 (C–H, aromatic), 2,942.6 (C–H), 1,690.6 (C=N), 1,624.7 (C=C), 1,328.0 (C–N), 1,266.5 (C–O), 736.4 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 3.36 (s, 3H), 3.90 (s, 7H), 4.20 (s, 2H), 7.14 (s, 3H), 7.49–7.54 (m, 2H), 7.71–7.74 (m, 2H), 7.80 (s, 1H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.08, 39.76, 39.86, 39.97, 40.07, 40.18, 56.49, 60.64, 108.84, 124.08, 127.16, 128.18, 130.87, 133.79, 134.58, 135.37, 137.75, 139.59, 150.56, 153.51, 193.82. LCMS (m/z): 393.25 [M+H]+. Anal. calcd. for C22H20N2O3S: C, 67.33; H, 5.14; N, 7.14. Anal. found: C, 67.21; H, 5.16; N, 7.16.
8-methoxy-5-(3,4,5-trimethoxyphenyl)-5,6-dihydroindeno[1,2-d]thiazolo[3,2-a]pyrimidine (9B)
Yellow crystals; yield: 66%; m.p. 208°C–210°C; Rf=0.68 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,060.8 (C–H, aromatic), 2,939.2 (C–H), 1,679.6 (C=N), 1,624.6 (C=C), 1,330.4 (C–N), 1,241.5 (C–O), 760.8 (C–S). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 3.35 (s, 3H), 3.74 (s, 6H), 3.91 (s, 4H), 4.15 (s, 2H), 7.04 (dd, 1H, J=8 Hz and J=8 Hz), 7.10 (s, 3H), 7.23 (s, 1H), 7.44 (s, 1H), 7.73 (d, 1H, J=8 Hz). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.22, 39.76, 39.86, 39.97, 40.07, 40.18, 56.31, 56.44, 60.63, 108.59, 110.41, 116.15, 125.87, 131.03, 132.40, 135.15, 139.32, 153.49, 165.38, 192.08. LCMS (m/z): 421.15 [M−H]−. Anal. calcd. for C23H22N2O4S: C, 65.38; H, 5.25; N, 6.63. Anal. found: C, 65.22; H, 5.26; N, 6.64.
5-(4-chlorophenyl)-3-methyl-5,6-dihydroindeno [1,2-d]thiazolo[3,2-a]pyrimidine (10B)
Yellow crystals; yield: 78%; m.p. 182°C–184°C; Rf=0.43 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,066.8 (C–H, aromatic), 3,030.5 (C–H), 1,695.5 (C=N), 1,623.9 (C=C), 1,326.7 (C–N), 729.1 (C–S), 1,090.1 (C–Cl). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 4.13 (s, 6H), 7.49–7.58 (m, 3H), 7.68 (d, 1H, J=8 Hz), 7.72 (d, 1H, J=8 Hz), 7.80–7.83 (m, 4H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.28, 39.76, 39.86, 39.97, 40.07, 40.18, 124.16, 127.19, 128.25, 129.51, 131.90, 132.88, 134.28, 134.93, 135.55, 136.25, 137.56, 150.52, 193.76. LCMS (m/z): 351.02 [M+]. Anal. calcd. for C20H15ClN2S: C, 68.46; H, 4.31; N, 7.98. Anal. found: C, 68.63; H, 4.30; N, 7.96.
5-(4-chlorophenyl)-8-methoxy-3-methyl-5,6-dihydroindeno[1,2-d]thiazolo[3,2-a]pyrimidine (11B)
Yellow crystals; yield: 65%; m.p. 186°C–188°C; Rf=0.48 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,019.3 (C–H, aromatic), 2,967.8 (C–H), 1,695.5 (C=N), 1,623.9 (C=C), 1,326.7 (C–N), 1,249.1 (C–O), 729.1 (C–S), 1,090.1 (C–Cl). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 3.90 (s, 5H), 4.07 (s, 4H), 7.03 (d, 1H, J=8 Hz), 7.18 (s, 1H), 7.44 (s, 1H), 7.56 (d, 2H, J=8 Hz), 7.73–7.79 (m, 3H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.35, 39.77, 39.87, 39.97, 40.08, 40.18, 56.30, 110.63, 115.96, 126.04, 129.47, 130.56, 130.89, 132.66, 134.44, 134.61, 136.85, 153.45, 165.51, 191.94. LCMS (m/z): 381.20 [M+]. Anal. calcd. for C21H17ClN2OS: C, 66.22; H, 4.50; N, 7.35. Anal. found: C, 66.38; H, 4.49; N, 7.34.
5-(4-chlorophenyl)-5,6-dihydroindeno[1,2-d] thiazolo[3,2-a]pyrimidine (12B)
Yellow crystals; yield: 65%; m.p. 176°C–178°C; Rf=0.51 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,067.4 (C–H, aromatic), 3,030.3 (C–H), 1,697.1 (C=N), 1,625.0 (C=C), 1,326.7 (C–N), 729.2 (C–S), 1,093.0 (C–Cl). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 4.13 (s, 3H), 7.50 (s, 1H), 7.54 (s, 1H), 7.57 (d, 2H, J=8 Hz), 7.68 (d, 1H, J=8 Hz), 7.72 (d, 1H, J=8 Hz), 7.80–7.83 (m, 4H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.28, 39.75, 39.86, 39.96, 40.07, 40.17, 124.17, 127.19, 128.26, 129.51, 131.91, 132.88, 134.28, 134.93, 135.55, 136.25, 137.55, 150.52, 193.77. LCMS (m/z): 339.15 [M+2]. Anal. calcd. for C19H13ClN2S: C, 67.75; H, 3.89; N, 8.32. Anal. found: C, 67.61; H, 3.90; N, 8.33.
5-(4-chlorophenyl)-8-methoxy-5,6-dihydroindeno [1,2-d]thiazolo[3,2-a]pyrimidine (13B)
Light brown crystals; yield: 60%; m.p. 184°C–186°C; Rf=0.47 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,058.1, 3,019.1 (C–H, aromatic), 2,943.5 (C–H), 1,688.0 (C=N), 1,622.1 (C=C), 1,304.0 (C–N), 1,249.4 (C–O), 765.2 (C–S), 1,088.4 (C–Cl). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 3.90 (s, 3H), 4.06 (s, 3H), 7.03 (d, 1H, J=8 Hz), 7.18 (s, 1H), 7.44 (s, 1H), 7.56 (d, 2H, J=8 Hz), 7.73–7.79 (m, 4H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.36, 39.77, 39.87, 39.98, 40.09, 40.19, 56.30, 110.63, 115.96, 126.03, 129.47, 130.55, 130.87, 132.65, 134.43, 134.61, 136.85, 153.44, 165.50, 191.93. LCMS (m/z): 339.05 [M+2]. Anal. calcd. for C20H15ClN2OS: C, 65.48; H, 4.12; N, 7.64. Anal. found: C, 65.63; H, 4.10; N, 7.63.
5-(4-chlorophenyl)-2-methyl-5,6-dihydroindeno [1,2-d]thiazolo[3,2-a]pyrimidine (14B)
Yellow crystals; yield: 68%; m.p. 180°C–182°C; Rf=0.51 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,031.5 (C–H, aromatic), 2,923.5 (C–H), 1,695.5 (C=N), 1,623.4 (C=C), 1,326.9 (C–N), 729.2 (C–S), 1,089.7 (C–Cl). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 4.13 (s, 6H), 7.49–7.59 (m, 3H), 7.68–7.73 (m, 2H), 7.80–7.84 (m, 4H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.28, 39.65, 39.86, 39.96, 40.07, 40.28, 124.17, 127.19, 128.26, 129.5, 131.9, 132.88, 134.28, 134.93, 135.55, 136.25, 137.55, 150.52, 193.77. LCMS (m/z): 349.0 [M−H]−. Anal. calcd. for C20H15ClN2S: C, 68.46; H, 4.31; N, 7.98. Anal. found: C, 68.69; H, 4.32; N, 7.99.
5-(4-chlorophenyl)-8-methoxy-2-methyl-5,6-dihydroindeno[1,2-d]thiazolo[3,2-a]pyrimidine (15B)
Yellow crystals; yield: 81%; m.p. 188°C–190°C; Rf=0.66 (SiO2, ethylacetate:n-hexane, 3:7, v/v). IR (KBr, cm−1): 3,019.2 (C–H, aromatic), 2,904.3 (C–H), 1,688.0 (C=N), 1,620.9 (C=C), 1,289.4 (C–N), 1,248.5 (C–O), 765.6 (C–S), 1,088.7 (C–Cl). 1H NMR (DMSO-d6, 800 MHz) δ ppm: 3.90 (s, 5H), 4.07 (s, 4H), 7.05 (d, 1H, J=8 Hz), 7.19 (s, 1H), 7.45 (s, 1H), 7.57 (d, 2H, J=8 Hz), 7.73–7.80 (m, 3H). 13C NMR (DMSO-d6, 200 MHz) δ ppm: 32.35, 39.66, 39.87, 39.97, 40.08, 40.29, 56.30, 110.64, 115.96, 126.04, 129.47, 130.56, 130.89, 132.66, 134.44, 134.61, 136.85, 153.45, 165.51, 191.95. LCMS (m/z): 382.95 [M+2]. Anal. calcd. for C21H17ClN2OS: C, 66.22; H, 4.50; N, 7.35. Anal. found: C, 66.28; H, 4.51; N, 7.33.
SRB assay
The Hep-G2 cells derived from human liver carcinoma were purchased from the National Cell Repository NCCS, Pune, India and later cultured in the lab of ACTREC, Tata Memorial Centre, Navi Mumbai, India for in vitro testing purposes. As per the instructions, the cells were grown in Roswell Park Memorial Institute medium 1640 containing 10% fetal bovine serum. Then, 100 μL cells containing media were seeded into 96-well plates at a density of 5×103 cells/well and incubated at 37°C in a humidified atmosphere containing 5% CO2 for 24 h prior to addition of the experimental drugs. Initially, all the experimental drugs were solubilized in DMSO at 100 mg/mL and further diluted to 1.0 mg/mL using water and stored frozen prior to use. Aliquots of 100 μL (containing 90 μL of medium) of various dilutions were added to appropriate wells, resulting in the required drug concentrations of 10, 20, 40, and 80 μg/mL, and were maintained at standard conditions for 48 h. For each of the experiments, a well-known anticancer drug Adriamycin (ADR) was used as a positive control at concentrations of 10, 20, 40, and 80 μg/mL. The assay was terminated by the gentle addition of 50 μL of cold 30% (w/v) trichloroacetic acid and the mixture was incubated for 60 min at 4°C. The supernatant was discarded and the plates were washed five times using tap water and then air dried. Next, SRB solution (50 μL) at 0.4% (w/v) in 1% acetic acid was added to each well and incubated for 20 min at room temperature. After staining, the unbound dye was recovered and the residual dye was removed by washing five times with 1% acetic acid and then air dried. Subsequently, the bound stain was eluted with 10 mM trizma base and the absorbance was examined at a wavelength of 540 nm on an enzyme-linked immunosorbent assay plate reader.
The growth inhibition of 50% (GI50) was calculated using the formula [(Ti-Tz)/(C-Tz)] ×100%, where Tz represents time zero growth, C is the control growth, and Ti is the test growth at four concentration levels in the presence of the drug.47,48
Molecular docking
The structures of the ligands were designed by Chem Draw Ultra 12.0, and the geometry was optimized six times through GaussView 5.0 (trial version). In addition, the National Centre for Biotechnology Information49 and the Protein Data Bank50 were utilized as chemical sources to obtain the established four homological cancer protein targets, that is, IL-2 (1Z92), IL-6 (1IL6), Caspase-3 (1QX3), and Caspase-8 (1IBC). DS Visualizer software51 was used to remove the co-crystal with the assigned target. Next, the active site was identified using the CASTp database.52 For a particular system, validation of the docking protocol was performed using re-docking studies, where we found excellent agreement between the localization of ligands upon docking, and from the crystal structure of the assigned targets. We demonstrated the reliability and quality of the docking method in reproducing experimentally observed binding-mode-assigned targets. In addition, the docking analyses of the test set were carried out using Autodock4.1,53 together with the Lamarkian genetic algorithm for automated flexible ligand docking, and the binding energy was estimated as negative kcal/mol. In addition, probable H-bonds and π-bonds were also assessed.54−56
Prediction of ADME properties
The ADME and drug-like properties of selected ligands were predicted by employing MedChem Designer and QikProp tools. Chemical structures were optimized with LigPrep. Additionally, the percentage absorbance (% ABS) and Lipinski’s violation were evaluated for this study.57
MD simulation
The nature of the inhibitor used on the active site domain of IL-6 was investigated by means of MD simulation studies. The energies of dock configurations were minimized to eliminate the unfavorable atomic contacts as starting conformations for dynamic simulation in Elmar Krieger MD simulation tools (trial version).58 An AMBER03 force field was assigned to execute a real-time MD simulation.59 The complex was solvated through an HOH model at density =0.997 g/L inside the 10Å simulation cell boundary and then adjusted to the physiologic pH at 7.4. Moreover, physiologic NaCl solution with 0.9% mass fraction Na+ and Cl− ion concentration was used to maintain and neutralize the simulation cell boundary. Then, the MD simulation was run for 3,000 ps at a temperature of 298 K and 1 bar pressure to obtain snapshot (sim) trajectories. Finally, the sim trajectories were analyzed and the resulting data were plotted using Sigma Plot 11.0 tools.
Results and discussion
Design
These two core structural motifs are formed by the fusion of three biodynamic privileged heterosystems in such a way that one nitrogen atom occupies a bridge head position, therefore being common to both the heterocyclic rings, that is, the thiazole and the pyrimidine rings, and possessing unique structural diversity. Thiazolo[2,3-b]quinazoline and thiazolo[3,2-a]pyrimidine, as heterocyclic skeletons, exhibit a diverse range of pharmacologic activities. In view of the great medicinal importance associated with these remarkable structures, we are interested in studying MDRs with the aim of evolving an efficient and highly convergent synthetic procedure for the library of densely functionalized 5H-benzo[h]thiazolo[2,3-b]quinazoline (1A−15A) and indeno[1,2-d]thiazolo[3,2-a]pyrimidine (1B−15B) analogs in a single operation.
We initiated our study of the proposed method with 10 mol% p-TSA as the acidic catalyst (Scheme 5) by investigating the conditions for the reaction comprising 5-methoxy-2,3-dihydro-1H-inden-1-one (1 mmol), 4-bromobenzaldehyde (1 mmol), 4-methylthiazol-2-amine (1 mmol), and EtOH (5.0 mL) at 60°C to yield the corresponding product (4B, 51% yield) as a model in 4.5 h (entry 1). With this finding in hand, when the acidic catalyst was changed from 10% to 20% and the temperature from 60°C to 80°C, the product yield increased to 69% after 3 h reaction time (entry 4). We examined the reaction in different organic solvents, including EtOH, methanol (MeOH), ethylacetate (EtOAc), acetonitrile (CH3CN), and toluene (PhCH3) under varied temperature conditions (Table 1). The proposed reaction is optimized for the amount of acidic catalyst required, as well as for temperature, to improve the yields of the desired molecules (entries 1–8), and the optimum amount was found to be 20 mol% p-TSA in EtOH at 80°C. The best results were obtained for 20 mol% p-TSA in 3 h at 80°C (entry 4, 69% yield).
Scheme 5.

Optimization of the model reaction.
Note: Substituted α-indanone (a′), substituted aromatic aldehydes (b) and distinctive 2-aminothiazoles (c).
Table 1.
Optimization of catalytic amount of p-TSA, nonaqueous solvents, and temperature in one-pot synthesis of the model reactiona
| Entry | p-TSA (mol%) | Solvents | Temperature (°C) | Time (h) | Yield (%)b |
|---|---|---|---|---|---|
| 1 | 10 | EtOH | 60 | 4.5 | 51 |
| 2 | 10 | EtOH | 80 | 4.0 | 56 |
| 3 | 20 | EtOH | 60 | 3.5 | 63 |
| 4 | 20 | EtOH | 80 | 3.0 | 69 |
| 5 | 10 | MeOH | 60 | 5.0 | 49 |
| 6 | 10 | MeOH | 80 | 4.5 | 54 |
| 7 | 20 | MeOH | 60 | 4.0 | 54 |
| 8 | 20 | MeOH | 80 | 3.5 | 60 |
| 9 | 20 | EtOAc | Reflux | 5.0 | 33 |
| 10 | 20 | CH3CN | Reflux | 5.0 | 29 |
| 11 | 20 | PhCH3 | Reflux | 5.0 | 27 |
Notes:
Reaction conditions: 5-methoxy-2,3-dihydro-1H-inden-1-one (1 mmol), 4-bromobenzaldehyde (1 mmol), 4-methylthiazol-2-amine (1 mmol), solvent (5.0 mL), and p-TSA.
Isolated pure yield. Bold entry signifies the optimized reaction conditions.
Abbreviations: CH3CN, acetonitrile; EtOAc, ethylacetate; EtOH, ethanol; MeOH, methanol; PhCH3, toluene; p-TSA, p-toluenesulfonic acid.
In order to explore the scope of these conditions, we embarked on the synthesis of a panel of novel 5H-benzo[h] thiazolo[2,3-b]quinazoline (1A–15A) and indeno[1,2-d] thiazolo[3,2-a]pyrimidine (1B–15B) analogs bearing a bridgehead nitrogen atom from a range of substrates by the advancement of facile and efficient MDRs of highly substituted α-tetralone (a) or α-indanone (a′) with some aromatic aldehydes (b) and distinctive 2-aminothiazoles (c) in EtOH in the presence of 20 mol% p-TSA. The α-tetralone, 6-methoxy-1-tetralone, 7-methoxy-1-tetralone or α-indanone, 5-methoxy-1-indanone, and the substituted benzaldehydes with 4-bromo, trimethoxy, 3-hydroxy-4-methoxy (vanillin), 4-chloro were reacted with distinctive 2-aminothiazole, 4-methylthiazol-2-amine, 5-methylthiazol-2-amine to afford a library of proposed target molecules, that is, (1A–15A) and (1B–15B), respectively.
The one-pot, three-component reaction went smoothly in EtOH in the presence of p-TSA and gave the targeted compounds (1A–15A) and (1B–15B) in impressive yields up to 86%. The various substitutions of synthesized derivatives are given in Table 2A and B. All the newly synthesized compounds were authenticated on the basis of Fourier transform infrared spectroscopy, LCMS, and 1H and 13C NMR spectral and elemental analyses.
Table 2.
Various substitutions and in vitro cytotoxicity data of synthesized derivatives against human hepatoma (Hep-G2) cell lines: A (1A–15A) and B (1B–15B)
| Comp code | X1 | X2 | R1 | R2 | R3 | Y1 | Y2 | *GI50 (µg/mL) | LC50 (µg/mL) | TGI (µg/mL) |
|---|---|---|---|---|---|---|---|---|---|---|
| A | ||||||||||
| 1A | –H | –H | –H | –Br | –H | –H | –H | >80 | NE | NE |
| 2A | –OCH3 | –H | –H | –Br | –H | –H | –CH3 | NE | NE | NE |
| 3A | –H | –OCH3 | –OCH3 | –OCH3 | –OCH3 | –H | –H | NE | NE | NE |
| 4A | –H | –OCH3 | –OCH3 | –OH | –H | –H | –H | <10 | >80 | >80 |
| 5A | –OCH3 | –H | –H | –Br | –H | –H | –H | >80 | >80 | >80 |
| 6A | –OCH3 | –H | –OCH3 | –OCH3 | –OCH3 | –H | –H | <10 | NE | NE |
| 7A | –H | –H | –H | –OH | –H | –H | –H | 76.1 | NE | NE |
| 8A | –H | –OCH3 | –H | –Cl | –H | –CH | –H | >80 | NE | NE |
| 9A | –OCH3 | –H | –H | –Cl | –H | –CH | –H | >80 | NE | NE |
| 10A | –H | –H | –H | –Cl | –H | –CH | –H | >80 | NE | NE |
| 11A | –H | –OCH3 | –H | –Cl | –H | –H | –H | >80 | NE | NE |
| 12A | –H | –H | –H | –Cl | –H | –H | –H | >80 | NE | NE |
| 13A | –H | –OCH3 | –H | –Cl | –H | –H | –CH | >80 | NE | NE |
| 14A | –OCH3 | –H | –H | –Cl | –H | –H | –CH | >80 | NE | NE |
| 15A | –H | –H | –H | –Cl | –H | –H | –CH | .80 | NE | NE |
|
| ||||||||||
| Comp code | X | R1 | R1 | R2 | R3 | Y1 | Y2 | *GI50 (µg/mL) | LC50 (µg/mL) | TGI (µg/mL) |
|
| ||||||||||
| B | ||||||||||
| 1B | –H | –H | –H | –Br | –H | –H | –H | >80 | NE | NE |
| 2B | –H | –Cl | –H | –H | –H | –H | –H | >80 | NE | NE |
| 3B | –H | –H | –H | –Br | –H | –H | –CH3 | >80 | NE | NE |
| 4B | –OCH3 | –H | –H | –Br | –H | –CH3 | –H | >80 | NE | NE |
| 5B | –H | –H | –H | –Br | –H | –CH3 | –H | >80 | NE | NE |
| 6B | –OCH3 | –H | –H | –Br | –H | –H | –H | >80 | NE | NE |
| 7B | –OCH | –H | –H | –Br | –H | –H | –CH3 | >80 | NE | NE |
| 8B | –H | –H | –OCH3 | –OCH3 | –OCH3 | –H | –H | >80 | NE | NE |
| 9B | –OCH3 | –H | –OCH3 | –OCH3 | –OCH3 | –H | –H | >80 | NE | NE |
| 10B | –H | –H | –H | –Cl | –H | –CH3 | –H | >80 | NE | NE |
| 11B | –OCH3 | –H | –H | –Cl | –H | –CH3 | –H | >80 | NE | NE |
| 12B | –H | –H | –H | –Cl | –H | –H | –H | >80 | NE | NE |
| 13B | –OCH3 | –H | –H | –Cl | –H | –H | –H | >80 | NE | NE |
| 14B | –H | –H | –H | –Cl | –H | –H | –CH3 | >80 | NE | NE |
| 15B | –OCH3 | –H | –H | –Cl | –H | –H | –CH3 | >80 | NE | NE |
| ADR | <10 | NE | <10 | |||||||
Notes:
GI50 value of ≤10 μg/mL (or 1 μM) is considered to demonstrate activity in case of pure compounds (synthetic compound). Bold values indicate the active compounds.
Abbreviations: ADR, Adriamycin, positive control compound; GI50, concentration of drug causing 50% inhibition of cell growth; LC50, concentration of drug causing 50% cell kill; NE, “not effective” even at the concentration >80 μg/mL; TGI, concentration of drug causing total inhibition of cell growth.
Desired compounds have also been synthesized by a multistep synthesis involving the reaction of distinctive 2-aminothiazoles with the appropriate benzylidene-tetralones/indanones achieved by the reaction of highly substituted α-tetralone or α-indanone with different aromatic aldehydes, piperidine and glacial acetic acid in EtOH. This reaction proceeds in two steps with a comparatively longer reaction time (18–20 h) and giving moderate yields (40%–50%).
Plausible mechanism
These reactions presumably proceed through a Knoevenagel condensation between substituted α-tetralone or α-indanone and some appropriate aromatic aldehydes in the first step to construct α,β-unsaturated ketones, respectively, which undergo a Michael-type addition approach with the nucleophilic endocyclic nitrogen of the distinctive 2-aminothiazole under the maintained reaction conditions. Then, successive intramolecular cyclization occurred with the loss of a water molecule to give 5H-benzo[h] thiazolo[2,3-b]quinazolines (1A−15A) and indeno[1,2-d] thiazolo[3,2-a]pyrimidines (1B−15B). In this setting, the domino approach and the reaction sequence of Knoevenagel condensation/Michael-type addition/intramolecular cyclization were done in a single step in a one-pot procedure in EtOH. The plausible mechanism for the reaction is delineated in Scheme 6.
Scheme 6.

Plausible scenario to account for the formation of (1A–15A) and same for the preparation of (1B–15B).
Note: Substituted α-tetralone (a), substituted aromatic aldehydes (b) and distinctive 2-aminothiazoles (c).
In vitro antitumor screening
A library of 30 5H-benzo[h]thiazolo[2,3-b]quinazoline and indeno[1,2-d]thiazolo[3,2-a]pyrimidine derivatives were screened against Hep-G2 cells. The inhibition activities (GI50) of the synthetic compounds 1A–15A, 1B–15B, and ADR on Hep-G2 cells are summarized in Table 2A and B. The effects of treatment with the most active members (4A and 6A) of this study and with ADR on Hep-G2 are shown by the plotted growth curve (Figure 1) and the microscopic images (Figure 2).
Figure 1.

Growth Curve of (1A–15A and 1B–15B): Human Hepatoma Cell Line (Hep-G2).
Abbreviation: ADR, Adriamycin.
Figure 2.

Microscopic pictures showing the effect of treatments with the active compounds (A) 4A and (B) 6A and the reference drug (C) ADR on Hep-G2 human liver cancer cell line.
Note: Scale bar 50 μm.
Abbreviation: ADR, Adriamycin.
Close examination of the activity data (Table 2A and B) revealed that the presence of –OH and –OCH3 groups in the phenyl ring, preferably at the R1, R2, and R3 positions, in conjugation with the introduction of the –OCH3 group in the tetralone ring at X1 or X2 is crucial for inhibitory activity. Substitution of 3-methoxy-4-hydroxy on the phenyl ring and 6-methoxy on the tetralone ring led to compound 4A, while substitutions of 3,4,5-trimethoxy on the phenyl ring and 7-methoxy on the tetralone ring led to compound 6A with better cytotoxicity (GI50 <10 μg/mL). In contrast, substitutions with –Br, –Cl, and –OH groups on the phenyl ring at the R2 position were ineffective against Hep-G2 cells. The methyl substitution on the thiazole ring did not contribute to producing inhibitory activity. Replacement of the tetralone ring with the indanone ring system, that is, 1B–15B, reduced the anticancer activity against Hep-G2 human liver cancer cell lines.
The growth curve of the in vitro findings suggested that the percentage growth inhibition values of the potent compounds 4A and 6A was ≤50% at 10 μg/mL concentration, but they did not move toward a negative value. Therefore, it might be expected in future that both compounds could lead to the death of cancerous cells while minimizing that of normal cells.
Molecular docking studies
Computational analysis was performed using various well-known liver cancer targets, specifically IL-2, IL-6, Caspase-3, and Caspase-8, through Autodoc 4.1, together with Lamarkian genetic algorithm for automated docking of flexible ligands. Molecular docking poses of both the potent compounds 4A and 6A with the related targets IL-2, IL-6, Caspase-3, and Caspase-8 are presented in Figure 3 which indicates the interactions of amino acids with ligands, H-bonds, and π-bonds as well as the bond lengths. The docking affinities (kcal/mol), numbers of H-bonds and π-bonds, and the interactions of amino acids of the potent members with them are given in Table 3. Compounds 4A and 6A exhibited strong affinity (binding interaction energy ranged from −6.1 to −7.87 kcal/mol), while others showed good binding affinity with preferred molecular targets. Compound 4A displayed excellent affinity with IL-6 (−7.87 kcal/mol, 2H and 4π-bonds), IL-2 (−7.68 kcal/mol, 5π-bonds), Caspase-3 (−6.69 kcal/mol, 1H and 7π-bonds), and Caspase-8 (−6.48 kcal/mol, 1π-bond). A similar pattern was observed for compound 6A with IL-2 (−7.63 kcal/mol, 1H and 1π-bonds), IL-6 (−7.43 kcal/mol, 2H and 4π-bonds), and Caspase-8 (−6.76 kcal/mol, 1H and 5π-bond), but with less affinity for Caspase-3 (−6.14 kcal/mol, 1H and 6π-bonds). Moreover, both the active members of the present study have strong affinity binding to IL-2, Caspase-3, Caspase-8, and the binding energies, particularly for the IL-6 receptor site, are predominantly high (from −7.43 to −7.87 kcal/mol). Accordingly, it might be assumed that the promising cytotoxic properties of these compounds, which were indicated by the in vitro antitumor activity on Hep-G2 cells, might be better mediated through an IL-6-dependent mechanism (Table 3).
Figure 3.

Docking images of active compounds (A, C, E, G) 4A and (B, D, F, H) 6A for caspase-3, caspase-8, IL-2 and IL-6.
Abbreviation: IL, interleukin.
Table 3.
Docking affinity of active compounds with assigned anticancer receptors
| Ligands | Receptors | Binding affinity (kcal/mol) | Amino acids involved in interaction | H-bonds | π-bonds |
|---|---|---|---|---|---|
| 4A | IL-2 | −7.680 | ARG A 38 THR A 41 ASP B 6 ALA B 13 THR B 14 PHE B 15 LYS B 16 TYR B 20 GLU B 106 THR B 115 GLU B 116 ARG B 117 ILE B 118 TYR B 119 PHE B 121 | 0 | 5 |
| IL-6 | −7.871 | ASN A 62 LEU A 63 PRO A 66 LYS A 67 MET A 68 ALA A 69 PHE A 75 LEU A 166 ARG A 169 SER A 170 GLU A 173 PHE A 174 SER A 177 | 2 | 4 | |
| Caspase-3 | −6.695 | THR A 62 SER A 65 TYR A 204 TRP A 206 ARG A 207 SER A 209 PHE A 250 SER A 251 PHE A 256 HOH A 645 HOH A 684 HOH A 696 HOH A 708 HOH A 733 HOH A 736 | 1 | 7 | |
| Caspase-8 | −6.480 | SER A 65 TYR A 204 TRP A 206 ARG A 207 ASN A 208 SER A 209 TRP A 214 GLU A 248 SER A 249 PHE A 250 SER A 251 | 0 | 1 | |
| 6A | IL-2 | −7.630 | ARG A 38 THR A 41 ASP B 6 GLU B 9 PHE B 15 LYS B 16 ALA B 17 TYR B 20 GLU B 113 THR B 115 GLU B 116 ARG B 117 ILE B 118 TYR B 119 PHE B 121 | 1 | 1 |
| IL-6 | −7.438 | GLU A 43 THR A 44 LYS A 47 SER A 48 MET A 50 ARG A 105 PHE A 106 GLU A 107 SER A 108 GLN A 157 TRP A 158 ASP A 161 THR A 164 | 0 | 3 | |
| Caspase-3 | −6.142 | SER A 65 TYR A 204 TRP A 206 ARG A 207 ASN A 208 SER A 209 TRP A 214 SER A 249 PHE A 250 SER A 251 PHE A 256 HOH A 645 HOH A 665 HOH A 684 HOH A 696 HOH A 736 | 1 | 6 | |
| Caspase-8 | −6.760 | SER A 63 ARG A 64 SER A 65 TYR A 204 SER A 205 TRP A 206 ARG A 207 ASN A 208 SER A 209 TRP A 214 GLU A 248 SER A 249 PHE A 250 SER A 251 | 1 | 5 |
Abbreviation: IL, interleukin.
Prediction of ADME properties
A study of pharmacokinetic parameters was carried out utilizing QikProp version 4.5 tools to predict the ADME properties of both series (1A–15A) and (1B–15B) and the ranges for the calculated properties of all members, along with their average values are summarized in Table 4. In addition, we also calculated % ABS, number of H-bond acceptors (n–OH), number of H–bond donors (n-OHNH), octanol/water partition coefficients (QPlogPo/w), and Lipinski’s violation. Interestingly, it was found that the % ABS obtained for all members was 100% and the QPlogPo/w prediction was found to be within the accepted range of −2.0 to 6.5. Moreover, all members followed the violated Lipinski parameters.
Table 4.
Pharmacokinetic parameters important for oral bioavailability and protein binding parameters of synthesized compounds
| Comp | % ABS | MW | Volume | Donor HB | Acceptor HB | Lipinski’s violation | QPlogPo/w |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Rule | <80% is high, >25% is poor | <500 | 500.0–2,000.0 | <5 | <10 | ≤1 | −2.0 to 6.5 |
| 1A | 100 | 395.316 | 1,041.638 | 0 | 1 | 1 | 6.493 |
| 2A | 100 | 439.369 | 1,168.198 | 0 | 1.75 | 1 | 6.799 |
| 3A | 100 | 436.525 | 1,261.8 | 0 | 4 | 1 | 6.007 |
| 4A | 100 | 392.472 | 1,138.921 | 1 | 3.25 | 1 | 5.196 |
| 5A | 100 | 425.342 | 1,105.697 | 0 | 1.75 | 1 | 6.476 |
| 6A | 100 | 436.525 | 1,261.747 | 0 | 4 | 1 | 6.006 |
| 7A | 100 | 332.419 | 1,011.802 | 1 | 1.75 | 1 | 5.108 |
| 8A | 100 | 394.92 | 1,041.638 | 0 | 1 | 1 | 6.493 |
| 9A | 100 | 394.92 | 1,159.867 | 0 | 1.75 | 1 | 6.724 |
| 10A | 100 | 364.892 | 1,065.077 | 0 | 1 | 1 | 6.576 |
| 11A | 100 | 380.891 | 1,097.431 | 0 | 1.75 | 1 | 6.402 |
| 12A | 100 | 350.865 | 1,033.256 | 0 | 1 | 1 | 6.419 |
| 13A | 100 | 394.918 | 1,159.867 | 0 | 1.75 | 1 | 6.724 |
| 14A | 100 | 394.918 | 1,159.815 | 0 | 1.75 | 1 | 6.724 |
| 15A | 100 | 364.892 | 1,095.69 | 0 | 1 | 1 | 6.741 |
| 1B | 100 | 381.289 | 972.937 | 0 | 1 | 1 | 6.036 |
| 2B | 100 | 336.838 | 942.479 | 0 | 1 | 1 | 5.672 |
| 3B | 100 | 395.316 | 1,037.84 | 0 | 1 | 1 | 6.374 |
| 4B | 100 | 425.342 | 1,080.745 | 0 | 1.75 | 1 | 6.247 |
| 5B | 100 | 395.316 | 1,016.635 | 0 | 1 | 1 | 6.264 |
| 6B | 100 | 411.315 | 1,037.038 | 0 | 1.75 | 1 | 6.018 |
| 7B | 100 | 425.342 | 1,101.954 | 0 | 1.75 | 1 | 6.357 |
| 8B | 100 | 392.472 | 1,122.39 | 0 | 3.25 | 1 | 5.516 |
| 9B | 100 | 422.498 | 1,177.838 | 0 | 4 | 1 | 5.445 |
| 10B | 100 | 350.865 | 1,008.558 | 0 | 1 | 1 | 6.191 |
| 11B | 100 | 380.891 | 1,072.663 | 0 | 1.75 | 1 | 6.174 |
| 12B | 100 | 336.838 | 964.548 | 0 | 1 | 1 | 5.961 |
| 13B | 100 | 366.864 | 1,028.656 | 0 | 1.75 | 1 | 5.943 |
| 14B | 100 | 350.865 | 1,029.454 | 0 | 1 | 1 | 6.3 |
| 15B | 100 | 380.891 | 1,093.555 | 0 | 1.75 | 1 | 6.282 |
Abbreviations: ABS, absorbance; QPlogPo/w, octanol/water partition coefficients; MW, molecular weight; HB, H-bonds.
MD simulation
MD simulation was performed on the active inhibitor 4A with IL-6 to explore the binding poses in depth. Compound 4A displayed high binding affinity (low docking energy) and stable complex for IL-6 receptor. Therefore, we decided to investigate the influence of compound 4A on the active site of IL-6 receptor. The stability of the system under simulation was assessed using the root-mean-square deviation (RMSD) of atomic positions of the backbone atoms, relative to the starting structures. The binding energy versus time plot indicates the change in conformation and, therefore, the binding energy, with respect to time. As shown in Figures 4 and 5, the RMSD, the binding energy, and the potential energy of IL-6 with compound 4A, including the complex, were computed using MD trajectory frames. Furthermore, we monitored the structural stability of the backbone structure throughout the process through a graphic profile. We observed a dramatic fluctuation in RMSD at 1,000 ps (time), whereas no significant fluctuations were observed after 1,500 ps (time), and it achieved an almost steady state, indicating the stability of the backbone structure with the ligand at about 1,500 ps time in the MD simulation. The binding energy and the potential energy of the complex were estimated as a function of time, which indicated that the potential energy (kJ/mol) did not show further fluctuations after 100 ps, whereas the average complex binding energy was observed to be about −0.9 kg/mol. The fluctuations in the residue of the backbone structure are illustrated in Figure 4. In summary, we examined these data, where we found the structural stability of compound 4A with the active site domain of IL-6 receptor.
Figure 4.
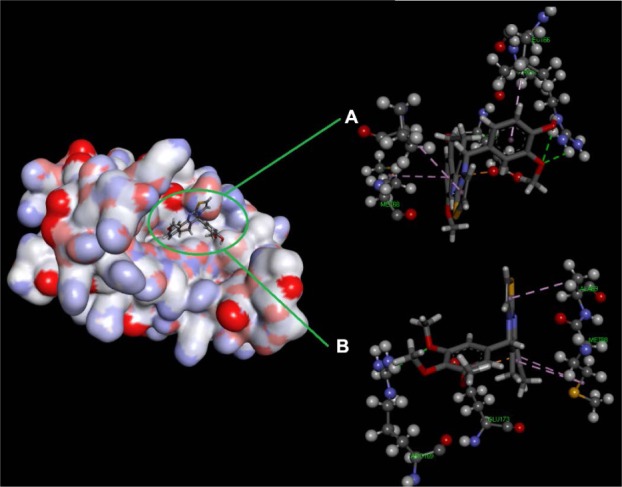
Docking complex of 4A with the IL-6 receptor.
Notes: Structural conformational changes (A) before MD simulation and (B) after MD simulation: backbone of the active site domain complex, which shows the contraction of ligand with amino acids residue.
Abbreviations: IL, interleukin; MD, molecular dynamic.
Figure 5.

The stability profile of ligand and protein complex under the MD simulation.
Notes: (A) Average RMSD versus time graph that shows the convergence of simulated structure toward an equilibrium state with respect to the reference structure (initial structure). (B) Potential energy of complex versus time graph that shows the stability of ligand and protein complex and (C) binding energy of complex versus time graph that also shows the stability of ligand and protein complex.
Abbreviations: MD, molecular dynamic; RMSD, root-mean-square deviation.
Conclusion
We have successfully devised a highly proficient and operationally simple metal-free, one-pot MDR for obtaining a series of novel, hitherto unreported, 5H-benzo[h]thiazolo[2,3-b] quinazoline (1A–15A) and indeno[1,2-d]thiazolo[3,2-a] pyrimidine (1B–15B) analogs displaying potent anticancer activity against Hep-G2 cells as an alternative approach for the treatment of HCC. Considering this library of novel compounds, we concluded that 5H-benzo[h]thiazolo[2,3-b] quinazoline, along with a substituted phenyl ring (hydrophobic side chain) establishes an important pharmacophoric structure, and the R1, R2, and R3 positions of the phenyl ring, as well as the X1 and X2 positions of the tetralone ring system, are the key reactive sites that could be modified with various groups to elicit greater antitumorigenic potential.
We have also demonstrated that substitutions with more electronegative groups (–OH, –OCH3) on the hydrophobic side chain directly linked to thiazolo[2,3-b]quinazoline led to active members 4A and 6A, eliciting enhanced anti-tumorigenic activity, with GI50 <10 μg/mL, which was confirmed by docking analyses. Additionally, 3-methoxy-4- hydroxyphenyl–substituted 5H-benzo[h]thiazolo[2,3-b] quinazoline led to 4A, which displayed excellent antitum-origenic activity among a library of 30 novel synthesized compounds with values of GI50 <10 μg/mL. Various computational approaches demonstrated effective oral absorption and protein binding. These compounds, therefore, might be stable in some form of pharmaceutical dosage. Additionally, MD simulation supported our hypothesis regarding the stability of compound 4A with IL-6 protein during the simulation run. The results from the MD simulation run of the active inhibitor showed very less fluctuation with the active site domain of IL-6 and it achieved an almost steady state. We concluded that the compounds would be bound stably to IL-6. Further studies on the biologic activities of the potent compounds are underway in our laboratory.
This approach, using a one-pot, multicomponent reaction sequence involving a domino Knoevenagel condensation/Michael-type addition followed by an intramolecular cyclization, where the desired molecules are obtained in a one-flask domino manner with atom and step economy in impressive yields (up to 86%) from readily available and low-priced starting materials, is a resource-effective and desirable route. Additionally, the target compounds were also synthesized by conventional two-step reactions, which were compared with this novel approach on the basis of obtained yields (40%–50%). Finally, we conclude that this newly developed method is both more useful and more profitable in each and every aspect, compared with the conventional route. Due to the importance of these two core structural motifs, that is, 5H-benzo[h]thiazolo[2,3-b]quinazoline and indeno[1,2-d] thiazolo[3,2-a]pyrimidine, especially in the areas of pharmaceutical and medicinal chemistry, we suggest that the protocols that we have outlined should open up new avenues of investigation, with enormous implications for achieving diversity in chemical synthesis.
Acknowledgments
The authors would like to express their gratitude to Babasaheb Bhimrao Ambedkar University (BBAU), Lucknow, India for providing the support and research facilities. The authors also express their sincere gratitude to the Centre of Biomedical Research (CBMR), Lucknow for providing the NMR facilities and the Advanced Centre for Treatment, Research and Education in Cancer (ACTREC), Tata Memorial Centre, Navi Mumbai, India for providing the in vitro SRB assay for anticancer screening of drugs. The authors also acknowledge Mr Pranesh Kumar for helping them during docking analysis.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.World Health Organization Cancer fact sheet, February 2017. [Accessed on May 19, 2017]. Available from: http://www.who.int/mediacentre/factsheets/fs297/en/
- 2.Zhang B, Wang N, Zhang C, et al. Novel multi-substituted benzyl acridone derivatives as survivin inhibitors for hepatocellular carcinoma treatment. Eur J Med Chem. 2017;129:337–348. doi: 10.1016/j.ejmech.2017.02.027. [DOI] [PubMed] [Google Scholar]
- 3.Eldehna WM, Fares M, Ibrahim HS, et al. Indoline ureas as potential anti-hepatocellular carcinoma agents targeting VEGFR-2: Synthesis, in vitro biological evaluation and molecular docking. Eur J Med Chem. 2015;100:89–97. doi: 10.1016/j.ejmech.2015.05.040. [DOI] [PubMed] [Google Scholar]
- 4.Gregoric T, Sedic M, Grbcic P, et al. Novel pyrimidine-2,4-dione 1,2,3-triazole and furo[2,3-d]pyrimidine-2-one 1,2,3-triazole hybrids as potential anti-cancer agents: Synthesis, computational and X-ray analysis and biological evaluation. Eur J Med Chem. 2017;125:1247–1267. doi: 10.1016/j.ejmech.2016.11.028. [DOI] [PubMed] [Google Scholar]
- 5.Qi X, Xiang H, Yang C. Synthesis of functionalized chromeno[2,3 b] pyrrol-4(1H) ones by silver-catalyzed cascade reactions of chromones/thiochromones and isocyanoacetates. Org Lett. 2015;17(22):5590–5593. doi: 10.1021/acs.orglett.5b02795. [DOI] [PubMed] [Google Scholar]
- 6.Nagaraju A, Ramula BJ, Shukla G, et al. A facile and highly convergent approach to thiazolo[3,2-a]pyridines via one-pot multicomponent domino reaction under metal-free and solvent-free conditions. Tetrahedron. 2015;71(21):3422–3427. [Google Scholar]
- 7.Kumar M, Sharma K, Sharma DK. Diversity oriented one-pot three-component sequential synthesis of annulated benzothiazoloquinazolines. Org Med Chem Lett. 2012;2(1):10. doi: 10.1186/2191-2858-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuramoto M, Sakata Y, Terai K, et al. Preparation of leukotriene B(4) inhibitory active 2- and 3-(2-aminothiazol-4-yl)benzo[b]furan derivatives and their growth inhibitory activity on human pancreatic cancer cells. Org Biomol Chem. 2008;6(15):2772–2781. doi: 10.1039/b803313g. [DOI] [PubMed] [Google Scholar]
- 9.Misra RN, Xiao HY, Kim KS, et al. N-(cycloalkylamino)acyl-2- aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarbox-amide (BMS-387032), a highly efficacious and selective antitumor agent. J Med Chem. 2004;47(7):1719–1728. doi: 10.1021/jm0305568. [DOI] [PubMed] [Google Scholar]
- 10.Patel AB, Chikhalia KH, Kumari P. An efficient synthesis of new thiazolidin-4-one fused s-triazines as potential antimicrobial and anticancer agents. J Saudi Chem Soc. 2014;18(5):646–656. [Google Scholar]
- 11.Rahman MU, Jeyabalan G, Saraswat P, Parveen G, Khan S, Yar MS. Quinazolines and anticancer activity: a current perspective. Synth Commun. 2016;47(5):379–408. [Google Scholar]
- 12.Monchaud D, Allain C, Teulade-Fichou MP. Development of a fluorescent intercalator displacement assay (G4-FID) for establishing quadruplex-DNA affinity and selectivity of putative ligands. Bioorg Med Chem Lett. 2016;26(18):4842–4845. doi: 10.1016/j.bmcl.2006.06.067. [DOI] [PubMed] [Google Scholar]
- 13.Chen X, Du Y, Sun H, Wang F, Kong L, Sun M. Synthesis and biological evaluation of novel tricyclic oxazine and oxazepine fused quinazolines. Part 1: erlotinib analogs. Bioorg Med Chem Lett. 2014;24(3):884–887. doi: 10.1016/j.bmcl.2013.12.079. [DOI] [PubMed] [Google Scholar]
- 14.Zahedifard M, Faraj FL, Paydar M, et al. Synthesis of apoptotic new quinazolinone-based compound and identification of its underlying mitochondrial signalling pathway in breast cancer cells. Curr Pharm Des. 2015;21(23):3417–3426. doi: 10.2174/1381612821666150326104952. [DOI] [PubMed] [Google Scholar]
- 15.Chandregowda V, Kush AK, Chandrasekara Reddy G. Synthesis and in vitro antitumor activities of novel 4-anilinoquinazoline derivatives. Eur J Med Chem. 2014;44(7):3046–3055. doi: 10.1016/j.ejmech.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 16.Alagarsamy V, Raja Solomon V, Dhanabal K. Synthesis and pharmacological evaluation of some 3-phenyl-2-substituted-3H-quinazolin- 4-one as analgesic, anti-inflammatory agents. Bioorg Med Chem. 2007;15(1):235–241. doi: 10.1016/j.bmc.2006.09.065. [DOI] [PubMed] [Google Scholar]
- 17.Nandy P, Vishalakshi MT, Bhat AR. Synthesis and antitubercular activity of Mannich bases of 2-methyl-3H-quinazolin-4-ones. Indian J Heterocycl Chem. 2006;15(3):293–294. [Google Scholar]
- 18.Georgey H, Abdel-Gawad N, Abbas S. Synthesis and anticonvulsant activity of some quinazolin-4-(3H)-one derivatives. Molecules. 2008;13(10):2557–2569. doi: 10.3390/molecules13102557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verhaeghe P, Azas N, Gasquet M, et al. Synthesis and antiplasmodial activity of new 4-aryl-2-trichloromethylquinazolines. Bioorg Med Chem Lett. 2008;18(1):396–401. doi: 10.1016/j.bmcl.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 20.Ismail MA, Barker S, Abau el-Ella DA, Abouzid KA, Toubar RA, Todd MH. Design and synthesis of new tetrazolyl- and carboxybiphe-nylylmethyl quinazoline derivatives as angiotensin II AT1 receptor antagonists. J Med Chem. 2006;49(5):1526–1535. doi: 10.1021/jm050232e. [DOI] [PubMed] [Google Scholar]
- 21.Malamas MS, Millen J. Quinazoline acetic acids and related analogues as aldose reductase inhibitors. J Med Chem. 1991;34(4):1492–1503. doi: 10.1021/jm00108a038. [DOI] [PubMed] [Google Scholar]
- 22.Roopan SM, Sompalle R. Synthetic chemistry of pyrimidines and fused pyrimidines: a review. Synth Commun. 2016;46(8):645–672. [Google Scholar]
- 23.Al-Omary FA, Hassan GS, El-Messery SM, El-Subbagh HI. Substituted thiazoles V. Synthesis and antitumor activity of novel thiazolo[2,3-b] quinazoline and pyrido[4,3-d]thiazolo[3,2-a]pyrimidine analogues. Eur J Med Chem. 2012;47(1):65–72. doi: 10.1016/j.ejmech.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 24.Keshari AK, Singh AK, Saha S. Bridgehead nitrogen thiazolo[3,2-a] pyrimidine: a privileged structural framework in drug discovery. Mini Rev Med Chem. 2017 Feb 16; doi: 10.2174/1389557517666170216142113. Epub. [DOI] [PubMed] [Google Scholar]
- 25.Lin R, Johnson SG, Connolly PJ, et al. Synthesis and evaluation of 2,7-diamino-thiazolo[4,5-d]pyrimidine analogues as anti-tumor epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. Bioorg Med Chem Lett. 2009;19(8):2333–2337. doi: 10.1016/j.bmcl.2009.02.067. [DOI] [PubMed] [Google Scholar]
- 26.Kolb S, Mondesert O, Goddard ML, et al. Development of novel thiazolopyrimidines as CDC25B phosphatase inhibitors. Chem Med Chem. 2009;4(4):633–648. doi: 10.1002/cmdc.200800415. [DOI] [PubMed] [Google Scholar]
- 27.Geist JG, Lauw S, Illarionova V, et al. Thiazolopyrimidine inhibitors of 2-methylerythritol 2,4-cyclodiphosphate synthase (IspF) from mycobacterium tuberculosis and plasmodium falciparum. Chem Med Chem. 2010;5(7):1092–1101. doi: 10.1002/cmdc.201000083. [DOI] [PubMed] [Google Scholar]
- 28.Zhao D, Chen C, Liu H, et al. Biological evaluation of halogenated thiazolo[3,2-a]pyrimidin-3-one carboxylic acid derivatives targeting the YycG histidine kinase. Eur J Med Chem. 2014;87:500–507. doi: 10.1016/j.ejmech.2014.09.096. [DOI] [PubMed] [Google Scholar]
- 29.Badawey E, Rida SM, Hazza AA, Fahmy HTY, Gohar YM. Potential anti-microbials. II. Synthesis and in vitro anti-microbial evaluation of some thiazolo[4,5-d]pyrimidines. Eur J Med Chem. 1993;28(2):97–101. [Google Scholar]
- 30.Bekhit AA, Fahmy HT, Rostom SA, Baraka AM. Design and synthesis of some substituted 1H-pyrazolyl-thiazolo[4,5-d]pyrimi dines as anti-inflammatory-antimicrobial agents. Eur J Med Chem. 2003;38(1):27–36. doi: 10.1016/s0223-5234(02)00009-0. [DOI] [PubMed] [Google Scholar]
- 31.Kuppast B, Spyridaki K, Lynch C, Hu YS, Liapakis G. Synthesis of new thiazolo[4,5-d] pyrimidines as corticotropin releasing factor modulators. Med Chem. 2015;11(1):50–59. doi: 10.2174/1573406410666140724115627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furrer H, Granzer E, Wagner R. A new class of potent hypolipemic agents raising high-density lipoproteins. Synthesis, reactions and pharmacological properties. Eur J Med Chem. 1994;29:819–829. [Google Scholar]
- 33.Rida SM, Habib NS, Badawey EA, Fahmy HT, Ghozlan HA. Synthesis of novel thiazolo[4,5-d]pyrimidine derivatives for antimicrobial, anti-HIV and anticancer investigation. Die Pharm. 1996;51(12):927–931. [PubMed] [Google Scholar]
- 34.Fahmy HT, Rostom SA, Saudi MN, Zjawiony JK, Robins DJ. Synthesis and in vitro evaluation of the anticancer activity of novel fluorinated thiazolo[4, 5-d]pyrimidines. Arch Pharm. 2003;336(4–5):216–225. doi: 10.1002/ardp.200300734. [DOI] [PubMed] [Google Scholar]
- 35.Jeanneau-Nicolle E, Benoit-Guyod M, Namil A, Leclerc G. New thiazolo[3,2-a] pyrimidine derivatives, synthesis and structure-activity relationships. Eur J Med Chem. 1992;27(2):115. [Google Scholar]
- 36.Varano F, Catarzi D, Vincenzi F, et al. Design, synthesis and pharmacological characterization of 2-(2 furanyl)thiazolo[5,4-d]pyrimidine-5,7-diamine derivatives: New highly potent A2A adenosine receptor inverse agonists with antinociceptive activity. J Med Chem. 2016;59(23):10564–10576. doi: 10.1021/acs.jmedchem.6b01068. [DOI] [PubMed] [Google Scholar]
- 37.Gali R, Banothu J, Porika M, Velpula R, Hnamte S. Indolylmethylene benzo[h]thiazolo[2,3-b]quinazolinones: Synthesis, characterization and evaluation of anticancer and antimicrobial activities. Bioorg Med Chem Lett. 2014;24(17):4239–4242. doi: 10.1016/j.bmcl.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 38.Gupta R, Chaudhary RP. X-ray, NMR and DFT studies on benzo[h] thiazolo[2,3-b] quinazoline derivatives. J Mol Struct. 2013;1049:189–197. [Google Scholar]
- 39.Sakram B, Sonyanaik B, Ashok K, Rambabu S, Johnmiya SK. Benzo[h] thiazolo[2,3-b] quinazolines by an efficient p-toluenesulfonic acid-catalyzed one-pot two-step tandem reaction. Res Chem Intermed. 2016;42(3):1699–1705. [Google Scholar]
- 40.Velpula R, Banothu J, Gali R, Sargam Y, Bavantula R. One-pot and solvent-free synthesis of 3-(9-hydroxy-3-methoxy-7-aryl-6,7,9,10- tetrahydro-5H-benzo[h]thiazolo [2,3-b]quinazolin-9-yl)-2H-chromen-2-ones and their antibacterial evaluation. Turk J Chem. 2015;39:620–629. [Google Scholar]
- 41.Gupta R, Chaudhary RP. Synthesis, spectroscopic characterization and DFT studies on the novel indeno-thiazolopyrimidine heterocyclic system. J Sulfur Chem. 2014;35(1):86–97. [Google Scholar]
- 42.Youssef MM, Amin MA. Microwave assisted synthesis of some new thiazolopyrimidine, thiazolodipyrimidine and thiazolopyrimidothiazo-lopyrimidine derivatives with potential antioxidant and antimicrobial activity. Molecules. 2012;17(8):9652–9667. doi: 10.3390/molecules17089652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohareb RM, Al-Omran F, Azzam RA. Heterocyclic ring extension of estrone: Synthesis and cytotoxicity of fused pyran, pyrimidine and thiazole derivatives. Steroids. 2014;84:46–56. doi: 10.1016/j.steroids.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 44.Al-Omary FA, Hassan GS, El-Messery SM, El-Subbagh HI. Substituted thiazoles V. Synthesis and antitumor activity of novel thiazolo[2,3-b] quinazoline and pyrido[4,3-d] thiazolo[3,2-a]pyrimidine analogues. Eur J Med Chem. 2012;47(1):65–72. doi: 10.1016/j.ejmech.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 45.Valderrama JV, Ríos D, Muccioli GG, Calderon PB, Brito I, Benites J. Hetero-annulation reaction between 2-acylnaphthoquinones and 2-aminobenzothiazoles. A new synthetic route to antiproliferative benzo[g]benzothiazolo[2,3-b]quinazoline-7,12-quinones. Tetrahedron Lett. 2015;56(36):5103–5105. [Google Scholar]
- 46.Rizzo S, Bartolini M, Ceccarini M, Piazzi L, Gobbi S. Targeting Alzheimer’s disease: Novel indanone hybrids bearing a pharmacophoric fragment of AP2238. Bioorg Med Chem. 2010;18(5):1749–1760. doi: 10.1016/j.bmc.2010.01.071. [DOI] [PubMed] [Google Scholar]
- 47.Vanicha V, Kanyawim K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 48.Skehn P, Storeng R, Scudiero A, et al. New colorimetric cytotoxicity assay for anticancer drug screening. J Natl Cancer Inst. 1990;82(13):1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 49.National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health [Accessed January 25, 2017]. Available from: https://www.ncbi.nlm.nih.gov/protein/
- 50.RCSB Protein Data Bank A structural view of biology. [Accessed January 25, 2017]. Available from: http://www.rcsb.org/pdb/home/home.do.
- 51.Dassault systemes DS Visualizer. [Accessed January 27, 2017]. Available from: http://accelrys.com/products/collaborative-science/biovia-discovery-studio/visualization-download.php.
- 52.CASTp Computed atlas of surface topography of proteins. [Accessed January 27, 2017]. Available from: http://sts.bioe.uic.edu/castp/
- 53.AutoDock 4.1 [Accessed December 19, 2016]. Available from: http://autodock.scripps.edu/
- 54.De Boer EC, De Jong WH, Steerenberg PA, et al. Induction of urinary interleukin-1 (IL-1), IL-2, IL-6, and tumour necrosis factor during intra-vesical immunotherapy with bacillus Calmette–Guérin in superficial bladder cancer. Cancer Immunol Immunother. 1992;34(5):306–312. doi: 10.1007/BF01741551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Du J, Yang H, Zhang D, et al. Structural basis for the blockage of IL-2 signaling by therapeutic antibody basiliximab. J Immunol. 2010;184(3):1361–1368. doi: 10.4049/jimmunol.0903178. [DOI] [PubMed] [Google Scholar]
- 56.Romanowski MJ, Scheer JM, O’Brien T, McDowell RS. Crystal structures of a ligand-free and malonate-bound human caspase-1: implications for the mechanism of substrate binding. Structure. 2004;12(8):1361–1371. doi: 10.1016/j.str.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 57.Lagorce D, Sperandio O, Galons H, Miteva MA, Villoutreix BO. FAF-drugs2: free ADME/tox filtering tool to assist drug discovery and chemical biology projects. BMC Bioinformatics. 2008;9:396. doi: 10.1186/1471-2105-9-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krieger E, Darden T, Nabuurs SB, Finkelstein A, Vriend G. Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins. 2004;57(4):678–683. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- 59.Best RB, Buchete NV, Hummer G. Are current molecular dyanamics force fields too helial? Biophys J. 2008;95(1):L07–L09. doi: 10.1529/biophysj.108.132696. [DOI] [PMC free article] [PubMed] [Google Scholar]